CHAPTER 83
Interstitial Lung Diseases
Interstitial lung disease (also called diffuse parenchymal or infiltrative lung disease) is a term used to describe a number of different disorders that affect the interstitial space. The interstitial space consists of the air sacs of the lungs (alveoli), the walls of alveoli, and the spaces around blood vessels and small airways. Interstitial lung diseases result in abnormal accumulation of inflammatory cells in lung tissue, cause shortness of breath and cough, and have similarities in their appearances on imaging studies but are otherwise unrelated. Some of these diseases are very unusual.
Early in the course of these diseases, white blood cells, macrophages, and protein-rich fluid accumulate in the interstitial space, causing inflammation. If the inflammation persists, scarring (fibrosis) may replace the normal lung tissue. As alveoli are progressively destroyed, thick-walled cysts (called honeycombing because they resemble the cells of a beehive) are left in their place. The condition resulting from these changes is called pulmonary fibrosis.
Although the various interstitial lung diseases are separate and have different causes, they have some similar features. All lead to a decreased ability to transfer oxygen to the blood, and all cause stiffening and shrinkage of the lungs, which makes breathing difficult and causes cough. However, the elimination of carbon dioxide from the blood is usually not a problem.
UNUSUAL INTERSTITIAL LUNG DISEASES

CAUSES OF INTERSTITIAL LUNG DISEASES
| TYPE | EXAMPLES |
| Autoimmune disorders | Ankylosing spondylitis, Behçet’s syndrome, Goodpasture’s syndrome, mixed connective tissue disease, polymyositis and dermatomyositis, relapsing polychondritis, rheumatoid arthritis, scleroderma, Sjögren’s syndrome, and systemic lupus erythematosus (lupus) |
| Infections | Fungal, mycoplasmal (a type of bacterial), parasitic, rickettsial, or viral infections and tuberculosis |
| Organic dust | Bird droppings and molds |
| Drugs | Amiodarone, bleomycin, busulfan, carbamazepine, chlorambucil, cocaine, cyclophosphamide, gold, methotrexate, nitrofurantoin, sulfasalazine, and sulfonamides |
| Gases, fumes, and vapors | Chlorine and sulfur dioxide |
| Therapeutic or industrial radiation | Radiation therapy for cancer |
| Idiopathic* interstitial pneumonias | Acute interstitial pneumonia, cryptogenic organizing pneumonia, desquamative interstitial pneumonia, idiopathic pulmonary fibrosis, lymphoid interstitial pneumonia, nonspecific interstitial pneumonia, and respiratory bronchiolitis-associated interstitial lung disease |
| Other disorders | Alveolar proteinosis, amyloidosis, chronic gastric microaspiration, lymphangiomyomatosis, neurofibromatosis, pulmonary Langerhans’ cell granulomatosis (histiocytosis), sarcoidosis, and vasculitic disorders (which cause inflammation of blood vessels) such as Churg-Strauss syndrome and Wegener’s granulomatosis |
| Idiopathic means with no known cause. | |
Diagnosis
Because interstitial lung diseases cause symptoms that are similar to those of much more common disorders (for example, pneumonia, chronic obstructive pulmonary disease), they may not be suspected at first. When interstitial lung disease is suspected, diagnostic testing is done. Testing can vary by the disease suspected but tends to be similar. Most people have a chest x-ray, computed tomography (CT), pulmonary function tests (see page 454), and often arterial blood gas analysis. CT is more sensitive than chest x-ray and helps doctors make a more specific diagnosis. CT is done using techniques that maximize resolution (high-resolution CT). Pulmonary function tests often show that the volume of air that the lungs can hold is abnormally small. In addition, the person’s response to exercise is commonly tested.
To confirm the diagnosis, doctors may remove a small sample of lung tissue for microscopic examination (lung biopsy) using a procedure called fiberoptic bronchoscopy. A lung biopsy done this way is called transbronchial lung biopsy (see page 457). Many times, a larger tissue sample is needed and must be removed surgically, sometimes with use of a thoracoscope (a procedure called video-assisted thoracoscopic lung biopsy.
Blood tests are usually done. They usually cannot confirm the diagnosis but are done as part of the search for other, similar disorders. Doctors may also order electrocardiography (ECG) or echocardiography to determine whether the heart has been affected by the lung disease.
Idiopathic Interstitial Pneumonias
Idiopathic interstitial pneumonias are interstitial lung diseases that have no known cause and that affect the lungs similarly.
 Some types of these pneumonias are much more serious than others.
Some types of these pneumonias are much more serious than others.
Diagnosis requires chest x-rays, computed tomography, and usually analysis of a sample of lung tissue (biopsy).
The word idiopathic means of unknown cause, so when the cause of interstitial lung disease is not identified, idiopathic interstitial pneumonia is diagnosed. Pneumonias are often thought of as infections, but these diseases do not appear to result from infection.
COMPARING TYPES OF IDIOPATHIC INTERSTITIAL PNEUMONIAS
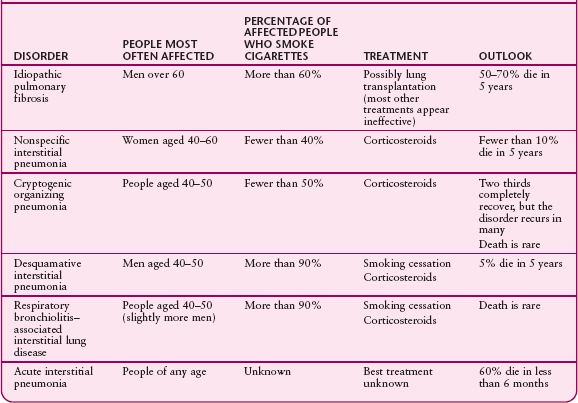
There are six types of idiopathic interstitial pneumonias. In decreasing order of frequency, they are
Idiopathic pulmonary fibrosis
Nonspecific interstitial pneumonia
Cryptogenic organizing pneumonia
Respiratory bronchiolitis-associated interstitial lung disease
Desquamative interstitial pneumonia
Acute interstitial pneumonia
Some experts also consider lymphoid interstitial pneumonia to be a type of idiopathic interstitial pneumonia.
All types cause shortness of breath and affect the lungs similarly. However, they differ in how quickly they develop, how they are treated, and how serious they are. For example, most diseases take weeks to months to develop, but idiopathic pulmonary fibrosis takes more than 12 months to fully develop. Acute interstitial pneumonia takes only 1 to 2 weeks.
Diagnosis
Chest x-rays, pulmonary function tests (see page 454), and computed tomography (CT) are done. CT may be diagnostic. If not, doctors remove a small sample of lung tissue for examination under a microscope (lung biopsy). Usually, biopsy is done surgically with use of a thoracoscope (see page 457).
Blood tests are usually done. They usually cannot confirm the diagnosis but are done as part of the search for other, similar disorders. Doctors may also order an electrocardiogram (ECG) or echocardiogram to determine whether the heart has been affected by the lung disease.
IDIOPATHIC PULMONARY FIBROSIS
Idiopathic pulmonary fibrosis is the most common form of idiopathic interstitial pneumonia.
Idiopathic pulmonary fibrosis affects mostly people in their 50s and 60s, usually men and usually smokers.
People may cough, lose weight, have difficulty breathing, and feel tired.
Lung transplantation may be the only effective treatment.
The lungs suffer progressive injury for a long period of time. The injury causes chronic inflammation that eventually leads to lung scarring (fibrosis).
Symptoms
Symptoms depend on the extent of the lung damage, the rate at which the disease progresses, and whether complications, such as lung infections and right-sided heart failure (cor pulmonale—see box on page 523) develop. The main symptoms start insidiously as shortness of breath on exertion, cough, and diminished stamina. Other common complaints include weight loss and fatigue. In most people, symptoms worsen over a period ranging from about 6 months to several years.
As the disease progresses, the level of oxygen in the blood decreases, and the skin may take on a bluish tinge (called cyanosis) and the ends of the fingers may become thick or club-shaped (see art on page 453). Strain on the heart may cause the right ventricle to enlarge, eventually resulting in right-sided heart failure. Through a stethoscope, doctors often hear crackling sounds. These sounds are called Velcro crackles, described as such because the sound is similar to that of Velcro when it is pulled apart.
Diagnosis
A chest x-ray may show widespread tiny white lines, often in a netlike pattern, most profuse in the lower parts of both lungs. Computed tomography (CT) typically shows a pattern of patchy, white lines in the lower lungs. In areas of more severe involvement, the thick scarring often creates a honeycombing appearance. Pulmonary function tests (see page 454) show that the amount of air the lungs can hold is below normal. Analysis of a blood sample or use of an oximeter (see page 455) shows a low level of oxygen with minimal exercise (walking at a normal pace) and, as the disease progresses, even when the person is resting.
To confirm the diagnosis, doctors may do a lung biopsy by using a procedure called bronchoscopy (see page 456). Many times, a larger tissue sample is needed and must be removed surgically, sometimes with use of a thoracoscope (see page 457).
Blood tests cannot confirm the diagnosis but are done as part of the search for other disorders that may cause a similar pattern of inflammation and scarring. For example, doctors do blood tests to screen for certain autoimmune disorders.
Prognosis and Treatment
Most people continue to get worse. On average, people live less than 3 years after diagnosis. A few people survive for more than 5 years after diagnosis. A few die within several months.
If a chest x-ray or lung biopsy shows that scarring is not extensive, the usual treatment is a corticosteroid (such as prednisone), with or without azathioprine, N-acetylcysteine, or both. Doctors evaluate the person’s response using chest x-rays, CT, and pulmonary function tests. High doses of prednisone are usually given for about 3 months, and then the dose is gradually reduced over another 3 months. Much lower doses are then continued for 6 more months. However, this combination treatment fails to help most people. Promising treatments that appear to decrease lung fibrosis and prolong survival include pirfenidone and bosentan.
Other treatments are aimed at relieving symptoms: pulmonary rehabilitation for improving ability to carry out activities of daily life (see page 459), oxygen therapy for low blood oxygen levels, antibiotics for infection, and drugs for the heart failure that is produced by cor pulmonale. Lung transplantation (often with a single lung) has been successful in some people with severe idiopathic pulmonary fibrosis.
RESPIRATORY BRONCHIOLITIS– ASSOCIATED INTERSTITIAL LUNG DISEASE AND DESQUAMATIVE INTERSTITIAL PNEUMONIA
Respiratory bronchiolitis-associated interstitial lung disease and desquamative interstitial pneumonia are rare conditions that cause chronic lung inflammation and occur mostly in current or former cigarette smokers.
These conditions have many similarities, so that some experts think they may be part of the same disorder. However, desquamative interstitial pneumonia is often more severe. Both disorders affect cigarette smokers in their 30s and 40s, with most people developing shortness of breath with even minimal exertion. Men are affected more often than women (ratio of almost 2:1).
A chest x-ray shows less severe changes than in idiopathic pulmonary fibrosis and may show no changes in up to 20% of people. Pulmonary function tests show a decline in the amount of air contained in the lungs. The amount of oxygen in a blood sample is low.
A lung biopsy is often needed to confirm the diagnosis.
About 70% of people who have respiratory bronchiolitis-associated interstitial lung disease or desquamative interstitial pneumonia survive for 10 years or longer. The response is even better when people stop smoking.
Some doctors give corticosteroids because they may be effective in other interstitial lung diseases, but the effectiveness is unknown.
CRYPTOGENIC ORGANIZING PNEUMONIA
Cryptogenic organizing pneumonia (also called bronchiolitis obliterans organizing pneumonia) is a rapidly developing idiopathic interstitial pneumonia characterized by lung inflammation and scarring that obstructs the small airways and air sacs of the lungs (alveoli).
The disease usually begins between the ages of 40 and 60 and affects men and women equally. Cigarette smoking does not appear to increase the risk of developing the disease.
Almost 75% of people have symptoms for less than 2 months before seeking medical attention. A flu-like illness, with a cough, fever, a feeling of illness (malaise), fatigue, and weight loss, heralds the onset in about 50% of people.
Diagnosis and Treatment
Doctors do not find any specific abnormalities on routine laboratory tests or on a physical examination, except for the frequent presence of crackling sounds (called Velcro crackles) when the doctor listens with a stethoscope. Pulmonary function tests usually show that the amount of air the lungs can hold is below normal. The amount of oxygen in the blood is often low at rest and is even lower with exercise.
The chest x-ray is distinctive with features that appear similar to an extensive pneumonia, with both lungs showing widespread white patches. The white patches may seem to migrate from one area of the lung to another as the disease persists or progresses. Computed tomography (CT) may be used and sometimes confirms the diagnosis. Often, the findings are typical enough to allow doctors to make a diagnosis without ordering additional tests.
In other cases, to confirm the diagnosis, doctors do a lung biopsy using a bronchoscope (see page 456). Many times, a larger sample is needed and must be removed surgically.
When treated with corticosteroids, about two thirds of people recover. However, symptoms may later return. If so, repeat treatment with corticosteroids is usually effective.
NONSPECIFIC INTERSTITIAL PNEUMONIA
Nonspecific interstitial pneumonia is an idiopathic interstitial pneumonia that occurs mainly in women, people who do not smoke, and people younger than 50 years.
Nonspecific interstitial pneumonia seems to be the second most common kind of idiopathic interstitial pneumonia. Most people are between the ages of 40 and 50. Most people have no known cause or risk factor. However, a similar process can develop in people with connective tissue disorders (in particular, systemic sclerosis and polymyositis or dermatomyositis), in some forms of drug-induced lung injury, and in people with hypersensitivity pneumonitis (see page 514).
A dry cough and shortness of breath develop over 6 to 18 months. Low-grade fever and a feeling of illness (malaise) may occur, but high fever, weight loss, and other general symptoms of illness are unusual.
Diagnosis and Treatment
As with other idiopathic interstitial pneumonias, chest x-rays are done, and CT is usually also done. Pulmonary function tests usually show that the amount of air the lungs can hold is below normal. The amount of oxygen in the blood is often low at rest and is even lower with exercise. Doctors often do bronchoscopy (see page 456) and wash segments of the lung with a salt-water solution and then collect the washings (bronchoalveolar lavage) for testing. More than half of people have more lymphocytes (a type of white blood cell) than normal in the washings. Lung biopsy is often necessary.
Corticosteroids are usually effective. More than 80% of people survive for more than 10 years after being diagnosed.
ACUTE INTERSTITIAL PNEUMONIA
Acute interstitial pneumonia (also called accelerated interstitial pneumonia or Hamman-Rich syndrome) is an idiopathic interstitial pneumonia that develops suddenly and is severe.
Acute interstitial pneumonia causes the same type of symptoms as the acute respiratory distress syndrome (see page 525). It tends to affect healthy men and women who are usually older than 40. Fever, cough, and difficulty breathing develop over 1 to 2 weeks, typically progressing to acute respiratory failure.
When possible, the diagnosis is confirmed with CT, lung biopsy, and pulmonary function tests.
Treatment aims to keep the person alive until the disorder resolves. Mechanical ventilation is needed if there is respiratory failure. Corticosteroids are generally used, but it is not clear whether they are effective.
More than 60% of affected people die within 6 months, usually from respiratory failure. In people who survive, lung function usually improves with time. However, the disease may recur.
Pulmonary Langerhans’ Cell Granulomatosis
Pulmonary Langerhans’ cell granulomatosis (histiocytosis or eosinophilic granuloma) is a disorder in which cells called histiocytes and eosinophils proliferate in the lung, often causing scarring.
People may have no symptoms or may cough and have difficulty breathing.
Diagnosis requires computed tomography and sometimes analysis of a sample of lung tissue (biopsy).
Whether and which treatments help are unknown.
Pulmonary Langerhans’ cell granulomatosis (histiocytosis) is one form of Langerhans’ cell granulomatosis, which can affect other organs (such as the pituitary gland and the white blood cells) as well as the lungs. The cause is unknown, and the disorder is rare. It occurs almost exclusively in whites aged 20 to 40 who smoke cigarettes. It starts with infiltration of the lung by histiocytes, which are cells that scavenge for foreign materials, and to a lesser extent by eosinophils, which are cells that are normally involved in allergic reactions.
Symptoms
About 15% of people have no symptoms and the disorder is first recognized when an imaging study of the chest is done for another reason. The remainder develop coughing, shortness of breath, fever, chest pain, fatigue, and weight loss. Pneumothorax is a common complication due to rupture of a lung cyst. It occurs in 15 to 25% of people with the disorder and may be the cause of the first symptoms that develop. Scarring makes the lungs stiff and impairs their ability to transfer oxygen into and out of the blood. A few people cough up blood (hemoptysis).
Some people have localized bone pain or a pathologic bone fracture (a fracture that occurs with only a minor injury because the bone has been thinned by a disorder). Diabetes insipidus occurs in about 15% of people. This condition results when histiocytes also affect the hypothalamus in the brain. The person makes excessive amounts of urine that is dilute. People with diabetes insipidus probably have a worse prognosis than those who do not.
Diagnosis
Chest x-rays show nodules, small lung cysts (honeycombing), and other changes that are typical of this disorder. Computed tomography (CT) may show these changes in enough detail to establish the diagnosis. X-rays may also show that the bones are affected. Pulmonary function tests show that the amount of air the lungs can hold is below normal. If CT is not diagnostic, biopsy is required. Biopsy can usually be done during bronchoscopy (see page 456).
Prognosis and Treatment
Half of people are alive more than 12 years after diagnosis. Death usually results from respiratory
failure or cor pulmonale (see box on page 523). When people stop smoking, improvement occurs in about one third of cases.
The disorder may be treated with corticosteroids and immunosuppressants such as cyclophosphamide, although no therapy is clearly beneficial.
Lymphoid Interstitial Pneumonia
Lymphoid interstitial pneumonia is an uncommon lung disease in which mature lymphocytes (a type of white blood cell) accumulate in the alveoli.
People usually have cough and difficulty breathing.
Diagnosis requires chest x-ray, computed tomography, pulmonary function tests, and often bronchoscopy.
Treatment involves corticosteroids, immunosuppressants, or both.
Lymphoid interstitial pneumonia can occur in children, usually those infected with the human immunodeficiency virus (HIV). Lymphoid interstitial pneumonia can also occur in adults, often those with autoimmune disorders such as plasma cell disorders, Sjögren’s syndrome (see page 577), Hashimoto’s thyroiditis, rheumatoid arthritis, and systemic lupus erythematosus (lupus). Average age of affected adults is 54.
Symptoms
Children develop wheezing, cough, and difficulty breathing, and they may not grow and gain weight. Adults develop difficulty breathing and cough over months or, in some cases, years. Less common symptoms include weight loss, fever, joint pain, and night sweats.
Diagnosis
Doctors can sometimes hear crackles in the lungs using a stethoscope.
Diagnosis requires chest x-ray, computed tomography (CT), and pulmonary function tests. Pulmonary function tests usually show a decrease in the amount of air the lungs can hold. Doctors often do bronchoscopy (see page 456) and wash segments of the lung with a salt-water solution and then collect the washings (bronchoalveolar lavage) for testing. Children may have abnormalities in their blood proteins that can help establish the diagnosis. If not, and for all adults, lung biopsy is usually necessary.
Prognosis and Treatment
The prognosis is difficult to predict. The disorder may resolve on its own or after treatment, or it may progress to lung fibrosis or lymphoma (a cancer). One half to two thirds of people are alive 5 years after diagnosis.
Treatment is with corticosteroids, other immunosuppressants, or both, but the effectiveness of these drugs is unknown.
Sarcoidosis
Sarcoidosis is a disease in which abnormal collections of inflammatory cells (granulomas) form in many organs of the body.
Sarcoidosis usually develops in people aged 20 to 40, most often people of Scandinavian ancestry and American blacks.
It can affect many organs, most commonly the lungs.
People cough and have difficulty breathing but can have various symptoms depending on which organs are affected.
Diagnosis usually requires chest x-ray, computed tomography, and analysis of a sample of tissue (biopsy), usually from the lungs.
Symptoms eventually subside without treatment in most people.
Treatment, when necessary, begins with corticosteroids.
The cause of sarcoidosis is unknown. It may result from an infection or from an abnormal response of the immune system. Inherited factors may be important. Sarcoidosis typically develops between the ages of 20 and 40. It is most common among people of Scandinavian ancestry and American blacks, although it can occur in anyone.
Sarcoidosis is characterized by the presence of collections of inflammatory cells (granulomas). The disease is primarily one of the lungs, but granulomas can also form in the lymph nodes, lungs, liver, eyes, and skin, and less often in the spleen, bones, joints, sinuses, skeletal muscles, kidneys, heart, reproductive organs, salivary glands, and nervous system. The granulomas may eventually disappear completely or become scar tissue.
Symptoms
Many people with sarcoidosis have no symptoms, and the disorder is discovered on a chest x-ray that is taken for other reasons. Most people develop minor symptoms that do not progress. Serious symptoms are rare.
The symptoms of sarcoidosis vary greatly according to the site and extent of the disease.
General: Fever, fatigue, vague chest pain, a feeling of illness (malaise), weight loss, and aching joints may be the first indications of a problem in about one third of people. Enlarged lymph nodes are common but do not often cause symptoms. Fever and night sweats may recur throughout the illness.
Lungs: The organ most affected by sarcoidosis is the lung. Enlarged lymph nodes at the place where the lungs meet the heart or to the right of the windpipe (trachea) may be seen on a chest x-ray. Sarcoidosis produces inflammation in the lungs that may eventually lead to scarring and the formation of cysts, which can cause coughing and shortness of breath. Fortunately, such progressive scarring occurs infrequently. Occasionally, the fungus Aspergillus can settle in (colonize) the lung cysts, grow, and cause bleeding. Breathing can become difficult, and sometimes the person may cough blood. Severe involvement of the lung by sarcoidosis can eventually strain the right side of the heart, causing right-sided heart failure (cor pulmonale—see box on page 523).
Skin: The skin is frequently affected by sarcoidosis. In people of Scandinavian ancestry, sarcoidosis often starts as raised, tender, red lumps, usually on the shins (erythema nodosum—see page 1292), accompanied by a fever and joint pain. This set of symptoms is less common in American blacks. Prolonged sarcoidosis may lead to the formation of flat patches (plaques), raised patches, or lumps just under the skin with discoloration of the nose, cheeks, lips, and ears (lupus pernio). Lupus pernio is most common in black women.
Liver and Spleen: About 70% of people with sarcoidosis have granulomas in their liver. These granulomas often produce no symptoms, and the liver seems to function normally. Fewer than 10% of people with sarcoidosis have an enlarged liver. Jaundice caused by liver malfunction is rare. The spleen also is enlarged in some people.
Eyes: The eyes are affected in 15% of people with sarcoidosis. Inflammation of certain internal eye structures (uveitis) makes the eyes red and painful and interferes with vision. Inflammation that persists for a long time may block fluid from draining from the eye, causing glaucoma (see page 1448), which can lead to blindness. Granulomas may form in the conjunctiva (the membrane over the eyeball and inside the eyelids). Such granulomas often do not cause symptoms, but the conjunctiva is an accessible site from which doctors can take tissue samples for examination. Some people with sarcoidosis complain of dry, sore, and red eyes, probably because sluggish tear glands that have been affected by the disorder no longer produce enough tears to keep the eyes lubricated.
Heart: Granulomas that form in the heart may cause chest pain (angina) or heart failure. Granulomas that form near the heart’s electrical conducting system can trigger potentially fatal irregularities in the heartbeat.
Joints and Bones: Inflammation can cause widespread pain in the joints. The joints in the hands and feet are most commonly affected. Cysts form in the bones and can make nearby joints swollen and tender.
Nervous System: Sarcoidosis can affect the cranial nerves (nerves of the head), causing double vision and making one side of the face droop. If the pituitary gland or the bones surrounding it are affected, diabetes insipidus (see page 986) may result. The pituitary gland stops producing vasopressin, a hormone needed by the kidney to concentrate urine, causing frequent urination and excessive amounts of urine.
High Calcium Levels: Sarcoidosis can cause high levels of calcium to accumulate in the blood and urine. These high levels occur because sarcoid granulomas produce activated vitamin D, which enhances calcium absorption from the intestine. High blood calcium levels lead to a loss of appetite, nausea, vomiting, thirst, and excessive urine production. If present for a long time, high blood calcium levels may lead to the formation of kidney stones or calcium deposits in the kidney and, eventually, to kidney failure.
Diagnosis
Doctors most often diagnose sarcoidosis by observing its distinctive changes, including enlarged lymph nodes and abnormal findings on a chest x-ray or on computed tomography (CT). When further testing is necessary, microscopic examination of a tissue sample showing inflammation and granulomas confirms the diagnosis. Bronchoscopy with transbronchial lung biopsy (see page 457) is 90% accurate and is the best procedure for people whose lungs are involved. Other possible sources of tissue specimens are skin abnormalities, enlarged lymph nodes close to the skin, and granulomas on the conjunctiva. A liver biopsy is rarely needed even if there is evidence that the liver is affected.
Tuberculosis can cause many changes similar to those caused by sarcoidosis. Therefore, doctors also do a tuberculin skin test (and sometimes a lung biopsy) to make sure the problem is not tuberculosis.
Other methods that can help doctors diagnose sarcoidosis or assess its severity include measuring the level of angiotensin-converting enzyme (ACE) in the blood, irrigating the lungs and examining the fluid, and using a whole-body gallium scan. In many people with sarcoidosis, the level of ACE in the blood is usually high, but this test is not always accurate. The washings from a lung with active sarcoidosis contain a large number of lymphocytes, but this finding is not unique to sarcoidosis. Because gallium scanning shows abnormal patterns in the lungs or lymph nodes of people with sarcoidosis in those places, this test is sometimes used when the diagnosis is uncertain.
In people with lung scarring, pulmonary function tests may show that the amount of air the lung can hold is below normal. Blood tests may reveal a low number of white blood cells, red blood cells, or platelets. Immunoglobulin levels are often high, especially in blacks. Levels of calcium in the blood may be high. The levels of liver enzymes, particularly alkaline phosphatase, may be high if the liver is affected.
Prognosis
Sarcoidosis improves or clears up spontaneously in nearly two thirds of people with lung sarcoidosis. Even enlarged lymph nodes in the chest and extensive lung inflammation may disappear in a few months or years. The course can be chronic or progressive in 10 to 30% of people. Serious involvement outside of the chest (for example, of the heart, nervous system, eyes, or liver) occurs in 4 to 7% of people at the beginning of their illness. The chance of involvement outside of the chest increases if lung disease persists.
People who have sarcoidosis that has not spread beyond the chest do better than those who also have sarcoidosis elsewhere in the body. People with enlarged lymph nodes in the chest but no sign of lung disease have a very good prognosis. People whose disease began with erythema nodosum and tender, swollen joints often have the best prognosis. About 50% of people who once had sarcoidosis have relapses.
About 10% of people with sarcoidosis develop a serious disability from damage to the eyes, respiratory system, or elsewhere. Lung scarring leading to respiratory failure and cor pulmonale is the most common cause of death, followed by bleeding from lung infection caused by Aspergillus.
Treatment
Most people with sarcoidosis do not need treatment. Corticosteroids are given to suppress severe symptoms such as shortness of breath, joint pain, and fever. These drugs also are given if
Tests show high levels of calcium in the blood, even if symptoms are mild.
Function of the heart, liver, or nervous system is affected.
Sarcoidosis causes disfiguring skin lesions or eye disease that corticosteroid eye drops do not cure.
Lung disease worsens.
People who have no symptoms should not take corticosteroids. Although corticosteroids control symptoms well, they do not prevent lung scarring over the years. About 10% of people who need treatment do not respond to corticosteroids alone, and they are also given methotrexate, which may be very effective. Hydroxychloroquine is sometimes helpful in eliminating disfiguring skin lesions.
The success of treatment can be monitored with chest x-rays, CT, pulmonary function tests, and measurements of calcium or ACE levels in the blood. These tests are repeated regularly to detect relapses after treatment stops.
Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis is a rare disorder in which the air sacs of the lungs (alveoli) become plugged with a protein-rich fluid.
Pulmonary alveolar proteinosis typically affects people who are aged 20 to 60 and who have not had lung disease.
People have difficulty breathing and cough.
Diagnosis is by computed tomography and testing a sample of lung fluid obtained using a bronchoscope.
If symptoms are severe, the lungs are washed out, one at a time.
The cause of pulmonary alveolar proteinosis is almost always unknown, but recent studies have linked it to production of an antibody directed against a protein that seems to be involved with the production or the breakdown of surfactant (a substance normally produced in the lungs). Occasionally, development of pulmonary alveolar proteinosis is related to exposure to toxic substances, such as inorganic dusts, infection with Pneumocystis jiroveci (see page 470), certain cancers, and immunosuppressants. Rarely, it occurs in newborns.
The protein in the lungs plugs up the alveoli and small airways. In rare instances, lung tissue becomes scarred. The disease may progress, remain stable, or disappear spontaneously.
Symptoms
When the alveoli are plugged, the transfer of oxygen to the blood from the lungs is severely impaired. Consequently, most people with pulmonary alveolar proteinosis experience shortness of breath when they exert themselves. Some people have severe difficulty breathing, even at rest. Other symptoms may include fatigue, weight loss, and low-grade fever. Most people also have a cough that often does not produce sputum, but occasionally people expectorate chunky gelatinous material. People often have severe disability from inadequate lung function. Lung infections may quickly worsen symptoms of shortness of breath and produce fever.
Diagnosis
A chest x-ray shows extensive dense white patches in both lungs, usually located centrally near the heart. Computed tomography (CT) shows this and other changes that suggest the disease. Pulmonary function tests (see page 454) reveal that the volume of air that the lungs can hold is abnormally small. Tests show low levels of oxygen in the blood, at first only during exercise but later also when the person is at rest. The elimination of carbon dioxide from the lungs may be impaired. Blood test results are not specific for the diagnosis, although levels of some substances (for example, lactic dehydrogenase, red blood cells, serum surfactant proteins, and gamma globulin) are often elevated.
To make a definitive diagnosis, doctors examine a sample of the fluid from the alveoli. To obtain a sample, doctors use a bronchoscope (see page 456) to wash segments of the lung with a salt-water solution and then collect the washings (bronchoalveolar lavage). The washings are often opaque or milky because the fluid is rich in protein and fats. Sometimes doctors obtain a lung tissue sample for microscopic examination (lung biopsy) during bronchoscopy. Occasionally, a larger sample is needed, which must be removed surgically.
Treatment
People who have few or no symptoms do not require treatment. For people with disabling symptoms, the protein-and fat-rich fluid in the alveoli can be washed out with a salt solution during bronchoscopy or through a special tube inserted through the mouth or through the windpipe (trachea) and into one lung. Sometimes only a small section of the lung must be washed, but if symptoms are severe and the levels of oxygen in the blood are very low, the person is given general anesthesia, so that one entire lung can be washed. About 3 to 5 days later, the other lung is washed, again after the person has been given general anesthesia. One washing is enough for some people, but others need washings every 6 to 12 months for many years.
Corticosteroids, such as prednisone, are not effective and may actually increase the chance of infection. Bacterial infections are treated with antibiotics, usually taken by mouth.
Some people with pulmonary alveolar proteinosis are short of breath indefinitely, but the disease is rarely fatal as long as they have regular lung washings.