6. The Feasibility of Reconstructing Neanderthal Demography as an Approach to Examining Extinction
Introduction
The Neanderthal extinction is a contentious issue, which has led to extensive discussion of the factors that might be behind it. It may be possible to map Neanderthal population change through time to identify the impact that the extent of climatic deterioration and the arrival of anatomically modern humans had upon Neanderthal population decline and, therefore, their contribution to their extinction. However, this requires a high quality and quantity of genetic data. Despite new Neanderthal data being continually produced, genetic data is still relatively limited. This paper examines the requirements needed to enable a reconstruction of Neanderthal population change against a chronology. This will be done through the use of bison and spotted hyaenas (Crocucta crocuta) as proxies to identify weaknesses in the Neanderthal genetic data.
Issues affecting Neanderthal Extinction
The two factors commonly invoked as leading to Neanderthal extinction have been climate deterioration (Dansgaard et al., 1993; Finlayson et al., 2008; Golonova et al. 2010; Hublin and Roebrooks 2009; Sepulchre et al. 2007; Van Andel et al. 2003) and the arrival of anatomically modern humans (Banks et al. 2003; D’Errico and Sánchez-Goni 2003). There has been a distinct dichotomy between these factors, where often one is cited as the trigger at the exclusion of the other (Golonova et al. 2010; Lowe et al. 2012; Sepulchre et al. 2007).
The question of whether anatomically modern humans played a role in Neanderthal extinction is centred on the contemporaneity of radiocarbon dates. While it is important to bear in mind the limitations inherent in radiocarbon dating, as outlined by Pettitt and Pike (2001) and Higham (2011), radiocarbon dates do suggest contemporaneity between the two hominin species (Pettitt 1999) and there are studies that suggest levels of co-existence. Finlayson et al. (2006) discuss Neanderthals in refugia in Gibraltar with modern human presence 100 kilometres eastwards. Szmidt et al. (2010) find a chronological overlap between faunal remains from Aurignacian levels at Isturitz, France with those from Châtelperronian sites. Similarly, Longo et al. (2012) find an overlap in Northern Italy between dates from the Mousterian level at Riparro Mezzena, and the Proto-Aurignacian level at Grotta di Fumane. Several explanations have been proposed as leading to Neanderthal extinction via interactions with anatomically modern humans: warfare (Pettitt 1999), diseases brought by modern humans (Stringer et al. 2003), Neanderthal cannibalism (Underwood 2008), or a modern human advantage over Neanderthals, such as cognitive ability (Klein 2000), diet (Hockett 2012; Hockett and Haws 2005; Richards and Trinkaus 2009), which has been argued against (Hardy 2010; Stringer et al. 2008), and differences in weaning resulting in differential fertility (Pettitt 2000; Skinner 1997). Given the possibility of interbreeding between Neanderthals and modern humans (Green et al. 2010; Hammer 2011; Wall et al. 2009; Yotova et al. 2011) it is possible that modern humans subsumed the Neanderthal population. Nevertheless, this depends on the extent of interbreeding and requires further analyses to confirm/refute this theory (Alves et al. 2012; Durand et al. 2011; Eriksson and Manica 2012; Hodgson et al. 2010).
It is possible that both climatic deterioration and modern human presence affected the Neanderthal population, but the extent of this is unclear. One method that can be used to examine this is to reconstruct the Neanderthal demographic model alongside a chronology. This will allow a correlation between Neanderthal population change and particular dated events, thereby identifying how the climate and modern human arrival affected Neanderthals.
Neanderthal Genetic Review
Reconstructing Neanderthal demography requires a genetic dataset sufficient in both quality and quantity. The mitochondrial hypervariable region 1 (HVR1) within the control region is the gene for which there is the most genetic data available. Table 6.1 provides a summary of all the Neanderthal genetic data up to present, including several autosomal genes (Krause et al. 2007; Lalueza-Fox et al. 2007, 2008, 2009; Lopez-Valenzuela et al. 2012; Wang et al. 2012) and three Neanderthal draft genomes (including both mitochondrial and autosomal). As shown in Table 6.1, there are eighteen Neanderthal HVR1 DNA sequences (italicised). Only fourteen of these have radiocarbon dates (there were only twelve at the time of this study). The (DNA) sequence lengths vary considerably from 31 base pairs (bp) to the complete HVR1.
Reconstructing the demographic model
Various software programmes with suitable genetic and genealogical input, can reconstruct demographic models. BEAST (Bayesian Evolutionary Analysis of Sampling Trees) is one such programme, classified as Bayesian Markov Chain Monte Carlo (Drummond and Rambaut 2007). By altering the input parameters, BEAST tests various evolutionary hypotheses. The resultant output is a series of parameter values that best describes the evolutionary (genetic mutation/substitution) and demographic model (Dodge 2012). BEAST also creates the Bayesian Skyline plot (BSP) (Drummond et al. 2005) – the demographic model showing the effective population size (Ne) through time. The construction of the BSP is rooted in the coalescent principle (Kingman 1982), whereby there will be a time in the past when the individuals will share a common ancestor. The number of individuals sharing this common ancestor decreases further back in time reflecting a relationship between population size and individuals of shared ancestry (Griffiths and Tavaré 1994).
Using animals as proxies: Significance and data used
Using BEAST, Shapiro et al. (2004) and Drummond et al. (2003) were able to show how climatic effects affected bison population. The bison genetic dataset is large, with 191 sequences with various sequence lengths from 313 bp (Shapiro et al. 2004, Supplementary table S1, 8–18), making it an ideal candidate as a proxy to examine the minimal requirements of the BEAST programme. This will be done by systematically reducing the data for input into BEAST and removing data by clades.
White and Pettitt (2011) suggest hyaenas and Neanderthals share features that might make them sympatric, making hyaenas a suitable proxy for Neanderthal presence. Genetic data from British spotted hyaenas (Dodge et al. 2012) was added to data from the European mainland (Hofreiter et al. 2004; Rohland et al. 2005) (Table 6.2). Analysis of the cytochrome b gene enabled the British sequences to be placed within the tree (Figure 6.1). By partitioning the data, this can determine how geographical sampling affects the demographic model.
Goals and Objectives
Given the current limitations of the Neanderthal genetic data, the goal here is to identify what is required to allow for demographic reconstruction. Therefore, the objectives are to:
1) Identify the minimum quantitative and qualitative data requirements to enable demographic model reconstruction.
2) Determine how geographical sampling can affect the demographic model.
3) Assess how using an animal as a proxy can complement the Neanderthal demographic model to determine the extinction trigger(s).
4) Assess if a demographic model can be retrieved from a simulated Neanderthal dataset.
Examination of the bison dataset
Quantitative Analysis-Identifying the minimum quantitative and qualitative data requirements
Method
Ten sequences were randomly selected and removed from the data file before input into BEAST (e.g. 191 sequences, 181, 171 etc.) until 31 sequences remained in the input file, at which point one sequence was removed each time until 13 remained. For each output, the evolutionary parameter values were recorded (mutation rate, model etc.), and the BSPs were reconstructed. For each of the genetic parameter values, the cut-off point was identified. This is the point at which the confidence intervals (CI) dramatically increase, thereby decreasing the resolution to such an extreme degree that the values no longer reflect the initial model parameters (see Dodge 2012). The cut-off points identified for each parameter can then be coalesced into a single number to provide the minimum amount of sequences required that still enables an accurate demographic reconstruction.
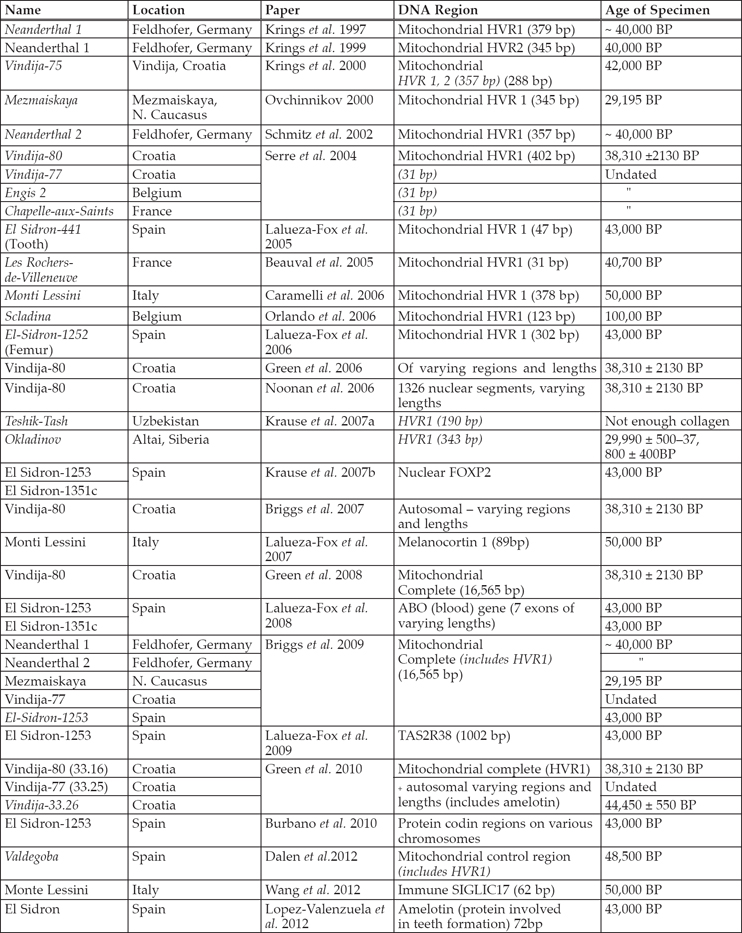
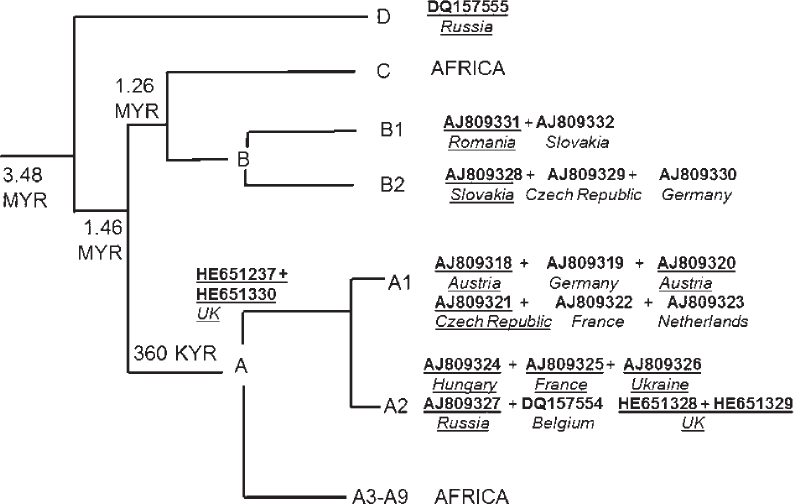
Figure 6.1. Spotted hyaena tree displaying the clades and which of the genetic sequences (with their accession numbers) from Rohland et al. (2005), Hofreiter et al. (2004) and Dodge et al. (2012) belong to which clade, using the mitochondrial cytochrome b gene. Underlined samples are those that have radiocarbon dates. The dates indicate the times to the most recent common ancestor. Clade C and A3–A9 show modern day African hyaena populations, while the remainder are Pleistocene spotted hyaenas.
Results
The results indicate that no less than 31 sequences should be used as the minimum data requirement for the evolutionary parameters (Dodge 2012). In the BSPs, the resolution of the population signal, as indicated by the CI, diminished when 51 sequences remained in the input file. This lack of resolution is reflected in the mean Ne at the mode of the time to most recent common ancestor (tMRCA), as shown by Dodge (2012, figure 11). It is clear there is a significant increase in the CI from 51 sequences in the input file to 41 sequences remaining in the input file.
Discussion
Dodge (2012) discusses several essential factors when interpreting the results. The first is the substitution rate, which provides the calibration. Substitution rates vary between genes and different species, due to gene conservation and inherited factors respectively (Ho 2009). There has also been considerable debate regarding the time dependency of the mutation rate on the age of the DNA sequences (Emerson 2007; Ho et al. 2005). The time dependency model posits that within one gene of one species, the age of the sequence can affect the estimated substitution rate. Further analyses, however, revealed that over-estimation of the substitution rate was largely due to sequences containing low genetic information and inaccurate prior parameters (Debruyne and Poinar 2009; Ho et al. 2007), which would currently mean that the Neanderthal data would yield erroneous evolutionary genetic parameter results.
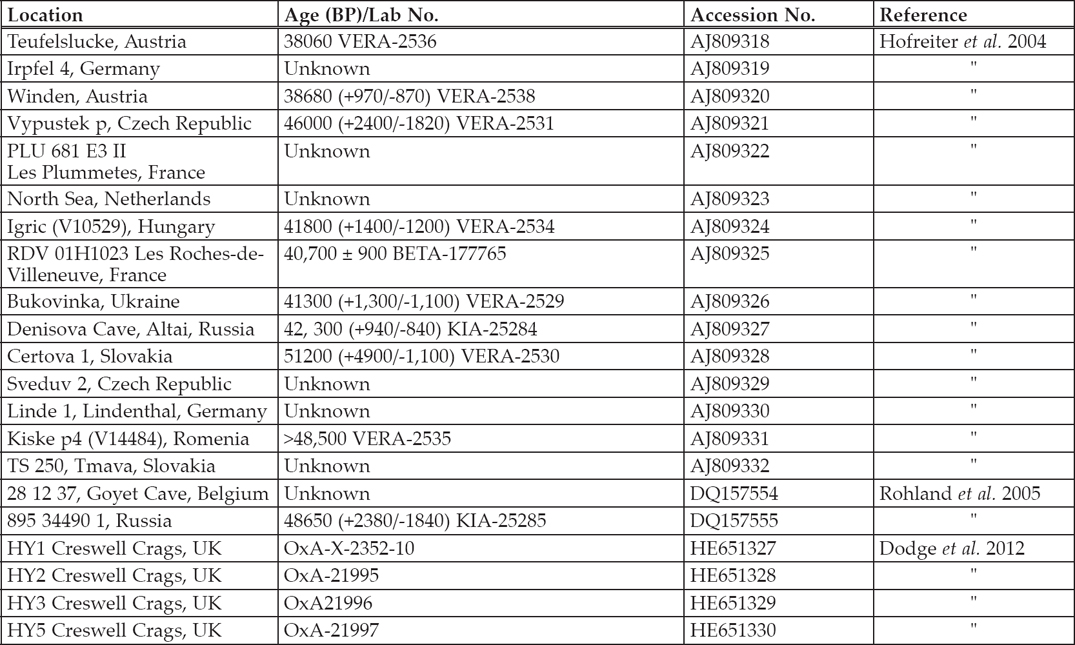
Table 6.2. Summary table of all Pleistocene spotted hyaena analyses to date.

Figure 6.2. Age distribution of radiocarbon dated samples in bins of 1000 for the bison molecular data used in this study.
Secondly, the change in parameter values as the data is reduced reflects how the information content is reduced; as the data is removed, so is the genetic variation. Consequently, the parameter values are specific only to the bison dataset, requiring that the results be viewed more as a guideline. While the bison results do not give an exact number of Neanderthal sequences to be input into BEAST, they do provide a minimum number: If the quantity and quality of the bison data means no less than 31 sequences should be used, the same can be said for a dataset that equals the quantity and quality of the bison sequences. Given the low quantity and quality of the Neanderthal data, no less than 31 Neanderthal sequences should be used.
The third factor regards the BSP. Because the coalescent principle is fundamentally rooted in the BSP construction (Drummond et al. 2002, 2005) any DNA damage could be mistakenly interpreted as false diversity. This can lead to erroneous calculations of the DNA substitution rate and/or the illusion of a more diverse species or node (Axelsson et al. 2008). Furthermore, false diversity can lead to an inaccurate tMRCA, which will impair the BSP.
Finally, just as with the genetic parameters, the removal of samples can affect the population signal due to the sample age distribution. Therefore, it becomes important to ensure that enough samples are used that span a range of dates. The full bison dataset, as shown in Figure 6.2, has a relatively uniform age distribution with at least one sample in every bin of 1,000 years, with a few exceptions, thus allowing for a population signal to be identified.
This discussion highlights the importance of obtaining good quality DNA to ensure accurate and valid evolutionary and demographic parameters.
Clade removal-Determining how geographical sampling can affect the demographic model
Method
The bison data consisted of four clades: Siberian (15 sequences), South of Ice (31 sequences), Eastern Beringia (106 sequences), and Edmonton (39 sequences) (Shapiro et al. 2004, Supplementary table S1, 8–18). This meant that removing a clade and comparing the parameter results here to the Quantitative Analysis results (where the input data between the Clade Removal and Quantitative Analysis is roughly equal) could test how geographical sampling affected the data. For example, removing the Siberian clade (15 sequences) left 176 sequences in the input file. This was compared to the Quantitative Analysis results of 171 sequences remaining in the input file. The same was done for the remaining data, so the data that had the South of Ice clade removed was compared with 161 sequences remaining in the input file from the Quantitative Analysis. The Eastern Beringia clade removal data was compared with 81 sequences remaining and the Edmonton clade removal data was compared with 151 sequences remaining. Significance testing using ‘R’ (R Foundation for Statistical Computing, 2001) was carried out on the genetic parameter values between the pairs. The result was expressed as a p-value where if p>0.05 then the result is not significant and removing the aforesaid clade made no difference to the genetic parameter being tested.
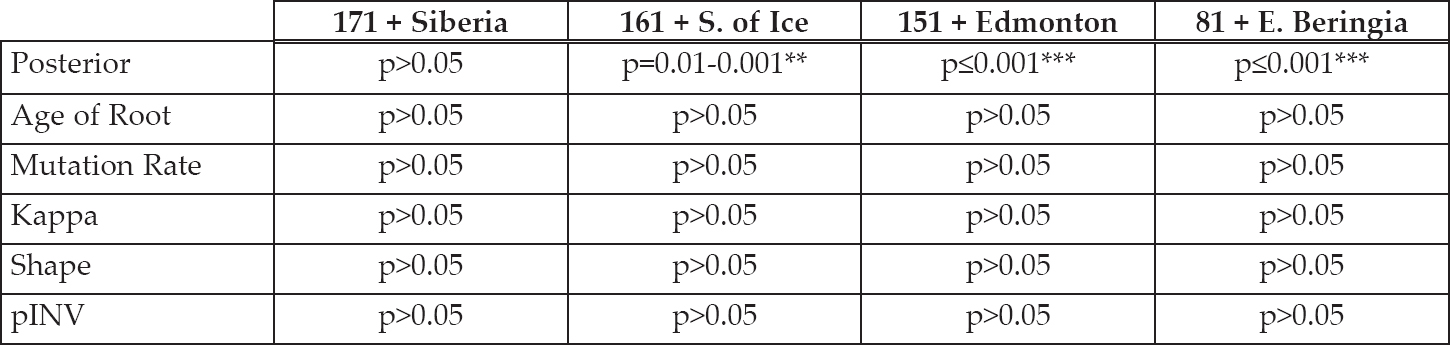
Table 6.3. Summary table showing the results of the significant tests between the Quantitative Analysis and Clade Removal. The posterior is the (normalised) product of the likelihood that the model fits the data. The age of root is the time to most recent common ancestor, while the mutation rate, kappa, shape parameter and the pINV (proportion of invariant sites) are characteristics of the DNA mutation model. See Strimmer and von Haessler (2003) for definitions of each genetic parameter and how they describe the mutation model. In this table, p>0.05 means not significant, p= 0.01–0.001** means significant and p≤0.001*** means highly significant.
Results
The general trend for all the evolutionary genetic parameters is no significance, as Table 6.3 shows. This indicates that removing a clade does not affect the results. The only parameter that is significant is the posterior. The posterior parameter is the normalised product of the likelihood that the model (i.e. evolutionary parameters and the tree) fit with the input prior data. This value reflects that the more data there is available, the more likely it is that the final model will be accurate. The pair of data that have the most input data in the file is 171 sequences with the Siberian clade removed. With most data remaining in the file after the Siberian clade is removed, there is no difference to the results when compared to the Quantitative Analysis (171) results: there is no significance (p>0.05). Therefore, removing a clade with few sequences as compared to removing data randomly (also of a few sequences) leads to similar results. However, as the data in the input file becomes less it appears that removing a clade does make a difference to the results. For example, p≤0.001 for 81 sequences with the Eastern Beringia clade removed. This suggests that geographical sampling does affect the evolutionary model, but only when the input data is already limited. In this case, the results start to become significant when the South of Ice clade is removed (31 sequences), which is less than 17% of the data. Selectively analysing parts of the data should be avoided if the data is already limited in quantity and quality. This could potentially reduce the resolution in the BSP.
Hyaena (Crocuta crocuta) analysis as a supplementary approach
Using hyaenas as a proxy to complement the Neanderthal demographic model
Archaeology and genetic analysis of the mitochondrial cytochrome b gene indicates three dispersal events of the spotted hyaena from Africa. Two of these reached Britain, the first prior to 700,000 (kyr) BP and the second after 360 kyr BP (Dodge et al. 2012; Hofreiter et al. 2004; Rohland et al. 2005). Genetic results from Dodge et al. (2012) support Turners (2009) theory of local extinction during the Ipswichian, and a second migration of hyaenas during the Devensian before their extinction around 30 kyr BP. Similar features in migration, distribution, and niche imply that hyaenas could be used as a proxy to infer Neanderthal migration and distribution. It is quite possible that hyaenas were drawn towards Neanderthal settlements just as modern hyaenas are drawn towards modern African settlements for scavenging opportunities (Kruuk 1972). This makes it likely that hyaenas were part of a suite of species that followed hominins as they migrated from Africa (Turner 1994). It may be possible to identify common environmental triggers from a hyaena demographic reconstruction that results in population decline and extinction in both Neanderthals and hyaenas. However, a later study refuted the idea that hominins were followed by a suite of species. O’Regan et al. (2011) examined large scale dispersals of species (including hyaenas) from Africa into other continents. The results suggested that, because of different environmental tolerances, species migrated of their own accord rather than following hominins. Lorenzen et al. (2011) identified a similar trend in the mega-fauna herbivores of Eurasia and North America, revealing that each species responded differently to an array of environmental triggers. These studies suggest that using animals as proxies may not be entirely plausible on a continental scale and that it may not be possible to identify common environmental triggers between hyaenas and Neanderthals. Nevertheless, similarities between them warrants further analysis of the potential to use hyaenas as a proxy to map distribution and migration regionally.
Hyaena data analysis in BEAST: determining how geographical sampling can affect the demographic model
Method
There were fourteen radiocarbon dated sequences, ten from the mainland and four from Britain, enabling data to be partitioned into four datasets for BEAST input: ‘All sequences with tree prior’, ‘All sequences without tree prior’, ‘Continental sequences with tree prior’, ‘Continental sequences without tree prior’ (where the tree prior refers to Figure 6.1). The input file for BEAST was adapted from Ho et al. (2007). By pairing the datasets appropriately, the following questions could be asked by conducting significance tests on the genetic parameter results between the pairs: 1) does the inclusion of the tree prior make a difference? 2) To what extent does omitting the British DNA sequences affect the results?
Results
Significance testing of all the pairs of data displayed no significance; i.e. inclusion of the tree prior or the British hyaena data did not make a difference. These results are a direct symptom of a large genetic parameter CI due to limited data, which is supported by the bison results. Where data is extremely limited, such partitioning for clade analyses will not yield accurate evolutionary parameters and a suitable BSP.
Neanderthal-Simulated data: can a demographic model be retrieved?
Method
DNA was simulated using Bayesian Serial Simcoal (BayeSSC) (Excoffier et al. 2000) to appear to be Neanderthal data. Sixty-two sequences were simulated at specifically dated times, and Briggs et al.’s (2009) substitution model was used to create a hypothetical HVR1 region, which was 278 bp long (the average length calculated from the twelve Neanderthal sequences that have dates and sequence lengths longer than 100 bp). Of the sixty-two sequences, twelve were specifically simulated at the time point where the twelve Neanderthal sequences have been dated at; the oldest at 100 kyr BP from Scladina (Orlando et al., 2006) to the youngest at 29.2 kyr BP from Mezmaiskaya (Ovchinnikov, 2000). This provided 2833 generations. The remaining fifty sequences were simulated between these time points, the frequency of which increases as time passes towards the youngest generation. This was to give the appearance of an aDNA dataset where younger samples were most likely to yield aDNA, assuming all other environmental conditions are equal. Sequences were simulated under two different demographic models – Crash and Decline – using the demographic priors from Fabre et al. (2009). The ‘Crash’ model postulates an initial Ne of 11,000 individuals at 100 kyr BP (generation 2832) remaining constant until 45,175 years BP (generation 640), whereupon the population declines to extinction; an Ne of two individuals at time 29,200 (generation 0). For the ‘Decline’ model, the Ne starts at 25,000 at generation 3102. The number of generations was extended so as to allow for sufficient Ne at the specific times when sequences are simulated. The population size was allowed to reduce at a steady rate until extinction at generation 0.
After simulation, half of the datasets from both of the models were randomly selected for BEAST input, as was the full ‘Decline’ model dataset. The same substitution model and demographic priors used for simulating were input into BEAST. The chain length, stating how long BEAST runs for and how many times the parameter values change, was set at 10 million iterations, where sampling occurred at every 20,000 iterations. Two further analyses were conducted on the ‘Decline’ halved data: one where the chain length was set at 20 million iterations, with sampling at every 20,000, and the other also set at 20 million iterations, but this time sampled at every 5,000. Additionally, for the full dataset the chain length remained at 20 million sampling at every 5,000.
Results
Tables 6.4 to 6.6 show the results of the age of root and the substitution rate of the three datasets. The age of root estimates here are very young and the corresponding CI are extremely small given the oldest sampled sequence is dated at 100 kyr BP. Due to the coalescent (Drummond and Rambaut 2007), this will adversely affect demographic model recovery as the BSP starts with Ne at the age of root estimate and not the actual age of root. Basu and Majumder (2003) identified factors that can result in severe age of root under-estimation: long sequence length (500+ bp), long evolutionary times (>500 generations), high mutation rates (mutation rate>10–5) and various population sizes used as prior information. For both models used here the sequence length was 278 bp, eliminating the effect of sequence length. The generation time could have affected the under-estimation, as in both models it was considerably longer than 500. In both sets of results, the mutation rate was low, although in the ‘Crash’ results the CI were small, while for the ‘Decline’ results the CI was large. It is likely that the wide CI is enabling an increase in the mean mutation rate. Consequently, given the possibility of the time-dependency model, the age of root will decrease, resulting in severe age of root under-estimation. Against the time-dependency model, Emerson (2007) suggests it is the associated dates of sequences that results in over-estimation of the rates and under-estimation of the age of root. Navascués and Emerson (2009) suggest the phylogenetic tree as prior information could also lead to inaccurate rate estimates. If aDNA sequences are being sampled from one clade that is reciprocally monophyletic, this could lead to increased rates. They also found that a demographic model that is too complex can affect rate estimates. For example, a strong bottleneck had a more negative affect on rate estimation. A more complex demographic model used as prior information can result in a phylogeny that is considerably different from the actual one. This occurs because the resulting phylogeny (used for the demographic reconstruction) is the one that has the highest likelihood, even though it may not necessarily be the most accurate (ibid.). This can therefore lead to age of root under-estimation additionally distorting mutation rates. The effective sample sizes (ESS) also supports that the complexity of the demographic model could affect the mutation rate and the age of root. The ESS is a measure of independent sampling, reflecting the extent of sampling where a value of <100 indicates that it is insufficient. In the case of the ‘Decline’ model, the values for the mean posterior, prior and skyline (Drummond and Rambaut 2007), and the ESS values were much lower than the recommended value of 100. To increase the ESS, further analyses were carried out by extending the chain length and sampling. Nevertheless, the values for those parameters did not increase sufficiently. With low ESS values, the ‘Decline’ model results cannot be included. The ‘Crash’ results did produce sufficient ESS values. This suggests that a difference in the prior demographic model could be affecting the parameter sampling and, therefore, the discrepancy in mutation rates and age of root.
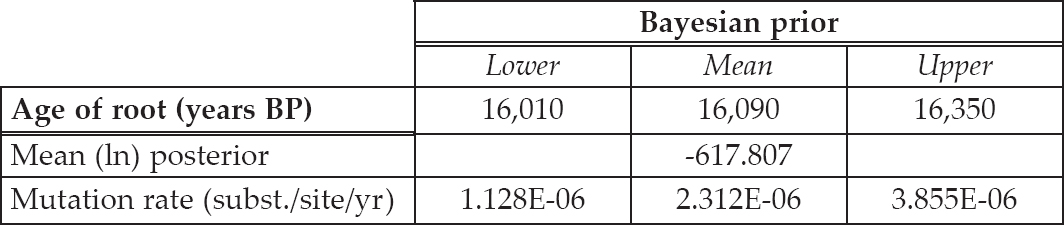
Table 6.4. Results from the Bayesian analyses on the thirty-one simulated sequences from the Crash model. The age of root estimate is measured as years before present (BP) and the mutation rate is measured as substitutions per site per year. The chain length was set at 10 million iterations, with sampling every 20,000 iterations.
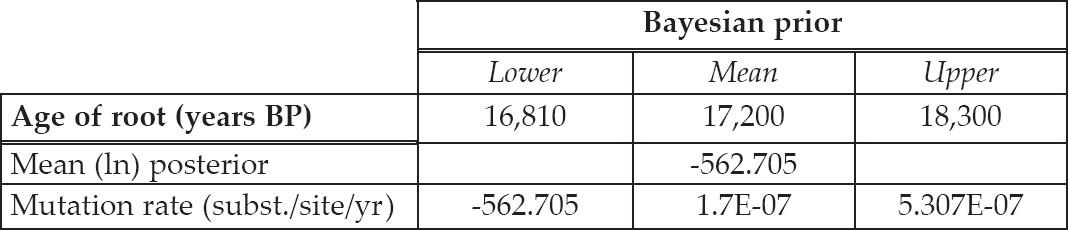
Table 6.5. Results from the Bayesian analyses on the thirty-one simulated sequences from the Decline model. The age of root estimate is measured as years before present (BP) and the mutation rate is measured as substitutions per site per year. The chain length was set at 10 million iterations, with sampling every 20,000 iterations.
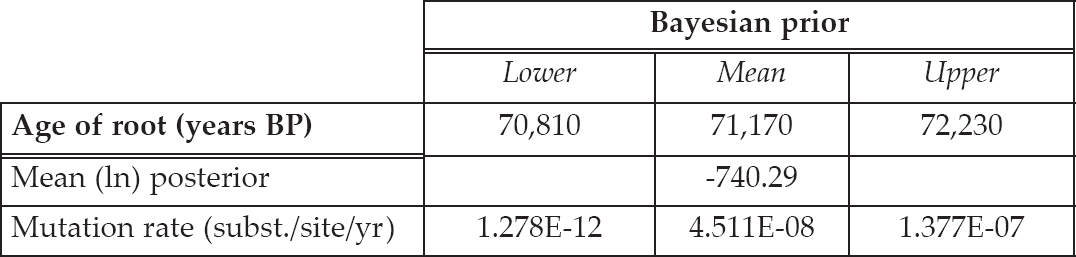
Table 6.6. Results from the Bayesian analyses on the complete simulated sequences from the Decline model. The age of root estimate is measured as years before present (BP) and the mutation rate is measured as substitutions per site per year. The chain length was set at 20 million and the frequency of sampling was every 5000 iterations.
It is clear that several factors are preventing a demographic model from being achieved. The final evolutionary parameters show it is the input information that is largely affecting the results; demographic priors and good quality DNA data.
Discussion
Establishing a set of criteria for genetic data
Three themes run concurrently in these results, highlighting their importance as criteria to consider that the genetic data must adhere to for demographic model reconstruction. The first is archaeological considerations; i.e. obtaining good quality DNA. DNA damage affects the results, producing false diversity and short sequence lengths, resulting in erroneous parameters, such as mutation rate, age of root, and invalid BSP. From all analyses, the second criterion is the importance of sampling across all time periods, geographic regions, and clades within a known phylogenetic tree. In addition, it must be ensured that samples are sufficient in quantity and have selected an appropriate gene. This has implications for the final criterion, which are Bayesian considerations. These concern the information that is known about a gene to input as a prior. Such information includes the mutation rate and the phylogenetic tree, which is dependent upon the gene that is sampled. Finally, it is important that complex demographic models be accounted for in order to input appropriate priors.
Application of criterion to the Neanderthal data
Archaeological Considerations
Despite technological advances, three issues still hinder the retrieval of good quality DNA: genome coverage (for the Neanderthal genome there is <2%), possibility of contamination by modern humans, and DNA degradation, which occurs post-mortem (Hofreiter et al. 2004; Pääbo et al. 2004). This study indicates that DNA degradation is the primary factor influencing the final results. Factors affecting the rate of degradation include age of the material, burial context (Eglinton et al. 1991), and environmental conditions, such as temperature (Smith et al. 2001) and pH (Pruvost et al. 2007). The biochemical damage incurred leads to nucleotide mis-incorporations and modified bases predisposing DNA to fragment, resulting in short sequence lengths (Pääbo et al. 2004). Models of DNA degradation can be incorporated into analyses as more is learnt. Before the introduction of high-throughput sequencing, Neanderthal genetic data was limited to mitochondrial DNA. The development of sequencing technology (Margulies et al. 2005) has led to a vast increase in the amount of Neanderthal genetic data available for use (Green et al. 2006, 2010; Noonan et al. 2006), including autosomal genes. The discovery of a new hominin species (Denisovans) (Krause et al. 2010; Reich et al. 2010) means that Neanderthal genetic data can be analysed in this context, providing more detail on Neanderthal evolutionary and phylogenetic history. Prevention of DNA contamination is an issue that starts at excavation (Brown 1998; Fortea et al. 2008; Pruvost et al. 2007) that can later be assessed (Green et al. 2010; Meyer et al. 2012), and a recent development in sequencing has enabled high coverage of ancient genomes to be obtained (Meyer et al. 2012).
Importance of Sampling
Bearing these archaeological considerations in mind, it is clear why the age distribution of the Neanderthal samples is not uniform (Figure 6.3). Enough samples need to be selected across a large geographical area that fall within the time range 60–100 kyr BP. If an additional five DNA samples (of sufficient quality and length) were in every bin (organised into every 5,000 years), this would yield a total of forty Neanderthal samples, which, when added to the current fourteen, provides fifty-four sequences – more than the bison minimum sequences required by BEAST to potentially provide a genetic dataset suitable for demographic analysis.
The accuracy of sample dates is important for calibration (Debruyne and Poinar 2009). However, when considering the time range for which samples need to be dated to and given the limits for radiocarbon dating, obtaining samples for the time period 60 kyr BP and beyond proves problematic. In this case, other dating methods need to be employed, such as thermoluminescence and electron spin resonance (Dreimanis et al. 1978; Grün and Stringer 1991). Dates near the 25 kyr BP mark do come with decreased reliability (Pettitt et al. 2003) and interpreting radiocarbon dates do come with considerations (Higham 2011).
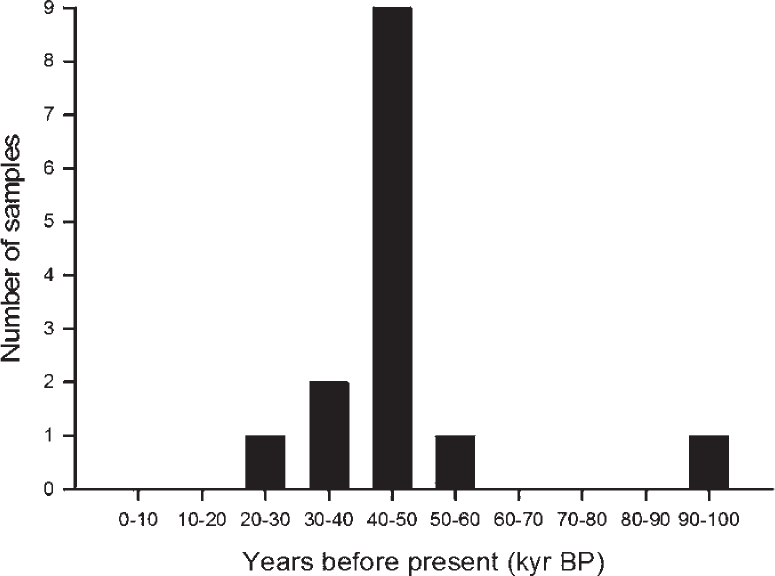
Figure 6.3. Age distribution of the dated Neanderthal samples that have had their mitochondrial HVR1 analysed. See Table 1 for details. The bar graph depicts the samples in bins of 10,000 years.
When selecting suitable genes, Ho and Shapiro (2011) suggest using loci with high evolutionary rates and high informative content, either in the form of sequence length or variable regions. Currently, the Neanderthal sequences have low information content (Debruyne and Poinar 2011). However, improved sequencing technology continues to yield a vast amount of Neanderthal DNA data, including autosomal and mitochondrial (Table 6.1) and, with it, better quality and higher coverage. In addition, using multi-locus data increases the accuracy of obtaining demographic histories (Ho and Shapiro 2011).
Bayesian Considerations
While BEAST does estimate a genealogy, evolutionary model and demographic history, the output is dependent on the priors. The accuracy of the priors, therefore, becomes paramount. Figure 1 in Dalen et al. (2012, 1894) is the most recent phylogenetic tree estimated using BEAST (Drummond et al. 2012), using the mitochondrial control region and the HKY substitution model (Strimmer and von Haessler 2003). The phylogenetic results produced by Dalen et al. (2012) are supported by Fabre et al. (2009), where the Neanderthal population is divided into east and west sub-populations. However, Fabre et al. (2009) do specify a third southern Neanderthal sub-population. Nevertheless, this should dictate thorough sampling within a tree. In a previous study, Briggs et al. (2009) analysed five complete mitochondrial genomes under the Kimura-2 substitution model. Knowledge of the selected gene(s) becomes important as this directs the substitution (evolutionary) model and the mutation rate used as input. In this case, it is worth considering that natural selection may be acting on the mitochondrial genome and to different extents within the genome (Endicott et al. 2010) which can be accounted for (Soares et al. 2009). Additionally, focusing only on the mitochondrial genome will produce a demographic history reflecting only a quarter of the true Ne due to mode of inheritance. This further emphasises the importance of using multi-locus data. A revision of the human mutation rate has implications in the calibration of Neanderthal and modern human divergence, and consequently Neanderthal mutation rate estimates (Scally and Durbin 2012).
Finally, developments in software programmes can provide for the complexity within demographic models, which is highlighted by Alves et al. (2012), who argue against Neanderthal and modern human interbreeding by invoking alternative demographic scenarios to explain the results. BayeSSC incorporates a variety of demographic scenarios, and BEAST now includes different demographic models and multi-locus datasets (Drummond and Rambaut 2012). This ensures a thorough analytical platform to enable demographic reconstruction.
Animals as proxies
In this study, bison were used, due to the large quantity and quality of the data, to systematically identify the minimal requirements for genetic data to obtain a demographic model. The results highlighted specific factors that confound the data. In this respect, the bison dataset by proxy has proved invaluable in identifying key criteria important for the Neanderthal genetic data.
In this paper, it was suggested that demographic models could be compared between Neanderthals and hyaenas due to possible sympatry. But studies continue to reveal species specific responses. Thus, even with a demographic model for the Neanderthals, using animals as proxies does not seem possible. But it can provide a general comparison for the Neanderthal demographic model within the topic of carnivore extinctions. Nevertheless, the hyaena analyses here do support results obtained from the bison analyses – appropriate sampling. Therefore, using animals as proxies can be a valuable tool depending on the context as long as limitations are acknowledged.
Currently, the Neanderthal genetic dataset lacks the quantity and quality to reconstruct a demographic model that can identify their extinction triggers. This study has used animal genetic data to identify what the Neanderthal dataset requires in terms of quality and quantity, sampling, and factors affecting demographic model retrieval. Using animals as proxies remains problematic, but it may be possible to utilise them in a wider context instead. The results here have highlighted three key considerations that are required of the Neanderthal genetic data for demographic reconstruction – archaeological, sufficient sampling, and Bayesian – thereby achieving the stated goal. Despite the limitations of the Neanderthal dataset, continued development in sequencing technology and analytical methods allows for improved genetic datasets and more accurate analyses providing a framework for demographic reconstruction allowing questions on Neanderthal extinction to be answered.
Acknowledgments
I would like to thank Paul Pettitt, Terry Brown, and Abigail Bouwman for their support. I am grateful for advice on the software programmes from Christian Anderson, Emma Finlayson, Sarah Ayling, Simon Ho, and Mark Thomas. Additionally, I would like to thank Andrew Chamberlain, Ian Barnes, and Beth Shapiro for useful comments and those who facilitated the sampling and 14C dating of the hyaena teeth: the NERC ORADS facility including Tom Higham, Diane Baker, and Mark White. I would also like to thank the reviewer.
Bibliography
Alves, T., Hanulova, A. S., Foll, M. and Excoffier, L. (2012) Genomic Data Reveal a Complex Making of Humans. PLoS Genetics 8(7), e1002837.
Axelsson, E., Willerslev, E., Gilbert, M. T. and Nielsen, R. (2008) The Effect of Ancient DNA Damage on Inferences of Demographic Histories. Molecular Biology and Evolution 25(10), 2181–187.
Banks, W. E., D’Errico, F., Peterson, A. T., Kageyama, M., Sima, A. and Sánchez-Goni, M. F. (2008) Neanderthal Extinction by Competitive Exclusion. PLoS One 3(12), e3972.
Basu, A. and Majumder, P. P. (2003) A Comparison of Two Population Statistical Methods for Estimating the Time to Most Recent Common Ancestor (TMRCA) from a Sample of DNA Sequences. Journal of Genetics 82, 7–12.
Beauval, C., Maureille, B., Lacrampe-Cuyabère, F., Serre, D., Peressinotto, D., Bordes, J.-G., Cochard, D., Couchoud, I., Dubrasquet, D., Laroulandie, V., Lenoble, A., Mallye, J.-B., Pasty, S., Primault, J., Rohland, N., Pääbo, S. and Trinkaus, E. (2005) A Late Neandertal Femur from Les Rochers-de-Villeneuve, France. Proceedings from the National Academy of Sciences 102(20), 7085–90.
Briggs, A.W., Stenzel, U., Johnson, P. L. F., Green, R. E., Kelso, J., Prüfer, K., Meyer, M., Krause, J., Ronan, M. T., Lachmann, M. and Pääbo, S. (2007) Patterns of Damage in Genomic DNA Sequences from a Neandertal. Proceedings of the National Academy of Sciences 104(37), 14616–621.
Briggs, A. W., Good, J. M., Green, R. E., Krause, J., Maricic, T., Stenzel, U., Lalueza-Fox, C., Rudan, P., Brajković, D., Kućan, Z., Gušic, I., Schmitz, R., Doronichev, V. B., Golovanova, L. V., de la Rasilla, M., Fortea, J., Rosas, A. and Pääbo, S. (2009) Targeted Retrieval and Analysis of Five Neandertal mtDNA Genomes. Science 325, 318–21.
Brown, K. (1998) Keeping it Clean: The collection and storage of ancient DNA samples from the field. The Archaeologist 33, 16–17.
Burbano, H. A., Hodges, E., Green, R. E., Briggs, A. W., Krause, J., Meyer, M., Good, J. M., Maricic, T., Johnson, P. L. F., Xuan, Z., Rooks, M., Bhattacharjee, A., Brizuela, L., Albert, F. W., de la Rasilla, M., Fortea, J., Rosas, A., Lachmann, M., Hannon, G. J. and Pääbo, S. (2010) Targeted Investigation of the Neandertal Genome by Array-Based Sequence Capture. Science 238, 723–25.
Caramelli, D., Lalueza-Fox, C., Condemi, S., Longo, L., Milani, L., Manfredini, A., de Saint Pierre, M., Adoni, F., Lari, M., Giunti, P., Ricci, S., Casoli, A., Calafell, F., Mallegni, F., Bertranpetit, J., Stanyon, R., Bertorelle, G. and Barbujani, G. (2006) A Highly Divergent mtDNA Sequence in a Neanderthal Individual from Italy. Current Biology 16(16), R630-R632.
Dalen, L., Orlando, L., Shapiro, B., Brandstrøm-Durling, M., Quam, R., Gilbert, M. T. P., Fernández-Lomana, J. C. D., Willerslev, E., Arsuaga, J. L. and Gütherström, A. (2012) Partial Genetic Turnover in Neandertals: Continuity in the east and population replacement in the west. Molecular Biology and Evolution 29(8), 1893–897.
Dansgaard, W., Johnsen, S. J., Clausen, H. B., Dahl-Jensen, D., Gundestrup, N. S., Hammer, C. U., Hvidberg, C. S., Steffensen, J. P., Svenbjörnsdottir, A. E., Jouzel, J. and Bond, G. (2003) Evidence for General Instability of Past Climate from a 250-kyr Ice Core Record. Nature 364, 218–20.
Debruyne, R. and Poinar, H. (2009) Time Dependency of Molecular Rates in Ancient DNA Data Sets, A Sampling Artifact? Systematic Biology 58(3), 48–360.
D’Errico, F. and Sánchez-Goni, M. F. (2003) Neandertal Extinction and the Millennial Scale Climatic Variability of OIS-3. Quaternary Science Reviews 22, 769–88.
Dodge, D. R. (2012) A Molecular Approach to Neanderthal Extinction. Quaternary International 259, 22–32.
Dodge, D. R., Bouwman, A. B., Pettitt, P. B. and Brown, T. A. (2012) Mitochondrial DNA Haplotypes of Devensian Hyaenas from Creswell Crags, England. Archaeological and Anthropological Sciences 4, 161–66.
Dreimanis, A., Hutt, G., Raukas, A. and Whippey, P. W. (1978) Dating Methods of Pleistocene Deposits and their Problems 1: Thermoluminescence dating. Geoscience Canada 52(2), 55–60.
Drummond, A. J. and Rambaut, A. (2007) BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evolutionary Biology 7, 214–29.
Drummond, A. J., Rambaut, A., Shapiro, B. and Pybus, O. (2005) Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Molecular Biology and Evolution 22(5), 1185–192.
Drummond, A. J., Suchard, M., Xie, D. and Rambaut, A. (2012) Bayesian Phylogenetics with BEAUTi and the BEAST 1.7. Molecular Biology and Evolution 29(8), 1969–973.
Drummond, A. J., Pybus, O. G., Rambaut, A., Forsberg R. and Rodrigo, A. G. (2003) Measurably Evolving Populations. Trends in Ecology and Evolution 18(9), 481–88.
Durand, E. Y., Patterson, N., Reich, D. and Slatkin, M. (2011) Testing for Ancient Admixture between Closely Related Populations. Molecular Biology and Evolution 28(8), 2239–252.
Eglinton, G., Logan, G. A., Ambler, R. P., Boon, J. J. and Perizonius, W. R. K. (1991) Molecular Preservation. Philosophical Transactions of the Royal Society, Series B: Biological Sciences 333, 315–28.
Emerson, B. C. (2007) Alarm Bells for the Molecular Clock? No support for Ho et al.’s model of time-dependent molecular rate estimates. Systematic Biology 56(2), 337–45.
Endicott, P. L., Ho, S. Y. W. and Stringer, C. B. (2010) Using Genetic Evidence to Evaluate Four Palaeoanthropological Hypotheses for the Timing of Neanderthal and Modern Human Origins. Journal of Human Evolution 59(1), 87–95.
Eriksson, A. and Manica, A. (2012) Effect of Ancient Population Structure on the Degree of Polymorphism Shared between Modern Human Populations and Ancient Hominins. Proceedings of the National Academy of Sciences 109(35), 13956–960.
Excoffier, L., Novembre, J. and Schneider, S. (2000) SimCoal: A general coalescent program for simulation of molecular data in interconnected populations with arbitrary demography. Journal of Heredity 91, 506–09.
Fabre, V., Condemi, S. and Degioanni, A. (2009) Genetic Evidence of Geographical Groups among Neanderthals. PLoS One 4(4), e5151.
Finlayson, C., Pacheco, F. G., Rodríguez-Vidal, J., Fa, D. A., López, J. M. G., Pérez, A. S., Finlayson, G., Allue, E., Preysler, J. B., Cáceres, I., Carrión, J. S., Jalvo, Y. F., Gleed-Owen, C. P., Espejo, F. J. J., López, P., Sáez, J. A. L., Cantal, J. A. R., Marco, A. S., Guzman, F. G., Brown, K., Fuentes, N., Valarion, C. A., Villalpando, A., Stringer, C. B., Ruiz, F. M. and Sakamoto, T. (2006) Late Survival of Neanderthals at the Southernmost Extreme of Europe. Nature 443, 850–53.
Finlayson, C., Fa, D. A., Jiménez-Espejo, F. J., Carrión, J. S., Finlayson, G., Pacheco, F. G., Vidal, J. R., Stringer, C. B. and Ruiz, F. M. (2008) Gorham’s Cave, Gibraltar – The persistence of a Neanderthal population. Quaternary International 181, 64–71.
Fortea, J., de la Rasilla, M., Garcia-Tabernero, A., Gigli, E., Rosas, A. and Lalueza-Fox, C. (2008) Excavation Protocol of Bone Remains for Neandertal DNA Analysis in El Sidrón Cave (Asturias, Spain). Journal of Human Evolution 55, 353–57.
Golanova, L. V., Doronichev, V. B., Cleghorn, N. E., Koulkova, A., Sapelko, T. V. and Shackley, M. S. (2010) Significance of Ecological Factors in the Middle to Upper Palaeolithic Transition. Current Anthropology 51(5), 655–91.
Green, R. E., Krause, J., Ptak, S. E., Briggs, A. W., Ronan, M. T., Simons, J. F., Du, L., Egholm, M., Rothberg J. M., Paunovic, M. and Pääbo, S. (2006) Analysis of One Million Base Pairs of Neanderthal DNA. Nature 444, 330–36.
Green, R. E., Malaspinas, A.-S., Krause, J., Briggs, A. W., Johnson, P. L. F., Uhler, C., Meyer, M., Good, J. M., Maricic, T., Stenzel, U., Prüer, K., Siebauer, M., Burbano, H. A., Ronan, M., Rotheberg, J. M., Egholm, M., Rudan P., Brajkovic, D., Kućan, Z., Gušić, I., Wikstrőm, M., Laakkonen, L., Kelso, J., Slatkin, M. and Pääbo, S. (2008) A Complete Neandertal Mitochondrial Genome Sequence Determined by High-Throughput Sequencing. Cell 134, 416–26.
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., Patterson, N., Li, H., Zhai, W., Fritz, M. H.-Y., Hansen, N. F., Durand, E. Y., Malaspinas, A.-S., Jensen, J. D., Marques-Bonet, T., Alkan, C., Prüfer, K., Meyer, M., Burbano, H. A., Good, J. M., Schultz, R., Aximu-Petri, A., Butthof, A., Höber, B., Höffner, B., Siegemund, M., Weihmann, A., Nusbaum, C., Lander, E. S., Russ, C., Novod, N., Affourtit, J., Egholm, M., Verna, C., Rudan, P., Brajkovic, D., Kucan, Z., Gušic, I., Doronichev, V. B., Golovanova, L. V., Lalueza-Fox, C., de la Rasilla, M., Fortea, J., Rosas, A., Schmitz, R.W., Johnson, P. L. F., Eichler, E. E., Falush, D., Birney, E., Mullikin, J. C., Slatkin, M., Nielsen, R., Kelso, J., Lachmann, M., Reich, D. and Pääbo., S. (2010) A Draft Sequence of the Neandertal Genome. Science 328, 710–22.
Griffiths, R. C. and Tavarè, S. (1994) Sampling Theory for Neutral Alleles in a Varying Environment. Philosophical Transactions of the Royal Society of London, Series B 344, 403–10.
Grün, R. and Stringer, C. B. (1991) Electron Spin Resonance Dating and the Evolution of Modern Humans. Archaeometry 33(2), 153–99.
Hammer, M. F., Woerner, A. E., Mendez, F. L., Watkins, J. C. and Wall J. D. (2011) Genetic Evidence for Archaic Admixture in Africa. Proceedings of the National Academy of Sciences 108(37), 15123–128.
Hardy, B. L. (2010) Climate Variability and Plant Food Distribution in Pleistocene Europe: Implications for Neanderthal diet and subsistence. Quaternary Science Reviews 29, 662–79.
Higham, T. (2011) European Middle and Upper Palaeolithic Radiocarbon Dates are often Older than they Look: Problems with previous dates and some remedies. Antiquity 85, 235–49.
Hockett, B. (2012) The Consequences of Middle Paleolithic Diets on Pregnant Neanderthal Women. Quaternary International 264, 78–82.
Hockett, B. and Haws, J. A. (2005) Nutritional Ecology and the Human Demography of Neandertal Extinction. Quaternary International 137, 21–34.
Hodgson, J. A., Bergey, C. M. and Disotell, J. R. (2010) Neandertal Genome: The ins and outs of African genetic diversity. Current Biology 20(12), 517–19.
Ho, S. Y. W. (2009) An Examination of Phylogenetic Methods of Substitution Rate Variation among Lineages. Biology Letters 5, 421–24.
Ho, S. Y. W., Phillips, M. J., Cooper, A. and Drummond, A. (2005) Time Dependency of Molecular Rate Estimates and Systematic Overestimation of Recent Divergence Times. Molecular Biology and Evolution 22(7), 1561–568.
Ho, S. Y. W., Kolokotronis, S.-O. and Allaby, R. G. (2007) Elevated Substitution Rates Estimated from Ancient DNA Sequences. Biology Letters 3, 702–05.
Ho, S. Y. W. and Shapiro, B. (2011) Skyline Plot Methods for Estimating Demographic History from Nucleotide Sequences. Molecular Ecology Resources 11, 423–34.
Hofreiter, M., Serre, D., Rohland, N., Rabeder, G., Nagel, D., Conard, N., Münzel, S. and Pääbo, S. (2004) Lack of Phylogeography in European Mammals before the Last Glaciation. Proceedings of the National Academy of Sciences 101, 12963–968.
Hublin, J.-J. and Roebroeks, W. (2009) Ebb and Flow or Regional Extinctions? On the character of Neandertal occupation of northern environments. Comptes Rendus Palevol 8, 503–09.
Kingman, J. F. C. (1982) On the Genealogy of Large Populations. Journal of Applied Probability (Essays in Statistical Science) 19, 27–43.
Klein, R. G. (2000) Archeology and the Evolution of Human Behavior. Evolutionary Anthropology 9(1), 17–36.
Krause, J., Fu, Q., Good, J. M., Viola, B., Shunkov, M. V., Derevianko, A. P. and Pääbo, S. (2010) The Complete Mitochondrial DNA Genome of an Unknown Hominin from Southern Siberia. Nature 264, 894–97.
Krause, J., Orlando, L., Serre, D., Viola, B., Prüfer, K., Richards, M. P., Hublin, J.-J., Hänni, C., Derevianko, A. P. and Pääbo, S. (2007a) Neanderthals in Central Asia and Siberia. Nature 449, 902–04.
Krause, R. E., Lalueza-Fox, C., Orlando, L., Enard, W., Green, R. E., Burbano, H. A., Hublin, J.-J., Hänni, C., Fortea, J., de la Rasilla, M., Betranpetit, J., Rosas, A. and Pääbo, S. (2007b) The Derived FOXP2 Variant of Modern Humans was Shared with Neandertals. Current Biology 17, 1–5.
Krings, M., Stone, A., Schmitz, R. W., Krainitzki, H., Stoneking M. and Pääbo, S. (1997) Neandertal DNA Sequences and the Origin of Modern Humans. Cell 90, 19–30.
Krings, M., Geisert, H., Schmitz, R. W., Krainitzki, H. and Pääbo, S. (1999) DNA Sequence of the Mitochondrial Hypervariable Region II from the Neandertal Type Specimen. Proceedings of the National Academy of Sciences 96, 581–85.
Krings, M., Capelli, C., Tschentscher, F., Geisert, H., Meyer, S., Von Haesler, A., Grosschmidt, K., Possnert, G., Paunovic, M. and Pääbo, S. (2000) A View of Neanderthal Genetic Diversity. Nature Genetics 26, 144–46.
Kruuk, H. (1972) The Spotted Hyaena: A study of predation and social behavior. Chicago: University of Chicago Press.
Lalueza-Fox, C., Gigli, E., de la Rasilla, M., Fortea, J., Rosas, A., Betranpetit, J. and Krause, J. (2008) Genetic Characterisation of the ABO Blood Group in Neandertals. BMC Biology 8, 342–46.
Lalueza-Fox, C., Gigli, E., de la Rasilla, M., Fortea, J. and Rosas, A. (2009) Bitter Taste Perception in Neanderthals through the Analysis of the TAS2R38 Gene. Biology Letters 5, 809–11.
Lalueza-Fox, C., Krause, J., Caramelli, D., Catalano, G., Milani, L., Sampietro, M. L., Calafell, F., Martìnez-Maza, C., Bastir, M., Garcìa-Tabernero, G., de la Rasilla, M., Fortea, J., Pääbo, S., Bertranpetit, J. and Rosas, A. (2006) Mitochondrial DNA of an Iberian Neandertal Suggests a Population Affinity with other European Neandertals. Current Biology 16, R629-R630.
Lalueza-Fox, C., Römpler, H., Caramelli, D., Stäubert, C., Catalano, G., Hughes, D., Rohland, N., Pilli, E., Longo, L., Condemi, S., de la Rasilla, M., Fortea, J., Rosas, A., Stoneking, M., Schöneberg, T., Betranpetit, J. and Hofreiter, M. (2007) A Melanocortin 1 Receptor Allele Suggests Varying Pigmentation among Neanderthals. Science 318, 1453–454.
Lalueza-Fox, C., Sampietro, M. L., Caramelli, D., Puder, Y., Lari, M., Calafell, F., Martínez-Maza, C., Bastir, M., Fortea, J., de la Rasilla, M., Bertranpetit, J. and Rosas, A. (2005) Neandertal Evolutionary Genetics: Mitochondrial DNA data from the Iberian peninsula. Molecular Biology and Evolution 22(4), 1077–81.
Longo, L., Boaretto, E., Caramelli, D., Giunti, P., Lari, M., Milani, L., Mannino, M. A., Sala, B., Hohenstein, U. T. and Condemi, S. (2012) Did Neandertals and Anatomically Modern Humans Coexist in Northern Italy during the Late MIS 3? Quaternary International 259, 102–12.
Lopez-Valenzuela, M., Ramírez, O., Rosas, A., García-Vargas, S., de la Rasilla, M., Lalueza-Fox, C. and Espinosa-Parrila, Y. (2012) An Ancestral miR-1304 Allele Present in Neanderthals Regulates Genes Involved in Enamel Formation and Could Explain Dental Differences with Modern Humans. Molecular Biology and Evolution 29(7), 1797–806.
Lorenzen, E. D., Nogués-Bravo, D., Orlando, L., Weinstock, J., Binladen, J., Marske, K. A., Ugan, A., Borregaard, M. K., Gilbert, M. T. P., Nielsen, R., Ho, S. Y. W., Goebel, T., Graf, K. E., Byers, D., Stenderup, J. T., Rasmussen, M., Campos, P. F., Leonard, J. A., Koepfli, K.-P., Froese, D., Zazula, G., Stafford Jr, T. W., Aaris-Sørensen, K., Batra, P., Haywood, A. M., Singarayer, J. S., Valdes, P. J., Boeskorov, G., Burns, J. A. B., Davydov, S. P., Haile, J., Jenkins, D. L., Kosintsev, P., Kuznetsova, T., Lai, X., Martin, L.D., McDonald, H.G., Mol, D., Meldgaard, M., Munch, K., Stephan, E., Sablin, M., Sommer, R. S., Sipko, T., Scott, E., Suchard, M. A., Tikhonov, A., Willerslev, R., Wayne, R. K., Cooper, A., Hofreiter, M., Sher, A., Shapiro, B., Rahbek, C. and Willerslev, E. (2011) Species-specific Responses of Late Quaternary Megafauna to Climate and Humans. Nature 479, 359–65.
Lowe, J., Barton, N., Blockley, S., Bronk-Ramsey, C., Cullen, V. L., Davies, W., Gamble, C., Grant, K., Hardiman, M., Housley, R., Lane, C. S., Lee, S., Lewis, M., MacLeod, A., Menzies, M., Müller, W., Pollard, M., Price, C., Roberts, A. P., Rohling, E. J., Satow, C., Smith, V. C., Stringer, C. B., Tomlinson, E. L., White, D., Albert, P., Arienzo, I., Barker, G., Borić, D., Carandente, A., Civetta, L., Ferrier, C., Guadelli, J.-L., Karkanas, P., Koumouzelis, M., Müller, U. C., Orsi, G., Pross, J., Rosi, M., Shalamanov-Korobar, L., Sirakov, N. and Tzedakis, P. C. (2012) Volcanic Ash Layers Illuminate the Resilience of Neanderthals and Early Modern Humans to Natural Hazards. Proceedings of the National Academy of Science 109(34), 13532–537.
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., Bemben, L. A., Berka, J., Braverman, M. S., Chen, Y.-J., Chen, Z., Dewell, S. B., Du, L., Fierro, J. M., Gomes, X. V., Godwin, B. C., He, W., Helgesen, S., Ho, C. H., Irzyk, G. P., Jando, S. C., Alenquer, M. L. I., Jarvie, T. P., Jirage, K. B., Kim, J.-B., Knight, J. R., Lanza., J. R., Leamon, J. H., Lefkowitz, S. M., Lei, M., Li, J. L., Lohman, K. L., Lu, H., Makhijani, V. B., McDade, K. E., McKenna, M. P., Myers, E. W., Nickerson, E., Nobile, J. R., Plant, R., Puc, B. P., Ronan, M. T., Roth, G. T., Sarkis, G. J., Simons, J. F., Simpson, J. W., Srinivasan, M., Tartaro, K. R., Tomasz, A., Vogt, K. A., Volkmer, G. A., Wang, S. H., Wang, Y., Weiner, M. P., Yu, P., Begley, R. F. and Rothberg, J. M. (2005) Genome Sequencing in Microfabricated High-density Picolitre Reactions. Nature 437, 376–80.
Meyer, M., Kircher, M., Gansauge, M.-T., Li, H., Racimo, F., Mallick, S., Schraiber, J. G., Jay, F., Prüfer, K., de Filippo, C., Sudmant, P. H., Alkan, C., Fu, Q., Do, R., Rohland, N., Tandon, A., Siebauer, M., Green, R. E., Bryc, K., Briggs, A. W., Stenzel, U., Dabney, J., Shendure, J., Kitzman, J., Hammer, M. F., Shunkov, M. V., Derevianko, A. P., Patterson, N., Andrés, A. M., Eichler, E. E., Slatkin, M., Reich, D., Kelso, J. and Pääbo, S. (2012). A High-coverage Genome Sequence from an Archaic Denisovan Individual. Science 338, 222–26.
Navascués, M. and Emerson, B. C. (2009) Elevated Substitution Rate Estimates from Ancient DNA: Model violation and bias of Bayesian models. Molecular Ecology 18, 4390-397.
Noonan, J. P., Coop, G., Kudaravalli, S., Smith, D., Krause, J., Alessi, J., Chen, F., Platt, D. and Pääbo, S. (2006) Sequencing and Analysis of Neanderthal Genomic DNA. Science 314, 1113–118.
O’Regan, H. J., Turner, A., Bishop, L. C., Elton, S. and Lamb, A. (2011) Hominins without Fellow Travellers? First appearances and inferred dispersals of Afro-Eurasian large mammals in the Plio-Pleistocene. Quaternary Science Reviews 30, 1343–352.
Orlando, L., Darlu, P., Toussaint, M., Bonjean, D., Otte, M. and Hänni, C. (2006) Revisiting Neandertal Diversity with a 100,000 Year Old mtDNA Sequence. Current Biology 16(11), 400–02.
Ovchinnikov, I. V., Götherström, A., Romanova, G. P., Kharitonov, V. M., Lidén, K. and Goodwin, W. (2000) Molecular Analysis of Neanderthal DNA from the Northern Caucasus. Nature 404, 490–93.
Pääbo, S., Poinar, H., Serre, D., Jaenicke-Deprés, V., Hebler, J., Rohland, N., Kuch, M., Krause, J., Vigilant, L. and Hofreiter, M. (2004) Genetic Analyses from Ancient DNA. Annual Review of Genetics 38, 645–79.
Pettitt, P. B. (1999) Disappearing from the World: An archaeological perspective on Neanderthal extinction. Oxford Journal of Archaeology 18(3), 217–38.
Pettitt, P. B. (2000) Neanderthal Lifecycles: Developmental and social phases in the lives of the last archaic. World Archaeology 31 (3), 351–66.
Pettitt, P. B., Davies, W., Gamble, C. S. and Richards, M. B. (2003) Palaeolithic Radiocarbon Chronology: Quantifying our confidence beyond two half lives. Journal of Archaeological Science 30, 1685–693.
Pettitt, P. B. and Pike, A. W. G. (2001) Blind in a Cloud of Data: Problems with the chronology of Neanderthal extinction and anatomically modern human expansion. Antiquity 75, 415–20.
Pruvost, M., Schwarz, R., Correia, V. B., Champlot, S., Braguier, S., Morel, N., Fernandez-Jalvo, Y., Grange, T. and Geigl, E.-M. (2007) Freshly Excavated Fossil Bones are Best for Amplification of Ancient DNA. Proceedings of the National Academy of Sciences USA 104(3), 739–44.
R Foundation for Statistical Computing (2009) R2.10.1: The R Project for Statistical Computing. Vienna University of Technology, Austria. Available from: <http://www.r-project.org/index.html> [Accessed 16/02/10]
Reich, D., Green, R.E., Kircher, M., Krause, J., Patterson, N., Durand, E. Y., Viola, B., Briggs, A. W., Stenzel, U., Johnson, P. L. F., Maricic, T., Good, J. M., Marques-Bonet, T., Altan, C., Fu, Q., Mallick, S., Li, H., Meyer, M., Eichler, E. E., Stoneking, M., Richards, M., Talamo, S., Shunkov, M. V., Derevianko, A. P., Hublin, J.-J., Kelso, J., Slatkin, M. and Pääbo, S. (2010) Genetic History of an Archaic Hominin Group from Denisova Cave in Siberia. Nature 468, 1053–60.
Richards, M. P. and Trinkaus, E. (2009) Isotopic Evidence for the Diets of European Neanderthals and Early Modern Humans. Proceedings of the National Academy of Sciences 106(38), 16034–039.
Rohland, N., Pollack, J. L., Nagel, D., Beauval, C. É., Airvaux, J., Pääbo, S. and Hofreiter, M. (2005) The Population History of Extant and Extinct Hyenas. Molecular Biology and Evolution 22(12), 2435–445.
Scally, A. and Durbin, R. (2012) Revising the Human Mutation Rate: Implications for understanding human evolution. Nature Reviews Genetics 13, 745–53.
Schmitz, R. W., Serre, D., Bonani, G., Feine, S., Hillgruber, F., Krainitzki, H., Pääbo, S. and Smith, F. (2002). The Neandertal Type Site Revisited: Interdisciplinary investigations of skeletal remains from the Neander Valley, Germany. Proceedings of the National Academy of Sciences 99(20), 13342–347.
Serre, D., Langaney, A., Chech, M., Teschler-Nicola, M., Paunovic, M., Mennecier, P., Hofreiter, M., Possnert, G. and Pääbo, S. (2004). No Evidence of Neandertal mtDNA Contribution to Early Modern Humans. PLoS Biology 2(3), 313–17.
Sepulchre, P., Ramstein, G., Kageyama, M., Vanhaeren, M., Krinner, G., Sánchez-Goni, M. F. and D’Errico, F. (2007) H4 Abrupt Event and Late Neanderthal Presence in Iberia. Earth, and Planetary Science Letters 258, 238–92.
Shapiro, B., Drummond, A. J, Rambaut, A., Wilson, M., Sher, A., Pybus, O. G., Gilbert, M. T. P., Barnes, I., Binladen, J., Willerslev, E., Hansen, A., Baryshnikov, G. F., Burns, J., Davydov, S., Driver, J., Gubin, S. V., Harington, C. R., Keddie, G., Kosintsev, P., Kunz, M. L., Martin, L. D., Stephenson, R., Storer, J., Tedford, R., Vorobiev, A., Zimov, S. and Cooper, A. (2004) Rise and Fall of the Beringian Steppe Bison. Science 306, 1561–564.
Skinner, M. (1997) Dental Wear in Immature Late Pleistocene European Hominines. Journal of Archaeological Sciences 24, 677–700.
Smith, C. I., Chamberlain, A., Riley, M. S., Cooper, A., Stringer, C. B. and Collins, M. J. (2001) Neanderthal DNA: Not just old, but old and cold? Nature 410, 771–72.
Soares, P., Ermini, L., Thomson, N., Mormina, M., Rito, T., Röhl, A., Salas, A., Oppenheimer, S., Macauley, V. and Richards, M. B. (2009) Correcting for Purifying Selection: An improved human mitochondrial molecular clock. American Journal of Human Genetics 84, 740–59.
Strimmer, K. and von Haeseler, A. (2003) Nucleotide Substitution Models. In: M. Salemi and A.-M. Vandamme (eds) The Phylogenetic Handbook, pp. 72–87. Cambridge: Cambridge University Press.
Stringer, C. B., Pälike, H., van Andel, T. H., Huntley, B., Valdes, P. and Allen, J. R. M. (2003) Climatic Stress and the Extinction of the Neanderthals. In: T. H. van Andel and W. Davies (eds) Neanderthals and Modern Humans in the European Landscape during the Last Glaciation, pp. 233–40. Cambridge: McDonald Institute Monographs for Archaeological Research.
Stringer, C. B., Finlayson, J. C., Barton, R. N. E., Fernández-Jalvo, Y., Cáceres, I., Sabin, R. C., Rhodes, E. J., Currant, A. P., Rodriguez-Vidal, J., Giles-Pacheco, F. and Riquelme-Cantel, J. A. (2008). Neanderthal Exploitation of Marine Mammals in Gibraltar. Proceedings of the National Academy of Sciences 105(38), 14319–324.
Szmidt, C. C., Normand, C., Burr, G. S., Hodgins, G. W. L. and LaMotta, S. (2010) AMS 14C Dating the Protoaurignacian/Early Aurignacian of Isturitz, France. Implications for Neanderthal-modern human interaction and the timing of technical and cultural innovations in Europe. Journal of Archaeological Sciences 37, 758–68.
Turner, A. (1984) Hominids and Fellow Travellers: Human migration into high latitudes as part of a large mammal community. In: R. Foley (ed.) Hominid Evolution and Community, pp. 193–217. London: Academic Press.
Turner, A. (2009) The Evolution of the Guild of Large Carnivora of the British Isles during the Middle and Late Pleistocene. Journal of Quaternary Science 24(8), 991–1005.
Underwood, S. (2008) A Potential Role for Transmissible Spongiform Encephalopathies in Neanderthal Extinction. Medical Hypotheses 71(1), 4–7.
van Andel, T. H. and Davies, W. (eds) (2003) Neanderthals and Modern Humans in the European Landscape during the Last Glaciation. Cambridge: McDonald Institute Monographs for Archaeological Research.
Yotova, V., Lefebvre, J.-F., Moreau, C., Gbeha, E., Hovhannesyan, K., Bourgeois, S., Bédarida, S., Azevedo, L., Amorim, A., Sarkisian, T., Avogbe, P. H., Chabi, N., Dicko, M. H., Amouzou, E. S. K., Sanni, A., Roberts-Thomson, J., Boettcher, B., Scott, R. J. and Labuda, D. (2011) An X-Linked Haplotype of Neandertal Origin is Present among all Non-African Populations. Molecular Biology and Evolution 28(7), 1957–962.
Wang, X., Mitra, N., Secundino, I., Band, K., Cruz, P., Padler-Karavani, V., Verhagen, A., Reid, C., Larie, M., Rizzif, E., Balsamo, C., Corti, G., De Bellis, G., Longo, L., NISC Comparative Sequencing Program, Beggs, W., Caramelli, D., Tishkoff, S.A., Hayakawa, T., Green, E. D., Mullikin, J. C., Nizet, V., Bui, J. and Varki, A. (2012) Specific Inactivation of Two Immunomodulatory SIGLEC Genes during Human Evolution. Proceedings of the National Academy of Sciences 109(25), 9935–940.
Wall, J. D. and Kim, S. K. (2007) Inconsistencies in Neanderthal Genomic DNA Sequences. PLoS Genetics 3(10), e175.
White, M. J. and Pettitt, P. B. (2011) The British Late Middle Palaeolithic: An interpretative synthesis of Neanderthal occupation at the northwestern edge of the Pleistocene world. Journal of World Prehistory 24, 25–97.