CHAPTER 46
Tubular and Cystic Kidney Disorders
The kidneys filter and cleanse the blood. They also maintain the body’s balance of water, dissolved salts (electrolytes, such as sodium, potassium, and calcium), and nutrients in the blood. The kidneys begin these tasks by filtering the blood as it flows through microscopic tufts of blood vessels with small pores (glomeruli). This process moves a large amount of water and electrolytes and other substances into small tubules. The cells lining these tubules reabsorb and return needed water, electrolytes, and nutrients (such as glucose and amino acids) to the blood. The cells also move waste products and drugs from the blood into the fluid (which becomes urine) as it flows through the tubules as well as add hormones that maintain blood supply (erythropoietin), blood pressure, and electrolyte balance.
Disorders that interfere with the function of the cells lining the tubules are called tubular disorders. Some conditions, called cystic disorders, interfere with these tubular cell functions by causing fluid-filled sacs (cysts) to form and replace or compress normal tubules. Many of these tubular and cystic disorders are hereditary. Of the hereditary disorders, some are detected at birth, and others are not obvious until years later.
Renal Tubular Acidosis
In renal tubular acidosis, the kidney tubules cannot adequately remove acids from the blood to excrete them in the urine.
 The tubules of the kidneys that remove acid from the blood are damaged when a person takes certain drugs or has another disorder that affects the kidneys.
The tubules of the kidneys that remove acid from the blood are damaged when a person takes certain drugs or has another disorder that affects the kidneys.
Typically muscle weakness and diminished reflexes occur when the disorder has been present for a long time.
Blood tests are done to detect high acid levels.
Some people drink a solution of baking soda every day to neutralize the acid.
Normally, the breakdown of food produces acids that circulate in the blood. The kidneys remove acids from the blood and excrete them in the urine. This function is predominantly performed by the kidney tubules. In renal tubular acidosis, the ability of the kidneys to excrete acids is partially impaired, and acid levels build up in the blood (metabolic acidosis). The balance of electrolytes is also affected. Renal tubular acidosis may lead to the following problems:
SOME TYPES OF RENAL TUBULAR ACIDOSIS
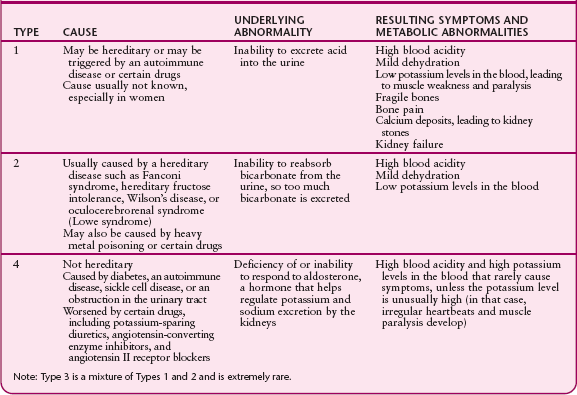
Low or high potassium levels in the blood
Calcium deposits in the kidneys, which may lead to kidney stones
Dehydration
Painful softening and bending of the bones (osteomalacia or rickets)
Renal tubular acidosis may be a permanent, inherited disorder. However, it may be an intermittent problem in people who have other disorders, such as diabetes mellitus, sickle cell disease, or an autoimmune disorder (such as systemic lupus erythematosus). Renal tubular acidosis may also be a temporary condition brought on by an obstruction of the urinary tract or by drugs, such as acetazolamide, amphotericin B, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), and diuretics that conserve the body’s potassium (so-called potassium-sparing diuretics).
There are four types of renal tubular acidosis, types 1 through 4. The types are distinguished by the particular abnormality in kidney function that causes acidosis. All four types are uncommon, but type 3 is extremely rare.
Symptoms and Diagnosis
Many people have no symptoms. Most others develop symptoms only after the disorder has been present for a long time. Which symptoms eventually develop depend on the type of renal tubular acidosis. When potassium levels in the blood are low, as occurs in types 1 and 2, neurologic problems may develop, including muscle weakness, diminished reflexes, and even paralysis. In type 4, potassium levels typically increase, although it is uncommon for the level to rise high enough to cause symptoms. If the level becomes too high, irregular heartbeats and muscle paralysis may develop. In type 1, kidney stones may develop, causing damage to kidney cells and, in some cases, chronic kidney failure.
A doctor considers the diagnosis of type 1 or type 2 renal tubular acidosis when a person has certain characteristic symptoms (such as muscle weakness and diminished reflexes) and when tests reveal high levels of acid and low levels of bicarbonate and potassium in the blood. Type 4 renal tubular acidosis is usually suspected when high potassium levels accompany high acid levels and low bicarbonate levels in the blood. Special tests help to determine the type of renal tubular acidosis.
Treatment
Treatment depends on the type. Types 1 and 2 are treated by drinking a solution of sodium bicarbonate (baking soda) every day to neutralize the acid that is produced from food. This treatment relieves the symptoms and prevents kidney failure and bone disease or keeps these problems from becoming worse. Other specially prepared solutions are available, and potassium supplements may also be required. In type 4, the acidosis is so mild that bicarbonate may not be needed. High potassium levels in the blood can usually be kept in check by restricting potassium intake, avoiding dehydration, and substituting different drugs or adjusting drug dosages.
Renal Glucosuria
In renal glucosuria (glycosuria), glucose (sugar) is excreted in the urine, despite normal or low glucose levels in the blood.
Normally, the body excretes glucose in the urine only when glucose levels in the blood are very high. In most healthy people, glucose that is filtered from the blood by the kidneys is completely reabsorbed back into the blood. In people with renal glucosuria, glucose may be excreted in the urine despite normal or low levels of glucose in the blood. This happens because of a defect in the tubular cells that decreases the reabsorption of glucose. Renal glucosuria may be a hereditary condition.
Renal glucosuria has no symptoms or serious consequences. A doctor makes the diagnosis when a routine urine test detects glucose in the urine even though glucose levels in the blood are normal. In a small number of people, renal glucosuria may be an early sign of diabetes mellitus. No treatment is needed.
Nephrogenic Diabetes Insipidus
In nephrogenic diabetes insipidus, the kidneys produce a large volume of dilute urine because they fail to respond to antidiuretic hormone and are unable to concentrate urine.
Often this disorder is hereditary, but it can be caused by drugs or disorders that affect the kidneys.
Symptoms include excessive thirst and excretion of large amounts of urine.
Diagnosis is based on tests of blood and urine.
Drinking large amounts of water, restricting salt in the diet, and sometimes taking drugs reduce urine volume.
Both diabetes insipidus and the better-known type of diabetes, diabetes mellitus, result in the excretion of large volumes of urine. Otherwise, the two types of diabetes are very different.
Two types of diabetes insipidus exist. In nephrogenic diabetes insipidus, the kidneys do not respond to antidiuretic hormone (vasopressin), so they continue to excrete a large amount of dilute urine. In the other, more common, type (central diabetes insipidus), the pituitary gland fails to secrete antidiuretic hormone (see page 986).
 Did You Know…
Did You Know…
Nephrogenic diabetes insipidus and diabetes mellitus are very different, except that both cause people to excrete large amounts of urine.
Causes
Normally, the kidneys adjust the concentration of urine according to the body’s needs. The kidneys make this adjustment in response to the level of antidiuretic hormone in the blood. Antidiuretic hormone, which is secreted by the pituitary gland, signals the kidneys to conserve water and concentrate the urine. In nephrogenic diabetes insipidus, the kidneys fail to respond to the signal.
Nephrogenic diabetes insipidus may be hereditary. The gene that causes the disorder is recessive and carried on the X chromosome, one of the two sex chromosomes, so usually only males develop symptoms. However, females who carry the gene can transmit the disease to their sons. In other people, nephrogenic diabetes insipidus may be caused by certain drugs that block the action of antidiuretic hormone, such as lithium. Also, high levels of calcium or low levels of potassium in the blood partially block the action of antidiuretic hormone. Nephrogenic diabetes insipidus can also occur if the kidney is affected by disorders such as polycystic kidney disease, sickle cell anemia, medullary sponge kidney, infections (pyelonephritis) that are severe, amyloidosis, Sjögren’s syndrome, or myeloma.
Symptoms and Diagnosis
The symptoms of nephrogenic diabetes insipidus are excessive thirst (polydipsia) and the excretion of large volumes of dilute urine (polyuria). When nephrogenic diabetes insipidus is hereditary, symptoms usually start soon after birth. Because infants cannot communicate thirst, they may become very dehydrated. They may develop a fever accompanied by vomiting and seizures.
If hereditary nephrogenic diabetes insipidus is not quickly diagnosed and treated, the brain may be damaged, leaving the infant with permanent mental retardation. Frequent episodes of dehydration can also retard physical development. With treatment, however, an infant who has this disorder is likely to develop normally.
Laboratory tests reveal high sodium levels in the blood and very dilute urine. A doctor may use a water deprivation test to help make the diagnosis (see page 987).
Prognosis and Treatment
The prognosis is good if nephrogenic diabetes insipidus is diagnosed before the person suffers severe episodes of dehydration. In cases in which the disorder is not inherited, correction of the underlying abnormality usually helps kidney function return to normal.
To prevent dehydration, people with nephrogenic diabetes insipidus must drink adequate amounts of water as soon as they feel thirsty. Infants, young children, and very sick older people must be given water often. People who drink enough water are not likely to become dehydrated, but several hours without water can lead to serious dehydration. A diet low in salt may help. Nonsteroidal anti-inflammatory drugs (NSAIDs) and thiazide diuretics are sometimes used to treat this disorder. NSAIDs and thiazide diuretics act by different mechanisms to increase the amounts of sodium and water that are reabsorbed by the kidney. These changes decrease the volume of urine.
Cystinuria
Cystinuria is a rare disorder that results in excretion of the amino acid cystine into the urine, often causing cystine stones to form in the urinary tract.
Cystinuria is caused by an inherited defect of the kidney tubules. The gene that causes cystinuria is recessive, so people with the disorder must have inherited two abnormal genes, one from each parent (see art on page 13). People who carry the gene but do not have the disorder have one normal and one abnormal gene. These people may excrete larger than normal amounts of cystine into the urine, but seldom enough to form cystine stones.
Cystine stones form in the bladder, renal pelvis (the area where urine collects and flows out of the kidney), or ureters (the long, narrow tubes that carry urine from the kidneys to the bladder). Occasionally, kidney failure develops.
Symptoms and Diagnosis
Symptoms usually start between the ages of 10 and 30. Often, the first symptom is intense pain caused by a spasm of the ureter where a stone becomes lodged. The stone may also become a site where bacteria collect and cause a serious infection.
A doctor tests for cystinuria when a person has recurring kidney stones. Cystine crystals may be seen during a microscopic examination of the urine (urinalysis), and high cystine levels are found in the urine.
Treatment
Treatment consists of preventing cystine stones from forming by keeping the concentration of cystine in the urine low. To keep the cystine concentration low, a person must drink enough fluids to produce at least 8 pints (4 liters) of urine each day. During the night, however, when the person is not drinking, less urine is produced and stone formation is more likely. This risk is reduced by drinking fluids before going to bed. Another treatment approach involves taking potassium citrate or sodium bicarbonate to make the urine more alkaline, because cystine dissolves more easily in alkaline urine than in acidic urine. Efforts to increase intake of water and make the urine more alkaline can lead to abdominal bloating, making the treatment difficult for some people to tolerate.
If stones continue to form despite these measures, drugs such as penicillamine, tiopronin, or captopril may be tried. These drugs react with cystine to keep it dissolved. Captopril is slightly less effective than the other drugs, but it has fewer serious side effects. Although the treatments are usually effective, there is a fairly high risk that stones will continue to form.
Fanconi Syndrome
Fanconi syndrome is a rare disorder of tubule function that results in excess amounts of glucose, bicarbonate, phosphates (phosphorus salts), uric acid, potassium, sodium, and certain amino acids being excreted in the urine.
Fanconi syndrome may be hereditary or may be caused by exposure to heavy metals or other chemical agents, vitamin D deficiency, kidney transplantation, multiple myeloma, or amyloidosis. Fanconi syndrome usually occurs with another hereditary disorder, such as cystinuria.
Symptoms and Diagnosis
In hereditary Fanconi syndrome, symptoms usually begin during infancy. A child with Fanconi syndrome may excrete a large amount of urine. Other symptoms include weakness and bone pain.
The symptoms and a test that shows a high level of acid in the blood may lead a doctor to suspect Fanconi syndrome. The diagnosis is confirmed when high levels of glucose, bicarbonate, phosphates, uric acid, potassium, and sodium are detected in the urine. Most often, some damage to bones or kidney tissue has occurred before the diagnosis is made.
Treatment
Fanconi syndrome cannot be cured, but it can be controlled with proper treatment. Effective treatment can keep the damage to bones and kidney tissue from getting worse and in some cases correct it. The high acid level of the blood (acidosis) may be neutralized by drinking sodium bicarbonate. People with low potassium levels in the blood may need to take potassium supplements by mouth. Bone disease requires treatment with phosphates and vitamin D supplements given by mouth. Kidney transplantation may be lifesaving if a child with the disorder develops kidney failure.
Hypophosphatemic Rickets
Hypophosphatemic rickets (previously called vitamin D–resistant rickets) is a disorder in which the bones become painfully soft and bend easily because the blood contains low levels of phosphate.
This very rare disorder is nearly always hereditary, passed as a dominant gene that is carried on the X chromosome, one of the two sex chromosomes. The genetic defect causes a kidney abnormality that allows an inappropriately high amount of phosphate to be excreted into the urine, resulting in low levels of phosphate in the blood. Because bones need phosphate for growth and strength, this deficiency causes defective bones. Females with hypophosphatemic rickets have less severe bone disease than do males. In rare cases, the disorder develops as a result of certain cancers, such as giant cell tumors of bone, sarcomas, prostate cancer, and breast cancer. Hypophosphatemic rickets is not the same as rickets caused by vitamin D deficiency (see page 929).
Symptoms and Diagnosis
Hypophosphatemic rickets usually begins to cause abnormalities in the first year of life. Abnormalities may be so mild that they cause no noticeable symptoms or so severe that they cause bowing of the legs and other bone deformities, bone pain, and a short stature. Bony outgrowth where muscles attach to bones may limit movement at those joints. The space between a baby’s skull bones may close too soon, leading to seizures. Laboratory tests show that calcium levels in the blood are normal but phosphate levels are low.
Treatment
The aim of treatment is to raise phosphate levels in the blood, which promotes normal bone formation. Phosphate can be taken by mouth and should be combined with calcitriol, the activated form of vitamin D. Taking vitamin D alone is not sufficient. The amounts of phosphate and calcitriol must be adjusted carefully because this treatment often leads to high levels of calcium in the blood, the accumulation of calcium in kidney tissue, or kidney stones. These effects can harm the kidneys and other tissues. In some adults, hypophosphatemic rickets resulting from cancer improves dramatically after the cancer is removed.
Hartnup Disease
Hartnup disease is a rare hereditary disorder that results in a skin rash and brain abnormalities because tryptophan and certain other amino acids are not well absorbed from the intestine and not well reabsorbed by the kidneys.
Hartnup disease occurs when a person inherits two copies of the abnormal gene for the disorder, one from each parent. The defective gene controls the absorption of certain amino acids from the intestine and the reabsorption of those amino acids in the kidneys. Consequently, a person with Hartnup disease cannot absorb amino acids properly from the intestine and cannot reabsorb them properly from tubules in the kidneys. Excessive amounts of amino acids, such as tryptophan, are excreted in the urine. The body is thus left with inadequate amounts of amino acids, which are the building blocks of protein. With too little tryptophan in the blood, the body is unable to make a sufficient amount of the B-complex vitamin niacinamide, particularly under stress when more vitamins are needed.
Symptoms
Hartnup disease is a disorder of amino acid transport in the intestine and kidneys; otherwise, the intestine and kidneys function normally, and the effects of the disease occur mainly in the brain and skin. Symptoms may begin in infancy or early childhood, but sometimes they begin as late as early adulthood. Symptoms may be triggered by sunlight, fever, drugs, or emotional or physical stress. A period of poor nutrition nearly always precedes an attack. The attacks usually become progressively less frequent with age. Most symptoms occur sporadically and are caused by a deficiency of niacinamide. A rash develops on parts of the body exposed to the sun. Mental retardation, short stature, headaches, an unsteady gait, and collapsing or fainting are common. Psychologic problems (such as anxiety, rapid mood changes, delusions, and hallucinations) may also result.
Diagnosis and Treatment
Laboratory tests performed on urine samples reveal abnormally high excretion of amino acids and their breakdown products.
People with Hartnup disease can prevent attacks by maintaining good nutrition and supplementing their diet with niacinamide or niacin, a B-complex vitamin very similar to niacinamide. A diet that is adequate in protein can overcome the deficiency caused by poor intestinal absorption and excess excretion of amino acids into the urine.
Bartter Syndrome
In Bartter syndrome, the kidneys excrete excessive amounts of electrolytes (potassium, sodium, and chloride), resulting in electrolyte abnormalities in the blood.
Bartter syndrome is usually hereditary and is caused by a recessive gene. Thus, a person with the disorder has inherited two recessive genes for the disorder, one from each parent. In affected people, the kidneys excrete excessive amounts of sodium, chloride, and potassium. The loss of sodium and chloride leads to excessive urine production and thus mild dehydration, which causes the body to produce more renin and aldosterone. The increase in aldosterone increases potassium and acid secretion in the kidneys, leading to low blood potassium (hypokalemia) and loss of acids in the blood that causes blood pH to be alkaline (metabolic alkalosis—see page 974).
Symptoms and Diagnosis
Children with Bartter syndrome grow slowly and appear malnourished. They may have muscle weakness and excessive thirst, may produce large amounts of urine, and may be mentally retarded. The loss of sodium and chloride leads to chronic mild dehydration. Abnormally low blood pressure (hypotension) may occur.
The diagnosis of Bartter syndrome in young children is based on a physical examination and on the characteristic abnormalities of electrolytes in blood and urine. Abnormal results are confirmed by finding high levels of certain hormones (renin, aldosterone) on blood tests. However, similar findings may occur when children with certain eating disorders, such as bulimia nervosa, self-induce vomiting and misuse diuretics.
Treatment
Many of the consequences of Bartter syndrome can be prevented by taking potassium supplements and a drug that reduces excretion of potassium into the urine, such as spironolactone (which also blocks the action of aldosterone), triamterene, amiloride, angiotensin-converting enzyme (ACE) inhibitors, or nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin. Drinking adequate amounts of fluids is necessary to compensate for the excessive fluid losses.
Liddle Syndrome
Liddle syndrome is a rare hereditary disorder in which the kidneys excrete potassium but retain too much sodium and water, leading to high blood pressure.
The gene that causes Liddle syndrome is dominant, meaning that children of a person with the disorder have a 50% chance of inheriting the defective gene. The disorder does not always cause symptoms. When it does, symptoms such as high blood pressure often begin during childhood. Some people have low levels of potassium in the blood.
The condition is effectively treated by drugs that increase sodium excretion and lessen potassium excretion, such as triamterene or amiloride. These drugs effectively lower the blood pressure. The prognosis is very good.
Polycystic Kidney Disease
Polycystic kidney disease is a hereditary disorder in which many fluid-filled sacs (cysts) form in both kidneys. The kidneys grow larger but have less functioning tissue.
Polycystic kidney disease is caused by an inherited gene defect.
Some people have such mild symptoms that they do not realize they have a disorder, but others have pain in the side, blood in the urine, and crampy pain caused by kidney stones.
Diagnosis is based on laboratory tests of kidney function and ultrasonography or computed tomography scans of the kidneys.
The kidney stones and infections are treated, but more than half of affected people eventually need dialysis or kidney transplantation.
There are several genetic defects that causes polycystic kidney disease. Several types are caused by dominant genes, and one rare type is caused by a recessive gene. That is, a person with the disease has inherited either one copy of a dominant gene from one parent or two copies of a recessive gene, one from each parent. People with dominant gene inheritance usually have no symptoms until adulthood. Those with recessive gene inheritance develop severe illness in childhood.
The genetic defect leads to the widespread formation of cysts in the kidneys. Gradual enlargement of the cysts with increasing age is accompanied by a reduction of blood flow and scarring within the kidneys. Kidney stones may develop. Kidney failure can occur eventually. The genetic defect may also cause cysts to develop in other parts of the body, such as the liver and pancreas.
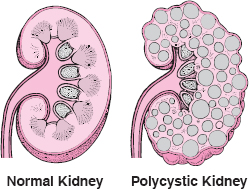
Polycystic Kidney Disease
In polycystic kidney disease, many cysts form in both kidneys. The cysts gradually enlarge, destroying some or most of the normal tissue in the kidneys.
Symptoms
In the rare, recessive form of this disease that begins during childhood, the cysts become very large and cause the abdomen to protrude. A severely affected newborn may die shortly after birth, because kidney failure can develop in the fetus, leading to poor development of the lungs. The liver is also affected, and at 5 to 10 years of age, a child with this disorder tends to develop high pressure in the blood vessels that connect the intestine and the liver (portal system). Eventually, liver failure and kidney failure occur.
In the more common, dominant form of polycystic kidney disease, the cysts develop slowly in number and size. Typically, symptoms begin in early or middle adulthood. Sometimes symptoms are so mild that people with the disease will live their whole life without ever having known that they had the disorder. Symptoms usually include discomfort or pain in the abdomen or side (flank), blood in the urine, frequent urination, and intense crampy (colicky) pain from kidney stones. In other cases, fatigue, nausea, and other consequences of slowly developing kidney failure may result because the person has less functioning kidney tissue. Repeated urinary tract infections can worsen the kidney failure. At least half of the people with polycystic kidney disease have high blood pressure by the time the disorder is recognized.
Complications: About one third of people who have the dominant form of polycystic kidney disease also have cysts in their liver, but these cysts do not affect liver function. As many as 10% of people have dilated blood vessels (aneurysms) in their brain. Usually, the dilated blood vessels cause headaches when they expand. Many of these brain aneurysms bleed and cause strokes.
Diagnosis, Prognosis, and Treatment
A doctor suspects this disease on the basis of family history and laboratory tests of kidney function. Ultrasonography and computed tomography (CT) reveal the characteristic appearance of cysts in the kidneys and liver.
Effective treatment of urinary tract infections and high blood pressure slows the rate of kidney destruction. However, more than half of the people who have this disease develop kidney failure at some time in their life. Without dialysis or kidney transplantation, kidney failure is fatal.
Genetic testing is available to help people with polycystic kidney disease understand the probability that their children will inherit the condition.
Nephronophthisis–Medullary Cystic Disease Complex
Nephronophthisis–medullary cystic disease complex is a group of disorders in which fluid-filled sacs (cysts) develop deep within the kidneys, leading to kidney failure.
Nephronophthisis–medullary cystic disease complex is a group of hereditary disorders that affect the development of microscopic tubules deep within the kidneys that concentrate the urine and reabsorb sodium. The damaged tubules become inflamed and scarred, eventually causing kidney failure.
Nephronophthisis is inherited as an autosomal recessive disease, so one defective gene must be received from each parent. It causes symptoms that usually begin during childhood or early adolescence and usually leads to kidney failure in early adolescence.
Medullary cystic disease is inherited as an autosomal dominant disorder, so a defective gene from only one parent is necessary, and it usually causes symptoms that begin in adulthood. Occasionally, the disorder occurs in a person with no family history of kidney disease. These people may have developed the gene defect as a new mutation (the gene becomes abnormal for no apparent reason) or the defect was present but not recognized in one or both parents.
Symptoms and Diagnosis
A person starts to produce excessive amounts of urine and becomes excessively thirsty because the kidneys become unable to concentrate urine and conserve sodium.
With nephronophthisis, the symptoms begin in children age one year or older and are associated with retarded growth. People with nephronophthisis may have eye disorders, liver disorders, and mental retardation. Later in childhood, kidney failure may cause anemia, nausea, and weakness.
With medullary cystic disease, the symptoms develop in adolescence or early adulthood. Excessive thirst and abnormal urine production are not as severe as with nephronophthisis. Other organs are not affected. Kidney failure occurs usually between the ages of 34 and 65. Some people develop gout.
Family history of this type of kidney disease is an important clue to the diagnosis. Laboratory tests indicate poor kidney function and a low level of sodium in the blood. Computed tomography (CT) is the imaging test that is most likely to detect cysts. In the future, genetic testing may become the most precise method of making the diagnosis.
Treatment
When kidney failure occurs, dialysis or kidney transplantation is needed. Particularly in nephronophthisis, large daily intake of fluids and salt (sodium) is needed to compensate for the excessive excretion of sodium and the production of large volumes of dilute urine.
Medullary Sponge Kidney
Medullary sponge kidney is an uncommon disorder in which the urine-containing tubules of the kidneys are dilated.
Medullary sponge kidney is usually caused by a non-genetic abnormality that occurs during development of the fetus. Much less often, the abnormality is hereditary. Medullary sponge kidney causes no symptoms most of the time, but a person with the disorder is prone to developing painful kidney stones, blood in the urine, and kidney infections. Calcium deposits in the kidneys occur in more than half of the people with the disorder. Calcium deposits may form kidney stones.
A doctor may suspect medullary sponge kidney based on the symptoms. X-rays of the kidneys reveal calcium deposits if there are any. The diagnosis is usually made by computed tomography (CT). Ultrasound scans may help but may not detect tiny fluid-filled sacs (cysts) lying deep within the kidneys.
Most people do well without treatment. Treatment may be necessary if infection develops or if medullary sponge kidney causes calcium to deposit and repeatedly form stones. Treatment for calcium stones is high fluid intake (more than 2 quarts [2 liters] per day) and a diet that is low in sodium, normal in calcium, and low to normal in protein. Sometimes a thiazide diuretic or amiloride is recommended. Surgery may be needed if the urinary tract becomes obstructed. Infections are treated with antibiotics.