Chikungunya and Zika Virus Vaccines
Stefan W. Metz⁎,†; Gorben P. Pijlman† ⁎ University of North Carolina, Chapel Hill, NC, United States
† Wageningen University, Wageningen, The Netherlands
Abstract
The spread of chikungunya and Zika viruses can be controlled by vaccination of human populations. Classical inactivated virus formulations and live attenuated viruses have demonstrated different rates of success. The molecular toolbox is now advanced enough to explore the potential of novel vaccine modalities that are in different stages of development and (pre)clinical testing. Here, we describe the state of the art of classical and next-generation vaccine platforms including viral vectored and chimeric vaccines, virus-like particles (VLPs), and nucleic acid (DNA, RNA), and subunit vaccines. We review their development, characterization, immunogenicity, and safety and we provide a future outlook for these vaccines to alleviate the burden of disease in areas where mosquitoes continue to spread these pathogens. Finally, we indicate particular challenges for chikungunya and Zika vaccines to proceed though clinical trials and become commercially available.
Keywords
Chikungunya; Zika; Arbovirus; Mosquito; Vaccines; Immunity; Animal models; Clinical trials
Introduction
Driven by the recent emergence of chikungunya virus (CHIKV) and Zika virus (ZIKV), there is great urgency toward the development of effective vaccines against both CHIKV and ZIKV infections. The spread of both of these viruses and consequences of infection can most effectively be controlled by vaccination of human populations that live in, or travel to/from, endemic areas. Many different vaccine platforms have been developed or are under development, with demonstrated different rates of success. In addition to tried and tested, well-established classical vaccine approaches, scientific progress has provided a more sophisticated molecular toolbox used to develop novel vaccine platforms, including chimeras, virus-like particles (VLPs), and synthetic nucleic acid vaccines. Although no CHIKV or ZIKV vaccines have been licensed at this point in time, several promising vaccine prototypes have been developed to undergo preclinical testing in animal models and enter clinical phase I and II trials in humans (Smalley et al., 2016; Fernandez and Diamond, 2017).
Since the onset of the Zika epidemic in South and Latin America in 2015, many vaccine candidates have been generated and are in different stages of development and/or are currently in (pre)clinical testing. Furthermore, the outbreak of CHIKV in the Caribbean in late 2013, although overshadowed in the media by the fierce Ebola outbreak in West Africa, has renewed the interest to finally bring a CHIKV vaccine to the market. In this chapter, we summarize the state of the art in the progress of different vaccine platforms, including inactivated virus formulations, live attenuated viruses, viral vectored vaccines, chimeric vaccines, and nucleic acid (DNA, RNA), and subunit vaccines (Fig. 1), all of which have previously been effective for other viral pathogens and are invented and developed by academia, research institutes, and pharma companies to fight CHIKV and ZIKV infections (Ahola et al., 2015; Tripp and Ross, 2016). We review the development, characterization, immunogenicity, safety, and challenges of these CHIKV and ZIKV vaccine candidates (Tables 1 and 2).
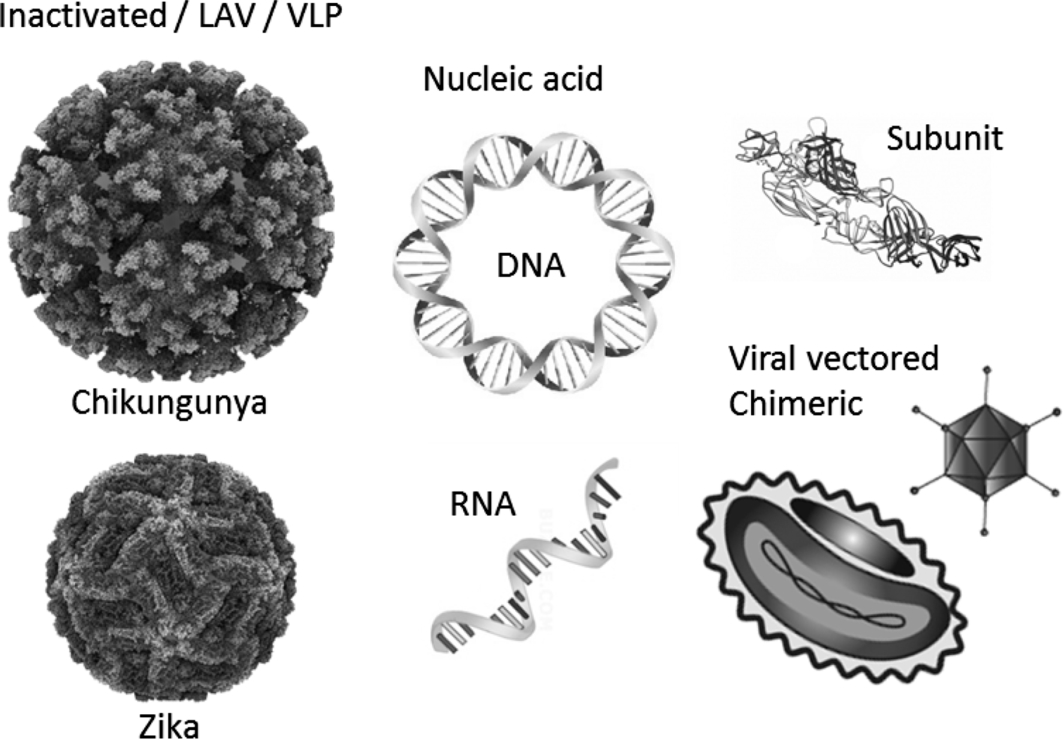
Table 1
| Vaccine platform | Species | Status | Ref. |
|---|---|---|---|
| Inactivated virus vaccine candidates | |||
| Formalin-inactivated whole virus | Swiss Bagg mice | Preclinical | Harrison et al. (1971) |
| Tween-ether-inactivated whole virus | Swiss Bagg mice | Preclinical | Eckels et al. (1970) |
| Vero-adapted, formalin-inactivated whole virus | Swiss albino mice | Preclinical | Tiwari et al. (2009) |
| BPL-formalin-inactivated whole virus | BALB/c mice | Preclinical | Kumar et al. (2012) |
| Formalin-inactivated whole virus | C57BL/6 mice | Preclinical | Gardner et al. (2010) |
| Live-attenuated virus vaccine candidates | |||
| Live-attenuated virus (181/25 strain) | Human | Phase II | Edelman et al. (2000) |
| Live-attenuated virus (IRES) | NHP (rhesus macaques) | Preclinical | Plante et al. (2011) and Roy et al. (2014) |
| Live-attenuated (6K, nsP3 mutants) | NHP (cynomolgus macaques) C57BL/6 mice | Preclinical | Hallengard et al. (2014) and Roques et al. (2017) |
| Live-attenuated (nucleolar C mutant) | C57BL/6 mice | Preclinical | Taylor et al. (2017) |
| Chimeric virus, viral vector vaccine candidates | |||
| Chimeric virus (VEEV, EEEV) | NHP (rhesus macaques) NIH Swiss and C57BL/6 mice | Preclinical | Mallilankaraman et al. (2011) |
| Adenovirus vector (C + E1 + E2) | CD-1 and C57BL/6 mice | Preclinical | Wang et al. (2011) |
| Rec. measles virus (structural polyprotein) | CD46 IFNAR mice | Phase II | Brandler et al. (2013) and Ramsauer et al. (2015) |
| rVSV (envelope polyprotein) | C57BL/6 mice | Preclinical | Chattopadhyay et al. (2013) |
| Rec. MVA (E2E3, structural polyprotein) | NHP (cynomolgus macaques) AG129 and C57BL/6 mice | Preclinical | van den Doel et al. (2014), Roques et al. (2017), and Garcia-Arriaza et al. (2014) |
| Nucleic acid vaccine candidates | |||
| DNA delivery (C-E2-E1) | C57BL/6 mice | Preclinical | Muthumani et al. (2008) |
| DNA delivery (E3-E2-E1) | BALB/c mice | Preclinical | Mallilankaraman et al. (2011) and Muthumani et al. (2016) |
| DNA delivery (E3-E2-E1-nsP2) | C57BL/6 mice | Preclinical | Bao et al. (2013) |
| DNA delivery (181/25 genome) | BALB/c mice | Preclinical | Tretyakova et al. (2014) |
| DNA delivery (RNA-replicon envelope polyprotein) | NHP (cynomolgus macaques) | Preclinical | Roques et al. (2017) |
| Subunit and VLP vaccine candidates | |||
| Bacterially expressed E1-E2 subunits | BALB/c mice | Preclinical | Khan et al. (2012) and Kumar et al. (2012) |
| Insect-cell-expressed E1-E2 subunits | AG129 mice | Preclinical | Metz et al. (2011, 2013b) |
| Insect-cell-produced virus-like particles | AG129 and C57BL/6 mice Guinea pigs | Preclinical | Metz et al. (2013a,b) and Wagner et al. (2014) |
| Mammalian-cell-expressed virus-like particles | Human NHP (rhesus macaques) BALB/c mice | Phase I | Akahata et al. (2010) and Chang et al. (2014) |
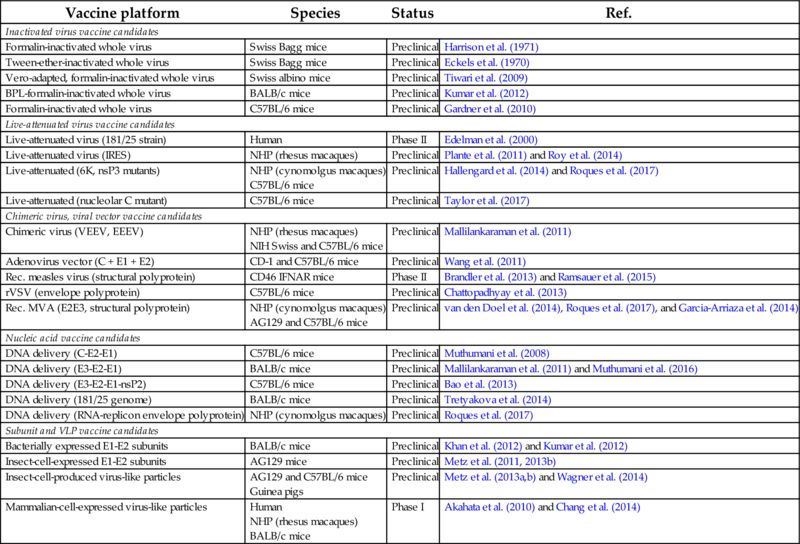
Vaccine candidates are described according to the platform used, tested species (NHP, nonhuman primates) and clinical status.
Table 2
| Vaccine platform | Species | Status | Ref. |
|---|---|---|---|
| Inactivated virus vaccine candidates | |||
| Formalin-inactivated whole virus | NHP (rhesus macaques) BALB/c mice | Preclinical | Larocca et al. (2016) and Abbink et al. (2016) |
| Live-attenuated virus vaccine candidates | |||
| Live-attenuated (DENV-ZIKV chimera) | NA | Preclinical | Kirkpatrick et al. (2015) |
| Live-attenuated (10 nt deletion in 3′ UTR) | A129 mice | Preclinical | Shan et al. (2017) |
| Viral vector vaccine candidates | |||
| Adenovirus vector-52 (prM-E) | NHP (rhesus macaques) | Preclinical | Abbink et al. (2016) |
| Adenovirus vector-5 (T4-fibritin/recE fusion) | C57BL/6 mice | Preclinical | Kim et al. (2016) |
| Nucleic acid vaccine candidates | |||
| DNA delivery (prM-E) | NHP (rhesus macaques) BALB/c mice | Preclinical-Phase I | Larocca et al. (2016), Abbink et al. (2016), and Dowd et al. (2016) |
| Nanoparticle delivery (mRNA of prM-E) | AG129, C57BL/6 and BALB/c mice | Preclinical-Phase I | Richner et al. (2017) and Pardi et al. (2017) |
| MDNP delivery (RNA of prM-E) | Mice | Preclinical | Chahal et al. (2017) |
| Subunit and VLP vaccine candidates | |||
| T4-fibritin/recE fusion subunit | C57BL/6 mice | Preclinical | Kim et al. (2016) |
| Virus-like particle (C-prM-E) | BALB/c mice | Preclinical | Garg et al. (2017) |
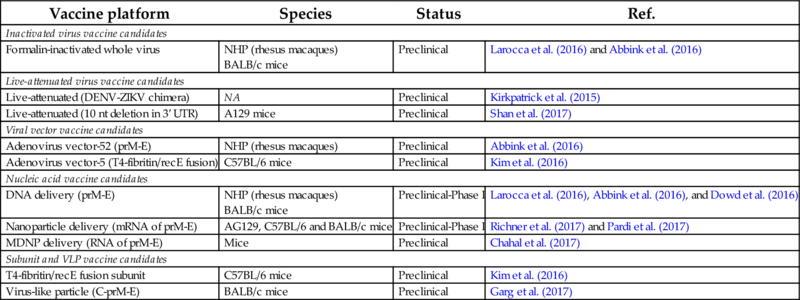
Vaccine candidates are described according to the platform used, tested species (NHP, nonhuman primates; NA, not assessed, but in progress) and clinical status.
CHIKV Whole Virus Formulations
The first CHIKV vaccine candidates that were developed were based on formalin or Tween-ether-inactivated whole-virus formulations of the CHIKV 168 strain grown on green monkey kidney (likely Vero) cells (Eckels et al., 1970; Harrison et al., 1971). In preclinical mouse vaccination studies (Swiss Bagg strain) and preliminary clinical trials, these vaccine candidates induced effective immune responses with limited side effects. Groups of mice (numbers not reported) were immunized intraperitoneally with a single dose (not quantified to μg) of inactivated vaccine and challenged intracerebrally after 14 days with 100–1000 LD50 units of the homologous CHIKV 168 strain. Postvaccination sera after a two-dose immunization scheme were tested in neutralization assays and were shown to effectively neutralize the homologous CHIKV 168 and the heterologous CHIKV strains C-226 and BAH-306, but not Mayaro virus or Semliki Forest virus (family Togaviridae, genus Alphavirus).
A more recent formalin-inactivated, alum-adjuvanted CHIKV virus vaccine candidate showed immunological potential in preclinical studies in mice. The vaccine was based on a sucrose-gradient purified CHIKV ECSA lineage grown on Vero cells. The vaccine induced effective neutralizing antibody titers and resulted in a balanced humoral and cell-mediated immune response (Tiwari et al., 2009). Swiss albino mice were immunized with relatively high doses of inactivated virus (10, 25, and 50 μg) with boosters after 14 and 28 days. Postvaccination sera had very high ELISA titers and could neutralize CHIKV but not Ross River virus (family Togaviridae, genus Alphavirus) or dengue virus (family Flaviviridae, genus Flavivirus) in an in vitro plaque-reduction neutralization test. In this study, the vaccinated mice were not challenged with CHIKV, but instead it was shown that postvaccination sera could protect mice in an in vivo neutralization test.
Other studies have inactivated the same ECSA CHIKV genotype with β-propiolactone (BPL)-formalin and showed promising results in terms of neutralization and protection in mice (Kumar et al., 2012). In this study, BALB/c mice were immunized twice intramuscularly with relatively high doses of 10, 20, or 50 μg of vaccine formulations with or without adjuvants. Alum was the superior adjuvant based on antibody titers. The booster vaccination was required for complete seroconversion upon immunization with the lower doses of 10 and 20 μg/mouse. An Asian CHIKV strain isolated in 1963 was used for heterologous challenge 20 weeks postbooster vaccination. Both the inactivated vaccines yielded complete protection.
Other CHIKV isolates have also been used in the development of an inactivated virus vaccine. An Asian lineage derived, vaccine rendered complete protection in adult wild-type mice against viremia and clinical symptoms upon subcutaneous challenge in the hind foot with the Reunion Island isolate (Gardner et al., 2010). Subcutaneous immunization in the tail base with a low dose of just one microgram unadjuvanted, inactivated, whole-virus vaccine was sufficient to protect mice against viremia and CHIKV-induced arthritis in C57BL/6 mice.
ZIKV Whole-Virus Formulations
Inactivated virus vaccines have successfully been developed for other flaviviruses such as Japanese encephalitis virus (JEV), Yellow fever virus (YFV), and tick-borne encephalitis virus (TBEV) (Halstead and Thomas, 2011; Jarmer et al., 2014). Similar approaches have now been initiated for ZIKV vaccine development. A Puerto Rico isolate was propagated and purified from mammalian cell lines and subsequently inactivated by formalin (Larocca et al., 2016). After a single intramuscular or subcutaneous inoculation of BALB/c mice with 1 μg of inactivated and alum-adjuvanted virus, a potent neutralizing antibody response was detected. The intramuscular route rendered complete protection, whereas subcutaneous immunization partially protected mice against viremia after parenteral ZIKV challenge (Larocca et al., 2016).
The same vaccine candidate was tested in rhesus (Macaca mulatta) monkeys. Here, animals were primed with 5 μg of inactivated virus and boosted with similar quantities 4 weeks later. All immunized animals induced high IgG antibody titers with neutralizing potency. Subsequent challenge with different ZIKV isolates indicated that the vaccine candidate induced complete protection against viremia and absence of viral RNA in different secretions (Abbink et al., 2016). This vaccine candidate is currently being tested in Phase 1 clinical trials.
CHIKV Live-Attenuated Virus Vaccines
Live-attenuated vaccines (LAV) are the most effective countermeasures to prevent or eradicate infectious diseases due to the very effective induction of immune memory. These vaccines are generally easy to produce at low production cost, which makes them very suitable for CHIKV-endemic low-income countries where recourses are limited. The first CHIKV LAV was developed and analyzed in 1986. The vaccine was composed of clone 181/25 of a highly MRC-5 (fetal human lung fibroblasts) cell-culture passaged Thailand isolate (Levitt et al., 1986). Preclinical studies showed that the 181/25 clone was able to replicate in the main CHIKV mosquito vectors Aedes aegypti and Aedes albopictus and induced protection against CHIKV infection in mice (ICR strain) and nonhuman primates (NHP; rhesus macaques) (Levitt et al., 1986).
In human safety and immunogenicity studies, the vaccine candidate was reported as highly immunogenic with side effects well within acceptable boundaries. During phase II clinical trials, excellent seroconversion was observed in 98% of the vaccinated subjects and 85% of the subject developed neutralizing antibody titers after a single dose (Edelman et al., 2000). Although generally regarded as safe, some safety issues such as the development of arthritis and transient arthralgia were reported. Although the vaccine was considered effective, further analysis and production of the 181/25 CHIKV vaccine was aborted due to the lack of urgency at the time of development. In subsequent studies, combinations of E2 mutations were found to highly attenuate the virus and reduce virulence in mice, allowing their use as future CHIK vaccine candidates (Gorchakov et al., 2012; Gardner et al., 2014).
Other mechanisms of viral attenuation have been used to generate CHIKV candidates, for example, the use of picornaviral internal ribosome entry sites (IRES) to drive the expression of the CHIKV structural proteins. In other studies, this technology has been used to attenuate the Murray Valley encephalitis virus (Flaviviridae; genus flavivirus) (Frese et al., 2014). For CHIKV, the 26S subgenomic promotor that regulates the expression of the structural polyprotein was replaced with the IRES of the encephalomyocarditis virus (Plante et al., 2011). The IRES-based CHIKV vaccine candidate was tested in several mice studies, clearly showing that the attenuated virus was highly immunogenic and efficacious after single-dose vaccination. Additionally, the IRES virus was unable to replicate in mosquito vectors. Similar studies have shown that immunization with the IRES-based CHIKV vaccines induces protection from challenge and viremia against the La Reunion strain, by the induction of a strong neutralizing antibody response in cynomolgus (Macaca fascicularis) macaques (Roy et al., 2014).
Recently, new methods of attenuation have been tested by removing the complete open reading frame of 6K and deleting parts of the hypervariable C-terminus of the nsp3 gene (Hallengard et al., 2014; Roques et al., 2017). These engineered, infectious clone-derived vaccine candidates induced a balanced immune response with high yields of neutralizing antibodies and a robust T-cell response. After a single dose, the vaccines protected mice and cynomolgus macaques against viremia and clinical manifestations.
Mutations in the nucleolar localization signal of the CHIKV capsid protein reduced nuclear import of capsid and attenuated virus replication both in mammalian and insect cells. C57BL/6 mice immunized with the capsid mutant strain showed no typical CHIKV disease symptoms and viremia was significantly decreased compared to wild-type virus. Mice that were challenged after immunization did not develop clinical symptoms or detectable viremia (Taylor et al., 2017).
ZIKV Live-Attenuated Virus Vaccines
The platforms that are being used for ZIKV vaccine development all derive from vaccine candidates that have previously been developed against other flaviviruses. The Laboratory of Infectious Diseases of the U.S. National Institute of Health (NIH) is using its tetravalent dengue vaccine candidate as a blueprint for ZIKV vaccine development. The ZIKV LAV is a chimera of ZIKV prM and E proteins and the nonstructural proteins and capsid gene of DENV-2. This tetravalent dengue LAV is currently being tested in a Phase 3 clinical trial in Brazil and has previously shown tetravalent responses in a high frequency of naïve vaccinees (Kirkpatrick et al., 2015). If the ZIKV vaccine candidate is proven to be effective in a flavivirus naïve population, it may progress into an efficacy evaluation in a human challenge model that the NIH is currently developing for DENV (Kirkpatrick et al., 2016). Additionally, the NIH is aiming to combine their DENV and ZIKV vaccine candidates in order to generate a pentavalent vaccine that can be administered in DENV and ZIKV endemic regions.
The first ZIKV LAV candidate that has been described contains a 10-nucleotide deletion of minimally 10 nt in the dumbbell region of the 3′ untranslated region (UTR). After a single-dose immunization in interferon receptor-deficient A129 mice, the vaccine candidate induced a highly robust and effective neutralizing antibody response. Mice were completely protected from viremia after challenge and developed a strong T-cell response. Importantly, the attenuated virus was not able to be transmitted to mosquitoes through infected blood meals, representing another safety feature when the vaccine will be used in the field (Shan et al., 2017). The live-attenuated vaccine is currently being evaluated in Phase 1 clinical trials.
Chimeric Virus and Viral Vectored CHIKV Vaccines
Vectored vaccines use attenuated and/or recombinant viral backbones to express heterologous viral genes in host cells. Being regarded as one of the best vaccine platforms to induce cellular immunity, these vaccine candidates also induce potent humoral responses (Trovato et al., 2012). These approaches for CHIKV vaccines have recently been reviewed (Ramsauer and Tangy, 2016). Attenuated strains of other alphaviruses such as Sindbis virus, Venezuelan equine encephalitis, and Eastern equine encephalitis have been used to express the structural polyprotein of CHIKV. These alphavirus chimeras replicate efficiently in vitro and generate robust neutralizing immune responses in NIH Swiss and C57BL/6 mice (Wang et al., 2008). Adenoviral vectors have been used extensively in vaccine development. A nonreplicating complex adenovirus vaccine expressing CHIKV capsid and envelope proteins elicited high neutralizing antibody titers in CD-1 outbred mice and C57BL/6 mice and protected vaccinated animals from challenge and arthritic disease (Wang et al., 2011).
A replicating recombinant measles virus expressing CHIKV virus-like particles protected immune-deficient mice (CD46-IFNAR) against lethal CHIKV challenge after a single immunization (Brandler et al., 2013). This vaccine completed phase I human trials, had an overall good safety profile, and showed that all vaccinees seroconverted after the second vaccination. The rate of adverse events increased with vaccine dose and volume, and no vaccination-related serious adverse events were reported (Ramsauer et al., 2015). In 2016, it was announced that a phase II clinical trial of this promising prophylactic vaccine candidate against CHIKV had commenced (Themis Bioscience GmbH, Vienna, Austria).
Other recombinant viral vectors including vesicular stomatitis virus (rVSV) and modified vaccinia virus Ankara (MVA), have also been used to express CHIKV structural proteins when inoculated in target hosts. An MVA expressing E3E2 or the complete structural cassette protected interferon-receptor deficient (AG129) mice against lethal challenge with CHIKV-S27 by the induction of high-neutralizing antibody titers (van den Doel et al., 2014). The same MVA-CHIKV chimera strategy was used to induce protection by immunization with 6KE1, but this did not result in full protection, indicating that vaccine responses should be targeted to E2 (van den Doel et al., 2014). Another MVA-based vaccine expressing all structural proteins raised high neutralizing antibody titers in C57BL/6 mice and conferred complete protection against viremia several weeks after the final boost (Garcia-Arriaza et al., 2014). The rVSV vector was also used to deliver the CHIKV structural cassette to mice. The recombinant VSV-CHIKV had impaired growth kinetics in tissue culture and induced neutralizing antibodies in C57BL/6 mice to protect against viremia after CHIKV challenge (Chattopadhyay et al., 2013).
Chimeric Virus and Viral Vectored ZIKV Vaccines
Several research groups are studying the efficacy of viral vectored ZIKV vaccine candidates in preclinical studies (Durbin, 2016; Tripp and Ross, 2016). Two groups have successfully engineered an adenovirus-based vaccine candidate expressing ZIKV envelope antigens. A novel rhesus adenovirus serotype 52 (RhAd52) vector was used to express ZIKV premembrane and envelope proteins, which should result in the formation of subviral particles (Abbink et al., 2015, 2016). A single intramuscular immunization in rhesus monkeys induced potent ZIKV specific neutralizing antibody responses, which rendered complete protection against subsequent subcutaneous ZIKV challenge (Abbink et al., 2016).
These initial findings served for further development of the adenoviral vector platform. An adenovirus serotype-5 vector, to which lower seroprevalence exists in the human population, was used to express the extracellular domain of the ZIKV envelope protein, fused to the T4 fibritin foldon trimerization domain Efl (Kim et al., 2016). C57BL/6 mice were subcutaneously immunized with 1011 vp of Ad5.ZIKV.Efl, which resulted in the induction of ZIKV-specific neutralizing antibodies. Pups born to immunized females were all protected against lethal ZIKV challenge and did not show weight loss or neurological signs. Thus, maternal immunization protected the suckling mice from clinical disease with a 100% survival rate. Further analysis showed high maternal IgG-ZIKV specific antibody titers in suckling mice that were nursed by immunized females (Kim et al., 2016).
CHIKV Nucleic Acid Vaccines
DNA vaccinology is based on the use of expression plasmids that express recombinant proteins in transfected cells in vivo. Here, CHIKV genes of interest are cloned into a variety of plasmid backbones and are administered through different routes of DNA vaccination. Once delivered to the cell, the expressed proteins are processed by the immune system and transported to lymph nodes by antigen-presenting cells, resulting in a very broad and balanced immune response (Garcia et al., 2015). One of the initial DNA vaccine candidates was based on a capsid, E1, and E2 consensus sequence strategy using molecular translational enhancers like a Kozak sequence and immunoglobulin E leader sequences (Muthumani et al., 2008). The vaccine candidate appeared highly immunogenic in C57BL/6 mice after intramuscular inoculation by inducing both humoral and cellular immune responses (Muthumani et al., 2008).
Using similar translation enhancers, other DNA vaccine prototypes focused on the expression of E1, E2, and E3. Vaccination protected mice against CHIKV challenge and induced potent humoral and cellular immunity. Additionally, the same vaccine induced humoral and cell mediated immunity in rhesus macaques. Protection data were missing in this study, but response kinetics and seroconvalescence appeared similar as in humans (Mallilankaraman et al., 2011). In subsequent studies in BALB/c mice, the immune responses were significantly increased by the adjuvant effect of coexpressed nsP2. Neutralizing antibody responses appeared more robust and specific compared to the previous prototype (Bao et al., 2013). Long-term immunity in BALB/c mice was obtained via electroporation-enhanced intramuscular injection of the DNA vaccines (Muthumani et al., 2016). Instead of expressing CHIKV subunits or single CHIKV polyproteins, DNA vaccines have been designed that initiate replication of the attenuated 181/25 CHIKV clone. In vivo murine studies (BALB/c mice) showed good seroconversion and protection against CHIKV challenge (Tretyakova et al., 2014).
Homologous prime-boost vaccination of a DNA-launched RNA replicon (DREP-E) encoding the CHIKV envelope cassette resulted in protection against wild-type virus in cynomolgus macaques and induced neutralizing antibodies against heterologous serotypes (Roques et al., 2017). Immunization with the same DREP-E vaccine candidate, followed by a boost with a recombinant MVA virus expressing the full-length structural polyprotein (MVA-CE) also generated a strong neutralizing antibody response that induced protection against subsequent challenge (Roques et al., 2017).
ZIKV Nucleic Acid Vaccines
Multiple DNA vaccines have been developed that typically express ZIKV antigens from an expression vector backbone. A plasmid DNA vaccine candidate expressing ZIKV prM and E was successfully tested in susceptible animal models (Larocca et al., 2016). BALB/c mice that were inoculated intramuscularly with 50 μg of the DNA vaccine induced high ZIKV specific neutralizing antibody titers and induced complete protection against two heterologous ZIKV strains. Interestingly, DNA vaccines that only encoded the ZIKV E protein, or prM with a truncated form of E were not able to induce neutralization and protection, although good seroconversion was detected (Larocca et al., 2016). Subsequent passive immunization of naïve mice with purified ZIKV specific IgG derived from immunized mice, resulted in complete protection against ZIKV challenge. CD44 + and CD8 + T-cell depletions indicated that neutralizing antibodies were sufficient for protection (Larocca et al., 2016). In another study, 5 mg of the same DNA vaccine candidate induced high neutralizing ZIKV specific antibody titers in rhesus monkeys and rendered complete protection against ZIKV challenge (Abbink et al., 2016). In addition, other DNA vaccines were developed where the ZIKV prM signal sequence and E stem/transmembrane domain were replaced by those of JEV. Immunization rendered protection in monkeys without detectable viremia. These candidates are being tested in Phase 1 clinical trials (Dowd et al., 2016).
These studies clearly illustrate that simply using the E protein or recombinant forms of the ZIKV E protein as a vaccine immunogen is not sufficient to confer protection. Expression of prM and E results in the formation of secreted subviral particles that display the surface E protein in higher-order structure that enables the formation of quaternary epitopes that are most likely essential for the induction of highly neutralizing antibodies (Fernandez and Diamond, 2017).
In line with the DNA vaccine principles, messenger RNA vaccines have been developed as vaccine candidates to protect against ZIKV infection. Lipid nanoparticles (LNPs) are used to deliver modified mRNA encoding ZIKV prM-E genes. One vaccine boost after an initial prime induced high neutralizing antibody titers and conferred sterilizing immunity in AG129, C57BL/6, and BALB/c mice (Richner et al., 2017). Interestingly, a modification in the conserved fusion-loop region still conferred complete protection, but diminished the production of antibodies that enhance DENV infection in vivo (Richner et al., 2017). Similar results were obtained by another mRNA vaccine delivered by lipid nanoparticles. A single low-dose vaccination protected nonhuman primates against challenge 5 weeks post immunization (Pardi et al., 2017).
Another RNA vaccine platform that has proven to induce protection against viral pathogens, such as Influenza and Ebola, has been applied to ZIKV (Chahal et al., 2016). This model is based upon a modified dendrimer nanoparticle (MDNP) that delivers a Venezuelan equine encephalitis virus (VEEV) replicon RNA encoding ZIKV prM and E. Two immunizations in C57BL/6 mice induced a ZIKV specific IgG response, though data on protection against ZIKV infection are missing. This model identified unique H-2Db-restricited epitopes that induced CD8+ T-cell responses in mice (Chahal et al., 2017).
CHIKV Subunit and Virus-Like Particle Vaccines
Protein subunits vaccine candidates based on proteinaceous portions of the CHIKV virion have been developed and their immunogenicity has been analyzed and compared to more complex subunit platforms, such as virus-like particles (VLPs). CHIKV subunits mainly represent envelope glycoprotein E1 and E2 protein variants, expressed with or without their native signals peptides E3 or 6K (Metz et al., 2011; Khan et al., 2012). The earliest subunit vaccine candidates date from 1972. This E2 subunit vaccine expressed in bacteria showed the induction of neutralizing antibodies and high levels of IFN-γ. After high-dose immunizations, mice were protected against viremia in several organs after CHIKV challenge (Salazar-Gonzalez et al., 2015). More recent studies also used bacterial expressed recombinant E1 and E2 proteins. These candidates, used as formulations of 40 μg in combination with different adjuvants (Freunds complete, Alum or Montanide ISA720), were able to induce both robust neutralizing antibody titers and cell-mediated immune responses in BALB/c mice (Khan et al., 2012). Similar results were obtained in another study with BALB/c mice immunized with vaccine doses of 10–50 μg per mouse in combination with Alum (Kumar et al., 2012).
Other groups have used insect cell expression systems to produce glycosylated CHIKV E1 and E2 subunits (adjuvanted with Matrix-M) and showed that two immunizations of with 2 μg of soluble E2 subunits per mouse induced potent neutralizing antibody titers in AG129 mice (Metz et al., 2013b). The subunits, however, only induced protection upon lethal CHIKV challenge in mice that seroconverted, showing one of the major disadvantages of the use of subunits. In the same study, the soluble E1 and E2 subunits were directly compared to insect cell–derived VLPs. This promising new generation of vaccine candidates resembles the wild-type virus structural morphology and has been successfully used as a vaccine platform against other viral pathogens (Lua et al., 2014). The insect cell expressed CHIKV VLPs outperformed the soluble subunits in terms of immunogenicity, neutralizing antibody induction, and conferred complete protection against arthritic disease and lethal challenge in immunecompetent C57BL/6 (nonadjuvanted VLPs, single shot of 1 μg) and immunocompromised AG129 (Matrix-M adjuvanted VLPs, two immunizations of 1 μg) mouse models (Metz et al., 2013a,b).
The first and most advanced VLP vaccine candidate uses mammalian cell (HEK293F) expression of the CHIKV structural polyprotein. In preclinical studies, this adjuvanted VLP vaccine prototype produced potent neutralizing antibody responses in both mice (BALB/c; two immunizations of 19 μg adjuvanted VLPs per mouse) and nonhuman primates (rhesus macaques; three immunizations with 20 μg nonadjuvanted VLPs per animal), inducing protection against viremia after challenge (Akahata et al., 2010). This VLP vaccine candidate was subsequently tested in a Phase I dose-escalation trial and the results showed great promise. Participants received different doses of the VLP antigen and according to analyses, all subjects developed long-lasting high-titer neutralizing antibody responses, with good safety profiles (Chang et al., 2014). Recent analysis showed that immunization with VLPs derived from one single CHIKV serotype, induced crossprotection against the other there circulating serotypes (Goo et al., 2016), suggesting that there is no need to generate vaccines against all different CHIKV serotypes.
ZIKV Subunit Vaccine Development
Recombinant ZIKV envelope proteins produced in either mammalian or insect cells have been reported to be used in preclinical ZIKV subunit vaccine development (Durbin, 2016; Tripp and Ross, 2016). In early stage studies, the ectodomain of ZIKV E was fused to the T4 fibritin foldon trimerization domain Efl and the protein vaccine was delivered by carboxymethylcellulose microneedle array (MNA) (Kim et al., 2016). Compared to same antigen that was delivered using an adenovirus vector, the subunit vaccine was considerably less immunogenic in C57BL/6 mice. Although the recipient mice did show humoral responses after immunization, the immune kinetics and absolute magnitude of the response were delayed and less than the adenovirus vector platforms (Kim et al., 2016). Additionally, the low yield protein production indicated that prM, absent in the tested construct, is most likely crucial for optimal protein stability, folding, and immunogenicity of E (Kennedy, 2016). The immunogenicity of ZIKV VLPs has been compared to that of a DNA vaccine expressing prM-E. The VLPs were produced by coexpressing ZIKV C-prM and E with the WNV NS2B-3 protease to cleave C from prM. The VLPs expressed in mammalian cells outcompeted the DNA vaccine in terms of neutralizing antibody titers and the presence of C is beneficial for VLP production and immunogenicity (Garg et al., 2017).
Challenges and Future Outlook for CHIKV and ZIKV Vaccines
Many years of fundamental and applied research on arbovirus vaccine design and development of small animal models for CHIKV and ZIKV disease have delivered potent vaccine candidates that are likely to move forward through clinical testing. For CHIKV, four vaccine candidates have been tested in humans in a phase I clinical trial (inactivated virus, LAV, VLP, and a measles viral vectored). At present, only the recombinant measles virus expressing CHIKV VLPs has entered phase II clinical trials. All CHIKV vaccines so far tested are focusing on protection against acute and not chronic disease and it remains to be seen what the longevity of the immune response in humans will be. Certainly, a registered human CHIKV vaccine would help to stop virus transmission and alleviate the burden of disease in areas where Aedes spp. mosquitoes continue to spread CHIKV, alongside the other major arboviruses DENV, YFV, and ZIKV. For ZIKV, tremendous progress has been made in understanding the disease itself and developing animal models to study ZIKV infection, transplacental transmission, and the immunogenicity of candidate vaccines. A challenge for a ZIKV vaccine is the safe use during pregnancy—ideally the vaccine should be nonreplicating to reduce vaccine-related effects on the mother and the fetus. Another key challenge in ZIKV vaccine design is the likely interaction of crossreactive ZIKV antibodies that may negatively impact the outcome of DENV infection (immune enhancement), and vice versa (Stettler et al., 2016; Bardina et al., 2017). One approach is to design ZIKV antigens in such a way that the production of crossreactive antibodies is diminished (Richner et al., 2017), but clearly more research is required to clarify this issue to guide the design of a truly safe, efficacious ZIKV vaccine. Changes in epidemiology and funding resources will have an effect on ZIKV vaccine development. Vaccine candidates such as the inactivated virus vaccine developed by a partnership between the U.S. Army and Sanofi Pasteur are terminated due to these reasons.