CHAPTER 26
Olanzapine
Amy L. Silberschmidt, M.D.
Jacob S. Ballon, M.D., M.P.H.
S. Charles Schulz, M.D.
History and Discovery
The story of specific antipsychotic medications for patients with schizophrenia and other severe psychiatric illnesses began in the early 1950s, when chlorpromazine was first given to psychotic patients in France (Delay and Bernitzer 1952). The antipsychotic qualities of this compound, as well as its “tranquilizing” effect, were dramatic and substantial. Studies performed around the world during the 1950s showed the usefulness of this new compound and of the others that followed. As is well known, multicenter trials of antipsychotic medications found that the approved medications were substantially and significantly better than placebo (Guttmacher et al. 1964). Furthermore, despite the range of chemical structures, the clinical effects were similar. In addition, the need to investigate the new medications for psychiatric illness led to improved clinical trial methodology for the field. During the 1960s, randomized and placebo-controlled trials became the standard for assessing the new medications for schizophrenia. These trials led to adoption of antipsychotic medications as the standard somatic treatment for schizophrenia.
However, over time, the adverse effects of these medications began to be recognized as more troublesome (Table 26–1). For example, many patients complained of medication-induced parkinsonism, dystonias, slowed thinking, blunted affect, akathisia, and tardive dyskinesia. These side effects were uncomfortable for patients taking the medications and, in many cases, led to poor treatment adherence.
Significant response in only 60%–70% of patients |
Movement disorder side effects |
Dystonia |
Parkinsonism |
Tardive dyskinesia |
Akathisia |
Slowed thinking (“cognitive parkinsonism”) |
Secondary negative symptoms |
Looking for ways to achieve the same treatment benefit with fewer side effects, investigators at Eli Lilly began to screen numerous compounds for potentially useful psychotropic properties. In 1990, the company applied for and received a patent for the compound olanzapine. It is interesting to note that the new compound had many structural similarities to clozapine, which in 1989 had been approved for use in treating refractory schizophrenia. Hailed as a novel second-generation antipsychotic drug, clozapine was thought to have potential for schizophrenia, mania, and anxiety. Clozapine was noted for its efficacy as well as its freedom from neurological side effects.
Olanzapine was first given to patients with schizophrenia in 1995 (Baldwin and Montgomery 1995). The patients in the study experienced a substantial decrease in their symptoms while receiving 5–30 mg/day of the compound. The study researchers noted a low degree of extrapyramidal side effects (EPS), although concern was raised regarding elevation of liver enzymes, as one patient had to discontinue the study for that reason.
The initial testing of olanzapine had useful results and led to a program of three pivotal trials of the drug. These first three controlled studies compared olanzapine (at two fixed dosages: 1 mg/day and 10 mg/day) against placebo (Beasley et al. 1996a); olanzapine at three fixed dosages (low, medium, or high) against olanzapine at 1.0 mg/day or haloperidol at 15 mg/day or placebo (Beasley et al. 1996b); and olanzapine against haloperidol in a large international study (Tollefson et al. 1997). The positive results for olanzapine led to U.S. Food and Drug Administration (FDA) approval in 1997 and subsequent widespread use in the United States and around the world.
Structure–Activity Relations
Olanzapine is a thiobenzodiazepine derivative that bears a close structural resemblance to clozapine. The formal chemical name of olanzapine is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b] [1,5]benzodiazepine. Structurally, olanzapine differs from clozapine by two additional methyl groups and the lack of a chloride moiety (Figure 26–1). The in vitro receptor binding profiles of olanzapine and clozapine are relatively similar. According to the package insert, olanzapine is known to have a high affinity for dopaminergic (D1–4), serotonergic (5-HT2A/2C, 5-HT6), histaminergic (H1), and α-adrenergic (α1) receptors, with moderate affinity for muscarinic (M1–5) receptors and weak activity at benzodiazepine, γ-amino-butyric acid type A (GABAA), and β-adrenergic receptors (Eli Lilly 2015).
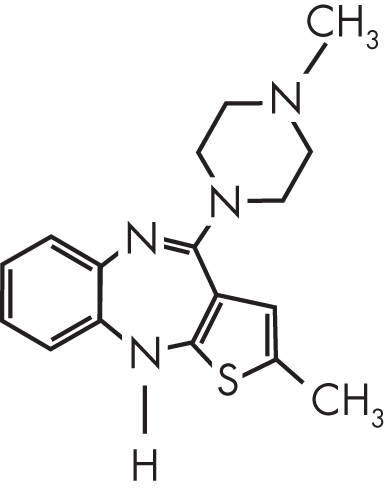
FIGURE 26–1. Chemical structure of olanzapine.
Pharmacological Profile
In vitro and preclinical behavioral studies of olanzapine predicted significant antipsychotic activity with a low propensity to induce EPS. Because clozapine is the prototype for second-generation antipsychotic action, it serves as the yardstick for “atypicality” of comparator compounds. Despite widespread general use of the term atypical to refer to any antipsychotic developed after clozapine, the term was originally coined to connote medications with an EPS risk no greater than that of placebo. While it is true that olanzapine carries a lower risk of tardive dyskinesia than do many other antipsychotics, its risk is still appreciably greater than that of placebo, and therefore the term atypical does not apply (Farah 2013).
One property that may lower a compound’s risk of EPS is nonselective binding of dopamine receptors. Classical antipsychotics selectively block dopamine D2-like (D2, D3, and D4) receptors over D1-like (D1 and D5) receptors—for example, haloperidol has a D2-to-D1 binding ratio of 25:1. Clozapine nonselectively binds all five dopamine receptor subtypes, with a D2-to-D1 ratio of 0.7:1, whereas olanzapine is only partially selective for the D2-like group, with a D2-to-D1 ratio of approximately 3:1, intermediate between those of haloperidol and clozapine.
In animal models predictive of antipsychotic efficacy, olanzapine produces effects indicating dopamine antagonism, with a low propensity to produce EPS. For example, in rats, olanzapine reduces climbing behavior induced by apomorphine and antagonizes stimulant-induced hyperactivity, both characteristic of antipsychotic effects. The ratio of the dose needed to produce catalepsy to the dose needed to inhibit conditioned avoidance, another model for atypical efficacy, is higher for olanzapine than for conventional agents (Moore 1999).
Another potential mechanism whereby dopamine antagonists may exert antipsychotic effects with minimal EPS is through selective activity in the A10 dopaminergic tracts from the ventral tegmentum to mesolimbic areas compared with effects antagonizing the A9 nigrostriatal projections that mediate EPS. Olanzapine in chronic administration, like clozapine, selectively inhibits firing of A10 neurons without significant inhibition of A9 tracts (Stockton and Rasmussen 1996a). Olanzapine shows increased c-fos activity in the nucleus accumbens relative to the dorsolateral striatum, thus demonstrating selective blockade of the mesolimbic dopamine tract compared with the nigrostriatal tract (Robertson and Fibiger 1996).
A leading theory regarding atypicality relates to the fleeting effects of atypical antipsychotics at the D2 receptor, coupled with regional selectivity of these compounds (Seeman 2002). Olanzapine’s D2 receptor occupancy saturation—which has been shown to be intermediate between that of clozapine and that of haloperidol—may be responsible for its decreased risk of EPS (Tauscher et al. 1999). However, because the current second-generation antipsychotic medications have substantially differing effects at many of the targets thought to play a role in atypicality, there is not yet consensus regarding the true rationale for atypicality in these agents compared with the first-generation antipsychotics (Farah 2005).
Amphetamine administration in rats is often used as a model for psychosis. The sympathomimetic activity and dopamine release provide a target for testing antipsychotic medications. Olanzapine disrupts the activity of amphetamines in rats (Gosselin et al. 1996). Olanzapine was shown in a rat model to decrease dopamine release in the A10 dopaminergic neurons of the ventral tegmentum greater than the A9 dopaminergic neurons of the striatum after chronic administration and after an amphetamine challenge (Stockton and Rasmussen 1996a, 1996b). Olanzapine does not induce catalepsy in rats at doses needed for antipsychotic efficacy.
Another model of psychosis in rats is produced by administration of the glutamatergic N-methyl-D-aspartate (NMDA) receptor antagonist phencyclidine (PCP). Chronic PCP use in humans is associated with symptoms similar to those in schizophrenia, including negative symptoms, thus making it a putative model for schizophrenia (Krystal et al. 1994). Olanzapine has been shown to decrease the hyperactivity of NMDA receptors under chronic PCP administration, which may have a bearing on its effect on negative symptoms (Ninan et al. 2003). With chronic administration, glutamatergic activity continues to be affected by olanzapine (Jardemark et al. 2000). Despite these findings, olanzapine has no direct affinity for the NMDA receptor (Stephenson and Pilowsky 1999).
Receptor-binding studies show that olanzapine has a broad range of neurotransmitter effects (Bymaster et al. 1996). Although olanzapine has potent muscarinic M1–5 receptor affinity in vitro (another contributor to putative anti-EPS effects), in practice few olanzapine-treated patients have anticholinergic side effects that are clinically significant. α1-Adrenergic and H1 histaminergic antagonism contribute to olanzapine’s adverse-effect profile of orthostatic hypotension (α1), sedation (H1), and possibly weight gain (H1). Olanzapine, like other second-generation antipsychotics, has a higher affinity for 5-HT2 receptors than for D2 receptors (Kapur et al. 1999). There is also indirect evidence that olanzapine blocks 5-HT2C receptors (Sharpley et al. 2000). Olanzapine has little or no effect on α2- and β-adrenergic, H2, nicotinic, GABA, opioid, sigma, or benzodiazepine receptors.
Pharmacokinetics and Disposition
Olanzapine is well absorbed after oral administration, with peak concentrations typically occurring 4–6 hours after ingestion (Kassahun et al. 1997). Approximately 40% of a given dose undergoes first-pass metabolism and therefore does not reach the systemic circulation, and food has little effect on olanzapine’s bioavailability (Callaghan et al. 1999; Eli Lilly 2015; Kassahun et al. 1997).
Two bioequivalent oral formulations of olanzapine are currently available: a standard oral tablet and an oral disintegrating tablet. The oral disintegrating tablets are intended for swallowing and absorption through the gut; however, sublingual administration has also been favored by some, as it allows the clinician to verify that the medication was taken. Markowitz et al. (2006) discovered that although the oral disintegrating preparation of olanzapine is absorbed more quickly than the standard oral tablet, this preparation’s onset of effect is approximately the same regardless of whether it is taken sublingually or conventionally swallowed. In either case, the onset of action with the oral dissolving tablet is faster than that with the standard oral tablet. After a 12.5-mg oral dose of 14C-labeled olanzapine, approximately 57% of the radiocarbon is recovered in urine and 30% in feces. In vitro studies suggest that olanzapine is approximately 93% protein bound, binding primarily to albumin and α1-acid glycoprotein (Kassahun et al. 1997).
Olanzapine is available as an intramuscular preparation, intended for treatment of the acute agitation typically seen in schizophrenia or in acute manic episodes of bipolar disorder. The peak plasma concentration is typically reached between 15 and 45 minutes after administration. The potency of intramuscular olanzapine is nearly five times greater than that of the orally administered drug, based on plasma levels. Clinical antipsychotic onset with intramuscular olanzapine is evident within 2 hours of administration, with benefits lasting for at least 24 hours (Kapur et al. 2005).
Olanzapine is also available in a long-acting injectable (LAI) preparation composed of a dihydrate form of olanzapine pamoate. As a dihydrate molecule, it is less soluble in water than a monohydrate and thus has the longer half-life required for a depot formulation. This formulation is designed to be dosed once every 4 weeks (Mamo et al. 2008).
Finally, olanzapine is available in a combined preparation with fluoxetine. The olanzapine–fluoxetine combination (OFC) tablet provides fixed doses of olanzapine and fluoxetine. Overall, few pharmacokinetic changes result from adding fluoxetine to olanzapine, and those that do occur are generally related to cytochrome P450 (CYP) 2D6 inhibition. There is no change in the overall half-life of olanzapine. Although minor yet statistically significant changes occur in the concentration of olanzapine when it is coadministered with fluoxetine, these changes are not clinically significant and do not alter the side-effect profile of olanzapine (Gossen et al. 2002).
Olanzapine is extensively metabolized to multiple metabolites, but primarily to 10-N-glucuronide and 4′-N-desmethylolanzapine (Macias et al. 1998). In vitro studies assessing the oxidative metabolism of olanzapine suggest that CYP1A2 is the enzyme primarily responsible for the formation of 4′-N-desmethylolanzapine, flavin-containing monooxygenase-3 (FMO3) is responsible for the formation of 4′-N-oxide olanzapine, and CYP2D6 is the primary enzyme responsible for the formation of 2-hydroxymethyl olanzapine (Ring et al. 1996b). Although CYP1A2 appears to be a major route of metabolism, olanzapine clearance in one study was not significantly correlated with salivary paraxanthine-to-caffeine ratio (thought to be a measure of CYP1A2 activity) (Hägg et al. 2001). Another analysis, however, found that the 4′-N-desmethylolanzapine–to–olanzapine plasma metabolic ratio significantly correlated with olanzapine clearance (Callaghan et al. 1999). Olanzapine pharmacokinetic parameters do not differ significantly between extensive and poor metabolizers of CYP2D6 (see Hägg et al. 2001).
Olanzapine shows linear pharmacokinetics within the recommended dosage range (Aravagiri et al. 1997; Bergstrom et al. 1995; Callaghan et al. 1999). Mean half-life is 36 hours, mean clearance is 29.4 L/hour, mean volume of distribution is 19.2 L/kg, and area under the concentration–time curve over 24 hours (AUC0–24) is 333 ng*hour/mL. The half-lives of the two major metabolites (4′-N-desmethylolanzapine and 10-N-glucuronide) are 92.6 and 39.6 hours, respectively (Macias et al. 1998). Other analyses also have found the mean half-life of olanzapine to be approximately 30 hours and the mean apparent clearance to be approximately 25 L/hour (Callaghan et al. 1999; Eli Lilly 2015; Kassahun et al. 1997). Once-daily administration of olanzapine produces steady-state concentrations in about a week that are approximately twofold higher than concentrations after a single dose (Callaghan et al. 1999).
Clearance of olanzapine is approximately 25%–30% lower in women than in men, based on results of population pharmacokinetic analyses (Callaghan et al. 1999; Patel et al. 1995, 1996). A study of 20 male and 7 female patients with schizophrenia receiving olanzapine also found that women had higher trough concentrations after receiving 1 week of olanzapine 12.5 mg/day (Kelly et al. 1999). Despite the differences in clearance and plasma levels, there is no difference between sexes in the incidence of EPS or other movement disorders (Aichhorn et al. 2006).
Olanzapine’s pharmacokinetics in the elderly and in children differ from those in adults. In the elderly, olanzapine clearance is approximately 30% lower than in younger individuals, and the half-life is approximately 50% longer (Callaghan et al. 1999; Patel et al. 1995). A study of eight children and adolescents (ages 10–18 years) found pharmacokinetic parameters similar to those reported in nonsmoking adults, with an average Tmax (time required to reach the maximal plasma concentration) of 4.7 hours, an average apparent oral clearance of 9.6 L/hour, and an average half-life of 37.2 hours (Grothe et al. 2000). The highest concentrations were seen when smaller-sized patients received dosages greater than 10 mg/day; therefore, dosing should take into consideration the size of the child.
Impairment in either hepatic or renal function has not been associated with altered olanzapine disposition. In a study of four healthy individuals and eight patients with hepatic cirrhosis, no significant differences in olanzapine pharmacokinetics were found, although urinary concentrations of olanzapine 10-N-glucuronide were increased in patients with cirrhosis (Callaghan et al. 1999). A study comparing olanzapine pharmacokinetics in six subjects with normal renal function, six subjects with renal failure who received an olanzapine dose 1 hour before hemodialysis, and six subjects with renal failure who received an olanzapine dose during their 48-hour interdialytic interval did not find any significant differences. These data suggest that olanzapine dosage does not need to be adjusted in patients with renal or hepatic disease (Callaghan et al. 1999).
Mechanism of Action
In discussing olanzapine’s mechanism of action in the treatment of schizophrenia, it should be noted that there is no established molecular mechanism that can unify the symptoms of schizophrenia. No precise animal or in vitro model for the illness exists, nor is there a consensus on its precise etiology or pathophysiology. Numerous neurochemical hypotheses have been proposed, including theories implicating abnormalities in dopaminergic, glutamatergic, serotonergic, and other systems, such as neurotensin (Boules et al. 2007) or neuregulin (Benzel et al. 2007). However, the discovery that multiple receptor types exist for each neurotransmitter has added many layers of complexity to the search for explanations regarding the root causes of schizophrenia. Thus, it is no longer possible to use broad terms such as “increased dopamine” when discussing ideas about the etiology of the disorder.
Despite the caveats mentioned above regarding our rudimentary knowledge of the nature of schizophrenia, it is important to note that all approved antipsychotic medications have a significant effect on the dopaminergic system, largely through the blockade of D2 receptors (Kapur and Remington 2001). Even though there are substantial differences in D2 receptor affinity among the traditional antipsychotics and the second-generation antipsychotics, they all are either full antagonists or partial agonists at the dopamine D2 receptor. Of interest is evolving research indicating the importance of multiple-receptor blockade to the effectiveness of the second-generation antipsychotic class. As the various neurotransmitter systems have been investigated in the neuropsychopharmacology of schizophrenia, evidence is emerging that the second-generation antipsychotics, and olanzapine in particular, may improve different schizophrenia symptom domains by means of effects on 5-HT receptors, on multiple-receptor binding, on region-specific and more fleeting binding to dopamine receptors, on glutamate neurotransmission, and perhaps on neuropeptide neurotransmitters. In the following paragraphs, we discuss each of these specific ideas about olanzapine’s mechanism of action in turn.
In clinical investigations with positron emission tomography (PET) imaging, Kapur et al. (1998) showed that olanzapine at a wide range of dosages blocks a high percentage (95% or greater) of 5-HT2A receptors and also blocks dopamine receptors in a dose-dependent fashion—crossing the putative antipsychotic blockade line at dosages commonly used to diminish psychotic symptoms of schizophrenia. This study indicated that olanzapine’s primary mechanism was related to the blockade of dopamine receptors and additionally noted that olanzapine showed stronger affinity for 5-HT2A receptors than for dopamine receptors at all dosage ranges.
A more compelling hypothesis regarding the mechanism of olanzapine’s effects emerged from in vivo PET scanning work performed in a series of experiments at the University of Toronto and in Sweden. Results of the initial PET scanning studies of patients receiving clozapine indicated that atypical dopamine D2 receptor binding was occurring (Farde and Nordström 1992; Farde et al. 1992; Kapur et al. 2000). The group subsequently found similar unusual D2 receptor binding with quetiapine and, to some degree, olanzapine (Kapur et al. 1998). The authors proposed that the so-called atypical antipsychotic effect—successful treatment of psychotic symptoms without induction of movement disorder side effects—may be the result of a “fast off” property of some second-generation agents, wherein the drug blocks the dopamine D2 receptor but leaves it quickly, a receptor occupancy pattern that effectively decreases psychosis yet causes minimal interference to the body’s own dopamine receptor activity. Thus, for olanzapine, this mechanism might help explain how the drug can exert strong therapeutic effects on schizophrenia symptoms yet cause few EPS at standard dosages. From a clinical viewpoint, it is important to note that at higher olanzapine dosages (30 mg/day), greater dopamine receptor blockade is seen, and movement disorder side effects, such as akathisia, are more likely to occur.
Over the past 30 years, there has been substantial interest in the role of glutamate, an excitatory neurotransmitter, in the pathophysiology of schizophrenia (see, e.g., Coyle 2006; Kim et al. 1980; Krystal et al. 1994; Moghaddam and Javitt 2012). Glutamate’s inclusion in theories about the development of schizophrenia is supported by the psychotomimetic properties of glutamate antagonists such as PCP and ketamine. These NMDA receptor antagonists induce a group of behaviors that often show closer parallels to schizophrenia than do those induced by dopamine sympathomimetic agents, in both mice and humans. Clinical trial evidence points to the potential usefulness of glutamatergic agonists (e.g., D-cycloserine) in treating schizophrenia (Kantrowitz et al. 2010). One way of examining the potential effectiveness of medication is to look at changes in deficits in prepulse inhibition (i.e., attenuation of the startle response), a measure of sensory motor gating that is diminished in patients with schizophrenia and can be similarly diminished pharmacologically through administration of PCP (Dulawa and Geyer 1996). In a study of rats with isolation-induced disruption of prepulse inhibition, both quetiapine and olanzapine successfully reversed the prepulse inhibition deficit (Bakshi et al. 1998).
Neurotensin receptors are collocated with and modulate mesolimbic dopaminergic neurons (Boules et al. 2014). Cerebrospinal fluid (CSF) concentrations of neurotensin are abnormal in some untreated patients with schizophrenia and have been found to normalize with antipsychotic administration. The degree of clinical improvement, particularly in negative symptoms, was found to correlate with the degree of increase in CSF neurotensin (Sharma et al. 1997). In rats, olanzapine administration was shown to increase extracellular neurotensin in the ventral striatum and medial prefrontal cortex acutely. Over time, chronic olanzapine administration decreased the concentration of neurotensin in the medial prefrontal cortex but increased the concentration in the ventral striatum. Furthermore, chronic olanzapine administration abolished the stimulatory effects of amphetamine administration in these regions (Gruber et al. 2011). Olanzapine does not bind to the neurotensin 1 (NT1) receptor in humans (Theisen et al. 2007) but may influence neurotensin through its action on other subtypes of neurotensin receptor or its downstream effects.
Neuregulins are a family of growth factors that stimulate the ERbB receptor tyrosine kinases and have been shown to play a role in the assembly of neural circuitry, myelination, neurotransmission, and synaptic plasticity. The neuregulin-1 gene (NRG1) has been associated with schizophrenia in several large genomewide association studies and meta-analyses (Mei and Nave 2014). In rats, (NRG1) expression increases in the hippocampus after 1 week of olanzapine administration; however, after 12 weeks, its expression decreases in the prefrontal cortex and cingulate cortex (Deng et al. 2015). In a study using immortalized lymphocytes from schizophrenia patients and control subjects from unrelated families, expression of the NRG1 glial growth factor isoform was found to be lower in schizophrenia patients than in control subjects, both before and after stimulation with olanzapine (Chagnon et al. 2008).
In summary, olanzapine works at least at the dopamine, 5-HT, and glutamate receptors. There is intriguing circumstantial evidence that olanzapine may modulate neurotensin and neuregulin. Current theories suggest that dopamine receptor–blocking capabilities are a necessary but not sufficient requirement for antipsychotic effectiveness. The other studied mechanisms, when taken in total, may be the factors leading to olanzapine’s broad efficacy and side-effect profile.
Indications and Efficacy
Olanzapine initially received FDA approval for the treatment of psychosis. Currently, it has multiple indications, including treatment of schizophrenia; acute treatment of manic or mixed states in bipolar disorder; and maintenance treatment of bipolar disorder, both as monotherapy and as an adjunct to mood stabilizer therapy. The OFC preparation has an FDA indication for bipolar depression and treatment-resistant depression. Intramuscular olanzapine carries an indication for acute agitation in schizophrenia and bipolar mania. Olanzapine is approved for adults and for children ages 13–18 years.
In addition to the approved indications, olanzapine has been studied—and at times used with limited evidence—in several other illnesses. In this section, we present the evidence base supporting the use of olanzapine for its FDA-indicated uses as well as for off-label uses.
Approved Indications
Schizophrenia
As noted earlier, olanzapine was originally developed as a medication with potential for treating schizophrenia, mania, and anxiety. To gain FDA approval for the treatment of schizophrenia, olanzapine was tested in four pivotal studies to assess the compound for efficacy, safety, and dosage range. The earliest testing of olanzapine was an assessment of olanzapine dosages of 5–30 mg/day following an initial starting dosage of 10 mg/day. Brief Psychiatric Rating Scale (BPRS) scores were reduced substantially, and EPS incidence was low (Baldwin and Montgomery 1995). These encouraging results led to further studies and pointed to a dosage range to be tested.
Efficacy studies. The first pivotal study compared two dosages of olanzapine (1 mg/day and 10 mg/day) with placebo in a large international sample. The 10 mg/day dosage was statistically significantly superior to placebo on objective rating scales (Beasley et al. 1996a). The second pivotal study was a dose-ranging comparison of olanzapine versus haloperidol and placebo, also in a large international sample. Three dosage ranges for olanzapine were included: 1) low (5±2.5 mg/day), 2) medium (10±2.5 mg/day), and 3) high (15±2.5 mg/day). Haloperidol was dosed to 15±2.5 mg/day. Olanzapine at the medium and high dosage ranges and haloperidol were associated with significant improvements compared with placebo (Beasley et al. 1996b). Beasley et al. (1997), using the same sample, further demonstrated that acute EPS were reported less frequently in all olanzapine dosage groups than in the haloperidol group. Tollefson and Sanger (1997) re-analyzed data from the first two studies and found that olanzapine had a direct therapeutic effect on negative symptoms that was not mediated by its effects on positive symptoms, EPS, or mood.
Another large international multicenter trial used a flexible dosing strategy to show olanzapine’s clinical superiority to haloperidol on positive symptoms, negative symptoms, comorbid depression, EPS, and overall drug safety. In this study, patients were started on olanzapine or haloperidol at dosages ranging from 5 to 20 mg/day. Ultimately, patients received olanzapine 13.2 mg/day compared with haloperidol 11.8 mg/day (Tollefson et al. 1997).
The group of studies described above led to olanzapine’s approval for psychosis (later changed to schizophrenia). Several other studies have confirmed olanzapine’s efficacy in the treatment of schizophrenia. With its added benefit of low movement disorder side effects, olanzapine has become an important addition to the armamentarium of the psychiatrist treating schizophrenia. Leucht et al. (1999) reported that olanzapine was statistically more effective than placebo (moderate effect) and also more effective than haloperidol (small effect) on global schizophrenia symptomatology.
A large National Institute of Mental Health (NIMH)–funded investigation sought to compare the second-generation antipsychotics olanzapine, risperidone, quetiapine, and ziprasidone with perphenazine in order to understand the efficacy and side-effect profiles of the newer versus older antipsychotic medications (Lieberman et al. 2005). This project, the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), was designed to provide a double-blind yet reasonably naturalistic setting for clinicians to treat patients, using time to discontinuation for any reason as the primary outcome variable. Although olanzapine had the longest time to all-cause discontinuation of any medication in the trial, it also had the highest rate of discontinuation due to metabolic complications such as weight gain, whereas perphenazine had the highest rate of discontinuation due to EPS. Overall, however, the discontinuation rate was high for all medications, with nearly 75% of patients changing medications within the 18-month study duration. Much discussion and written commentary has ensued since the initial findings were published, including analyses of cost-effectiveness (Rosenheck et al. 2006), of the effects of antipsychotic medication on psychosocial functioning (Swartz et al. 2007), and of the impact of switching medications (Essock et al. 2006), as well as numerous editorials about the treatment implications of the study.
Schizophrenia is often not fully responsive to antipsychotic treatment. Assessment of olanzapine in patients with treatment-refractory illness did not support its usefulness in a stringent nonresponder protocol (Conley et al. 1998). However, in an Eli Lilly–sponsored double-blind noninferiority multicenter trial, olanzapine’s effect in lowering Positive and Negative Syndrome Scale (PANSS) scores was comparable to that of clozapine (Tollefson et al. 2001). A second phase of the CATIE project examined the use of clozapine in patients with treatment-refractory illness and found, based on the time to discontinuation, that clozapine was a superior treatment for patients who had not responded to other second-generation medications (McEvoy et al. 2006).
Although early research noted olanzapine’s beneficial effects on negative symptoms and cognitive symptoms of schizophrenia, these effects were not as strong as the drug’s effects on positive symptoms. Olanzapine’s efficacy in negative symptoms was first reported by Tollefson et al. (1997) following completion of a large double-blind trial of olanzapine and haloperidol. When all symptom improvements were taken into account, olanzapine-treated subjects showed greater improvement than haloperidol-treated patients in negative symptoms (as measured by improvement in scores on both the Scale for the Assessment of Negative Symptoms [SANS] and the BPRS negative symptom subscale). In a flexible-dose comparison between risperidone and olanzapine in patients followed for 1 year, olanzapine-treated patients showed significantly greater improvement on SANS scores than did risperidone-treated patients (Alvarez et al. 2006). Interestingly, in a study by researchers from Eli Lilly that followed patients taking either quetiapine or olanzapine for 6 months, the two groups showed similar improvements on the SANS at study completion (Kinon et al. 2006). In a meta-analysis of 50 studies of second-generation antipsychotics (Komossa et al. 2010), no difference in short-, medium-, or long-term improvement of negative symptoms (as measured by PANSS negative, BPRS negative, and/or SANS total scores) was found between olanzapine and the other medications, which included amisulpride, clozapine (1 of 12 studies found clozapine to be superior as measured by SANS total scores), quetiapine, and ziprasidone (1 of 2 studies found olanzapine to be superior on PANSS negative scores, but overall no difference was found). The same study found olanzapine to outperform risperidone on improvement of negative symptoms (as measured by SANS total score) (Komossa et al. 2010).
Cognitive functioning is the most important indicator of schizophrenia prognosis (Green 2006). In a 1-year comparison of olanzapine with risperidone, both groups showed modest benefits on a cognitive function battery (Gurpegui et al. 2007). In a study conducted over a 1-year period by Eli Lilly researchers in Spain, olanzapine-treated patients showed greater benefit in social functioning than did risperidone-treated patients, as assessed by scores on the Social Functioning Scale (SFS). The greatest difference was in occupation/employment, but improvements were also seen on measures of independence, social engagement, and recreation (Ciudad et al. 2006).
In an 8-week double-blind, placebo-controlled study, the LAI formulation of olanzapine showed a statistically significant separation from placebo (as measured by decreases in PANSS total scores) that was evident by day 7 (Lauriello et al. 2008). Another study of olanzapine LAI that involved a longer period of treatment—24 weeks—found that a high percentage of subjects remained free of psychotic exacerbation (Kane et al. 2010). The published report of this study also discussed the relationship between oral and LAI olanzapine dosing and noted that the potential for accidental intravascular injection—which can cause sedation and/or delirium—requires that patients be observed for 3 hours after injection.
Studies have investigated olanzapine’s efficacy in the treatment of schizophrenia spectrum and other psychotic disorders in adolescents. In a systematic review of the literature in children, second-generation antipsychotics were shown to be beneficial overall in improving psychotic symptoms (Jensen et al. 2007). Olanzapine’s safety profile in adolescents is similar to its profile in adults, although its adverse effects on weight and prolactin are more significant in young people (Kryzhanovskaya et al. 2009a). A double-blind, flexible-dose study demonstrated comparable efficacy for risperidone, olanzapine, and haloperidol in psychotic young people (Sikich et al. 2004). A similar trial of olanzapine, risperidone, and quetiapine in early psychosis found equivalent rates of discontinuation for the three drugs over a 52-week period, with olanzapine and risperidone showing slightly greater reductions on PANSS positive scores compared with quetiapine (McEvoy et al. 2007). In late 2009, olanzapine received FDA approval for the treatment of schizophrenia in teenagers, but with the recommendation that clinicians first consider the use of other agents, given concerns about metabolic consequences (for further discussion, see section titled “Side Effects and Toxicology” later in this chapter).
Studies have shown that approximately 25% of individuals with early signs of schizophrenia will ultimately develop the disease, although in the past, rates of conversion were estimated as being much higher (40%–50%; see Kaur and Cadenhead 2010 for a review). Olanzapine’s use in a population considered to be at high risk for schizophrenia but not meeting full symptom criteria was evaluated in a double-blind multicenter study (McGlashan et al. 2006). The olanzapine group did not show statistically significant reductions in the rate of conversion to psychosis compared with the placebo group. However, a number of factors, including high dropout rates in both groups, the lack of a systematic method for diagnosing clinical psychiatric disorders, and problems regarding selection and classification of patients, limited the generalizability and reliability of the findings (McGlashan et al. 2006). The number needed to treat (NNT), a measure of effect size, was 4.5 in this study; thus, whereas early treatment with olanzapine may benefit some individuals, the increased risk of metabolic side effects associated with use of antipsychotics in adolescents mandates that an abundance of caution be exercised before making the decision to administer these medications to young people who are not yet ill. Olanzapine treatment was not found to improve cognition in adolescents identified as being at risk of psychosis, and olanzapine treatment did not significantly alter neuropsychological deficits either at baseline or after conversion to psychosis (Hawkins et al. 2008). Given the long-term side-effect consequences, antipsychotic medication is not indicated in people with subthreshold levels of psychosis.
At the other end of the age spectrum, olanzapine has been investigated in the treatment of psychotic disorders in the elderly. Olanzapine, like all antipsychotic medications, carries an FDA black box warning regarding an increased risk of stroke associated with its use in elderly individuals with dementia-related psychosis. (The use of olanzapine in dementia is discussed in greater detail in a separate subsection; see “Dementia-Related Agitation and Psychosis” later in this chapter.) A study of olanzapine versus haloperidol in a group of older patients with chronic schizophrenia found a statistical advantage for olanzapine (Barak et al. 2002). In a study of olanzapine in 94 acutely ill inpatients age 65 years or older, some with psychosis, Hwang et al. (2003) reported mean reductions from baseline of greater than 50% on BPRS scores, thus illustrating olanzapine’s usefulness in treating a broad range of psychotic symptoms in older patients. As with any treatment used in elderly persons, special care must be taken to avoid cardiovascular complications. With olanzapine, orthostatic hypotension, oversedation, and thus the risk of falls must be factored into the dosing decision (Gareri et al. 2006).
Treatment approaches. Early studies of olanzapine assessed dosages ranging from 5 to 30 mg/day. When olanzapine was initially released, it was recommended that the medication be started at a dosage of 10 mg/day, often given as a bedtime dose. Subsequently, clinicians have used average dosages higher than 10 mg/day (e.g., approximately 13 mg/day). In the flexible-dosing segment of CATIE, in which the available dosages were 7.5, 15.0, 22.5, and 30.0 mg/day), the average dosage was 20.1 mg/day (Lieberman et al. 2005). For inpatient treatment, clinicians often will give patients 5 mg of olanzapine in the morning and 10 mg at bedtime (Schulz 1999). Because some patients appear to have an inadequate response to olanzapine at the recommended dosages, clinicians have assessed the usefulness of olanzapine at dosages above the recommended 20 mg/day. Many hospital clinicians employ a loading strategy with olanzapine, particularly in patients presenting with acute agitation, using dosages up to 40 mg/day for the first 2 days and gradually decreasing to a target dosage of 20–30 mg/day (Baker et al. 2003; Brooks et al. 2008). Although sedation and hypotension must be watched for in any individual patient, increased rates of those side effects were not seen in a study comparing the loading-dose strategy with conventional 10-mg/day dosing (Baker et al. 2003).
Typically, agitated patients are best treated with the rapid-dissolving preparation of olanzapine. Given its faster onset of action and the decreased risk of “cheeking” of medication, the rapid-dissolving tablet formulation is preferable to the conventional pill in the acute setting, particularly when some sedation is also needed. When patients are severely agitated, use of injectable olanzapine is often necessary. The intramuscular preparation has a rapid onset of action, similar to that of dissolvable tablets, as well as a certainty of delivery that is imperative in an emergency. The injectable preparation has been shown to be superior to placebo at doses of 10 mg and as effective as haloperidol, with significantly fewer side effects (Breier et al. 2002). However, case reports caution against use of olanzapine in conjunction with intramuscular lorazepam because of the risk of hypotension (Zacher and Roche-Desilets 2005).
For patients who require long-term treatment, olanzapine LAI can be used on a once-monthly administration schedule. Because of concerns regarding cases of delirium and even coma reported in early studies, patients receiving olanzapine LAI must be observed for 3 hours after the injection. This monitoring requirement has significantly limited the use of the LAI form.
Bipolar Disorder and Treatment-Resistant Depression
Before the introduction of second-generation antipsychotic medications, it was well known that traditional antipsychotic medications were useful in the treatment of mania. Findings from a study assessing the effects of olanzapine versus haloperidol on symptoms of schizoaffective disorder provided a rationale for studying olanzapine in bipolar disorder. Compared with the schizoaffective disorder patients who received haloperidol in this study, the patients who received olanzapine showed superior outcomes on many, but not all, measures (Tran et al. 1999).
The first controlled study of olanzapine in bipolar disorder was a 21-day comparison of olanzapine with placebo (Tohen et al. 1999). Significantly greater improvement in manic symptoms (as assessed by mean change from baseline to endpoint on Young Mania Rating Scale [YMRS] total score) was observed for patients taking olanzapine compared with those taking placebo. No difference was seen in the outcomes for depression. In this study, EPS were not more frequent in the olanzapine-treated patients than in the patients taking placebo (Tohen et al. 1999). These findings were confirmed in a second pivotal study that also showed an advantage for olanzapine over placebo (Tohen et al. 2000). An open-label follow-up (49 weeks) added valuable information, especially noting that decreases in YMRS scores continued. For the longer term, depression scores also improved. Importantly, for the patients who were exposed to olanzapine at a mean dosage of approximately 14 mg/day, no cases of tardive dyskinesia occurred (Sanger et al. 2001).
An important question of practical interest is how olanzapine compares with conventional mood stabilizers. In a double-blind trial conducted by Eli Lilly, olanzapine was compared with lithium in the maintenance treatment of bipolar disorder (Tohen et al. 2005). In the study, patients were stabilized on a combination of lithium and olanzapine and then randomly assigned to receive one or the other for 52 weeks. In the noninferiority analysis, olanzapine was shown to be as effective as lithium in preventing depression relapse, and in fact the olanzapine-treated patients had lower rates of mixed or manic symptom relapse than did the lithium-treated patients over the 52-week follow-up. Weight gain was higher in the olanzapine group (Tohen et al. 2005). Further studies confirmed the equivalent efficacy of olanzapine and the most widely used anticonvulsant mood stabilizer, divalproex (Tohen et al. 2002).
This line of research led to FDA approval of olanzapine for the treatment of manic symptoms. The mean dosage used in olanzapine monotherapy for mania is similar to that used in schizophrenia: 13 mg/day. For acute mania in agitated bipolar patients, intramuscular olanzapine has been shown to be effective (Meehan et al. 2001).
The treatment of bipolar depression is often complex. Monotherapy with antidepressants is associated with an increased risk of switching into mania. Olanzapine has been shown to be efficacious in both manic and—in combination with fluoxetine (OFC)—depressive phases of bipolar illness (see Schulz and Cornelius 2010 for a review). In an 8-week double-blind trial conducted by Eli Lilly, OFC was compared against olanzapine monotherapy and placebo in patients with bipolar I disorder in a depressed phase. Although both treatments were more effective than placebo, OFC was significantly more effective than olanzapine in treating depressive symptoms. OFC-treated patients showed greater improvement in mood compared with olanzapine-treated patients by the fourth week of the study (Tohen et al. 2003). Subjects’ health-related quality of life improved as well (Shi et al. 2004). Brown et al. (2006) also compared OFC with lamotrigine in a 7-week study. Although OFC demonstrated a statistical separation from lamotrigine by the first week, it is difficult to make a full comparison in such a short study. Lamotrigine requires slow titration to minimize the risk of serious rash; thus, it was received at the target dosage (200 mg/day) only for the last 2 weeks of the study, whereas the OFC dosage could be titrated to therapeutic levels much more quickly. Although the rapid titration of OFC is helpful when a more urgent approach is required, further study is needed to determine whether OFC’s greater benefits versus olanzapine monotherapy persist once lamotrigine has had an opportunity to remain at a therapeutic dose for a longer period of time. Rates of treatment-emergent mania with OFC were low and did not significantly differ from rates with placebo or olanzapine monotherapy (Amsterdam and Shults 2005; Tohen et al. 2003).
In bipolar I adolescents, Tohen et al. (2007) reported significantly greater improvement in mania ratings with olanzapine than with placebo. Scientists at Eli Lily demonstrated that OFC was superior to placebo in bipolar adolescents in a double-blind randomized controlled trial (Detke et al. 2015). Side effects were similar to those in adults, although the magnitude of changes in lipids was somewhat higher and that of changes in glucose somewhat lower. Both olanzapine and OFC have been FDA approved for bipolar depression in adolescents.
OFC has been studied in treatment-refractory major depressive disorder. Thase et al. (2007) conducted a study comparing OFC with olanzapine or fluoxetine monotherapy in patients who had not shown adequate response to at least two prior trials of antidepressants. In the pooled analysis (Thase et al. 2007), OFC produced greater improvement versus olanzapine monotherapy or fluoxetine monotherapy (as assessed by scores on the Montgomery-Åsberg Depression Rating Scale [(MADRS]). In a double-blind trial sponsored by Eli Lilly, olanzapine, fluoxetine, OFC, and venlafaxine showed similar rates of efficacy (Corya et al. 2006).
The second-generation antipsychotic medications have found a role in treating not only mania but also depression in bipolar disorder. Olanzapine monotherapy has demonstrated efficacy in both acute and maintenance phases of bipolar disorder, and when combined with fluoxetine in the OFC formulation, it has shown benefit in treating depression in bipolar I disorder. Particularly in cases of mania in which psychosis is prominent, olanzapine is a reasonable first-line agent, although consideration must be given to the potential for metabolic consequences. In depression, olanzapine has been studied primarily in—and is FDA approved only for—treatment-refractory illness. Given the drug’s metabolic side-effect profile, it is appropriate to restrict its use to cases of resistant depression.
Off-Label Uses
Dementia-Related Agitation and Psychosis
Olanzapine does not have an FDA-approved indication for the treatment of dementia. Because elderly people are generally more sensitive to the EPS and tardive dyskinesia associated with first-generation antipsychotic medications, the second-generation medications are often preferred when antipsychotics are needed.
A large placebo-controlled trial of olanzapine in Alzheimer’s patients showed that the lower dosages of olanzapine (5–10 mg/day) were significantly better than placebo in treating target symptoms of agitation, hallucinations, and delusions (Street et al. 2000, 2001). The FDA placed a black box warning on the prescribing information of antipsychotic medications calling attention to the increased risk of death, primarily from cardiovascular and infectious complications. According to the warning, second-generation antipsychotic use over a 10-week period carries a 1.6- to 1.7-fold increased risk of mortality based on data from 17 placebo-controlled trials of second-generation antipsychotics in dementia-related psychosis. Ultimately, clinical judgment and thorough documentation are important, as in certain situations the hazards of untreated psychotic agitation may outweigh the potential risks of treatment.
Several studies have examined olanzapine in the treatment of dementia without agitation (Brooks and Hoblyn 2007). A placebo-controlled multicenter trial conducted by researchers at Eli Lilly evaluated olanzapine at low fixed dosages (1.0, 2.5, 5.0, and 7.5 mg/day) in the treatment of dementia-related psychosis (De Deyn et al. 2004). Although olanzapine did not separate from placebo on the primary outcome measure, Hallucinations and Delusions items of the Neuropsychiatric Inventory—Nursing Home edition (NPI/NH), improvements were seen in each of the dosage groups studied. All patients who received dosages of 2.5 mg/day or greater were initially started on 2.5 mg/day, with the dosage titrated upward by 2.5 mg/week (as indicated based on their assigned study group), and there was an overall difference from placebo in the acute phase of the study, suggesting that a 2.5-mg dose was an effective starting dose in the more acute setting. On some secondary outcome measures, the greatest improvement was seen with the highest olanzapine dosage (7.5 mg/day), suggesting that for some patients, an increase to 7.5 mg/day is beneficial. Because no higher dosages were used in the study, it is unclear whether continuing to increase the dosage would lead to greater efficacy (De Deyn et al. 2004).
Acetylcholine has been the focus of treatments aimed at slowing the rate of cognitive deterioration among individuals with dementia. Cholinesterase inhibitors have been used on that basis. Olanzapine may have beneficial effects on prefrontal cortex cholinergic and serotonergic neurons that may facilitate acetylcholine release to that region. However, in a double-blind study conducted by researchers at Eli Lilly, olanzapine was shown to worsen cognitive functioning, as assessed on the Alzheimer’s Disease Assessment Scale for Cognition (ADAS-Cog), and there was no statistical difference between the olanzapine and placebo groups in scores on the Clinician’s Interview-Based Impression of Change (CIBIC) scale (Kennedy et al. 2005). Patients in the olanzapine group also showed cognitive worsening on the Mini-Mental State Examination (MMSE). Previous studies found little or no benefit on cognition from olanzapine treatment in nonagitated patients with dementia (De Deyn et al. 2004; Street et al. 2000).
The CATIE study also had an Alzheimer’s disease component in which olanzapine, risperidone, and quetiapine were compared with placebo in the treatment of psychosis and agitation in outpatients (Schneider et al. 2006b). Patients were included if they had psychotic symptoms and resided either in an assisted living facility or at home, but they were excluded if they had skilled nursing needs or primary psychotic disorders. Patients who were to receive cholinesterase inhibitors or antidepressants were also excluded from the study. As in the schizophrenia portion of CATIE, the primary outcome variable was time to discontinuation. No difference was found among the groups in time to discontinuation, and no benefit was seen on the Alzheimer’s Disease Cooperative Study—Clinical Global Impression of Change (ADCS-CGIC). The average time to discontinuation ranged from 5 to 8 weeks among the treatments. Discontinuation due to lack of efficacy occurred sooner for patients receiving placebo or quetiapine than for those receiving risperidone or olanzapine. Side effects such as parkinsonism, sedation, and increased body mass index occurred more frequently with the study medications than with placebo (Schneider et al. 2006b).
Overall, there are limited data to support the effectiveness of second-generation antipsychotics in the treatment of dementia, and the available evidence does not support olanzapine as the first choice in this medication class. Risks for worsened cognitive function and metabolic concerns must be considered when use of antipsychotic medications is contemplated. Nonetheless, there are times when behavioral consequences and patient safety require more aggressive treatment, and antipsychotic medication may be warranted. Olanzapine is considered an intermediate-risk antipsychotic in this population (Kales et al. 2014). Ultimately, a painstaking evaluation of the risk–benefit ratio of antipsychotic medication use must precede any decision to prescribe these agents, in both the acute and the long-term time frames. Further study is needed, however, regarding the use of second-generation antipsychotic medications in this population (Schneider et al. 2006a).
Borderline Personality Disorder
Borderline personality disorder is a severe psychiatric illness that afflicts nearly 1% of the population (Torgersen et al. 2001). Based on earlier studies indicating that low dosages of traditional antipsychotic medications might be useful for psychosis spectrum and overall symptoms in borderline personality disorder (Goldberg et al. 1986; Soloff et al. 1986), Schulz et al. (1999) conducted an open-label study and found that olanzapine led to substantial improvement in psychoticism, depression, interpersonal sensitivity, and anger (as measured by Hopkins Symptom Checklist–90 ratings), as well as improvement on objective measures of impulsivity and aggression. Of the 11 women enrolled, 9 (82%) completed the study. The design of the trial allowed for early flexible dosing, and the subjects ended the 8-week trial taking olanzapine at an average dosage of approximately 7.5 mg/day, usually at bedtime. In an extension of this open-label trial, Zanarini and Frankenburg (2001 showed olanzapine’s superiority to placebo over a longer (26-week) study period. This interesting study indicated that lower dosages (5 mg/day) of olanzapine can be useful in this patient population.
In a study comparing olanzapine, fluoxetine, and OFC in women with borderline personality disorder, olanzapine monotherapy was found to be more effective (as assessed on the MADRS) than either fluoxetine or OFC in treating the depressive symptoms of the disorder. Additionally, olanzapine was superior to fluoxetine in treating symptoms of impulsivity and aggression, as measured by the Overt Aggression Scale (OAS). Weight gain was seen in a greater percentage of olanzapine-treated patients than of fluoxetine-treated patients (Zanarini et al. 2004).
Two large placebo-controlled studies evaluating olanzapine in borderline personality disorder yielded mixed results. Schulz et al. (2008) reported no significant advantage for olanzapine compared with placebo in a flexible dosing design, whereas Zanarini et al. (2011) noted a statistical advantage for higher-dosage olanzapine in a fixed-dose trial comparing low-dosage olanzapine, higher-dosage olanzapine, and placebo for 12 weeks. In an open-label 12-week extension phase of the same study, Zanarini et al. (2012) found that patients who had been treated with olanzapine in the initial study maintained and continued the modest improvements in their symptoms, while patients who were treated with placebo in the initial 12 weeks and started on olanzapine in the extension phase achieved a level of symptom relief similar to that of those who had started with olanzapine. These mixed results have led to controversy in the field about the potential use of antipsychotic medications in borderline personality disorder.
Dialectical behavioral therapy (DBT) is a mainstay of current treatment for borderline personality disorder. In a double-blind, placebo-controlled trial, olanzapine was studied as an adjunctive agent in patients receiving DBT. Impulsive and aggressive behaviors were found to be lower in the group that received olanzapine than in the placebo group. The average olanzapine dosage in the trial was 8.8 mg/day. Statistically significant increases in weight gain and dyslipidemia were observed in the olanzapine group compared with the placebo group (Soler et al. 2005). Therefore, with consideration for side effects, olanzapine may be helpful for a broader range of illnesses, particularly when used in conjunction with psychotherapy.
Anorexia Nervosa
Anorexia nervosa is a common and severe psychiatric illness that may well have the highest mortality rate of any mental disorder. Severe restriction of food intake, leading to low weight, is a primary feature of the illness; however, patients also have psychotic-like disturbances in self-perceived body size or shape, as well as unusual ideas about food and metabolism. Some investigators have begun to explore the possibility that olanzapine may help with this patient group. Initial reports were largely from pilot studies, including case series, but data are now emerging from small controlled trials.
In an open-label trial, 17 patients hospitalized for anorexia nervosa were given olanzapine in conjunction with concurrent cognitive-behavioral therapy (CBT) and DBT group treatment (Barbarich et al. 2004). Olanzapine was initiated at a dosage of 1.25–5.00 mg/day, with upward titration as needed, balancing sedation and side effects against efficacy. Although patients showed improvement in weight as well as in Beck Depression Inventory (BDI) and Spielberger State-Trait Anxiety Inventory (STAI) scores, the lack of a control group limited the validity of these results (Barbarich et al. 2004). A trial of 15 women with anorexia nervosa randomly assigned to either olanzapine or chlorpromazine in a balanced block design found that olanzapine reduced anorexic ruminations (as measured by the impaired control over mental activities subscale of the Padua Inventory). There was no difference in weight gain between the two groups. However, this study was somewhat limited by its lack of blinding (Mondraty et al. 2005). A case series evaluating low-dosage olanzapine treatment in 13 adolescent girls with restricting-type anorexia nervosa (Leggero et al. 2010) found improvements in weight and reductions in hyperactivity in the 7 girls who were olanzapine responders (defined as improvement of at least 50% in Eating Attitudes Test–26 scores).
Results of randomized double-blind, placebo-controlled trials have been mixed. In a 10-week trial of 34 women with anorexia nervosa, Bissada et al. (2008) found significant increases in weight and reductions in obsessive symptoms among those treated with olanzapine. Similarly, in an 8-week study of 23 outpatient women with anorexia nervosa, end-of-treatment body mass index was greater in women receiving olanzapine as compared with placebo. Psychological symptoms improved equally in both groups (Attia et al. 2011). However, a trial of olanzapine versus placebo in 20 adolescent girls with anorexia found no difference in median body weight from baseline at either week 5 or week 10. The two groups showed similar improvements in eating attitudes, psychological functioning, and resting energy expenditure (Kafantaris et al. 2011). A planned fourth clinical trial of adolescent girls was discontinued owing to inability to adequately recruit subjects, primarily because potential subjects did not meet study criteria (71% of those screened) and eligible subjects declined participation due to concerns about medication use (74% of those eligible) (Norris et al. 2010).
Given that weight gain is a prominent side effect of olanzapine, studies have begun to examine the mechanisms underlying this effect and the possibility that olanzapine might be useful as a weight-gain agent. In a double-blind, placebo-controlled trial investigating whether olanzapine might induce weight gain through modulation of ghrelin and leptin (hormones associated with satiety), patients with anorexia received olanzapine concurrently with CBT, with levels of ghrelin and leptin assessed over 3 months. Although both the olanzapine patients and the placebo patients gained weight, there was no statistical difference between the groups in amount of weight gained or in leptin or ghrelin levels, which remained unchanged over the course of the study (Brambilla et al. 2007).
The role of olanzapine as an augmentation to psychotherapy in anorexia nervosa is limited as best. The majority of studies using the most rigorous methods did not find psychological improvement for patients after olanzapine augmentation. However, in patients for whom timely weight gain is medically imperative, there may be a limited role for olanzapine. Trials have been small, with mixed results; further research is required to clarify the potential benefits and risks for patients.
Posttraumatic Stress Disorder
Posttraumatic stress disorder (PTSD) is characterized by disproportionate and disturbing symptoms of fear persisting for longer than 1 month after a severe trauma. For some patients, psychosis can occur, leading to intense feelings of horror and helplessness. This phenomenon led investigators to consider a potential role for olanzapine in a broader treatment plan for PTSD patients.
A limited number of small studies have examined olanzapine’s potential utility in the treatment of PTSD. In a double-blind, placebo-controlled augmentation trial of olanzapine in 19 patients with PTSD who were minimally responsive to 12 weeks of SSRI treatment, olanzapine produced limited symptomatic improvements compared with placebo on measures of PTSD, depression, and sleep (Stein et al. 2002). However, overall clinical improvement with olanzapine was no different from that with placebo, and patients receiving olanzapine had an average weight gain of 13 pounds. In a 6-week open-trial comparison of olanzapine versus fluphenazine as monotherapy in male patients with combat-related PTSD and psychosis, olanzapine was significantly more efficacious than fluphenazine in reducing symptoms on the PANSS (negative, general, psychopathology subscale, supplementary items), Watson’s PTSD subscales (avoidance, increased arousal), and a variety of global impression scales (Clinical Global Impression Severity Scale [CGI-S], Clinical Global Impression Improvement Scale [CGI-I], Patient Global Impression Improvement Scale [PGI-I]) when evaluated at 3 and 6 weeks. The two medications produced similar improvements on PANSS positive and Watson’s trauma re-experiencing subscale scores. An additional 3 weeks of treatment did not increase the efficacy of either drug (Pivac et al. 2004). In a small double-blind, randomized, placebo-controlled trial in patients with non-combat-related PTSD, olanzapine-treated patients demonstrated significantly greater improvement on the Clinician Administrated PTSD Scale over 8 weeks of treatment compared with those treated with placebo (Carey et al. 2012).
Obsessive-Compulsive Disorder
Obsessive-compulsive disorder (OCD) is characterized by recurrent, unwanted, and anxiety-inducing thoughts and repetitive behaviors that a person feels driven to perform in response to those thoughts. At times, obsessive thoughts can become sufficiently divorced from reality that they resemble or overlap with psychosis.
A few double-blind, placebo-controlled clinical trials have examined olanzapine’s potential efficacy in augmentation of traditional SSRI treatment in OCD patients. The first such trial enrolled 26 patients who had not responded to traditional SSRI therapy. Patients received olanzapine or placebo augmentation for 6 weeks, with biweekly assessment on the Yale-Brown Obsessive Compulsive Scale (Y-BOCS). Six (46%) of the 13 olanzapine-treated patients showed symptom response (defined as a 25% or greater reduction on Y-BOCS scores), compared with none of the placebo-treated patients (Bystritsky et al. 2004). In the second trial, partial responders or nonresponders to fluoxetine received an additional 6 weeks of either olanzapine or placebo augmentation. Both groups improved, but there were no differences between groups in the extent or timing of improvement as measured by Y-BOCS scores (Shapira et al. 2004). Findings of more recent studies provide further confirmation that olanzapine is effective in only a limited set of OCD patients, such as those with primarily hoarding or symmetry-based symptoms; the data do not support the drug’s first-line use in OCD overall (Matsunaga et al. 2009). Given the mixed results and small sample sizes of the existing studies, it is clear that further research is needed to establish the benefit, if any, of olanzapine augmentation in the treatment of OCD patients.
Side Effects and Toxicology
Neurological and Extrapyramidal Adverse Effects
Adverse effects of olanzapine reported in clinical use are consistent with findings in preclinical studies, which predicted few neurological effects. In Phase II and III clinical trials, olanzapine-treated patients generally showed an improvement in EPS from baseline, reflecting the fact that most of the subjects had previously taken first-generation antipsychotics. In a large multinational comparison study (Tollefson et al. 1997), olanzapine produced fewer treatment-emergent neurological adverse effects than haloperidol, including parkinsonism (14% vs. 38%) and akathisia (12% vs. 40%). In a comprehensive meta-analysis, the relative risk (RR) of EPS for olanzapine was 0.39 compared with haloperidol (Leucht et al. 2009).
Antiparkinsonian medication is sometimes required when patients are treated with olanzapine, although the need for such medication may be lower with olanzapine than with antipsychotics having greater potency at the dopamine D2 receptor. In one study (Volavka et al. 2002), antiparkinsonian agents were prescribed to 13% of both clozapine-treated subjects and olanzapine-treated subjects, compared with 32% of risperidone-treated patients. In a meta-analysis of randomized controlled trials, Komossa et al. (2010) found that olanzapine incurred a modestly higher risk of EPS (as measured by use of antiparkinsonian medication) compared with quetiapine (RR=2.05) but a lower EPS risk compared with risperidone (RR=0.78) and ziprasidone (RR=0.70).
Evidence now exists for phenotypic variation in the risk of EPS among olanzapine-treated patients. A meta-analysis of 4,831 subjects compared the incidence of olanzapine-induced EPS in schizophrenia versus bipolar patients. Parkinsonism was significantly higher among schizophrenia patients (13.9% vs. 3.1%), and this difference remained significant after adjustment for gender. There were no differences between groups on measures of akathisia or use of antiparkinsonian medication (Moteshafi et al. 2012).
The reduction of EPS is predictive of decreased risk of tardive dyskinesia, the most problematic of the common adverse effects of classic antipsychotics. The accumulated experience with second-generation antipsychotics indicates that tardive dyskinesia is 10- to 15-fold less common with these agents than with conventional agents, with an annual rate of 0.52% for olanzapine-treated patients compared with 7.45% for haloperidol-treated patients, based on pooled data from long-term comparison trials (Beasley et al. 1999). Miller et al. (2008) examined data from the previously described CATIE study. After exclusion of subjects who met criteria for tardive dyskinesia at baseline, there was no difference in incidence of tardive dyskinesia over 1 year between patients treated with olanzapine and patients treated with any other antipsychotic (perphenazine, quetiapine, risperidone, or ziprasidone).
Weight Gain and Other Metabolic Effects
Weight gain and associated dyslipidemia are among the most significant major adverse effects found during treatment with olanzapine. Weight gain is a serious concern, because persons with schizophrenia are more likely than the general population to be obese, and weight gain may contribute to nonadherence to antipsychotic treatment, leading to increased risk for relapse. With the reduced risk of neurological side effects attached to second-generation antipsychotic agents, metabolic effects have emerged as a major risk for patients and a focus of consideration for clinicians.
The relative degree of weight gain associated with first- and second-generation antipsychotics was studied in a comprehensive meta-analysis by Allison et al. (1999). Estimates of weight change associated with standardized dosages over 10 weeks were calculated from published data on 81 studies. Clozapine produced the greatest weight gain (4.45 kg), followed by olanzapine (4.15 kg). By comparison, risperidone was associated with a gain of 2.1 kg and haloperidol was associated with a gain of 1.08 kg, whereas patients lost 0.74 kg while taking placebo. In long-term treatment, 30%–50% of patients gain more than 7% of body weight, with low pretreatment weight and good clinical response associated with more weight gain (Russell and Mackell 2001). In a large database of British patients, use of olanzapine conferred a fivefold increase in the risk of dyslipidemia over an untreated control condition and a threefold increase over conventional antipsychotics, whereas risperidone did not increase the risk (Koro et al. 2002b).
The risk for metabolic dysfunction appears to be present even before patients receive antipsychotics, and risk is further compounded by antipsychotic medication use. In a sample of 404 patients with first-episode psychosis enrolled in community health centers, patients with no previous exposure to antipsychotics had lower levels of non–high-density lipoprotein (HDL) cholesterol but were more likely to carry diagnoses of obesity and hypertension compared with patients with any lifetime exposure to antipsychotic medication (average exposure = 47.3 days) (Correll et al. 2014). Furthermore, longer duration of antipsychotic treatment was associated with greater abnormalities in triglycerides and cholesterol (both HDL and non-HDL) and lower systolic blood pressure. In this study, which allowed treatment with any antipsychotic or no medication, patients receiving olanzapine had a significantly increased risk for elevated triglycerides, insulin, and homeostasis model assessment for insulin resistance (HOMA-IR) compared with patients receiving other antipsychotic medications (Correll et al. 2014).
Studies using metformin, an agent known to decrease hepatic glucose output, have tested its potential to help patients either lose weight or remain at the same weight while receiving olanzapine or other second-generation antipsychotics. In a double-blind, placebo-controlled trial (Baptista et al. 2006), patients were given 10 mg of olanzapine and randomly assigned to receive either metformin or placebo for 14 weeks. No differences between groups were seen in body mass index or waist circumference. There was a modest improvement in overall glucose levels and in HOMA-IR, but no change was seen in lipid levels. A follow-up study conducted by the same group (Baptista et al. 2007) demonstrated similar results, although in the second study, small differences in weight gain between the groups were found, with the metformin group losing an average of 1.5 kg and showing decreased leptin levels, whereas the placebo group maintained a consistent weight.
Investigators from the Use of Metformin in the Treatment of Antipsychotic-Induced Weight Gain in Schizophrenia (METS) Study randomly assigned 148 clinically stable overweight outpatients with schizophrenia or schizoaffective disorder to up to 2,000 mg of metformin or placebo daily (Jarskog et al. 2013). Patients were taking either one or two of any of the FDA-approved antipsychotic medications. All patients received weekly diet and exercise counseling. After 16 weeks, metformin-treated patients had lost significantly more weight than placebo-treated patients (6.6 lbs. vs. 2.2 lbs.). The metformin group also showed significant reductions in triglyceride levels (−7.0 mg/dL vs. +13.2 mg/dL) as well as modest reductions in hemoglobin A1c, a measure of elevated blood glucose (−0.06% vs. +0.011%), with a time-by-treatment interaction, suggesting that the benefits of metformin increase with time. There were no significant differences in fasting glucose or insulin levels (Jarskog et al. 2013).
In a double-blind, placebo-controlled trial in adolescents who had gained weight after 1 year of treatment with a second-generation antipsychotic (olanzapine, risperidone, or quetiapine), the addition of metformin resulted in statistical reductions in waist circumference and body mass index and stabilization of weight gain (Klein et al. 2006). HOMA-IR scores were significantly decreased, and the number of subjects requiring referral for a glucose tolerance test was reduced, among the subjects who received metformin.
Interestingly, a recent small study of first-episode schizophrenia patients started on olanzapine found that daily supplementation with melatonin attenuated increases in weight and abdominal obesity as compared with olanzapine and placebo, and additionally improved scores on the PANSS. There was also a trend for attenuation of hypertriglyceridemia (Modabbernia et al. 2014).
Weight gain is an even greater concern in the treatment of children and adolescents, who may be exposed to medication for a longer time than adults. In a study evaluating weight gain among hospitalized adolescents after 12 weeks of treatment with olanzapine (n=21), risperidone (n=21), or haloperidol (n=8), patients in the olanzapine group gained an average of 7.2±6.3 kg, approximately twice the amount gained in the risperidone group (3.9±4.8 kg) and more than 6 times the amount gained in the haloperidol group (1.1±3.3 kg) (Ratzoni et al. 2002). In a double-blind multisite trial comparing first- and second-generation antipsychotic medications among children and adolescents, the olanzapine arm was discontinued because of weight gain without evidence of improved efficacy over risperidone or molindone (Sikich et al. 2008). A further study of olanzapine in adolescents demonstrated a statistically significant difference in symptom improvement ratings for olanzapine over placebo. As in previous studies, significantly more weight gain occurred in the adolescents on olanzapine (Kryzhanovskaya et al. 2009b).
Changes in weight, lipids, and insulin metabolism raise serious concerns for diabetes and related complications. Cases reported to the FDA Drug Surveillance System and published cases of olanzapine-associated diabetes and hyperglycemia were reviewed by Koller and Doraiswamy (2002). Two hundred eighty-nine cases were identified, of which 225 (78%) involved new-onset diabetes and 100 (35%) involved ketosis or acidosis; in 23 (8%) cases, the patient died. Most cases developed within 6 months of initiation of olanzapine therapy. Many cases occurred during the first month of therapy, indicating that weight gain alone did not mediate the occurrence of diabetes-related problems. On the basis of the temporal relation between the appearance of metabolic changes and the introduction and withdrawal of olanzapine, the young age of patients affected, and the number of reports, the authors concluded that the data suggested that olanzapine was causally related to the development or worsening of diabetes. A similar conclusion about clozapine and diabetes had been reported previously (Koller et al. 2001). Because case studies and reports by clinicians to regulatory agencies may reflect reporting bias, controlled studies comparing the development of metabolic disorders are needed to clarify whether these are related to the underlying psychosis, causally related to drug treatment in general, or specifically related to individual agents.
Studies using large health system databases have linked antipsychotic treatment with subsequent diagnoses of diabetes or use of hypoglycemic agents. These studies show an increased risk of development of type 2 diabetes among individuals using olanzapine or clozapine relative to those using risperidone or typical antipsychotics or compared with matched untreated persons (Gianfrancesco et al. 2002; Koro et al. 2002a; Sernyak et al. 2002).
In addition to olanzapine’s association with weight gain and type 2 diabetes, it has also been linked to diabetic ketoacidosis (DKA), a condition more often associated with type 1 than type 2 diabetes mellitus. Reports of this connection first appeared in the literature in 1999 (Gatta et al. 1999; Goldstein et al. 1999; Lindenmayer and Patel 1999). In a review of California Medicaid data on cases of risperidone- and olanzapine-associated DKA, Ramaswamy et al. (2007) found a higher incidence of DKA with olanzapine treatment than with risperidone treatment and noted that the risk increased with duration of treatment with olanzapine. By contrast, a multicenter retrospective cohort study using administrative health data (encompassing the records of 725,489 patients) from Canada and the United Kingdom (Lipscombe et al. 2014) found no increased risk of hyperglycemic emergency associated with initiation of olanzapine versus risperidone.
Other Side Effects
Among other side effects reported with olanzapine, sedation is frequent at the start of therapy but diminishes as patients develop tolerance for this side effect. In long-term treatment, the incidence of sedation is about 15%, similar to that of haloperidol. Prolactin elevations observed during olanzapine treatment occur early in the course of treatment, and levels are much lower than those seen with risperidone or typical antipsychotics. Leukopenia is rare and occurs at a rate similar to that seen with other first- and second-generation antipsychotics, but olanzapine does not cause agranulocytosis, even in patients who developed this effect while taking clozapine. In animal toxicology studies and in clinical trials of olanzapine, no QTc prolongation was observed, and other cardiovascular effects are rarely of clinical importance (Cadario 2000).
Safety in Overdose
Reviews of case series and reports of olanzapine toxicity in adults describe predominantly circulatory and central nervous system effects, including coma, psychomotor agitation, hypertension and hypotension, tachycardia and bradycardia, hyperthermia and hypothermia, miosis and mydriasis, and QTC prolongation (QTC prolongation only with >1,040-mg ingestion and in mixed overdoses) (Ciszowski et al. 2011; Tan et al. 2009). Olanzapine overdose was rarely fatal in the aforementioned case reports, and a systematic study of second-generation antipsychotic overdose found that of 422 olanzapine overdoses, there were no fatalities and 14% required ventilation (Berling et al. 2016). There are very few data describing olanzapine overdose in children. Available case report data are consistent with adult presentations of EPS and elevated prolactin levels (Tanoshima et al. 2013) and circulatory and neurological symptoms (McAllister et al. 2012; Singh et al. 2012; Theisen et al. 2005).
Use in Pregnancy
Exposure to second-generation antipsychotics was associated with gestational diabetes in a Swedish birth cohort study (Bodén et al. 2012). The risk was similar for medications known to have severe anabolic effects (olanzapine and clozapine) and other antipsychotic medications. Fetuses exposed to antipsychotic medication in utero were more likely than those not exposed to be born small for gestational age, but this association was not robust to adjustment for maternal factors. Because of the difficulty in studying medication use during pregnancy, data on antipsychotic use during pregnancy are extremely sparse. No specific fetal abnormalities have been reported with olanzapine, although there is some evidence suggesting an association between antipsychotic use in pregnancy and neonatal respiratory distress and withdrawal symptoms (Kulkarni et al. 2015). Further research is needed to aid clinicians in maximizing infant and maternal well-being.
Drug–Drug Interactions
Olanzapine is metabolized primarily via glucuronidation and via oxidation by CYP1A2 (see section “Pharmacokinetics and Disposition” earlier in this chapter). Other drugs that affect the activity of these metabolic pathways would therefore be expected to affect olanzapine pharmacokinetics. Indeed, drugs that inhibit CYP1A2 activity have been shown to decrease olanzapine clearance, thereby increasing olanzapine plasma concentrations.
Fluvoxamine, a known inhibitor of CYP1A2, has been shown to inhibit olanzapine metabolism in several studies. In an 11-day study of 10 healthy male smokers, coadministration of fluvoxamine (50–100 mg/day) with olanzapine (2.5–7.5 mg/day for 8 days [beginning on day 4]) resulted in an 84% increase in olanzapine peak serum concentrations (Cmax) and a 119% increase in AUC0–24 compared with placebo coadministered with olanzapine. No change in half-life was observed in either olanzapine or 4′-N-desmethylolanzapine, suggesting that fluvoxamine inhibited olanzapine’s first-pass metabolism (Maenpaa et al. 1997).
Fluoxetine and imipramine, although not known to be significant inhibitors of CYP1A2, when coadministered with olanzapine have been associated with small but statistically significant changes in olanzapine pharmacokinetics. Coadministration of fluoxetine with olanzapine resulted in a 15% decrease in olanzapine clearance and an 18% increase in Cmax, with no significant difference in olanzapine half-life (Callaghan et al. 1999). Coadministration of imipramine with olanzapine resulted in an approximately 14% increase in olanzapine Cmax and a non–statistically significant increase in olanzapine AUC of 19% (Callaghan et al. 1997).
Inducers of the CYP1A2 enzyme increase olanzapine clearance, thereby decreasing olanzapine systemic exposure. Carbamazepine, an inducer of several CYP enzymes (including 1A2), affects olanzapine disposition. A study in healthy volunteers reported that a single 10-mg dose of olanzapine taken after 2 weeks of pretreatment with carbamazepine (200 mg twice daily) had significantly higher clearance and apparent volume of distribution—but significantly lower Cmax, AUC, and half-life—compared with a single 10-mg dose taken before carbamazepine pretreatment (Lucas et al. 1998).
Smoking, also known to induce CYP1A2, can affect olanzapine disposition. A study comparing 19 male smokers with 30 male nonsmokers found that olanzapine clearance in smokers was 23% higher than that in nonsmokers (Callaghan et al. 1999). A population pharmacokinetic analysis of 910 patients receiving olanzapine found that clearance among nonsmokers was 37% lower in men and 48% lower in women than it was in the corresponding group of smokers (Patel et al. 1996). A smaller analysis of healthy volunteers also found higher drug clearances among smokers (Patel et al. 1995). The polycyclic aromatic hydrocarbons in cigarette smoke are responsible for inducing the aryl hydrocarbon hydroxylases, thereby leading to enzymatic induction (Desai et al. 2001). For this reason, dosage adjustments may be needed when a patient who smokes is placed in a smoke-free inpatient unit, even if adequate nicotine replacement is provided. Conversely, when there is a sudden increase in cigarette consumption (often associated with stress or impending relapse), a dosage increase may be needed to prevent an abrupt worsening of symptoms.
In vitro studies suggest that olanzapine does not significantly inhibit the activity of CYP 1A2, 3A, 2D6, 2C9, or 2C19 (Ring et al. 1996a). In vivo studies suggest that olanzapine does not affect the disposition of aminophylline (Macias et al. 1998), diazepam, alcohol, imipramine (Callaghan et al. 1997), warfarin, biperiden, or lithium (Callaghan et al. 1999; Demolle et al. 1995).
Conclusion
From review of the research focused on olanzapine, it is clear that this compound, which has been approved for use in the United States since 1997, has wide utility and some distinct advantages compared with traditional antipsychotics. In addition to its positive effects on a broad group of symptoms in schizophrenia, olanzapine is approved for the treatment of mania, both acute and long term, in bipolar disorder. Recent research has shown that olanzapine may be beneficial for disorders beyond psychosis (e.g., borderline personality disorder, anorexia nervosa, PTSD, OCD). The extension of olanzapine’s uses is in many ways made possible by its low rate of movement disorders. Olanzapine’s low risk of dystonia, parkinsonism, and tardive dyskinesia has led to its greater acceptability in chronic schizophrenia and has encouraged clinicians to prescribe it for other conditions, although concerns about metabolic side effects limit the drug’s overall utility.
As noted in the earlier section “Side Effects and Toxicology,” olanzapine is not free of adverse effects. Weight gain and metabolic disturbances are of significant concern and are the focus of intense research in both pathophysiology and prevention/treatment. Because of these concerns, the FDA does not recommend olanzapine as a first-line agent in young people.
In addition to providing better treatment for schizophrenia and other disorders, olanzapine promotes actions in the brain that have revealed new directions for research on the pathophysiological mechanisms underlying psychiatric disease. As noted previously, investigation of olanzapine’s effects on glutamate, neurotensin, neuregulin, and other neurotransmitters may improve our understanding of the pathophysiology of these diseases and open new avenues of treatment.
References
Aichhorn W, Whitworth AB, Weiss EM, Marksteiner J: Second-generation antipsychotics: is there evidence for sex differences in pharmacokinetic and adverse effect profiles? Drug Saf 29(7):587–598, 2006 16808551
Allison DB, Mentore JL, Heo M, et al: Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry 156(11):1686–1696, 1999 10553730
Alvarez E, Ciudad A, Olivares JM, et al: A randomized, 1-year follow-up study of olanzapine and risperidone in the treatment of negative symptoms in outpatients with schizophrenia. J Clin Psychopharmacol 26(3):238–249, 2006 16702888
Amsterdam JD, Shults J: Comparison of fluoxetine, olanzapine, and combined fluoxetine plus olanzapine initial therapy of bipolar type I and type II major depression—lack of manic induction. J Affect Disord 87(1):121–130, 2005 15923042
Aravagiri M, Ames D, Wirshing WC, Marder SR: Plasma level monitoring of olanzapine in patients with schizophrenia: determination by high-performance liquid chromatography with electrochemical detection. Ther Drug Monit 19(3):307–313, 1997 9200772
Attia E, Kaplan AS, Walsh BT, et al: Olanzapine versus placebo for out-patients with anorexia nervosa. Psychol Med 41(10):2177–2182, 2011 21426603
Baker RW, Kinon BJ, Maguire GA, et al: Effectiveness of rapid initial dose escalation of up to forty milligrams per day of oral olanzapine in acute agitation. J Clin Psychopharmacol 23(4):342–348, 2003 12920409
Bakshi VP, Swerdlow NR, Braff DL, Geyer MA: Reversal of isolation rearing-induced deficits in prepulse inhibition by Seroquel and olanzapine. Biol Psychiatry 43(6):436–445, 1998 9532349
Baldwin DS, Montgomery SA: First clinical experience with olanzapine (LY 170053): results of an open-label safety and dose-ranging study in patients with schizophrenia. Int Clin Psychopharmacol 10(4):239–244, 1995 8748045
Baptista T, Martínez J, Lacruz A, et al: Metformin for prevention of weight gain and insulin resistance with olanzapine: a double-blind placebo-controlled trial. Can J Psychiatry 51(3):192–196, 2006 16618011
Baptista T, Rangel N, Fernández V, et al: Metformin as an adjunctive treatment to control body weight and metabolic dysfunction during olanzapine administration: a multicentric, double-blind, placebo-controlled trial. Schizophr Res 93(1–3):99–108, 2007 17490862
Barak Y, Shamir E, Zemishlani H, et al: Olanzapine vs. haloperidol in the treatment of elderly chronic schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry 26(6):1199–1202, 2002 12452546
Barbarich NC, McConaha CW, Gaskill J, et al: An open trial of olanzapine in anorexia nervosa. J Clin Psychiatry 65(11):1480–1482, 2004 15554759
Beasley CM Jr, Sanger T, Satterlee W, et al: Olanzapine versus placebo: results of a double-blind, fixed-dose olanzapine trial. Psychopharmacology (Berl) 124(1–2):159–167, 1996a 8935812
Beasley CM Jr, Tollefson G, Tran P, et al: Olanzapine versus placebo and haloperidol: acute phase results of the North American double-blind olanzapine trial. Neuropsychopharmacology 14(2):111–123, 1996b 8822534
Beasley CM Jr, Hamilton SH, Crawford AM, et al: Olanzapine versus haloperidol: acute phase results of the international double-blind olanzapine trial. Eur Neuropsychopharmacol 7(2):125–137, 1997 9169300
Beasley CM, Dellva MA, Tamura RN, et al: Randomised double-blind comparison of the incidence of tardive dyskinesia in patients with schizophrenia during long-term treatment with olanzapine or haloperidol. Br J Psychiatry 174:23–30, 1999 10211147
Benzel I, Bansal A, Browning BL, et al: Interactions among genes in the ErbB-Neuregulin signalling network are associated with increased susceptibility to schizophrenia. Behav Brain Funct 3:31, 2007 17598910
Bergstrom RF, Callaghan JT, Cerimele BJ, et al: Pharmacokinetics of olanzapine in elderly and young (abstract). Pharm Res 12 (9 suppl):S358, 1995
Berling I, Buckley NA, Isbister GK: The antipsychotic story: changes in prescriptions and overdose without better safety. Br J Clin Pharmacol 82(1):249–254, 2016 26945707
Bissada H, Tasca GA, Barber AM, Bradwejn J: Olanzapine in the treatment of low body weight and obsessive thinking in women with anorexia nervosa: a randomized, double-blind, placebo-controlled trial. Am J Psychiatry 165(10):1281–1288, 2008 18558642
Bodén R, Lundgren M, Brandt L, et al: Antipsychotics during pregnancy: relation to fetal and maternal metabolic effects. Arch Gen Psychiatry 69(7):715–721, 2012 22752236
Boules M, Shaw A, Fredrickson P, Richelson E: Neurotensin agonists: potential in the treatment of schizophrenia. CNS Drugs 21(1):13–23, 2007 17190526
Boules MM, Fredrickson P, Muehlmann AM, Richelson E: Elucidating the role of neurotensin in the pathophysiology and management of major mental disorders. Behav Sci (Basel) 4(2):125–153, 2014 25379273
Brambilla F, Monteleone P, Maj M: Olanzapine-induced weight gain in anorexia nervosa: involvement of leptin and ghrelin secretion? Psychoneuroendocrinology 32(4):402–406, 2007 17395395
Breier A, Meehan K, Birkett M, et al: A double-blind, placebo-controlled dose-response comparison of intramuscular olanzapine and haloperidol in the treatment of acute agitation in schizophrenia. Arch Gen Psychiatry 59(5):441–448, 2002 11982448
Brooks JO, Hoblyn JC: Neurocognitive costs and benefits of psychiatric medication in older adults: invited review. J Geriatr Psychiatry Neurol 20(4):199–214, 2007 18004007
Brooks JO, Karnik N, Hoblyn JC: High initial dosing of olanzapine for stabilization of acute agitation: a retrospective case series. J Pharm Technol 24(1):7–11, 2008 doi: 10.1177/875512250802400103
Brown EB, McElroy SL, Keck PE Jr, et al: A 7-week, randomized, double-blind trial of olanzapine/fluoxetine combination versus lamotrigine in the treatment of bipolar I depression. J Clin Psychiatry 67(7): 1025–1033, 2006 16889444
Bymaster FP, Calligaro DO, Falcone JF, et al: Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology 14(2):87–96, 1996 8822531
Bystritsky A, Ackerman DL, Rosen RM, et al: Augmentation of serotonin reuptake inhibitors in refractory obsessive-compulsive disorder using adjunctive olanzapine: a placebo-controlled trial. J Clin Psychiatry 65(4):565–568, 2004 15119922
Cadario B: Olanzapine (Zyprexa): suspected serious reactions. CMAJ 163(1):85–86, 89–90, 2000 10920744
Callaghan JT, Cerimele BJ, Kassahun KJ, et al: Olanzapine: interaction study with imipramine. J Clin Pharmacol 37(10):971–978, 1997 9505989
Callaghan JT, Bergstrom RF, Ptak LR, Beasley CM: Olanzapine. Pharmacokinetic and pharmacodynamic profile. Clin Pharmacokinet 37(3):177–193, 1999 10511917
Carey P, Suliman S, Ganesan K, et al: Olanzapine monotherapy in posttraumatic stress disorder: efficacy in a randomized, double-blind, placebo-controlled study. Hum Psychopharmacol 27(4):386–391, 2012 22730105
Chagnon YC, Roy MA, Bureau A, et al: Differential RNA expression between schizophrenic patients and controls of the dystrobrevin binding protein 1 and neuregulin 1 genes in immortalized lymphocytes. Schizophr Res 100(1–3): 281–290, 2008 18234478
Ciszowski K, Sein Anand J, Wilimowska J, Jawień W: [The clinical picture of acute olanzapine poisonings] (in Polish). Przegl Lek 68(8):426–433, 2011 22010430
Ciudad A, Olivares JM, Bousoño M, et al: Improvement in social functioning in outpatients with schizophrenia with prominent negative symptoms treated with olanzapine or risperidone in a 1 year randomized, open-label trial. Prog Neuropsychopharmacol Biol Psychiatry 30(8): 1515–1522, 2006 16820255
Conley RR, Tamminga CA, Bartko JJ, et al: Olanzapine compared with chlorpromazine in treatment-resistant schizophrenia. Am J Psychiatry 155(7):914–920, 1998 9659857
Correll CU, Robinson DG, Schooler NR, et al: Cardiometabolic risk in patients with first-episode schizophrenia spectrum disorders: baseline results from the RAISE-ETP study. JAMA Psychiatry 71(12):1350–1363, 2014 25321337
Corya SA, Williamson D, Sanger TM, et al: A randomized, double-blind comparison of olanzapine/fluoxetine combination, olanzapine, fluoxetine, and venlafaxine in treatment-resistant depression. Depress Anxiety 23(6):364–372, 2006 16710853
Coyle JT: Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol 26(4–6):365–384, 2006 16773445
De Deyn PP, Carrasco MM, Deberdt W, et al: Olanzapine versus placebo in the treatment of psychosis with or without associated behavioral disturbances in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 19(2):115–126, 2004 14758577
Delay J, Bernitzer P: Le traitment des psychoses par une methode neuroleptique derivee de l’hibernotherapie, in Congres de Medecins Alienistes et Neurologistes de France. Edited by Ossa PC. Paris, Masson, 1952, pp 497–502
Demolle D, Onkelinx C, Müller-Oerlinghausen B: Interaction between olanzapine and lithium in healthy male volunteers (abstract 486). Therapie 50 (suppl), 1995
Deng C, Pan B, Hu CH, et al: Differential effects of short- and long-term antipsychotic treatment on the expression of neuregulin-1 and ErbB4 receptors in the rat brain. Psychiatry Res 225(3):347–354, 2015 25576368
Desai HD, Seabolt J, Jann MW: Smoking in patients receiving psychotropic medications: a pharmacokinetic perspective. CNS Drugs 15(6):469–494, 2001 11524025
Detke HC, DelBello MP, Landry J, Usher RW: Olanzapine/fluoxetine combination in children and adolescents with bipolar I depression: a randomized, double-blind, placebo-controlled trial. J Am Acad Child Adolesc Psychiatry 54(3):217–224, 2015 25721187
Dulawa SC, Geyer MA: Psychopharmacology of prepulse inhibition in mice. Chin J Physiol 39(3):139–146, 1996 8955560
Eli Lilly: Zyprexa (olanzapine) tablets: prescribing information. Indianapolis, IN, Eli Lilly, 2015. Available at: http://pi.lilly.com/us/zyprexa-pi.pdf. Accessed August 2, 2016.
Essock SM, Covell NH, Davis SM, et al: Effectiveness of switching antipsychotic medications. Am J Psychiatry 163(12):2090–2095, 2006 17151159
Farah A: Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry 7(6):268–274, 2005 16498489
Farah A: Atypicality of atypical antipsychotics revisited. Curr Psych Reviews 9(4):316–324, 2013
Farde L, Nordström AL: PET analysis indicates atypical central dopamine receptor occupancy in clozapine-treated patients. Br J Psychiatry Suppl (17):30–33, 1992 1358126
Farde L, Nordström AL, Wiesel FA, et al: Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch Gen Psychiatry 49(7):538–544, 1992 1352677
Gareri P, De Fazio P, De Fazio S, et al: Adverse effects of atypical antipsychotics in the elderly: a review. Drugs Aging 23(12): 937–956, 2006 17154659
Gatta B, Rigalleau V, Gin H: Diabetic ketoacidosis with olanzapine treatment. Diabetes Care 22(6):1002–1003, 1999 10372259
Gianfrancesco FD, Grogg AL, Mahmoud RA, et al: Differential effects of risperidone, olanzapine, clozapine, and conventional antipsychotics on type 2 diabetes: findings from a large health plan database. J Clin Psychiatry 63(10):920–930, 2002 12416602
Goldberg SC, Schulz SC, Schulz PM, et al: Borderline and schizotypal personality disorders treated with low-dose thiothixene vs placebo. Arch Gen Psychiatry 43(7):680–686, 1986 3521531
Goldstein LE, Sporn J, Brown S, et al: New-onset diabetes mellitus and diabetic ketoacidosis associated with olanzapine treatment. Psychosomatics 40(5):438–443, 1999 10479950
Gosselin G, Oberling P, Di Scala G: Antagonism of amphetamine-induced disruption of latent inhibition by the atypical antipsychotic olanzapine in rats. Behav Pharmacol 7(8):820–826, 1996 11224476
Gossen D, de Suray JM, Vandenhende F, et al: Influence of fluoxetine on olanzapine pharmacokinetics. AAPS PharmSci 4(2):E11, 2002 12102620
Green MF: Cognitive impairment and functional outcome in schizophrenia and bipolar disorder. J Clin Psychiatry 67(10): e12, 2006 17107235
Grothe DR, Calis KA, Jacobsen L, et al: Olanzapine pharmacokinetics in pediatric and adolescent inpatients with childhood-onset schizophrenia. J Clin Psychopharmacol 20(2):220–225, 2000 10770461
Gruber SH, Angelucci F, Nomikos GG, Mathé AA: Effects of olanzapine on extracellular concentrations and tissue content of neurotensin in rat brain regions. Eur Neuropsychopharmacol 21(12):918–927, 2011 21316929
Gurpegui M, Alvarez E, Bousoño M, et al: Effect of olanzapine or risperidone treatment on some cognitive functions in a one-year follow-up of schizophrenia outpatients with prominent negative symptoms. Eur Neuropsychopharmacol 17(11):725–734, 2007 17543505
Guttmacher MS, Goldberg SC, Klerman GL: Phenothiazine in treatment of acute schizophrenia. Arch Gen Psychiatry 10(3):246–261, 1964 14089354
Hägg S, Spigset O, Lakso HA, Dahlqvist R: Olanzapine disposition in humans is unrelated to CYP1A2 and CYP2D6 phenotypes. Eur J Clin Pharmacol 57(6–7):493–497, 2001 11699614
Hawkins KA, Keefe RS, Christensen BK, et al: Neuropsychological course in the prodrome and first episode of psychosis: findings from the PRIME North America Double Blind Treatment Study. Schizophr Res 105(1–3):1–9, 2008 18774696
Hwang JP, Yang CH, Lee TW, Tsai SJ: The efficacy and safety of olanzapine for the treatment of geriatric psychosis. J Clin Psychopharmacol 23(2):113–118, 2003 12640211
Jardemark KE, Liang X, Arvanov V, Wang RY: Subchronic treatment with either clozapine, olanzapine or haloperidol produces a hyposensitive response of the rat cortical cells to N-methyl-D-aspartate. Neuroscience 100(1):1–9, 2000 10996453
Jarskog LF, Hamer RM, Catellier DJ, et al; METS Investigators: Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry 170(9):1032–1040, 2013 23846733
Jensen PS, Buitelaar J, Pandina GJ, et al: Management of psychiatric disorders in children and adolescents with atypical antipsychotics: a systematic review of published clinical trials. Eur Child Adolesc Psychiatry 16(2):104–120, 2007 17075688
Kafantaris V, Leigh E, Hertz S, et al: A placebo-controlled pilot study of adjunctive olanzapine for adolescents with anorexia nervosa. J Child Adolesc Psychopharmacol 21(3):207–212, 2011 21663423
Kales HC, Kim HM, Zivin K, et al: Risk of mortality among individual antipsychotics in patients with dementia. Am J Psychiatry 169(1):71–79, 2014 22193526
Kane JM, Detke HC, Naber D, et al: Olanzapine long-acting injection: a 24-week, randomized, double-blind trial of maintenance treatment in patients with schizophrenia. Am J Psychiatry 167(2): 181–189, 2010 20008947
Kantrowitz JT, Malhotra AK, Cornblatt B, et al: High dose D-serine in the treatment of schizophrenia. Schizophr Res 121(1–3): 125–130, 2010 20541910
Kapur S, Remington G: Dopamine D(2) receptors and their role in atypical antipsychotic action: still necessary and may even be sufficient. Biol Psychiatry 50(11): 873–883, 2001 11743942
Kapur S, Zipursky RB, Remington G, et al: 5-HT2 and D2 receptor occupancy of olanzapine in schizophrenia: a PET investigation. Am J Psychiatry 155(7):921–928, 1998 9659858
Kapur S, Zipursky RB, Remington G: Clinical and theoretical implications of 5-HT2 and D2 receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatry 156(2):286–293, 1999 9989565
Kapur S, Zipursky R, Jones C, et al: A positron emission tomography study of quetiapine in schizophrenia: a preliminary finding of an antipsychotic effect with only transiently high dopamine D2 receptor occupancy. Arch Gen Psychiatry 57(6):553–559, 2000 10839333
Kapur S, Arenovich T, Agid O, et al: Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry 162(5):939–946, 2005 15863796
Kassahun K, Mattiuz E, Nyhart E Jr, et al: Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Drug Metab Dispos 25(1):81–93, 1997 9010634
Kaur T, Cadenhead KS: Treatment implications of the schizophrenia prodrome. Curr Top Behav Neurosci 4:97–121, 2010 21312398
Kelly DL, Conley RR, Tamminga CA: Differential olanzapine plasma concentrations by sex in a fixed-dose study. Schizophr Res 40(2):101–104, 1999 10593449
Kennedy J, Deberdt W, Siegal A, et al: Olanzapine does not enhance cognition in non-agitated and non-psychotic patients with mild to moderate Alzheimer’s dementia. Int J Geriatr Psychiatry 20(11): 1020–1027, 2005 16250069
Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmüller B: Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett 20(3):379–382, 1980 6108541
Kinon BJ, Noordsy DL, Liu-Seifert H, et al: Randomized, double-blind 6-month comparison of olanzapine and quetiapine in patients with schizophrenia or schizoaffective disorder with prominent negative symptoms and poor functioning. J Clin Psychopharmacol 26(5):453–461, 2006 16974184
Klein DJ, Cottingham EM, Sorter M, et al: A randomized, double-blind, placebo-controlled trial of metformin treatment of weight gain associated with initiation of atypical antipsychotic therapy in children and adolescents. Am J Psychiatry 163(12):2072–2079, 2006 17151157
Koller EA, Doraiswamy PM: Olanzapine-associated diabetes mellitus. Pharmacotherapy 22(7):841–852, 2002 12126218
Koller E, Schneider B, Bennett K, Dubitsky G: Clozapine-associated diabetes. Am J Med 111(9):716–723, 2001 11747852
Komossa K, Rummel-Kluge C, Hunger H, et al: Olanzapine versus other atypical antipsychotics for schizophrenia. Cochrane Database Syst Rev (3):CD006654, 2010 20238348
Koro CE, Fedder DO, L’Italien GJ, et al: Assessment of independent effect of olanzapine and risperidone on risk of diabetes among patients with schizophrenia: population based nested case-control study. BMJ 325(7358):243, 2002a 12153919
Koro CE, Fedder DO, L’Italien GJ, et al: An assessment of the independent effects of olanzapine and risperidone exposure on the risk of hyperlipidemia in schizophrenic patients. Arch Gen Psychiatry 59(11):1021–1026, 2002b 12418935
Krystal JH, Karper LP, Seibyl JP, et al: Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51(3):199–214, 1994 8122957
Kryzhanovskaya LA, Robertson-Plouch CK, Xu W, et al: The safety of olanzapine in adolescents with schizophrenia or bipolar I disorder: a pooled analysis of 4 clinical trials. J Clin Psychiatry 70(2):247–258, 2009a 19210948
Kryzhanovskaya L, Schulz SC, McDougle C, et al: Olanzapine versus placebo in adolescents with schizophrenia: a 6-week, randomized, double-blind, placebo-controlled trial. J Am Acad Child Adolesc Psychiatry 48(1):60–70, 2009b 19057413
Kulkarni J, Storch A, Baraniuk A, et al: Antipsychotic use in pregnancy. Expert Opin Pharmacother 16(9):1335–1345, 2015 26001182
Lauriello J, Lambert T, Andersen S, et al: An 8-week, double-blind, randomized, placebo-controlled study of olanzapine long-acting injection in acutely ill patients with schizophrenia. J Clin Psychiatry 69(5):790–799, 2008 18452346
Leggero C, Masi G, Brunori E, et al: Low-dose olanzapine monotherapy in girls with anorexia nervosa, restricting subtype: focus on hyperactivity. J Child Adolesc Psychopharmacol 20(2):127–133, 2010 20415608
Leucht S, Pitschel-Walz G, Abraham D, Kissling W: Efficacy and extrapyramidal side-effects of the new antipsychotics olanzapine, quetiapine, risperidone, and sertindole compared to conventional antipsychotics and placebo. A meta-analysis of randomized controlled trials. Schizophr Res 35(1):51–68, 1999 9988841
Leucht S, Corves C, Arbter D, et al: Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 373(9657):31–41, 2009 19058842
Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators: Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353(12):1209–1223, 2005 16172203
Lindenmayer JP, Patel R: Olanzapine-induced ketoacidosis with diabetes mellitus. Am J Psychiatry 156(9):1471, 1999 10484968
Lipscombe LL, Austin PC, Alessi-Severini S, et al; Canadian Network for Observational Drug Effect Studies (CNODES) Investigators: Atypical antipsychotics and hyperglycemic emergencies: multicentre, retrospective cohort study of administrative data. Schizophr Res 154(1–3):54–60, 2014 24581419
Lucas RA, Gilfillan DJ, Bergstrom RF: A pharmacokinetic interaction between carbamazepine and olanzapine: observations on possible mechanism. Eur J Clin Pharmacol 54(8):639–643, 1998 9860152
Macias WL, Bergstrom RF, Cerimele BJ, et al: Lack of effect of olanzapine on the pharmacokinetics of a single aminophylline dose in healthy men. Pharmacotherapy 18(6):1237–1248, 1998 9855322
Maenpaa J, Wrighton S, Bergstrom R, et al: Pharmacokinetic (PK) and pharmacodynamic (PD) interactions between fluvoxamine and olanzapine (abstract). Clin Pharmacol Ther 61(2):225, 1997
Mamo D, Kapur S, Keshavan M, et al: D2 receptor occupancy of olanzapine pamoate depot using positron emission tomography: an open-label study in patients with schizophrenia. Neuropsychopharmacology 33(2):298–304, 2008 17443131
Markowitz JS, DeVane CL, Malcolm RJ, et al: Pharmacokinetics of olanzapine after single-dose oral administration of standard tablet versus normal and sublingual administration of an orally disintegrating tablet in normal volunteers. J Clin Pharmacol 46(2):164–171, 2006 16432268
Matsunaga H, Nagata T, Hayashida K, et al: A long-term trial of the effectiveness and safety of atypical antipsychotic agents in augmenting SSRI-refractory obsessive-compulsive disorder. J Clin Psychiatry 70(6):863–868, 2009 19422759
McAllister RK, Tutt CD, Colvin CS: Lipid 20% emulsion ameliorates the symptoms of olanzapine toxicity in a 4-year-old. Am J Emerg Med 30(6):1012.e1–1012e2, 2012 21641141
McEvoy JP, Lieberman JA, Stroup TS, et al; CATIE Investigators: Effectiveness of clozapine versus olanzapine, quetiapine, and risperidone in patients with chronic schizophrenia who did not respond to prior atypical antipsychotic treatment. Am J Psychiatry 163(4):600–610, 2006 16585434
McEvoy JP, Lieberman JA, Perkins DO, et al: Efficacy and tolerability of olanzapine, quetiapine, and risperidone in the treatment of early psychosis: a randomized, double-blind 52-week comparison. Am J Psychiatry 164(7):1050–1060, 2007 17606657
McGlashan TH, Zipursky RB, Perkins D, et al: Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 163(5):790–799, 2006 16648318
Meehan K, Zhang F, David S, et al: A double-blind, randomized comparison of the efficacy and safety of intramuscular injections of olanzapine, lorazepam, or placebo in treating acutely agitated patients diagnosed with bipolar mania. J Clin Psychopharmacol 21(4):389–397, 2001 11476123
Mei L, Nave KA: Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron 83(1):27–49, 2014 24991953
Miller DD, Caroff SN, Davis SM, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators: Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry 193(4):279–288, 2008 18827289
Modabbernia A, Heidari P, Soleimani R, et al: Melatonin for prevention of metabolic side-effects of olanzapine in patients with first-episode schizophrenia: randomized double-blind placebo-controlled study. J Psychiatr Res 53:133–140, 2014 24607293
Moghaddam B, Javitt D: From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37(1):4–15, 2012 21956446
Mondraty N, Birmingham CL, Touyz S, et al: Randomized controlled trial of olanzapine in the treatment of cognitions in anorexia nervosa. Australas Psychiatry 13(1):72–75, 2005 15777417
Moore NA: Behavioural pharmacology of the new generation of antipsychotic agents. Br J Psychiatry Suppl 174 (38 suppl 138):5–11, 1999 10884895
Moteshafi H, Zhornitsky S, Brunelle S, Stip E: Comparing tolerability of olanzapine in schizophrenia and affective disorders: a meta-analysis. Drug Saf 35(10):819–836, 2012 22967188
Ninan I, Jardemark KE, Wang RY: Olanzapine and clozapine but not haloperidol reverse subchronic phencyclidine-induced functional hyperactivity of N-methyl-D-aspartate receptors in pyramidal cells of the rat medial prefrontal cortex. Neuropharmacology 44(4):462–472, 2003 12646283
Norris ML, Spettigue W, Buchholz A, et al: Factors influencing research drug trials in adolescents with anorexia nervosa. Eat Disord 18(3):210–217, 2010 20419525
Patel BR, Nyhart EH, Callaghan JT, et al: Combined population pharmacokinetic analysis of olanzapine in healthy volunteers (abstract). Pharm Res 12 (9 suppl):S360, 1995
Patel BR, Kurtz DL, Callaghan JT, et al: Effects of smoking and gender on population pharmacokinetics of olanzapine (OL) in a phase III clinical trial (abstract). Pharm Res 13 (9 suppl):S408, 1996
Pivac N, Kozaric-Kovacic D, Muck-Seler D: Olanzapine versus fluphenazine in an open trial in patients with psychotic combat-related post-traumatic stress disorder. Psychopharmacology (Berl) 175(4):451–456, 2004 15064916
Ramaswamy K, Kozma CM, Nasrallah H: Risk of diabetic ketoacidosis after exposure to risperidone or olanzapine. Drug Saf 30(7):589–599, 2007 17604410
Ratzoni G, Gothelf D, Brand-Gothelf A, et al: Weight gain associated with olanzapine and risperidone in adolescent patients: a comparative prospective study. J Am Acad Child Adolesc Psychiatry 41(3):337–343, 2002 11886029
Ring BJ, Binkley SN, Vandenbranden M, Wrighton SA: In vitro interaction of the antipsychotic agent olanzapine with human cytochromes P450 CYP2C9, CYP2C19, CYP2D6 and CYP3A. Br J Clin Pharmacol 41(3):181–186, 1996a 8866916
Ring BJ, Catlow J, Lindsay TJ, et al: Identification of the human cytochromes P450 responsible for the in vitro formation of the major oxidative metabolites of the antipsychotic agent olanzapine. J Pharmacol Exp Ther 276(2):658–666, 1996b 8632334
Robertson GS, Fibiger HC: Effects of olanzapine on regional C-Fos expression in rat forebrain. Neuropsychopharmacology 14(2):105–110, 1996 8822533
Rosenheck RA, Leslie DL, Sindelar J, et al; CATIE Study Investigators: Cost-effectiveness of second-generation antipsychotics and perphenazine in a randomized trial of treatment for chronic schizophrenia. Am J Psychiatry 163(12): 2080–2089, 2006 17151158
Russell JM, Mackell JA: Bodyweight gain associated with atypical antipsychotics: epidemiology and therapeutic implications. CNS Drugs 15(7):537–551, 2001 11510624
Sanger TM, Grundy SL, Gibson PJ, et al: Long-term olanzapine therapy in the treatment of bipolar I disorder: an open-label continuation phase study. J Clin Psychiatry 62(4):273–281, 2001 11379842
Schneider LS, Dagerman K, Insel PS: Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry 14(3):191–210, 2006a 16505124
Schneider LS, Tariot PN, Dagerman KS, et al; CATIE-AD Study Group: Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med 355(15):1525–1538, 2006b 17035647
Schulz SC: Pharmacologic treatment of schizophrenia. Psychiatr Clin North Am 6:51–71, 1999
Schulz SC, Cornelius K: Statement 6: Second-generation antipsychotics are effective mood stabilizers. Curr Psychiatry 9 (suppl):S37–S43, 2010
Schulz SC, Camlin KL, Berry SA, Jesberger JA: Olanzapine safety and efficacy in patients with borderline personality disorder and comorbid dysthymia. Biol Psychiatry 46(10):1429–1435, 1999 10578457
Schulz SC, Zanarini MC, Bateman A, et al: Olanzapine for the treatment of borderline personality disorder: variable dose 12-week randomised double-blind placebo-controlled study. Br J Psychiatry 193(6):485–492, 2008 19043153
Seeman P: Atypical antipsychotics: mechanism of action. Can J Psychiatry 47(1):27–38, 2002 11873706
Sernyak MJ, Leslie DL, Alarcon RD, et al: Association of diabetes mellitus with use of atypical neuroleptics in the treatment of schizophrenia. Am J Psychiatry 159(4): 561–566, 2002 11925293
Shapira NA, Ward HE, Mandoki M, et al: A double-blind, placebo-controlled trial of olanzapine addition in fluoxetine-refractory obsessive-compulsive disorder. Biol Psychiatry 55(5):553–555, 2004 15023585
Sharma RP, Janicak PG, Bissette G, Nemeroff CB: CSF neurotensin concentrations and antipsychotic treatment in schizophrenia and schizoaffective disorder. Am J Psychiatry 154(7):1019–1021, 1997 9210757
Sharpley AL, Vassallo CM, Cowen PJ: Olanzapine increases slow-wave sleep: evidence for blockade of central 5-HT(2C) receptors in vivo. Biol Psychiatry 47(5):468–470, 2000 10704958
Shi L, Namjoshi MA, Swindle R, et al: Effects of olanzapine alone and olanzapine/fluoxetine combination on health-related quality of life in patients with bipolar depression: secondary analyses of a double-blind, placebo-controlled, randomized clinical trial. Clin Ther 26(1):125–134, 2004 14996525
Sikich L, Hamer RM, Bashford RA, et al: A pilot study of risperidone, olanzapine, and haloperidol in psychotic youth: a double-blind, randomized, 8-week trial. Neuropsychopharmacology 29(1):133–145, 2004 14583740
Sikich L, Frazier JA, McClellan J, et al: Double-blind comparison of first- and second-generation antipsychotics in early-onset schizophrenia and schizo-affective disorder: findings from the treatment of early-onset schizophrenia spectrum disorders (TEOSS) study. Am J Psychiatry 165(11):1420–1431, 2008 18794207
Singh LK, Praharaj SK, Sahu M: Nonfatal suicidal overdose of olanzapine in an adolescent. Curr Drug Saf 7(4):328–329, 2012 23062245
Soler J, Pascual JC, Campins J, et al: Double-blind, placebo-controlled study of dialectical behavior therapy plus olanzapine for borderline personality disorder. Am J Psychiatry 162(6):1221–1224, 2005 15930077
Soloff PH, George A, Nathan RS, et al: Progress in pharmacotherapy of borderline disorders. A double-blind study of amitriptyline, haloperidol, and placebo. Arch Gen Psychiatry 43(7):691–697, 1986 3521532
Stein MB, Kline NA, Matloff JL: Adjunctive olanzapine for SSRI-resistant combat-related PTSD: a double-blind, placebo-controlled study. Am J Psychiatry 159(10): 1777–1779, 2002 12359687
Stephenson CM, Pilowsky LS: Psychopharmacology of olanzapine. A review. Br J Psychiatry Suppl 174(38):52–58, 1999 10884900
Stockton ME, Rasmussen K: Electrophysiological effects of olanzapine, a novel atypical antipsychotic, on A9 and A10 dopamine neurons. Neuropsychopharmacology 14(2):97–105, 1996a 8822532
Stockton ME, Rasmussen K: Olanzapine, a novel atypical antipsychotic, reverses D-amphetamine-induced inhibition of midbrain dopamine cells. Psychopharmacology (Berl) 124(1–2):50–56, 1996b 8935800
Street JS, Clark WS, Gannon KS, et al; The HGEU Study Group: Olanzapine treatment of psychotic and behavioral symptoms in patients with Alzheimer disease in nursing care facilities: a double-blind, randomized, placebo-controlled trial. Arch Gen Psychiatry 57(10):968–976, 2000 11015815
Street JS, Clark WS, Kadam DL, et al: Long-term efficacy of olanzapine in the control of psychotic and behavioral symptoms in nursing home patients with Alzheimer’s dementia. Int J Geriatr Psychiatry 16 (suppl 1):S62–S70, 2001 11748789
Swartz MS, Perkins DO, Stroup TS, et al; CATIE Investigators: Effects of antipsychotic medications on psychosocial functioning in patients with chronic schizophrenia: findings from the NIMH CATIE study. Am J Psychiatry 164(3):428–436, 2007 17329467
Tan HH, Hoppe J, Heard K: A systematic review of cardiovascular effects after atypical antipsychotic medication overdose. Am J Emerg Med 27(5):607–616, 2009 19497468
Tanoshima R, Chandranipapongse W, Colantonio D, et al: Acute olanzapine overdose in a toddler: a case report. Ther Drug Monit 35(5):557–559, 2013 24052061
Tauscher J, Küfferle B, Asenbaum S, et al: In vivo 123I IBZM SPECT imaging of striatal dopamine-2 receptor occupancy in schizophrenic patients treated with olanzapine in comparison to clozapine and haloperidol. Psychopharmacology (Berl) 141(2):175–181, 1999 9952042
Thase ME, Corya SA, Osuntokun O, et al: A randomized, double-blind comparison of olanzapine/fluoxetine combination, olanzapine, and fluoxetine in treatment-resistant major depressive disorder. J Clin Psychiatry 68(2):224–236, 2007 17335320
Theisen FM, Grabarkiewicz J, Fegbeutel C, et al: Olanzapine overdose in children and adolescents: two case reports and a review of the literature. J Child Adolesc Psychopharmacol 15(6):986–995, 2005 16379519
Theisen FM, Haberhausen M, Firnges MA, et al: No evidence for binding of clozapine, olanzapine and/or haloperidol to selected receptors involved in body weight regulation. Pharmacogenomics J 7(4):275–281, 2007 16983399
Tohen M, Sanger TM, McElroy SL, et al; Olanzapine HGEH Study Group: Olanzapine versus placebo in the treatment of acute mania. Am J Psychiatry 156(5):702–709, 1999 10327902
Tohen M, Jacobs TG, Grundy SL, et al; The Olanzapine HGGW Study Group: Efficacy of olanzapine in acute bipolar mania: a double-blind, placebo-controlled study. Arch Gen Psychiatry 57(9):841–849, 2000 10986547
Tohen M, Baker RW, Altshuler LL, et al: Olanzapine versus divalproex in the treatment of acute mania. Am J Psychiatry 159(6):1011–1017, 2002 12042191
Tohen M, Vieta E, Calabrese J, et al: Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry 60(11): 1079–1088, 2003 14609883
Tohen M, Greil W, Calabrese JR, et al: Olanzapine versus lithium in the maintenance treatment of bipolar disorder: a 12-month, randomized, double-blind, controlled clinical trial. Am J Psychiatry 162(7):1281–1290, 2005 15994710
Tohen M, Kryzhanovskaya L, Carlson G, et al: Olanzapine versus placebo in the treatment of adolescents with bipolar mania. Am J Psychiatry 164(10):1547–1556, 2007 17898346
Tollefson GD, Sanger TM: Negative symptoms: a path analytic approach to a double-blind, placebo- and haloperidol-controlled clinical trial with olanzapine. Am J Psychiatry 154(4):466–474, 1997 9090332
Tollefson GD, Beasley CM Jr, Tran PV, et al: Olanzapine versus haloperidol in the treatment of schizophrenia and schizoaffective and schizophreniform disorders: results of an international collaborative trial. Am J Psychiatry 154(4):457–465, 1997 9090331
Tollefson GD, Birkett MA, Kiesler GM, Wood AJ; Lilly Resistant Schizophrenia Study Group: Double-blind comparison of olanzapine versus clozapine in schizophrenic patients clinically eligible for treatment with clozapine. Biol Psychiatry 49(1):52–63, 2001 11163780
Torgersen S, Kringlen E, Cramer V: The prevalence of personality disorders in a community sample. Arch Gen Psychiatry 58(6):590–596, 2001 11386989
Tran PV, Tollefson GD, Sanger TM, et al: Olanzapine versus haloperidol in the treatment of schizoaffective disorder. Acute and long-term therapy. Br J Psychiatry 174:15–22, 1999 10211146
Volavka J, Czobor P, Sheitman B, et al: Clozapine, olanzapine, risperidone, and haloperidol in the treatment of patients with chronic schizophrenia and schizoaffective disorder. Am J Psychiatry 159(2):255–262, 2002 11823268
Zacher JL, Roche-Desilets J: Hypotension secondary to the combination of intramuscular olanzapine and intramuscular lorazepam. J Clin Psychiatry 66(12):1614–1615, 2005 16401168
Zanarini MC, Frankenburg FR: Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo-controlled pilot study. J Clin Psychiatry 62(11):849–854, 2001 11775043
Zanarini MC, Frankenburg FR, Parachini EA: A preliminary, randomized trial of fluoxetine, olanzapine, and the olanzapine-fluoxetine combination in women with borderline personality disorder. J Clin Psychiatry 65(7):903–907, 2004 15291677
Zanarini MC, Schulz SC, Detke HC, et al: A dose comparison of olanzapine for the treatment of borderline personality disorder: a 12-week randomized, double-blind, placebo-controlled study. J Clin Psychiatry 72(10):1353–1362, 2011 21535995
Zanarini MC, Schulz SC, Detke H, et al: Open-label treatment with olanzapine for patients with borderline personality disorder. J Clin Psychopharmacol 32(3):398–402, 2012 22544004