After Chapter 4.2, you will be able to:
Almost all reactions in organic chemistry can be divided into one of two groups: oxidation–reduction reactions or nucleophile–electrophile reactions. Nucleophiles, electrophiles, and leaving groups are particularly important to the reactions of alcohols and carbonyl-containing compounds, which we will look at in depth in later chapters. Let’s take a look at how each of these terms is defined.
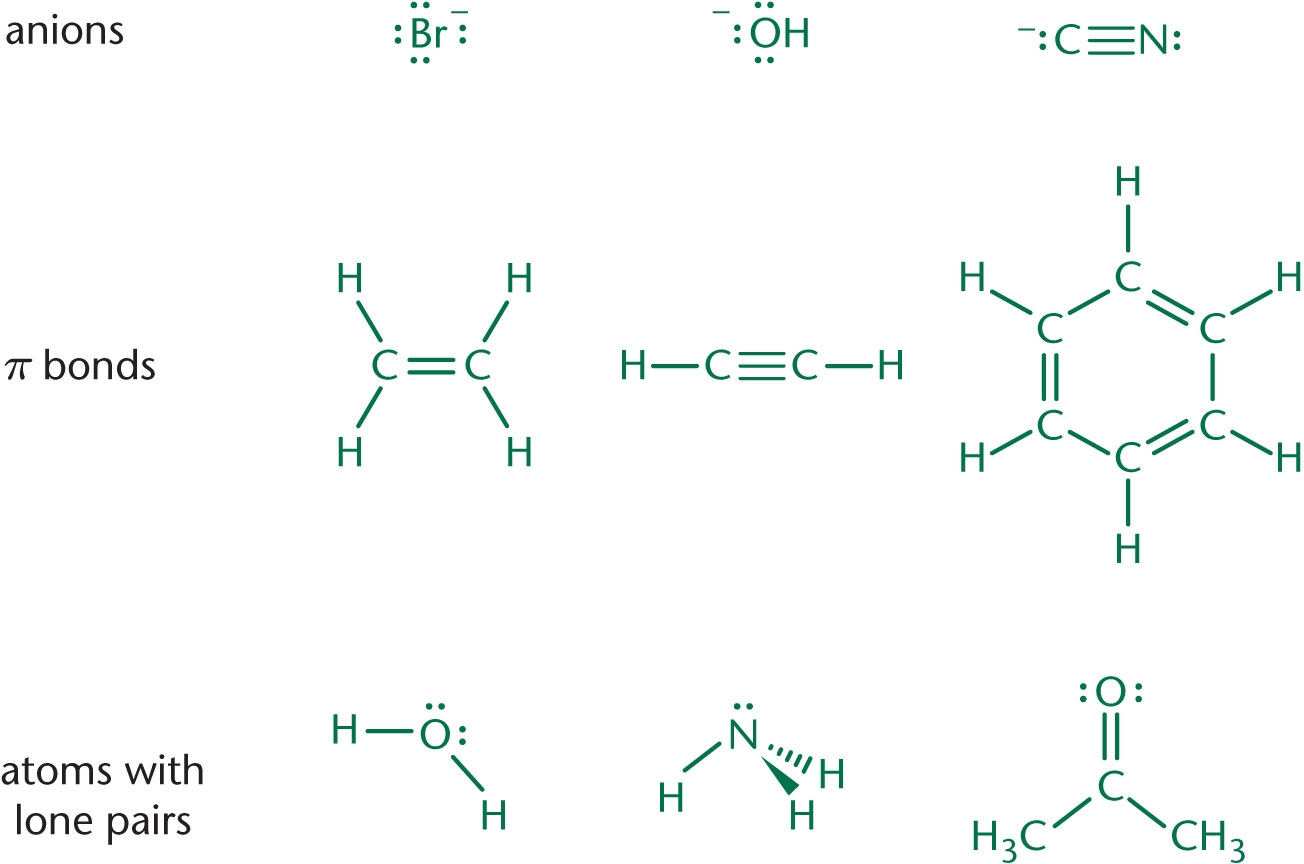
Nucleophiles are defined as “nucleus-loving” species with either lone pairs or π bonds that can form new bonds to electrophiles. You may have noted that nucleophilicity and basicity appear to have similar definitions—and this is true! Good nucleophiles tend to be good bases. There is, however, a distinction between the two. Nucleophile strength is based on relative rates of reaction with a common electrophile—and is therefore a kinetic property. Base strength is related to the equilibrium position of a reaction—and is therefore a thermodynamic property. Some common examples of nucleophiles are shown in Figure 4.2.
Nucleophiles tend to have lone pairs or π bonds that can be used to form covalent bonds to electrophiles. On Test Day, look for carbon, hydrogen, oxygen, or nitrogen (CHON) with a minus sign or lone pair to identify most nucleophiles.
As long as the nucleophilic atom is the same, the more basic the nucleophile, the more reactive it is. This also holds when comparing atoms in the same row of the periodic table, but not when proceeding down a column in the periodic table. Nucleophilicity is determined by four major factors:
The solvent consideration is worth spending a bit more time on. In polar protic solvents, nucleophilicity increases down the periodic table. In polar aprotic solvents, nucleophilicity increases up the periodic table. Examples of both types of solvents are shown in Figure 4.3.
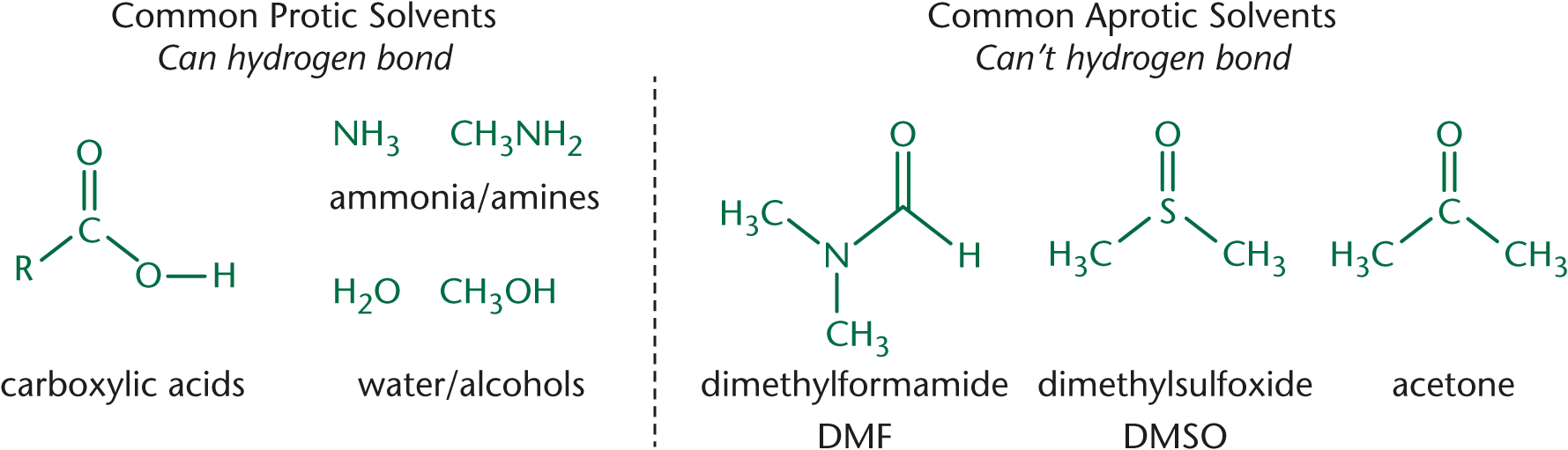
The halogens are good examples of the effects of the solvent on nucleophilicity. In protic solvents, nucleophilicity decreases in the order:
This is because the protons in solution will be attracted to the nucleophile. F– is the conjugate base of HF, a weak acid. As such, it will form bonds with the protons in solution and be less able to access the electrophile to react. I–, on the other hand, is the conjugate base of HI, a strong acid. As such, it is less affected by the protons in solution and can react with the electrophile.
If a solvent is not given on Test Day, assume that the reaction occurs in a polar solvent. Polar solvents—whether protic or aprotic—can dissolve nucleophiles and assist in any reaction in which electrons are moved. Organic chemistry is all about moving electrons, so it’s less common for these reactions to be performed in nonpolar solvents.
In aprotic solvents, on the other hand, nucleophilicity decreases in the order:
This is because there are no protons to get in the way of the attacking nucleophile. In aprotic solvents, nucleophilicity relates directly to basicity.
We won’t use nonpolar solvents with this type of reaction because we need our nucleophile
to dissolve. Because charged molecules are polar by nature, a polar solvent is required
to dissolve the nucleophile as well because like dissolves like
. Examples of strong nucleophiles include HO–, RO–, CN–, and
 . NH3 and
. NH3 and
 are fair nucleophiles, and H2O, ROH, and RCOOH are weak or very weak nucleophiles. As far as functional groups
go, amine groups tend to make good nucleophiles.
are fair nucleophiles, and H2O, ROH, and RCOOH are weak or very weak nucleophiles. As far as functional groups
go, amine groups tend to make good nucleophiles.
We can’t use nonpolar solvents in these nucleophile–electrophile reactions because our reactants are polar—they wouldn’t dissolve!
Electrophiles are defined as “electron-loving” species with a positive charge or positively polarized atom that accepts an electron pair when forming new bonds with a nucleophile. Again, this definition brings to mind Lewis acids. The distinction, as with nucleophiles and bases above, is that electrophilicity is a kinetic property, whereas acidity is a thermodynamic property. Practically, however, electrophiles will almost always act as Lewis acids in reactions. A greater degree of positive charge increases electrophilicity, so a carbocation is more electrophilic than a carbonyl carbon. Some comparisons between electrophiles are drawn in Figure 4.4. Additionally, the nature of the leaving group influences electrophilicity in species without empty orbitals; better leaving groups make it more likely that a reaction will happen. If empty orbitals are present, an incoming nucleophile can make a bond with the electrophile without displacing the leaving group.
Electrophilicity and acidity are effectively identical properties when it comes to reactivity. Just as alcohols, aldehydes and ketones, carboxylic acids, and their derivatives act as acids, they also act as electrophiles and can make good targets for nucleophilic attack.

The carboxylic acid derivatives are often ranked by electrophilicity. Anhydrides are the most reactive, followed by carboxylic acids and esters, and then amides. In practical terms, this means that derivatives of higher reactivity can form derivatives of lower reactivity but not vice versa, similar to the acid–base reactions described previously.
Leaving groups are the molecular fragments that retain the electrons after heterolysis. Heterolytic reactions are essentially the opposite of coordinate covalent bond formation: a bond is broken and both electrons are given to one of the two products. The best leaving groups will be able to stabilize the extra electrons. Weak bases are more stable with an extra set of electrons and therefore make good leaving groups. By this logic, the conjugate bases of strong acids (like I–, Br–, and Cl–) tend to make good leaving groups. Leaving group ability can be augmented by resonance and by inductive effects from electron-withdrawing groups: these help delocalize and stabilize negative charge.
Alkanes and hydrogen ions will almost never serve as leaving groups because they form very reactive, strongly basic anions. We can think of leaving groups and nucleophiles as serving opposite functions. In substitution reactions, the weaker base (the leaving group) is replaced by the stronger base (the nucleophile).
Just like acid–base reactions, nucleophilic attack will only occur if the reactants are more reactive than the products. Thus, the nucleophile must be more reactive than the leaving group.
Nucleophilic substitution reactions are perfect examples for demonstrating nucleophile–electrophile reactions. In both SN1 and SN2 reactions, a nucleophile forms a bond with a substrate carbon and a leaving group leaves.
Unimolecular nucleophilic substitution (SN1) reactions contain two steps. The first step is the rate-limiting step in which the leaving group leaves, generating a positively charged carbocation. The nucleophile then attacks the carbocation, resulting in the substitution product. This mechanism is shown in Figure 4.5 below.
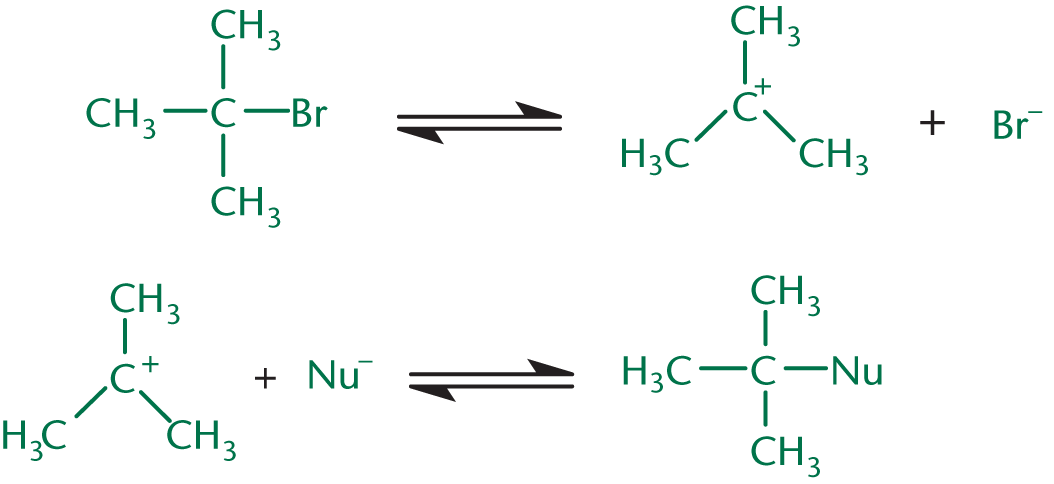
The more substituted the carbocation, the more stable it is because the alkyl groups act as electron donors, stabilizing the positive charge. Because the formation of the carbocation is the rate-limiting step, the rate of the reaction depends only on the concentration of the substrate: rate=k[R–L], where R–L is an alkyl group containing a leaving group. This is a first-order reaction; anything that accelerates the formation of the carbocation will increase the rate of an SN1 reaction.
Because SN1 reactions pass through a planar intermediate before the nucleophile attacks, the product will usually be a racemic mixture. The incoming nucleophile can attack the carbocation from either side, resulting in varied stereochemistry.
Bimolecular nucleophilic substitution (SN2) reactions contain only one step, in which the nucleophile attacks the compound at the same time as the leaving group leaves. Because this reaction has only one step, we call it a concerted reaction. The reaction is called bimolecular because this single rate-limiting step involves two molecules.
In SN2 reactions, the nucleophile actively displaces the leaving group in a backside attack. For this to occur, the nucleophile must be strong, and the substrate cannot be sterically hindered. Therefore, the less substituted the carbon, the more reactive it is in SN2 reactions. Note that this is the opposite of the trend for SN1 reactions. The one-step mechanism is shown in Figure 4.6.
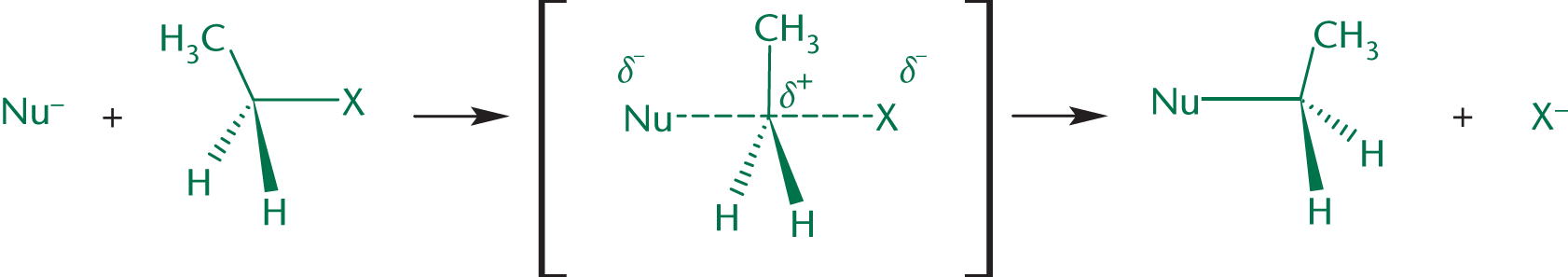
The single step of an SN2 reaction involves two reacting species: the substrate (often an alkyl halide, tosylate, or mesylate) and the nucleophile. Therefore, the concentrations of both have a role in determining the rate: rate = k[Nu:][R–L]
SN2 reactions are accompanied by an inversion of relative configuration. Much like an umbrella being turned inside out on a blustery day, the position of substituents around the substrate carbon will be inverted. If the nucleophile and leaving group have the same priority in their respective molecules, this inversion will also correspond to a change in absolute configuration from (R) to (S) or vice-versa. This is an example of a stereospecific reaction, one in which the configuration of the reactant determines the configuration of the product due to the reaction mechanism.