CHAPTER 14
Citalopram and Escitalopram
Patrick H. Roseboom, Ph.D.
Ned H. Kalin, M.D.
Citalopram (Celexa) and its pharmacologically active enantiomer, escitalopram (Lexapro), are among the most selective serotonin reuptake inhibitors available. Both drugs are widely prescribed and have been shown in large-scale controlled trials to be effective in the treatment of depression; escitalopram has also been shown to be effective in the treatment of anxiety disorders. Generic formulations of both drugs are now available. Both drugs are well tolerated in patients and show a low potential for pharmacokinetic drug–drug interactions. Citalopram and escitalopram have similar effectiveness in the treatment of depression, although some studies suggest a modest superiority of escitalopram over citalopram on some measures of efficacy, including a possibly faster onset of therapeutic effect for escitalopram. Antagonism of the effects of escitalopram by R-citalopram has been invoked to explain the purported therapeutic differences between the two drugs. Also, the affinity of citalopram for histamine receptors appears to reside in the R-enantiomer, suggesting that escitalopram has a decreased potential for antihistaminergic side effects. Finally, in terms of cardiac safety, citalopram has a greater potential to prolong the electrocardiogram (ECG) QT interval and produce potentially serious arrhythmias.
History and Discovery
The pharmacology of citalopram was first described in 1977 (Christensen et al. 1977; Hyttel 1977). Citalopram was shown to be a very potent inhibitor of serotonin (5-HT) reuptake in both in vitro and in vivo models (Hyttel 1977, 1978). It was subsequently discovered that all of the inhibitory activity of citalopram on 5-HT reuptake resides in the S-(+)-enantiomer (escitalopram) (Hyttel et al. 1992). Originally introduced in Denmark in 1989, citalopram was approved by the U.S. Food and Drug Administration (FDA) for the treatment of depression in July 1998. Escitalopram received FDA approval for the treatment of major depressive disorder in August 2002 and for the treatment of generalized anxiety disorder (GAD) in December 2003. Escitalopram also received FDA approval in March 2009 for the treatment of major depressive disorder in adolescents ages 12–17 years.
Structure–Activity Relations
Citalopram has a single chiral center (Figure 14–1). A chiral center is an atom surrounded by an asymmetrical arrangement of atoms such that the three-dimensional configuration is not superimposable on its mirror image. At this chiral center, there are two possible stereoisomers. Often, drugs are produced as a mixture of both stereoisomers, referred to as the racemate. However, because desired pharmacological activity or unwanted toxicity may reside in only one of the stereoisomers, a stereoisomer-selective formulation may be superior (Agranat et al. 2002). Citalopram was originally characterized and marketed as the racemate, but subsequently the single stereoisomer of citalopram, escitalopram (Figure 14–2), was developed for the treatment of depression and other psychiatric disorders. Preclinical studies indicate that inhibition of 5-HT transporter activity resides in the S-enantiomer (Hyttel et al. 1992), with escitalopram being 30 times more potent than R-citalopram at inhibiting 5-HT transport (Owens et al. 2001).
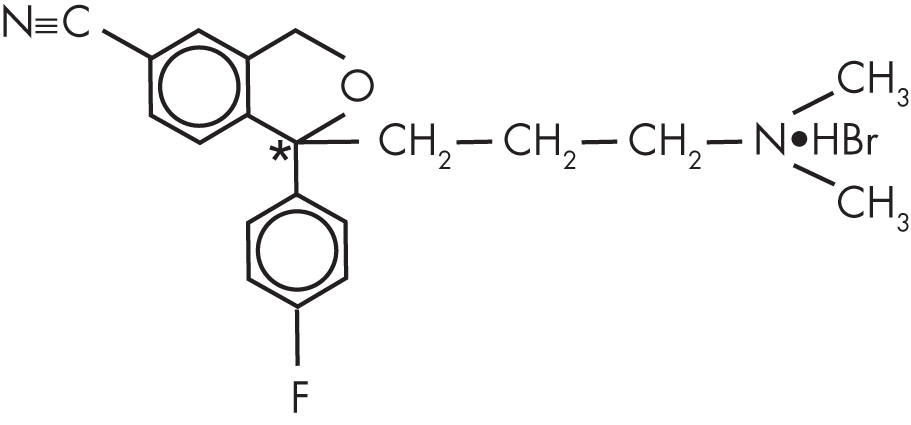
FIGURE 14–1. Chemical structure of citalopram hydrobromide.
*Indicates chiral center.
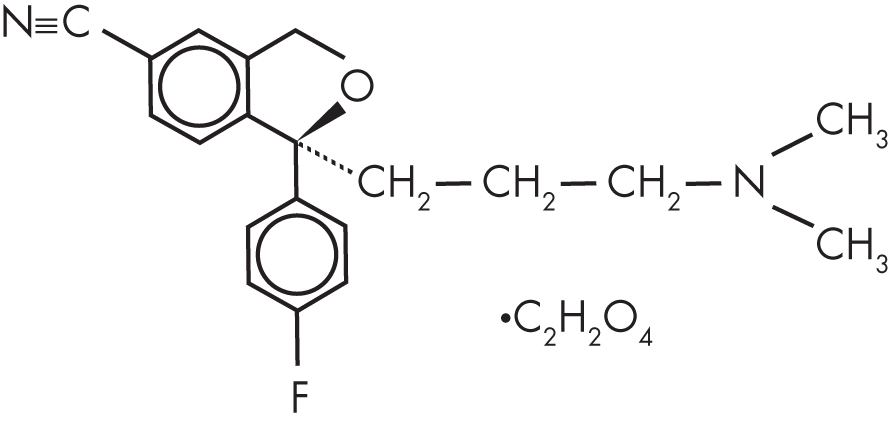
FIGURE 14–2. Chemical structure of escitalopram oxalate.
A possible explanation for the postulated modest superiority of escitalopram over citalopram in some measures of antidepressant efficacy is provided by the evidence that the R-enantiomer of citalopram may interfere with the activity of the S-enantiomer, as evidenced in several behavioral and physiological assays (for a review, see Sánchez 2006). This antagonism has been hypothesized to result from a kinetic interaction at the level of the 5-HT transporter (Stórustovu et al. 2004).
Pharmacological Profile
Citalopram
Of the selective serotonin reuptake inhibitors (SSRIs) approved to date, citalopram is one of the most selective, with a 524-fold lower potency for inhibiting the human norepinephrine (NE) transporter and a >10,000-fold lower potency for inhibiting the human dopamine transporter (Owens et al. 2001). In addition, citalopram has low affinity for a wide variety of neurotransmitter receptors (for a review, see Hyttel et al. 1995). Citalopram has been reported to have submicromolar affinity for the histamine type 1 (H1) receptor (Hyttel 1994; Richelson and Nelson 1984), but this appears to be true only for the R-enantiomer (Owens et al. 2001). Citalopram does not show significant inhibition of monoamine oxidase (MAO) (Hyttel 1977). Behavioral studies in rats and mice have shown citalopram to be a potent and selective inhibitor of 5-HT reuptake (Christensen et al. 1977; Hyttel 1994). In contrast, citalopram is ineffective in models that reflect in vivo inhibition of dopamine and NE reuptake (Hyttel 1994). Citalopram is active in various behavioral models related to antidepressant activity (Martin et al. 1990; Sánchez and Meier 1997) and anxiolytic activity (Inoue 1993; Sánchez 1995).
Escitalopram
Escitalopram is also a highly potent inhibitor of 5-HT reuptake, with a Ki for binding to the human 5-HT transporter of 1.1 nM compared with a Ki of 1.9 nM for citalopram and 36 nM for R-citalopram (Owens et al. 2001). Escitalopram is the most selective SSRI approved for clinical use, with a 2,600-fold lower potency for inhibiting the human NE transporter and a >45,000-fold lower potency for inhibiting the human dopamine transporter. Escitalopram has no appreciable binding affinity for a large number of other neurotransmitter receptors (Owens et al. 2001; Sánchez et al. 2003). Escitalopram shows potent activity in various in vivo paradigms, including a model of 5-HT reuptake inhibition and behavioral models of antidepressant, antiaggressive, and anxiolytic activity (for a review, see Sánchez et al. 2003). In these in vivo paradigms, escitalopram’s potency ranges from being similar to that of citalopram to being approximately twofold greater than that of citalopram. In contrast, in the majority of these paradigms, R-citalopram is severalfold less potent than either escitalopram or citalopram.
Pharmacokinetics and Disposition
Citalopram
Citalopram is well absorbed after oral administration, with an absolute bioavailability of 80% for citalopram tablets (Joffe et al. 1998). The peak plasma concentration is normally observed 2–4 hours following an oral dose (Kragh-Sørensen et al. 1981). The bioavailability of citalopram is not affected by food (Baumann 1992), and it is subject to very little first-pass metabolism (Kragh-Sørensen et al. 1981). The apparent volume of distribution is 12–16 L/kg (Fredricson Overø 1982; Kragh-Sørensen et al. 1981), which indicates that the drug distributes widely. There is a linear relationship between steady-state plasma concentration and dose (Bjerkenstedt et al. 1985), and plasma protein binding is approximately 80% (Baumann 1992). Systemic clearance of citalopram is 0.3–0.4 L/minute (Baumann 1992), and renal clearance of citalopram is approximately 0.05–0.08 L/minute (Sindrup et al. 1993).
Racemic citalopram undergoes N-demethylation by the hepatic cytochrome P450 (CYP) system to the major metabolite monodesmethylcitalopram (DCT). CYP enzymes 2C19, 3A4, and 2D6 all contribute approximately equally to the formation of DCT (Kobayashi et al. 1997; Rochat et al. 1997; von Moltke et al. 1999). DCT also undergoes N-demethylation to the minor metabolite didesmethylcitalopram (DDCT) by the actions of CYP2D6 (Sindrup et al. 1993; von Moltke et al. 2001). Clinical studies indicate that the half-lives for citalopram, DCT, and DDCT are approximately 36 hours, 50 hours, and 100 hours, respectively (Dalgaard and Larsen 1999; Fredricson Overø 1982; Kragh-Sørensen et al. 1981). Citalopram is metabolized by human cytochromes that display genetic polymorphisms, and metabolism of citalopram and DCT is impaired in subjects who show poor metabolism via the CYP2C19 and CYP2D6 pathways (Baumann et al. 1996; Sindrup et al. 1993; Yu et al. 2003).
Escitalopram
The clinical pharmacokinetics of escitalopram, reviewed by Rao (2007), are similar to those described for citalopram. The pharmacokinetic characteristics of escitalopram are essentially the same regardless of whether patients are given a single oral dose of 20 mg of escitalopram or 40 mg of racemic citalopram (which contains 20 mg of escitalopram); this indicates that there is no pharmacokinetic interaction or interconversion between R-citalopram and escitalopram (Rao 2007).
Mechanism of Action
The majority of studies on mechanism of action have focused on citalopram, with a relatively limited number of studies using escitalopram. Because the antidepressant activity of citalopram results from escitalopram, the majority of the conclusions from these studies pertain to both citalopram and escitalopram.
Citalopram is a potent and selective inhibitor of 5-HT reuptake and acts by binding directly to the 5-HT transporter. Citalopram selectively inhibits radioligand binding to the 5-HT transporter (Ki=0.75 nM) versus the NE transporter (Ki=3,042 nM) in rat cortical membranes. A similar selectivity was found for inhibiting the binding of the same radioligands to the cloned human 5-HT and NE transporters expressed in transfected cells and for inhibiting [3H]5-HT (Ki=8.9 nM) and [3H]NE (Ki=30,285 nM) reuptake into these transfected cells (Owens et al. 1997).
Several studies have described how repeated dosing alters the effects of citalopram on serotonergic neuronal function. As with other SSRIs, the ability of citalopram to inhibit the firing of 5-HT neurons in the dorsal raphe nucleus is greatly reduced after 14 days of repeated administration (Chaput et al. 1986). This change is associated with an increase in the ability of citalopram to elevate the extracellular levels of 5-HT in the cortex (Invernizzi et al. 1994). These two effects appear to result from a desensitization of 5-HT1A autoreceptors (Chaput et al. 1986; Cremers et al. 2000; Invernizzi et al. 1994). This adaptive change of 5-HT1A receptors following repeated administration of citalopram, or other SSRIs, has been postulated to underlie the slow onset of antidepressant efficacy that is observed clinically (Blier and de Montigny 1994).
Evidence suggests that a variety of antidepressants, including those that block monoamine reuptake or metabolism, produce their therapeutic response in part by overcoming depression-associated decreases in neurogenesis and synaptogenesis, possibly through effects on brain-derived neurotrophic factor (BDNF) expression. Citalopram, like several other antidepressants, has been shown to increase the levels of BDNF messenger RNA (mRNA) in various subregions of the rat ventral hippocampus (Russo-Neustadt et al. 2004) and to induce signaling through the BDNF receptor tyrosine kinase receptor B (TrkB) (Rantamäki et al. 2007). Interestingly, this effect of antidepressants on TrkB appears to be independent of BDNF release and 5-HT transporter blockade and does not involve a direct binding of the antidepressant to TrkB (Rantamäki et al. 2011). Escitalopram treatment has been demonstrated to affect circulating BDNF levels. A study in depressed subjects (n=18) showed elevated plasma levels of BDNF and decreased platelet levels of BDNF compared with healthy control subjects (n=14), and these differences were normalized with 24 weeks of escitalopram (10–40 mg/day) treatment (Serra-Millàs et al. 2011). Additionally, there are preliminary clinical data suggesting that a polymorphism within the coding region for BDNF (Val66Met) is associated with therapeutic response to citalopram (Choi et al. 2006).
A number of studies have implicated the 5-HT2A receptor in a variety of neuropsychiatric disorders (Norton and Owen 2005), and there is evidence implicating the 5-HT2A receptor in the mechanism of action of antidepressants, including citalopram (Chen and Lawrence 2003; Peremans et al. 2005). Also, a large-scale clinical study involving 1,953 patients who participated in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial identified a significant association between a polymorphism contained in the second intron of the gene for the 5-HT2A receptor (rs7997012) and treatment response to citalopram (McMahon et al. 2006). While perhaps relevant to clinical work, the functional significance of this intronic polymorphism on the 5-HT2A receptor has yet to be determined.
Several cellular and in vivo animal model studies have shown that antidepressants can increase glucocorticoid receptor (GR) translocation, induce GR downregulation, and decrease GR agonist–mediated effects (Anacker et al. 2011; Carvalho and Pariante 2008), and there is evidence suggesting that similar effects occur in vivo in humans. In a double-blind, placebo-controlled, crossover study in healthy men, treatment for 4 days with citalopram 20 mg/day was associated with a diminished ability of cortisol to increase electroencephalogram alpha power and to impair working memory (Pariante et al. 2012). These results suggest that GR activation by antidepressants does occur in the human brain.
Corticotropin-releasing factor (CRF) mediates many of the effects of psychological stress and is postulated to play a role in the pathophysiology of depression and anxiety disorders (Arborelius et al. 1999). In rats, lentiviral-mediated chronic elevation of amygdala CRF reproduces many of the behavioral and endocrine consequences of chronic stress that are consistent with a depressive or anxious phenotype (Flandreau et al. 2012). Interestingly, chronic escitalopram treatment of these CRF-overexpressing rats for 4 weeks reversed some but not all of the CRF-induced anxiety-like and depressive-like behavioral alterations (Flandreau et al. 2013).
Indications and Efficacy
Depression
Citalopram
The efficacy of citalopram (dosage range 20–80 mg/day) in the treatment of depression has been shown in at least 11 placebo-controlled clinical trials (for a review, see Keller 2000). In addition, meta-analyses of multiple placebo-controlled studies reported similar findings (Bech and Cialdella 1992; Montgomery et al. 1994). In the United States, three large multicenter clinical trials have demonstrated citalopram’s efficacy in the treatment of major depression (Feighner and Overø 1999; Mendels et al. 1999; Stahl 2000). More recently, primary care patients in the United Kingdom were given citalopram 20 mg/day (n=274) or the NE reuptake inhibitor reboxetine 4 mg twice daily (n=272), which were found to be equally effective in the treatment of severe depression (defined as scores ≥15 on the Beck Depression Inventory) (Wiles et al. 2012).
STAR*D trial. The effectiveness of citalopram has also been demonstrated in a study designed to simulate real-world conditions for the treatment of depression. The STAR*D trial was a large-scale multicenter study that enrolled 4,041 patients with nonpsychotic major depression in a test of various antidepressant therapies (Rush et al. 2004). The mean daily dose of citalopram at study exit was 41.8 mg, the remission rates were 28% (when remission was defined as a score of ≤7 on the 17-item version of the Hamilton Rating Scale for Depression [Ham-D]) and 33% (when defined as a score of ≤5 on the 16-item Quick Inventory of Depressive Symptomatology, Self-Report [QIDS-SR]), and the response rate was 47% (when response was defined as a ≥50% reduction in QIDS-SR score) (Trivedi et al. 2006). These response and remission rates are comparable to those found in 8-week controlled clinical trials examining the efficacy of acute antidepressant treatment.
Long-term treatment with citalopram. Two placebo-controlled studies indicate that citalopram may be effective in continuation therapy to prevent depression relapse. Both studies showed that for patients with an acute therapeutic response to citalopram, continuation of citalopram therapy at the same dosage (20, 40, or 60 mg/day) for an additional 24 weeks significantly decreased the relapse rate compared with placebo (Montgomery et al. 1993; Robert and Montgomery 1995). Two additional studies suggest that citalopram may be beneficial in patients with a history of recurrent depression. Long-term (at least 48 weeks) administration of citalopram at the same fixed dosage at which patients initially showed therapeutic response (20–60 mg/day) can significantly increase the time before depression recurs in adult patients (ages 18–64 years) and in elderly patients (ages 65 years and older) (Hochstrasser et al. 2001; Klysner et al. 2002).
Escitalopram
A number of placebo-controlled clinical trials and retrospective analyses have demonstrated the efficacy of escitalopram (dosage range 10–20 mg/day) in the treatment of major depression (Burke et al. 2002; Lepola et al. 2003; Montgomery et al. 2001b; Wade et al. 2002). Also, a large number of retrospective pooled analyses have compared escitalopram with other SSRIs (Einarson 2004; Kennedy et al. 2006; Llorca et al. 2005). In general, escitalopram was at least as effective as other widely used antidepressants, and in some clinical trials it has been suggested to be superior to other antidepressants based on modestly greater score changes on various depression rating scales, especially in patients with severe depression. In addition, escitalopram has been suggested in a few clinical trials to possibly have a faster onset of therapeutic effect (based on changes in depression rating scale scores), occurring as early as week 1. It remains to be demonstrated whether the modest differences between escitalopram and other antidepressants in controlled clinical trials translate into a therapeutically meaningful difference in the treatment of depression in psychiatric practice.
Major depressive disorder with severe asthma. Major depressive disorder is often seen in asthmatic individuals, and depression may be a risk factor for asthma-related morbidity. In a placebo-controlled, randomized, double-blind proof-of-concept trial (Brown et al. 2012), escitalopram 10–20 mg/day was evaluated for the treatment of depression in patients (n=25) with asthma. Improvement in depression symptoms was seen in both placebo and escitalopram groups from week 1 to study exit at week 12, with a trend favoring escitalopram for depression remission based on changes in the Ham-D score (Brown et al. 2012).
Long-term treatment with escitalopram. In a 36-week placebo-controlled clinical trial, subjects given escitalopram 10–20 mg/day (n=181) were less likely to experience relapse following resolution of a depressive episode than were those given placebo (n=93) (Rapaport et al. 2004). Additionally, the effectiveness of long-term escitalopram therapy in the prevention of recurrence of depression was also demonstrated in a group of patients who had been diagnosed with recurrent major depressive disorder (Kornstein et al. 2006). Patients given escitalopram 10 or 20 mg/day (n=73) in this 52-week study had significantly prolonged time to recurrence compared with those given placebo (n=66).
iSPOT-D trial. Treatment of depression will be significantly improved with the discovery of biomarkers that can identify which patients will respond best to which treatments. For example, depression is associated with impairments in a wide range of cognitive and emotional functioning (Snyder 2013). The focus of the International Study to Predict Optimized Treatment in Depression (iSPOT-D) trial was to determine whether laboratory measures of impaired functioning can be used to predict antidepressant treatment outcomes. In one report, medication-free outpatients with nonpsychotic major depression (n=665 completers) were assessed before treatment with 13 computerized tests of cognitive and emotional functioning, and their performance was compared with that of healthy controls (n=336). Patients were then randomly assigned to receive 8 weeks of treatment with escitalopram, sertraline, or extended-release venlafaxine (Etkin et al. 2015). Approximately one-quarter of the patients had significant impairment across most cognitive tests relative to the healthy controls, and these patients had poorer treatment outcomes. For this subset of patients, better task performance predicted remission (based on scores on the 16-item QIDS-SR) with 72% accuracy for treatment with escitalopram but not for treatment with the other antidepressants. Among the patients predicted to be nonresponders, the greatest impairments were on tests of attention, decision speed, working memory, and speed of emotion identification.
Neuroimaging studies have also focused on identifying patterns of brain activation that may be predictive of antidepressant treatment outcomes. Previous functional magnetic resonance imaging (fMRI) studies demonstrated that activation of the right lateral and medial prefrontal cortices and limbic regions during inhibitory responses on the go/no-go task predicted a greater treatment response to escitalopram (Langenecker et al. 2007). A similar approach was described in a study from the iSPOT-D trial that involved 80 medication-free outpatients with major depression and 34 matched healthy controls (Gyurak et al. 2016). During the fMRI scans, subjects completed three tasks to assess core domains of cognitive function. Subjects then received 8 weeks of antidepressant treatment, fMRI scans were repeated, and depression remission was assessed with the Ham-D. Intact activation of the frontoparietal network during inhibitory “no-go” responses predicted remission, particularly for subjects given the SSRIs escitalopram and sertraline. During the “no-go” responses, remitters showed the same pretreatment dorsolateral prefrontal cortex activation as control subjects, and nonremitters showed hypoactivation relative to control subjects. These study findings hold promise for identifying biomarkers that can help predict which patients are most likely to respond to citalopram and escitalopram treatment.
GENDEP study. A genome-based approach to identifying genes associated with therapeutic response to escitalopram has also yielded interesting findings. As part of the Genome-Based Therapeutic Drugs for Depression (GENDEP) study, genomewide association analysis examined 539,391 single-nucleotide polymorphisms (SNPs) to identify genes that underlie individual differences in antidepressant treatment response. The partially randomized, single-blind study examined DNA from unrelated patients of European ancestry who were assigned to receive 12 weeks of treatment with either escitalopram 10–30 mg/day (n=394) or nortriptyline 50–150 mg/day (n=312). Although no marker was associated at a genomewide level of significance, escitalopram response was predicted at a suggestive level of significance (P=0.00049) by a synonymous SNP (rs1126757) in the gene for interleukin 11 (IL11) (Uher et al. 2010). In addition, a targeted analysis of 72 a priori selected candidate genes identified an association between escitalopram response and a SNP (rs7801617) in the gene encoding interleukin 6 (IL6), a close homolog of IL11. A follow-up epigenetic study examined variations in DNA methylation of IL11 as a predictor of antidepressant treatment response using a subset of subjects who were randomly selected from the GENDEP study and received either escitalopram (n=80) or nortriptyline (n=33) (Powell et al. 2013b). This study found that DNA methylation levels on some CpG islands predicted antidepressant treatment response and that an interaction between CpG island methylation and genotype at rs1126757 could also predict treatment response. These studies are some of the first to provide evidence that a genomic approach may be fruitful for identifying molecular biomarkers that can predict which patients will best respond to specific antidepressants.
Another investigation from the GENDEP study sought to identify gene expression differences present prior to treatment that could predict treatment response. This study examined leukocyte mRNA expression in healthy controls (n=34) and depressed patients (n=74) before and after 8 weeks of treatment with escitalopram or nortriptyline (Cattaneo et al. 2013). The analysis was limited to 15 candidate genes that belonged to one of three groups previously implicated in the pathogenesis of depression or putative mechanisms of action of antidepressants (glucocorticoid receptor function, inflammation, and neuroplasticity). In regard to predictors present prior to treatment, nonresponders had higher mRNA levels for interleukin 1β (IL1β; +33%), macrophage inhibiting factor (MIF; +48%), and tumor necrosis factor (TNF; +39%). A similar study using a nonoverlapping set of GENDEP subjects and focusing only on escitalopram treatment also found higher mRNA levels for TNF (+20%) in nonresponders (Powell et al. 2013a).
A second aim of the Cattaneo et al. (2013) study was to identify changes in gene expression occurring during the course of successful treatment that could point to potential targets of drug action. Successful antidepressant treatment was associated with modest reductions in mRNA levels for IL6 (−9%) and FK506 binding protein 5 (FKBP5) (−11%), as well as increases in mRNA levels for BDNF (+48%) and VGF nerve growth factor inducible (VGF) (+20%). Taken together, these data indicate that the genes thought to serve as predictors of antidepressant response may be separate from the genes hypothesized to be altered by successful treatment. The authors also noted that with the exception of IL6, all gene expression differences were in the same direction for both escitalopram and nortriptyline despite their different mechanisms of action, suggesting that these gene expression changes reflect alterations in pathways common to the two antidepressants and not pathways selective to serotonergic or adrenergic neurotransmission (Cattaneo et al. 2013).
Depression in Children and Adolescents
Fluoxetine is currently the only FDA-approved medication for the treatment of depression in both children and adolescents (ages 8 years and older). Escitalopram was approved in 2009 for the treatment of depression in adolescents (ages 12–17 years) but not in younger children. In addition, the FDA has mandated the placement of black box warnings on product labels to indicate the potential for increased suicidality associated with the use of SSRIs in patients younger than 25 years. Importantly, a meta-analysis of pediatric trials conducted between 1988 and 2006 indicated that the benefits of antidepressant treatment of the young outweigh the risks (Bridge et al. 2007).
Citalopram
Only a limited number of clinical studies have examined the effectiveness of citalopram in the treatment of depression in youth. In one double-blind trial involving 174 children and adolescents (ages 7–17 years), citalopram (20 mg/day) showed a modest superiority over placebo in the treatment of depression (Wagner et al. 2004). Conversely, citalopram (10–40 mg/day) was not superior to placebo in a clinical trial involving 244 adolescents (ages 13–18 years) receiving treatment for 12 weeks (von Knorring et al. 2006). Clearly, additional clinical trials are required to establish the efficacy and safety of citalopram in the treatment of childhood depression.
Escitalopram
Three studies have addressed the effectiveness of escitalopram in the pediatric population. Escitalopram was shown to be effective in the treatment of depression in adolescents (ages 12–17 years) in a randomized, double-blind, placebo-controlled multicenter clinical trial (Emslie et al. 2009). In this study, the group given escitalopram 10–20 mg/day (n=155), compared with the group given placebo (n=157), had greater improvement based on change from baseline to week 8 in scores on the Children’s Depression Rating Scale—Revised (CDRS-R). In another randomized, double-blind trial investigating treatment of major depressive disorder in children and adolescents (ages 6–17 years), those given escitalopram 10–20 mg/day (n=131) did not improve significantly more than those given placebo (n=133). However, in a post hoc analysis of just the adolescents (ages 12–17 years), those given escitalopram (n=80), compared with those given placebo (n=77), had significantly improved CDRS-R scores from baseline to week 8 (Wagner et al. 2006). Finally, the long-term benefits of escitalopram treatment for depression in adolescents were demonstrated in an extension trial that enrolled a subset of the sample participating in the aforementioned Emslie et al. (2009) study. This double-blind 16- to 24-week trial (Findling et al. 2013) showed that in comparison with subjects given placebo (n=40), those given escitalopram 10–20 mg/day (n=37) had a modest but statistically significantly greater improvement in CDRS-R score from baseline of the lead-in study to treatment week 24 (8-week lead-in study plus 16-week extension) (P=0.005).
Generalized Anxiety Disorder
Citalopram
No large-scale randomized, double-blind clinical trials have examined the effectiveness of citalopram in the treatment of GAD. As described in the next subsection, all large-scale trials have focused on the use of escitalopram in the treatment of GAD.
Escitalopram
The efficacy of escitalopram in the treatment of GAD has been established in several randomized controlled clinical trials (Baldwin and Nair 2005). Escitalopram’s effectiveness in acute treatment of GAD was shown in three double-blind, placebo-controlled clinical trials that were also subjected to pooled analysis. These studies demonstrated that escitalopram (10–20 mg/day administered for 8–12 weeks) was superior to placebo (Davidson et al. 2004; Goodman et al. 2005; Stein et al. 2005). The efficacy of escitalopram (10–20 mg/day) in the long-term treatment of GAD was demonstrated in two 24-week controlled clinical trials, one open label (Davidson et al. 2005) and the other double blind (Bielski et al. 2005). Escitalopram also showed efficacy in the prevention of GAD relapse for an additional 24–76 weeks (Allgulander et al. 2006).
Panic Disorder
Citalopram
Few well-controlled studies have evaluated the effectiveness of citalopram in the treatment of panic disorder. In a 1-year placebo-controlled, double-blind study of 279 patients who agreed to continue treatment after an acute treatment period during which they had been randomly assigned to receive citalopram (20 or 30 mg/day, or 40 or 60 mg/day), clomipramine (60 or 90 mg/day), or placebo, all drug-treated groups showed significantly greater improvement compared with placebo on a variety of anxiety rating instruments, including the Clinical Anxiety Scale (CAS) panic attack item. The authors concluded that citalopram at a dosage range of 20–60 mg/day was an effective long-term therapy for the management of panic disorder (Lepola et al. 1998).
Escitalopram
Escitalopram 5–10 mg/day was shown to be effective in the treatment of panic disorder in a 10-week randomized, double-blind, placebo-controlled, flexible-dosage study in patients with a diagnosis of panic disorder with or without agoraphobia (Stahl et al. 2003). The relative panic attack frequency was significantly lower in the escitalopram group (n=125) than in the placebo group (n=114), and at the end of the study, a greater proportion of patients had zero panic attacks in the escitalopram group (50%) than in the placebo group (38%) (P=0.051).
Social Anxiety Disorder (Social Phobia)
Citalopram
A large-scale, double-blind, placebo-controlled study demonstrated that paroxetine is effective in the treatment of social anxiety disorder, suggesting that some SSRIs may be effective in the treatment of this disorder (Stein et al. 1998). Despite the lack of large-scale, placebo-controlled studies using citalopram, case reports indicate that citalopram may have effectiveness in the treatment of social anxiety disorder (Bouwer and Stein 1998; Lepola et al. 1994, 1996; Simon et al. 2001). In addition, one 12-week small-scale (n=21), flexible-dose, open-label study demonstrated the effectiveness of citalopram (20–60 mg/day) in relieving the symptoms of social anxiety disorder in patients with comorbid depression (Schneier et al. 2003).
Escitalopram
Two large-scale, multinational, multicenter clinical trials have demonstrated the effectiveness of escitalopram in the treatment of social anxiety disorder. In a 24-week fixed-dosage trial in patients with a diagnosis of social anxiety disorder, escitalopram at three dosages—5 mg/day (n=167), 10 mg/day (n=167), or 20 mg/day (n=170)—and paroxetine 20 mg/day (n=169) each showed a statistically superior therapeutic effect compared with placebo (n=166) by week 12 (Lader et al. 2004). Further improvement was seen by week 24 for all dosages of escitalopram and for paroxetine, and escitalopram 20 mg/day was superior to paroxetine 20 mg/day. In a 12-week study, escitalopram 10–20 mg/day (n=181) produced a superior therapeutic response compared with placebo (n=177) based on mean change from baseline in the Liebowitz Social Anxiety Scale score (Kasper et al. 2005). In a long-term study, escitalopram treatment at dosages of 10 or 20 mg/day for up to 24 weeks was shown to be effective in preventing relapse of social anxiety disorder following successful short-term therapy, with relapse being 2.8 times more likely with placebo treatment (n=181) than with escitalopram treatment (n=190) (Montgomery et al. 2005).
Anxiety Associated With Major Depressive Disorder
Citalopram
A retrospective study of 2,000 depressed patients enrolled in eight double-blind, placebo-controlled clinical trials revealed that citalopram was effective in relieving the symptoms of anxiety in depressed patients based on a greater decrease in the anxiety factor of the Ham-D (Flicker et al. 1998). Another double-blind, placebo-controlled study in 323 patients with a diagnosis of major depression revealed a significant antianxiety effect of citalopram 20–60 mg/day compared with placebo based on decreases in the Hamilton Anxiety Scale (Ham-A) score (Stahl 2000).
Escitalopram
The effectiveness of escitalopram in the treatment of anxiety symptoms associated with major depressive disorder was evaluated in a pooled analysis of five clinical trials (Bandelow et al. 2007). These placebo-controlled trials were originally designed to examine the effectiveness of escitalopram in treating major depression. In the pooled analysis, escitalopram 10–20 mg/day (n=850) was consistently superior to placebo (n=737) in relieving the anxious symptoms associated with depression, as evaluated with several different assessments of anxiousness. The analyses presented in this pooled study indicate that escitalopram is effective in relieving anxiety symptoms in depressed patients.
Obsessive-Compulsive Disorder
Citalopram
To date, a limited number of clinical studies have evaluated the effectiveness of citalopram in the treatment of obsessive-compulsive disorder (OCD). In the only published large-scale (N=401) double-blind, placebo-controlled study of citalopram in the treatment of OCD, all three dosages of citalopram (20, 40, and 60 mg/day) given for 12 weeks were significantly more effective than placebo in relieving the symptoms of OCD (Montgomery et al. 2001a). Citalopram appears to be effective in treating both obsessions and compulsions.
Escitalopram
The effectiveness of escitalopram in the treatment of OCD was demonstrated in a 24-week double-blind, placebo-controlled multicenter clinical trial (Stein et al. 2007). In this study, escitalopram 20 mg/day (n=116) and paroxetine 40 mg/day (n=119) produced significant improvement compared with placebo, and by week 24, all treatments, including escitalopram at 10 mg/day (n=116), were superior to placebo (n=115). Long-term treatment with escitalopram was shown in one large-scale study to prevent relapse of OCD in patients who had responded to initial treatment (Fineberg et al. 2007). Patients treated with escitalopram 10 or 20 mg/day (n=163) showed a significantly greater time to relapse compared with those receiving placebo (n=157).
Investigational Uses
Stress-Induced Myocardial Ischemia
Escitalopram
Substantial evidence implicates emotional stress as a trigger of acute coronary syndromes, and this effect can be reproduced in laboratory settings; studies show that up to 70% of patients with stable coronary heart disease display mental stress–induced myocardial ischemia (MSIMI) (Strike and Steptoe 2003). The Responses of Mental Stress Induced Myocardial Ischemia to Escitalopram Treatment (REMIT) study was designed to determine whether SSRI treatment could improve heart function following mental stress in patients with laboratory-diagnosed MSIMI. At the end of this 6-week randomized, double-blind, placebo-controlled study, significantly more of the patients receiving escitalopram treatment (n=56; 5 mg/day titrated to 20 mg/day over 3 weeks) than of those receiving placebo (n=56) (odds ratio, 2.62 [95% confidence interval, 1.06–6.44]; P=0.04) showed no MSIMI during three mental stressor tasks (Jiang et al. 2013). There was no statistically significant effect for exercise-induced ischemia. These results offer a preliminary indication of the potential utility of SSRIs in preventing mental stress–induced acute coronary syndromes.
Alzheimer’s Disease
Citalopram
Brain accumulation of amyloid plaques formed by the aggregation of the amyloid-β peptide (Aβ) is thought to be central to the pathophysiology of Alzheimer’s disease (AD) (Holtzman et al. 2011). The aggregation of Aβ into plaques is concentration dependent (Bero et al. 2011; Lomakin et al. 1997); therefore, methods to decrease Aβ levels may be therapeutic. Previous studies have shown that serotonin signaling suppresses the generation of Aβ in vitro and in animal models of AD (Cirrito et al. 2011; Nitsch et al. 1996). In an aged transgenic AD mouse model (APP/PS1 plaque-bearing mice), acute citalopram treatment led to a dose-dependent decrease in brain interstitial fluid Aβ levels, and chronic citalopram treatment arrested the growth of preexisting plaques and reduced the appearance of new plaques by 78% (Sheline et al. 2014). In addition, in a double-blind study, acute administration of citalopram (60 mg/day) in 23 healthy humans (ages 18–50 years, 11 females) resulted in lower cerebrospinal fluid Aβ production (37%) and concentration (38%) compared with placebo (Sheline et al. 2014). To the extent that AD involves the accumulation of Aβ, these results suggest a potential role for SSRI treatment in the prevention of AD, and future studies should be aimed at defining the mechanism by which citalopram treatment lowers Aβ levels.
Side Effects and Toxicology
Citalopram
In a meta-analysis of 746 depressed patients involved in several short-term clinical trials, the most common adverse events associated with citalopram were nausea and vomiting (20%), increased sweating (18%), and dry mouth and headache (17%) (Baldwin and Johnson 1995). Analysis of an integrated safety database, which includes data from 3,107 patients enrolled in 24 clinical trials, indicated that in placebo-controlled trials, nausea, dry mouth, somnolence, increased sweating, tremor, diarrhea, and ejaculatory failure of mild to moderate severity occurred with significantly greater frequency in patients given citalopram than in those given placebo (Muldoon 1996). The incidences of these adverse events with citalopram were less than 10% above those seen with placebo and were comparable to those reported with other SSRIs. Citalopram had a tolerability that was superior to that of the tricyclic antidepressants, with the exception that nausea and ejaculatory failure occurred with a 5% greater frequency in patients given citalopram (Keller 2000).
Escitalopram
In general, the side effects associated with escitalopram are similar to those observed with citalopram. In three placebo-controlled clinical trials performed with escitalopram, rates of discontinuation due to adverse events did not differ for patients given a dosage of 10 mg/day versus patients in the placebo group (Burke et al. 2002; Montgomery et al. 2001b; Wade et al. 2002). In the trial that included an escitalopram dosage of 20 mg/day, the rate of discontinuation was 10.4% for the group given escitalopram versus 2.5% for the group given placebo (Burke et al. 2002). In addition, the rate of adverse events overall in the group receiving escitalopram 20 mg/day (85.6%) was significantly greater than the rate in the placebo group (70.5%). Regardless of dosage, the adverse events that have been reported to occur more frequently with escitalopram compared with placebo are nausea, diarrhea, insomnia, dry mouth, and ejaculatory disorder, with nausea being reported most frequently, at a rate of 15% (McRae 2002). No published studies have reported clinically significant findings in laboratory test values, vital signs, weight gain or loss, or ECG values.
Specific Effects and Syndromes
QT Interval Prolongation
Citalopram has been shown to be associated with a dose-dependent prolongation of the corrected QT (QTc) interval in the ECG, which can increase the risk of a potentially fatal abnormal heart rhythm called torsades de pointes. As a result, the FDA recommends that citalopram not be prescribed at dosages greater than 40 mg/day (The Medical Letter on Drugs and Therapeutics 2013; U.S. Food and Drug Administration 2013). In addition, citalopram use should be avoided in populations at increased risk of QTc interval prolongation, such as patients with congenital long QT syndrome. For at-risk patients for whom no satisfactory alternatives to citalopram are available, a low dosage of citalopram should be used, and ECG and/or electrolytes should be monitored. Citalopram should be discontinued in patients with a QTc interval greater than 500 msec. Finally, for patients older than 60 years, dosages greater than 20 mg/day are not recommended. As in any clinical situation, the clinician must weigh the risk–benefit ratio for the treatment, and in some cases it may be reasonable to use dosages that exceed the FDA guidelines. Importantly, escitalopram at dosages therapeutically equivalent to those of citalopram has not been demonstrated to significantly impact the QTc interval.
Hyponatremia
Citalopram and escitalopram have been shown to produce hyponatremia in case reports involving elderly patients, and this information has been reviewed elsewhere (Jacob and Spinler 2006). In addition to advanced age, other factors that may increase the likelihood of hyponatremia include female gender, concurrent diuretic use, low body weight, and recent pneumonia. Treatment of SSRI-induced hyponatremia usually involves fluid restriction and/or administration of a loop diuretic such as furosemide and may include discontinuation of the SSRI.
Discontinuation Syndrome
The abrupt cessation of antidepressant therapy can result in a discontinuation syndrome characterized by dizziness, nausea and vomiting, lethargy, and flulike symptoms. This syndrome is more common with short-half-life SSRIs such as paroxetine and less common with long-half-life SSRIs such as fluoxetine. The data obtained from clinical trials suggest that the adverse events associated with discontinuation of citalopram or escitalopram tend to be mild and transient (Baldwin et al. 2007; Markowitz et al. 2000; Montgomery et al. 1993). Dose tapering is recommended for patients discontinuing treatment.
Treatment-Emergent Suicidal Ideation and Suicide
Considerable attention has been focused in recent years on the possibility that antidepressant drugs, especially SSRIs, may lead to treatment-emergent suicidal ideation (TESI) and an increased risk of suicide in some patients, particularly at the onset of therapy (Jick et al. 2004). This issue is of great concern in young patients and was described earlier in the chapter (see earlier section “Depression in Children and Adolescents”). In the case of citalopram, analyses of data obtained from 17 controlled clinical trials involving 5,000 patients indicate that the group of patients receiving citalopram had the lowest rate of suicide compared with the groups receiving placebo, tricyclic antidepressants, or other SSRIs (Nemeroff 2003). The risk of suicide associated with escitalopram was evaluated from data contained in the Summary Basis of Approval reports obtained from the FDA (Khan and Schwartz 2007). This study did not detect a significantly greater rate of suicide in the escitalopram group compared with either the citalopram or the placebo group. A meta-analysis of the escitalopram clinical trials database—consisting of 2,277 escitalopram-treated patients and 1,814 placebo-treated patients—also yielded no indication that escitalopram increased suicidal behavior in major depressive disorder and anxiety disorders (Pedersen 2005).
Although the data do not indicate a significantly greater risk of suicide for patients receiving either citalopram or escitalopram compared with those receiving placebo, most antidepressant studies show that a limited number of patients will exhibit TESI on initiation of therapy. Family and twin studies provide some evidence of a genetic influence on suicidal behavior, and it has been postulated that TESI may also be genetically influenced. Interestingly, a study in patients participating in the STAR*D trial demonstrated a significant association between citalopram-induced TESI in men and polymorphisms near the gene encoding the transcription factor cyclic adenosine monophosphate (cAMP) response-element binding (CREB) protein (Perlis et al. 2007). This protein is of great interest because it mediates the effects of second messengers on new gene transcription and has been implicated in antidepressant action and suicide (Dowlatshahi et al. 1998; Dwivedi et al. 2003a). An analysis of nine candidate genes in patients from the GENDEP study who received either escitalopram or nortriptyline identified polymorphisms in BDNF as being associated with TESI, and there was also a significant interaction between variants in BDNF and TRKB, the gene encoding the BDNF receptor (Perroud et al. 2009). A proposed role for the BDNF pathway in TESI is consistent with previous reports of lower levels of BDNF mRNA and protein in the hippocampus and prefrontal cortex of suicide victims and lower levels of BDNF protein in the plasma of suicide attempters (Dwivedi et al. 2003b; Karege et al. 2005; Kim et al. 2007). In addition, CREB is thought to control BDNF gene transcription, providing a functional link between these two genetic associations and TESI (Dwivedi et al. 2003a; Finkbeiner 2000).
Finally, in a genomewide association study also using subjects from the GENDEP study, the strongest association with TESI was with a single-nucleotide polymorphism located 30 kilobytes downstream of the gene encoding guanine deaminase (GDA). This study also found two suggestive escitalopram-specific associations with TESI that were contained in the gene for Kv channel-interacting protein 4 (KCNIP4) and near the gene for elongation protein 3 homolog (ELP3) (Perroud et al. 2012). Although no definitive conclusions can be drawn from this study or other genetic studies until they are replicated with larger samples, the findings point to the possibility that in the future, genetic markers may be used to predict not only treatment response but also vulnerabilities to the adverse effects of specific drugs.
Drug–Drug Interactions
Even though the majority of a dose of citalopram is metabolized in the liver (75%), because multiple P450 enzymes (CYP2C19, CYP3A4, and CYP2D6) contribute equally to the metabolism of citalopram and escitalopram, inhibition of any one of these enzymes by another drug is unlikely to significantly impact the overall metabolism of citalopram or escitalopram. Consistent with this, there are relatively few reports in the literature of drug–drug interactions involving citalopram or escitalopram. Because of the possibility of a potentially fatal pharmacodynamic interaction resulting in the serotonin syndrome, neither citalopram nor escitalopram should be administered with an MAO inhibitor or within 14 days of discontinuing an MAO inhibitor.
Conclusion
Citalopram and escitalopram are highly selective 5-HT reuptake inhibitors that are well tolerated and effective for the treatment of depression, with escitalopram also having proven efficacy in large-scale clinical trials in the treatment of GAD. Although the use of citalopram and escitalopram in the treatment of other psychiatric conditions has not been as thoroughly studied, the few well-controlled trials that have been completed suggest that both drugs may have a significant role in treating a wide range of psychiatric illnesses, including panic disorder, social anxiety disorder, anxiety associated with depression, and OCD. An advantage of citalopram and escitalopram compared with some other common SSRIs is a relatively weak inhibition of liver CYP450 enzymes, which reduces the potential for adverse pharmacokinetic drug–drug interactions. In addition, because escitalopram does not share with citalopram a modest affinity for the histamine H1 receptor, it may have a lower potential for antihistaminergic side effects compared with citalopram, a difference that has yet to be demonstrated in a clinical trial. In terms of effects on cardiac function, escitalopram’s risk of producing QT interval prolongation appears to be lower than that of citalopram, and careful monitoring must accompany use of citalopram in patients at risk for QT prolongation. There are a number of clinical trials comparing escitalopram with a variety of other antidepressants, including citalopram and venlafaxine, that suggest that escitalopram may have a faster onset of antidepressant efficacy and modest superiority in the treatment of individuals who are severely depressed. However, a clinically significant superiority of escitalopram over citalopram in the “real world” of psychiatric practice remains to be definitely established.
References
Agranat I, Caner H, Caldwell J: Putting chirality to work: the strategy of chiral switches. Nat Rev Drug Discov 1(10):753–768, 2002 12360254
Allgulander C, Florea I, Huusom AK: Prevention of relapse in generalized anxiety disorder by escitalopram treatment. Int J Neuropsychopharmacol 9(5):495–505, 2006 16316482
Anacker C, Zunszain PA, Cattaneo A, et al: Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry 16(7):738–750, 2011 21483429
Arborelius L, Owens MJ, Plotsky PM, et al: The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 160(1):1–12, 1999 9854171
Baldwin D, Johnson FN: Tolerability and safety of citalopram. Rev Contemp Pharmacother 6:315–325, 1995
Baldwin DS, Nair RV: Escitalopram in the treatment of generalized anxiety disorder. Expert Rev Neurother 5(4):443–449, 2005 16026227
Baldwin DS, Montgomery SA, Nil R, et al: Discontinuation symptoms in depression and anxiety disorders. Int J Neuropsychopharmacol 10(1):73–84, 2007 16359583
Bandelow B, Andersen HF, Dolberg OT: Escitalopram in the treatment of anxiety symptoms associated with depression. Depress Anxiety 24(1):53–61, 2007 16937393
Baumann P: Clinical pharmacokinetics of citalopram and other selective serotonergic reuptake inhibitors (SSRI). Int Clin Psychopharmacol 6 (suppl 5):13–20, 1992 1431018
Baumann P, Nil R, Souche A, et al: A double-blind, placebo-controlled study of citalopram with and without lithium in the treatment of therapy-resistant depressive patients: a clinical, pharmacokinetic, and pharmacogenetic investigation. J Clin Psychopharmacol 16(4):307–314, 1996 8835706
Bech P, Cialdella P: Citalopram in depression—meta-analysis of intended and unintended effects. Int Clin Psychopharmacol 6 (suppl 5):45–54, 1992 1431021
Bero AW, Yan P, Roh JH, et al: Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci 14(6):750–756, 2011 21532579
Bielski RJ, Bose A, Chang CC: A double-blind comparison of escitalopram and paroxetine in the long-term treatment of generalized anxiety disorder. Ann Clin Psychiatry 17(2):65–69, 2005 16075658
Bjerkenstedt L, Flyckt L, Overø KF, et al: Relationship between clinical effects, serum drug concentration and serotonin uptake inhibition in depressed patients treated with citalopram. A double-blind comparison of three dose levels. Eur J Clin Pharmacol 28(5):553–557, 1985 3899675
Blier P, de Montigny C: Current advances and trends in the treatment of depression. Trends Pharmacol Sci 15(7):220–226, 1994 7940983
Bouwer C, Stein DJ: Use of the selective serotonin reuptake inhibitor citalopram in the treatment of generalized social phobia. J Affect Disord 49(1):79–82, 1998 9574863
Bridge JA, Iyengar S, Salary CB, et al: Clinical response and risk for reported suicidal ideation and suicide attempts in pediatric antidepressant treatment: a meta-analysis of randomized controlled trials. JAMA 297(15):1683–1696, 2007 17440145
Brown ES, Howard C, Khan DA, et al: Escitalopram for severe asthma and major depressive disorder: a randomized, double-blind, placebo-controlled proof-of-concept study. Psychosomatics 53(1):75–80, 2012 22221724
Burke WJ, Gergel I, Bose A: Fixed-dose trial of the single isomer SSRI escitalopram in depressed outpatients. J Clin Psychiatry 63(4):331–336, 2002 12000207
Carvalho LA, Pariante CM: In vitro modulation of the glucocorticoid receptor by antidepressants. Stress 11(6):411–424, 2008 19065455
Cattaneo A, Gennarelli M, Uher R, et al: Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 38(3):377–385, 2013 22990943
Chaput Y, de Montigny C, Blier P: Effects of a selective 5-HT reuptake blocker, citalopram, on the sensitivity of 5-HT autoreceptors: electrophysiological studies in the rat brain. Naunyn Schmiedebergs Arch Pharmacol 333(4):342–348, 1986 3022157
Chen F, Lawrence AJ: The effects of antidepressant treatment on serotonergic and dopaminergic systems in Fawn-Hooded rats: a quantitative autoradiography study. Brain Res 976(1):22–29, 2003 12763618
Choi MJ, Kang RH, Lim SW, et al: Brain-derived neurotrophic factor gene polymorphism (Val66Met) and citalopram response in major depressive disorder. Brain Res 1118(1):176–182, 2006 16979146
Christensen AV, Fjalland B, Pedersen V, et al: Pharmacology of a new phthalane (Lu 10–171), with specific 5-HT uptake inhibiting properties. Eur J Pharmacol 41(2):153–162, 1977 12988
Cirrito JR, Disabato BM, Restivo JL, et al: Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A 108(36):14968–14973, 2011 21873225
Cremers TI, Spoelstra EN, de Boer P, et al: Desensitisation of 5-HT autoreceptors upon pharmacokinetically monitored chronic treatment with citalopram. Eur J Pharmacol 397(2–3):351–357, 2000 10844134
Dalgaard L, Larsen C: Metabolism and excretion of citalopram in man: identification of O-acyl- and N-glucuronides. Xenobiotica 29(10):1033–1041, 1999 10574684
Davidson JR, Bose A, Korotzer A, et al: Escitalopram in the treatment of generalized anxiety disorder: double-blind, placebo controlled, flexible-dose study. Depress Anxiety 19(4):234–240, 2004 15274172
Davidson JR, Bose A, Wang Q: Safety and efficacy of escitalopram in the long-term treatment of generalized anxiety disorder. J Clin Psychiatry 66(11):1441–1446, 2005 16420082
Dowlatshahi D, MacQueen GM, Wang JF, et al: Increased temporal cortex CREB concentrations and antidepressant treatment in major depression. Lancet 352(9142):1754–1755, 1998 9848357
Dwivedi Y, Rao JS, Rizavi HS, et al: Abnormal expression and functional characteristics of cyclic adenosine monophosphate response element binding protein in postmortem brain of suicide subjects. Arch Gen Psychiatry 60(3):273–282, 2003a 12622660
Dwivedi Y, Rizavi HS, Conley RR, et al: Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry 60(8):804–815, 2003b 12912764
Einarson TR: Evidence based review of escitalopram in treating major depressive disorder in primary care. Int Clin Psychopharmacol 19(5):305–310, 2004 15289704
Emslie GJ, Ventura D, Korotzer A, et al: Escitalopram in the treatment of adolescent depression: a randomized placebo-controlled multisite trial. J Am Acad Child Adolesc Psychiatry 48(7):721–729, 2009 19465881
Etkin A, Patenaude B, Song YJ, et al: A cognitive-emotional biomarker for predicting remission with antidepressant medications: a report from the iSPOT-D trial. Neuropsychopharmacology 40(6):1332–1342, 2015 25547711
Feighner JP, Overø K: Multicenter, placebo-controlled, fixed-dose study of citalopram in moderate-to-severe depression. J Clin Psychiatry 60(12):824–830, 1999 10665628
Findling RL, Robb A, Bose A: Escitalopram in the treatment of adolescent depression: a randomized, double-blind, placebo-controlled extension trial. J Child Adolesc Psychopharmacol 23(7):468–480, 2013 24041408
Fineberg NA, Tonnoir B, Lemming O, et al: Escitalopram prevents relapse of obsessive-compulsive disorder. Eur Neuropsychopharmacol 17(6–7):430–439, 2007 17240120
Finkbeiner S: Calcium regulation of the brain-derived neurotrophic factor gene. Cell Mol Life Sci 57(3):394–401, 2000 10823240
Flandreau EI, Ressler KJ, Owens MJ, et al: Chronic overexpression of corticotropin-releasing factor from the central amygdala produces HPA axis hyperactivity and behavioral anxiety associated with gene-expression changes in the hippocampus and paraventricular nucleus of the hypothalamus. Psychoneuroendocrinology 37(1):27–38, 2012 21616602
Flandreau EI, Bourke CH, Ressler KJ, et al: Escitalopram alters gene expression and HPA axis reactivity in rats following chronic overexpression of corticotropin-releasing factor from the central amygdala. Psychoneuroendocrinology 38(8): 1349–1361, 2013 23267723
Flicker C, Hakkarainen H, Tanghoj P: Citalopram in anxious depression: anxiolytic effects and lack of activation. Biol Psychiatry 43 (8 suppl 1):106S, 1998 doi: http://dx.doi.org/10.1016/S0006-3223(98)90799-5
Fredricson Overø K: Kinetics of citalopram in man; plasma levels in patients. Prog Neuropsychopharmacol Biol Psychiatry 6(3):311–318, 1982 6959195
Goodman WK, Bose A, Wang Q: Treatment of generalized anxiety disorder with escitalopram: pooled results from double-blind, placebo-controlled trials. J Affect Disord 87(2–3):161–167, 2005 15982747
Gyurak A, Patenaude B, Korgaonkar MS, et al: Frontoparietal activation during response inhibition predicts remission to antidepressants in patients with major depression. Biol Psychiatry 79(4):274–281, 2016 25891220
Hochstrasser B, Isaksen PM, Koponen H, et al: Prophylactic effect of citalopram in unipolar, recurrent depression: placebo-controlled study of maintenance therapy. Br J Psychiatry 178:304–310, 2001 11282808
Holtzman DM, Morris JC, Goate AM: Alzheimer’s disease: the challenge of the second century. Sci Transl Med 3(77):77sr1, 2011 21471435
Hyttel J: Neurochemical characterization of a new potent and selective serotonin uptake inhibitor: Lu 10–171. Psychopharmacology (Berl) 51(3):225–233, 1977 403537
Hyttel J: Effect of a specific 5-HT uptake inhibitor, citalopram (Lu 10–171), on 3H-5-HT uptake in rat brain synaptosomes in vitro. Psychopharmacology (Berl) 60(1):13–18, 1978 104340
Hyttel J: Pharmacological characterization of selective serotonin reuptake inhibitors (SSRIs). Int Clin Psychopharmacol 9 (suppl 1):19–26, 1994 8021435
Hyttel J, Bøgesø KP, Perregaard J, et al: The pharmacological effect of citalopram residues in the (S)-(+)-enantiomer. J Neural Transm 88(2):157–160, 1992 1632943
Hyttel J, Arnt J, Sanchez C: The pharmacology of citalopram. Rev Contemp Pharmacother 6:271–285, 1995
Inoue T: [Effects of conditioned fear stress on monoaminergic systems in the rat brain]. Hokkaido Igaku Zasshi 68(3):377–390, 1993 7686527
Invernizzi R, Bramante M, Samanin R: Chronic treatment with citalopram facilitates the effect of a challenge dose on cortical serotonin output: role of presynaptic 5-HT1A receptors. Eur J Pharmacol 260(2–3):243–246, 1994 7988650
Jacob S, Spinler SA: Hyponatremia associated with selective serotonin-reuptake inhibitors in older adults. Ann Pharmacother 40(9):1618–1622, 2006 16896026
Jiang W, Velazquez EJ, Kuchibhatla M, et al: Effect of escitalopram on mental stress-induced myocardial ischemia: results of the REMIT trial. JAMA 309(20):2139–2149, 2013 23695483
Jick H, Kaye JA, Jick SS: Antidepressants and the risk of suicidal behaviors. JAMA 292(3):338–343, 2004 15265848
Joffe P, Larsen FS, Pedersen V, et al: Single-dose pharmacokinetics of citalopram in patients with moderate renal insufficiency or hepatic cirrhosis compared with healthy subjects. Eur J Clin Pharmacol 54(3):237–242, 1998 9681666
Karege F, Vaudan G, Schwald M, et al: Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136(1–2):29–37, 2005 15893584
Kasper S, Stein DJ, Loft H, et al: Escitalopram in the treatment of social anxiety disorder: randomised, placebo-controlled, flexible-dosage study. Br J Psychiatry 186:222–226, 2005 15738503
Keller MB: Citalopram therapy for depression: a review of 10 years of European experience and data from U.S. clinical trials. J Clin Psychiatry 61(12):896–908, 2000 11206593
Kennedy SH, Andersen HF, Lam RW: Efficacy of escitalopram in the treatment of major depressive disorder compared with conventional selective serotonin reuptake inhibitors and venlafaxine XR: a meta-analysis. J Psychiatry Neurosci 31(2):122–131, 2006 16575428
Khan A, Schwartz K: Suicide risk and symptom reduction in patients assigned to placebo in duloxetine and escitalopram clinical trials: analysis of the FDA summary basis of approval reports. Ann Clin Psychiatry 19(1):31–36, 2007 17453659
Kim YK, Lee HP, Won SD, et al: Low plasma BDNF is associated with suicidal behavior in major depression. Prog Neuropsychopharmacol Biol Psychiatry 31(1):78–85, 2007 16904252
Klysner R, Bent-Hansen J, Hansen HL, et al: Efficacy of citalopram in the prevention of recurrent depression in elderly patients: placebo-controlled study of maintenance therapy. Br J Psychiatry 181:29–35, 2002 12091260
Kobayashi K, Chiba K, Yagi T, et al: Identification of cytochrome P450 isoforms involved in citalopram N-demethylation by human liver microsomes. J Pharmacol Exp Ther 280(2):927–933, 1997 9023308
Kornstein SG, Bose A, Li D, et al: Escitalopram maintenance treatment for prevention of recurrent depression: a randomized, placebo-controlled trial. J Clin Psychiatry 67(11):1767–1775, 2006 17196058
Kragh-Sørensen P, Overø KF, Petersen OL, et al: The kinetics of citalopram: single and multiple dose studies in man. Acta Pharmacol Toxicol (Copenh) 48(1):53–60, 1981 6939299
Lader M, Stender K, Bürger V, et al: Efficacy and tolerability of escitalopram in 12- and 24-week treatment of social anxiety disorder: randomised, double-blind, placebo-controlled, fixed-dose study. Depress Anxiety 19(4):241–248, 2004 15274173
Langenecker SA, Kennedy SE, Guidotti LM, et al: Frontal and limbic activation during inhibitory control predicts treatment response in major depressive disorder. Biol Psychiatry 62(11):1272–1280, 2007 17585888
Lepola U, Koponen H, Leinonen E: Citalopram in the treatment of social phobia: a report of three cases. Pharmacopsychiatry 27(5):186–188, 1994 7838888
Lepola U, Leinonen E, Koponen H: Citalopram in the treatment of early onset panic disorder and school phobia. Pharmacopsychiatry 29(1):30–32, 1996 8852532
Lepola UM, Wade AG, Leinonen EV, et al: A controlled, prospective, 1-year trial of citalopram in the treatment of panic disorder. J Clin Psychiatry 59(10):528–534, 1998 9818634
Lepola UM, Loft H, Reines EH: Escitalopram (10–20 mg/day) is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol 18(4):211–217, 2003 12817155
Llorca PM, Azorin JM, Despiegel N, et al: Efficacy of escitalopram in patients with severe depression: a pooled analysis. Int J Clin Pract 59(3):268–275, 2005 15857321
Lomakin A, Teplow DB, Kirschner DA, et al: Kinetic theory of fibrillogenesis of amyloid beta-protein. Proc Natl Acad Sci U S A 94(15):7942–7947, 1997 9223292
Markowitz JS, DeVane CL, Liston HL, et al: An assessment of selective serotonin reuptake inhibitor discontinuation symptoms with citalopram. Int Clin Psychopharmacol 15(6):329–333, 2000 11110008
Martin P, Soubrié P, Puech AJ: Reversal of helpless behavior by serotonin uptake blockers in rats. Psychopharmacology (Berl) 101(3):403–407, 1990 2362957
McMahon FJ, Buervenich S, Charney D, et al: Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet 78(5):804–814, 2006 16642436
McRae AL: Escitalopram. Curr Opin Investig Drugs 3(8):1225–1229, 2002 12211420
The Medical Letter on Drugs and Therapeutics: Citalopram, Escitalopram, and the QT interval. Med Lett Drugs Ther 55(1421):59, 2013 23863918
Mendels J, Kiev A, Fabre LF: Double-blind comparison of citalopram and placebo in depressed outpatients with melancholia. Depress Anxiety 9(2):54–60, 1999 10207659
Montgomery SA, Rasmussen JG, Tanghøj P: A 24-week study of 20 mg citalopram, 40 mg citalopram, and placebo in the prevention of relapse of major depression. Int Clin Psychopharmacol 8(3):181–188, 1993 8263316
Montgomery SA, Pedersen V, Tanghøj P, et al: The optimal dosing regimen for citalopram—a meta-analysis of nine placebo-controlled studies. Int Clin Psychopharmacol 9 (suppl 1):35–40, 1994 8021436
Montgomery SA, Kasper S, Stein DJ, et al: Citalopram 20 mg, 40 mg and 60 mg are all effective and well tolerated compared with placebo in obsessive-compulsive disorder. Int Clin Psychopharmacol 16(2):75–86, 2001a 11236072
Montgomery SA, Loft H, Sánchez C, et al: Escitalopram (S-enantiomer of citalopram): clinical efficacy and onset of action predicted from a rat model. Pharmacol Toxicol 88(5):282–286, 2001b 11393591
Montgomery SA, Nil R, Dürr-Pal N, et al: A 24-week randomized, double-blind, placebo-controlled study of escitalopram for the prevention of generalized social anxiety disorder. J Clin Psychiatry 66(10): 1270–1278, 2005 16259540
Muldoon C: The safety and tolerability of citalopram. Int Clin Psychopharmacol 11 (suppl 1):35–40, 1996 8732443
Nemeroff CB: Overview of the safety of citalopram. Psychopharmacol Bull 37(1):96–121, 2003 14561952
Nitsch RM, Deng M, Growdon JH, et al: Serotonin 5-HT2a and 5-HT2c receptors stimulate amyloid precursor protein ectodomain secretion. J Biol Chem 271(8):4188–4194, 1996 8626761
Norton N, Owen MJ: HTR2A: association and expression studies in neuropsychiatric genetics. Ann Med 37(2):121–129, 2005 16026119
Owens MJ, Morgan WN, Plott SJ, et al: Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. J Pharmacol Exp Ther 283(3):1305–1322, 1997 9400006
Owens MJ, Knight DL, Nemeroff CB: Second-generation SSRIs: human monoamine transporter binding profile of escitalopram and R-fluoxetine. Biol Psychiatry 50(5):345–350, 2001 11543737
Pariante CM, Alhaj HA, Arulnathan VE, et al: Central glucocorticoid receptor-mediated effects of the antidepressant, citalopram, in humans: a study using EEG and cognitive testing. Psychoneuroendocrinology 37(5):618–628, 2012 21958534
Pedersen AG: Escitalopram and suicidality in adult depression and anxiety. Int Clin Psychopharmacol 20(3):139–143, 2005 15812263
Peremans K, Audenaert K, Hoybergs Y, et al: The effect of citalopram hydrobromide on 5-HT2A receptors in the impulsive-aggressive dog, as measured with 123I-5-I-R91150 SPECT. Eur J Nucl Med Mol Imaging 32(6):708–716, 2005 15739093
Perlis RH, Purcell S, Fava M, et al: Association between treatment-emergent suicidal ideation with citalopram and polymorphisms near cyclic adenosine monophosphate response element binding protein in the STAR*D study. Arch Gen Psychiatry 64(6):689–697, 2007 17548750
Perroud N, Aitchison KJ, Uher R, et al: Genetic predictors of increase in suicidal ideation during antidepressant treatment in the GENDEP project. Neuropsychopharmacology 34(12):2517–2528, 2009 19641488
Perroud N, Uher R, Ng MY, et al: Genome-wide association study of increasing suicidal ideation during antidepressant treatment in the GENDEP project. Pharmacogenomics J 12(1):68–77, 2012 20877300
Powell TR, Schalkwyk LC, Heffernan AL, et al: Tumor necrosis factor and its targets in the inflammatory cytokine pathway are identified as putative transcriptomic biomarkers for escitalopram response. Eur Neuropsychopharmacol 23(9):1105–1114, 2013a 23142150
Powell TR, Smith RG, Hackinger S, et al: DNA methylation in interleukin-11 predicts clinical response to antidepressants in GENDEP. Transl Psychiatry 3:e300, 2013b 24002086
Rantamäki T, Hendolin P, Kankaanpää A, et al: Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology 32(10):2152–2162, 2007 17314919
Rantamäki T, Vesa L, Antila H, et al: Antidepressant drugs transactivate TrkB neurotrophin receptors in the adult rodent brain independently of BDNF and monoamine transporter blockade. PLoS One 6(6):e20567, 2011 21666748
Rao N: The clinical pharmacokinetics of escitalopram. Clin Pharmacokinet 46(4):281–290, 2007 17375980
Rapaport MH, Bose A, Zheng H: Escitalopram continuation treatment prevents relapse of depressive episodes. J Clin Psychiatry 65(1):44–49, 2004 14744167
Richelson E, Nelson A: Antagonism by antidepressants of neurotransmitter receptors of normal human brain in vitro. J Pharmacol Exp Ther 230(1):94–102, 1984 6086881
Robert P, Montgomery SA: Citalopram in doses of 20–60 mg is effective in depression relapse prevention: a placebo-controlled 6 month study. Int Clin Psychopharmacol 10 (suppl 1):29–35, 1995 7622809
Rochat B, Amey M, Gillet M, et al: Identification of three cytochrome P450 isozymes involved in N-demethylation of citalopram enantiomers in human liver microsomes. Pharmacogenetics 7(1):1–10, 1997 9110356
Rush AJ, Fava M, Wisniewski SR, et al; STAR*D Investigators Group: Sequenced treatment alternatives to relieve depression (STAR*D): rationale and design. Control Clin Trials 25(1):119–142, 2004 15061154
Russo-Neustadt AA, Alejandre H, Garcia C, et al: Hippocampal brain-derived neurotrophic factor expression following treatment with reboxetine, citalopram, and physical exercise. Neuropsychopharmacology 29(12):2189–2199, 2004 15199375
Sánchez C: Serotonergic mechanisms involved in the exploratory behaviour of mice in a fully automated two-compartment black and white text box. Pharmacol Toxicol 77(1):71–78, 1995 8532615
Sánchez C: The pharmacology of citalopram enantiomers: the antagonism by R-citalopram on the effect of S-citalopram. Basic Clin Pharmacol Toxicol 99(2):91–95, 2006 16918708
Sánchez C, Meier E: Behavioral profiles of SSRIs in animal models of depression, anxiety and aggression. Are they all alike? Psychopharmacology (Berl) 129(3):197–205, 1997 9084057
Sánchez C, Bergqvist PB, Brennum LT, et al: Escitalopram, the S-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. Psychopharmacology (Berl) 167(4):353–362, 2003 12719960
Schneier FR, Blanco C, Campeas R, et al: Citalopram treatment of social anxiety disorder with comorbid major depression. Depress Anxiety 17(4):191–196, 2003 12820174
Serra-Millàs M, López-Vílchez I, Navarro V, et al: Changes in plasma and platelet BDNF levels induced by S-citalopram in major depression. Psychopharmacology (Berl) 216(1):1–8, 2011 21308467
Sheline YI, West T, Yarasheski K, et al: An antidepressant decreases CSF Abeta production in healthy individuals and in transgenic AD mice. Sci Transl Med 6:1–8, 2014 24828079
Simon NM, Sharma SG, Worthington JJ, et al: Citalopram for social phobia: a clinical case series. Prog Neuropsychopharmacol Biol Psychiatry 25(7):1469–1474, 2001 11513360
Sindrup SH, Brøsen K, Hansen MG, et al: Pharmacokinetics of citalopram in relation to the sparteine and the mephenytoin oxidation polymorphisms. Ther Drug Monit 15(1):11–17, 1993 8451774
Snyder HR: Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: a meta-analysis and review. Psychol Bull 139(1):81–132, 2013 22642228
Stahl SM: Placebo-controlled comparison of the selective serotonin reuptake inhibitors citalopram and sertraline. Biol Psychiatry 48(9):894–901, 2000 11074227
Stahl SM, Gergel I, Li D: Escitalopram in the treatment of panic disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 64(11):1322–1327, 2003 14658946
Stein DJ, Andersen HF, Goodman WK: Escitalopram for the treatment of GAD: efficacy across different subgroups and outcomes. Ann Clin Psychiatry 17(2):71–75, 2005 16075659
Stein DJ, Andersen EW, Tonnoir B, et al: Escitalopram in obsessive-compulsive disorder: a randomized, placebo-controlled, paroxetine-referenced, fixed-dose, 24-week study. Curr Med Res Opin 23(4): 701–711, 2007 17407626
Stein MB, Liebowitz MR, Lydiard RB, et al: Paroxetine treatment of generalized social phobia (social anxiety disorder): a randomized controlled trial. JAMA 280(8): 708–713, 1998 9728642
Stórustovu Sí, Sánchez C, Pörzgen P, et al: R-citalopram functionally antagonises escitalopram in vivo and in vitro: evidence for kinetic interaction at the serotonin transporter. Br J Pharmacol 142(1):172–180, 2004 15037515
Strike PC, Steptoe A: Systematic review of mental stress-induced myocardial ischaemia. Eur Heart J 24(8):690–703, 2003 12713764
Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team: Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 163(1):28–40, 2006 16390886
Uher R, Perroud N, Ng MYM, et al: Genome-wide pharmacogenetics of antidepressant response in the GENDEP project. Am J Psychiatry 167(5):555–564, 2010 20360315
U.S. Food and Drug Administration: Revised recommendations for Celexa (citalopram hydrobromide) related to a potential risk of abnormal heart rhythms with high doses. FDA Drug Safety Communication 2013. Available at http://www.fda.gov/Drugs/DrugSafety/ucm297391.htm. Accessed March 1, 2016.
von Knorring AL, Olsson GI, Thomsen PH, et al: A randomized, double-blind, placebo-controlled study of citalopram in adolescents with major depressive disorder. J Clin Psychopharmacol 26(3):311–315, 2006 16702897
von Moltke LL, Greenblatt DJ, Grassi JM, et al: Citalopram and desmethylcitalopram in vitro: human cytochromes mediating transformation, and cytochrome inhibitory effects. Biol Psychiatry 46(6):839–849, 1999 10494454
von Moltke LL, Greenblatt DJ, Giancarlo GM, et al: Escitalopram (S-citalopram) and its metabolites in vitro: cytochromes mediating biotransformation, inhibitory effects, and comparison to R-citalopram. Drug Metab Dispos 29(8):1102–1109, 2001 11454728
Wade A, Michael Lemming O, Bang Hedegaard K: Escitalopram 10 mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol 17(3):95–102, 2002 11981349
Wagner KD, Robb AS, Findling RL, et al: A randomized, placebo-controlled trial of citalopram for the treatment of major depression in children and adolescents. Am J Psychiatry 161(6):1079–1083, 2004 15169696
Wagner KD, Jonas J, Findling RL, et al: A double-blind, randomized, placebo-controlled trial of escitalopram in the treatment of pediatric depression. J Am Acad Child Adolesc Psychiatry 45(3):280–288, 2006 16540812
Wiles NJ, Mulligan J, Peters TJ, et al: Severity of depression and response to antidepressants: GENPOD randomised controlled trial. Br J Psychiatry 200(2):130–136, 2012 22194183
Yu BN, Chen GL, He N, et al: Pharmacokinetics of citalopram in relation to genetic polymorphism of CYP2C19. Drug Metab Dispos 31(10):1255–1259, 2003 12975335