
Alkyl halides and other substituted carbon molecules can take part in reactions known as nucleophilic substitutions. Substitution reactions involve removing an atom or a functional group from a molecule and replacing it with another. Substitution reactions can occur in many different types of molecules; however, this chapter will specifically discuss substitution reactions of alkanes. In all substitution reactions, identifying the nucleophile and the leaving group are critical to an understanding of the mechanism.
Nucleophiles are molecules that are attracted to positive charge, as seen by their name: nucleophile means “nucleus lover.” Nucleophiles are electron-rich species that are often but not always negatively charged. Nucleophiles are attracted to atoms with partial or full positive charges.
If a group of nucleophiles are based on the same atom (for example, oxygen) then nucleophilicity is roughly correlated to basicity. In other words, the stronger the base, the stronger the nucleophile. This is because bases act as electron donors, and stronger nucleophiles are also better electron donors. For example, nucleophilic strength decreases in the order:
If a series of nucleophiles is based on different atoms, nucleophilic ability doesn’t necessarily correlate to basicity. In a protic solvent (a solvent that is able to form hydrogen bonds), large atoms or ions tend to be better nucleophiles. Larger ions more easily shed their solvent molecules and are more polarizable. Hence, nucleophilic strength decreases in the order:
In contrast, in an aprotic solvent (a solvent that cannot form hydrogen bonds) the nucleophiles are “naked”; they are not solvated. In this case, nucleophilic strength is directly related to basicity. For example, in DMSO (an aprotic solvent), the order of nucleophilic strength is the same as base strength:
The ease with which nucleophilic substitution takes place is dependent on the nature of the leaving group. The best leaving groups are those that are weak bases, as these can accept a negative charge and dissociate to form a stable ion in solution. In the case of the halogens, therefore, this is the opposite of base strength:
Other leaving groups besides halogens can be used for substitution reactions. For example, an −OH group that is a poor leaving group can be protonated to form −H2O+, which leaves as a water molecule.
SN1 designates a unimolecular nucleophilic substitution reaction. It is unimolecular because the rate of the reaction is dependent upon only one molecule in the reaction; in other words, the rate expression is first order. The rate-determining step of an SN1 reaction is the dissociation of the substrate (the starting molecule) to form a stable, positively charged ion called a carbocation. The formation and stabilization of the carbocation determine all other aspects of SN1 reactions.
SN1 reactions involve two steps: the dissociation of a substrate molecule into a carbocation and a leaving group, followed by the combination of the carbocation with a nucleophile to form the substituted product.
In the first step, a carbocation intermediate is formed. Carbocations are stabilized by polar solvents that have lone electron pairs available to donate (e.g., water or ethyl alcohol). Carbocations are also stabilized by charge delocalization throughout the molecule. More highly substituted carbocations are more stable, because hydrocarbon substituent groups donate electron density toward the positive charge. The order of stability for carbocations is:
To drive the reaction forward, the original leaving group should be a weaker nucleophile than the replacement nucleophile. The second step, in which the nucleophile combines with the carbocation, occurs very rapidly compared to the first step, and is essentially irreversible.
The rate at which a reaction occurs can never be greater than the rate of its slowest step. Such a step is termed the rate-limiting or rate-determining step of the reaction (see Chapter 30, Chemical Kinetics, for a more detailed discussion of rate expressions). In an SN1 reaction, the slowest step is the dissociation of the molecule to form a carbocation intermediate, a step that is energetically unfavorable. The formation of a carbocation is therefore the rate-limiting step of an SN1 reaction. The only reactant in this step is the original substrate molecule, and so the rate of the entire reaction depends only on the concentration of the substrate (a so-called first-order reaction): rate = k[substrate]. The rate is not dependent on the concentration or the nature of the nucleophile, because it plays no part in the rate-limiting step.
The rate of an SN1 reaction can be increased by anything that promotes the formation and stability of the carbocation. The most important factors are as follows:
a. Structural factors: Highly substituted alkanes allow for distribution of the positive charge over a greater number of carbon and hydrogen atoms, and thus form the most stable carbocations. The order of reactivity of substrates for SN1 reactions is tertiary > secondary > primary > methyl; in general, primary and methyl substrates do not react by the SN1 mechanism.
b. Solvent effects: Highly polar solvents are better at surrounding and isolating ions than are less polar solvents. Polar protic solvents such as water or alcohols work best for two reasons. Protic solvents can form hydrogen bonds with the leaving group, solvating it and preventing it from returning to the carbocation. Also, lone electron pairs on oxygen or nitrogen atoms in the solvent molecule can stabilize the carbocation intermediate.
c. Nature of the leaving group: Weak bases dissociate more easily from the alkyl chain and thus make better leaving groups, increasing the rate of carbocation formation.
d. Nature of the nucleophile: SN1 reactions do not require a strong nucleophile. SN1 reactions run equally well with either strong (fully charged) or weak (electron-rich but uncharged) nucleophiles.
SN1 reactions involve carbocation intermediates, which are sp2 hybridized and have trigonal planar geometry. The attacking nucleophile can approach the carbocation from either above or below with equal probability, and thus create either the (R) or (S) enantiomer with equal probability.

If the original compound is optically active because of the presence of a chiral center, then a racemic mixture will be produced. In some cases, the nucleophile may react so rapidly that it approaches the carbocation from the opposite side of the leaving group with greater frequency; in this case, the product will be characterized as a partial racemate.
SN2 designates a bimolecular nucleophilic substitution reaction. SN2 reactions involve a nucleophile pushing its way into a compound while simultaneously displacing the leaving group. Its rate-determining and only step involves two molecules: the substrate and the nucleophile.

SN2 reactions are concerted reactions, meaning that the entire mechanism occurs in single coordinated process. The nucleophile attacks the reactant from the backside of the leaving group, forming a trigonal bipyramidal transition state. As the reaction progresses, the bond to the nucleophile strengthens while the bond to the leaving group weakens. The leaving group is displaced as the bond to the nucleophile becomes complete.
The single step of an SN2 reaction involves two reacting species: the substrate and the nucleophile. The concentrations of both therefore play a role in determining the rate of an SN2 reaction; the two species must “meet” in solution, and raising the concentration of either will make such a meeting more likely. Since the rate of the SN2 reaction depends on the concentration of two reactants, it follows second-order kinetics. The rate expression for an SN2 reaction is: rate = k[substrate][nucleophile].
Other factors that can affect the rate of SN2 reactions include:
The single step of an SN2 reaction involves a chiral transition state. Since the nucleophile attacks from one side of the central carbon and the leaving group departs from the opposite side, the reaction “flips” the bonds attached to the carbon.

If the reactant is chiral, optical activity will be retained, but will invert between (R) and (S) as long as the nucleophile and leaving group have the same priority relative to the other groups in the substrate. If the nucleophile and leaving group have different priorities compared to the rest of the molecule, however, the absolute configuration must be determined for the substituted product.
Certain reaction conditions favor one substitution mechanism over the other, and provide distinctive “fingerprints” that allow the determination of whether SN1 or SN2 will proceed. Sterics, nucleophilic strength, leaving group ability, reaction conditions, and solvent effects are all important in determining which reaction will occur.
| SN1 | SN2 | |
| Substrate reactivity | 3° > 2° > 1° > CH3 | CH3 > 1° > 2° > 3° |
| Leaving group |
Cl−, Br−, I−, weak bases −OH if protonated to form OH2+ |
Cl−, Br−, I−, weak bases |
| Nucleophile | Any nucleophile | Strong (charged) nucleophile |
| Solvent | Polar protic | Polar aprotic |
| Reaction mechanism | 2-step: carbocation, then nucleophilic attack | Concerted 1-step |
| Rate law |
1st order: rate = k[substrate] |
2nd order: rate = k[substrate][nucleophile] |
| Stereochemistry | Chiral → racemic mix | Inversion of R/S |
Alkanes can react by a free radical substitution mechanism in which one or more hydrogen atoms are replaced by Cl, Br, or I atoms. These reactions involve three steps:

Propagation—A propagation step is one in which a radical produces another radical that can continue the reaction. A free radical reacts with an alkane, removing a hydrogen atom to form HX, and creating an alkyl radical. The alkyl radical can then react with X2 to form an alkyl halide (the substiuted product) and generate another X•, thus propagating the radical.

Termination—Two free radicals combine with one another to form a stable molecule.

A single free radical can initiate many reactions before the reaction chain is terminated.
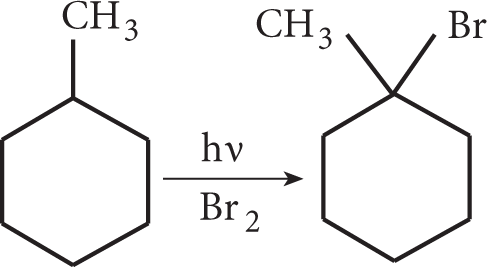
Larger alkanes have many hydrogens that the free radical can attack. Bromine radicals react fairly slowly, and primarily attack the hydrogens on the carbon atom that can form the most stable free radical, i.e., the most substituted carbon atom.
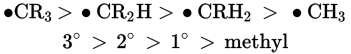
Thus, a tertiary radical is the most likely to be formed in a free-radical bromination reaction. As a result, the substitution product will have the bromine on the most highly substituted carbon. Note the same pattern of stability and the same resulting product compared to a carbocation-based mechanism.
Free-radical chlorination is a more rapid process and depends on both the stability of the intermediate and on the number of hydrogens present. Free-radical chlorination reactions are likely to replace primary hydrogens that are found abundantly in most molecules, despite the relative instability of primary radicals. Unfortunately, free-radical chlorination reactions produce mixtures of products, and are useful only when a single type of hydrogen is present.