1 Einleitung
Die Elemente der siebten Nebengruppe (Mangan, Technetium, Rhenium und Bohrium) sind zueinander physikalisch und chemisch relativ ähnlich. Bei Technetium und Rhenium zeigen sich zwar noch die Auswirkungen der Lanthanoidenkontraktion, aber nicht mehr so deutlich wie bei den Elementen der vierten bis sechsten Nebengruppe. In ihren physikalischen Eigenschaften unterscheiden sich Technetium und Rhenium schon erheblich, wenngleich das Technetium dem Rhenium wesentlich näher steht als dem Mangan. Die Elemente dieser Gruppe können maximal sieben äußere Valenzelektronen (jeweils zwei s- und fünf d-Elektronen) abgeben, um eine stabile Elektronenkonfiguration zu erreichen. Bei Chrom ist die Oxidationsstufe +3 die stabilste, bei Technetium und Rhenium sind es die Stufen +4 und +7.
Die Entdeckung des Mangans erfolgte gegen Ende des 18. Jahrhunderts, die des Rheniums Mitte der 1920er-Jahre, die des Technetiums 1937, und die ersten Isotope des Bohriums wurden 1981 erzeugt. Sie finden alle Elemente im unten stehenden Periodensystem in Gruppe 7 (VII B).
Elemente werden eingeteilt in Metalle (z. B. Natrium, Calcium, Eisen, Zink), Halbmetalle wie Arsen, Selen, Tellur sowie Nichtmetalle wie beispielsweise Sauerstoff, Chlor, Iod oder Neon. Die meisten Elemente können sich untereinander verbinden und bilden chemische Verbindungen; so wird z. B. aus Natrium und Chlor die chemische Verbindung Natriumchlorid, also Kochsalz).

Periodensystem der Elemente
Die Einzeldarstellungen der insgesamt vier Vertreter der Gruppe der Elemente der siebten Nebengruppe enthalten dabei alle wichtigen Informationen über das jeweilige Element, sodass ich nur eine sehr kurze Einleitung vorangestellt habe.
2 Vorkommen
Mangan ist in der Erdhülle mit einem relativ hohen Anteil von 850 ppm vertreten, wogegen Rhenium mit einem Anteil von 0,001 ppm (!) äußerst selten vorkommt. Technetium und Bohrium sind beide nur durch künstliche Kernreaktionen erhältlich. Technetium erzeugt man in größerem Maßstab in Kernreaktoren, wogegen Bohrium nur durch Kernfusion und dann in Mengen weniger Atome zugänglich ist.
3 Herstellung
Mangan gewinnt man entweder durch Elektrolyse seiner Salzlösungen oder aluminothermisch aus Braunstein, Technetium und Rhenium bevorzugt durch Reduktion der Pertechnetate bzw. Perrhenate mit Wasserstoff. Rhenium zeigt dabei Parallelen zum Wolfram, das man ebenfalls auf diese Weise erzeugt.
4 Eigenschaften
4.1 Physikalische Eigenschaften
Die physikalischen Eigenschaften sind auch in dieser Gruppe mit nur wenigen Ausnahmen regelmäßig nach steigender Atommasse abgestuft. In Analogie zu den Nachbarelementen der sechsten und achten Nebengruppe nehmen vom Mangan zum Rhenium Dichte, Schmelzpunkte und -wärmen sowie Siedepunkte und Verdampfungswärmen zu, die chemische Reaktionsfähigkeit geht dagegen zurück. Der bei den Elementen der ersten bis dritten Hauptgruppe zu beobachtende Effekt der Schrägbeziehung erscheint bei sämtlichen Nebengruppenelementen, also auch in dieser Gruppe, nicht. Das Element Mangan leitet in seinen Eigenschaften also nicht zum Ruthenium über; die beiden Elemente unterscheiden sich sogar stark.
4.2 Chemische Eigenschaften
Die Elemente der Mangangruppe sind teils reaktiv (Mangan), Technetium und Rhenium verhalten sich dagegen meist reaktionsträge. An der Luft lagernd, schützt sie eine sehr dünne, passivierende Oxidschicht vor weiterer Korrosion durch Luftsauerstoff, und auch in Säuren sind sie nur vereinzelt und auch dann nur unter Anwendung drastischer Methoden löslich. Mit vielen Nichtmetallen (Halogene, Sauerstoff, auch Stickstoff und Kohlenstoff) reagieren sie aber bei erhöhter Temperatur, Mangan ist nach Scandium sogar das reaktionsfähigste Metall der ersten Periode der Übergangsmetalle. Mangan-II-oxid (MnO) reagiert schwach basisch, alle Dioxide der Gruppe (Mn/Tc/ReO2) amphoter, und die Dimetallheptoxide (Mn/Tc/Re2O7) sauer.
5 Einzeldarstellungen
Im folgenden Teil sind die Elemente der Mangangruppe (siebte Nebengruppe) jeweils einzeln mit ihren wichtigen Eigenschaften, Herstellungsverfahren und Anwendungen beschrieben.
5.1 Mangan
Das Pigment Braunstein (Mangan-IV-oxid) kommt in der Natur vor und wird schon seit der Steinzeit verwendet (Chalmin et al. 2003, 2006). Zur Herstellung von Glas setzte man Verbindungen des Mangans schon ab 400 v. Chr. zu deren Herstellung ein. Braunstein färbt das Glas intensiv braun-violett, wogegen Verbindungen des Mn-III Glas entfärben, da sie das grüne Fe-II zum gelben Fe-III oxidieren, woraus in der Überlagerung der Farben mit dem Violett des Mangans ein graues, fast farbloses Aussehen resultiert (Sayre und Smith 1961; McCray 1998).
1770 erhielt Kaim durch Reduktion von Braunstein mit Aktivkohle wohl erstmalig metallisches Mangan; 1774 reduzierte Gahn auf Scheeles Hinweis hin ebenfalls Braunstein mit Kohlenstoff zu Mangan (Rancke-Madsen 1975).
Im 19. Jahrhundert entdeckte man, dass zulegiertes Mangan die Verformbarkeit von Eisen verbessert, weshalb es um 1860 schon in großen Mengen zur Produktion von Stahl eingesetzt wurde (Corathers und Machamer 2006). Braunstein selbst wurde seit 1866 im Weldon-Prozess zur Gewinnung von Chlor aus Salzsäure verwendet (Brock 1997, S. 182).
Der schwedische Chemiker Johan Gottlieb Gahn (* 19. August 1745 Voxnabruk; † 8. Dezember 1818 Falun) studierte ab 1762 Bergwerkskunde in Uppsala und war nach Abschluss des Studiums ab 1770 am Bergwerkskollegium als Ingenieur angestellt. Er begegnete Scheele (Kurzbiografie siehe „Molybdän“), der in Uppsala als Apotheker tätig war, und arbeitete hiernach mit ihm eng zusammen.
Gahn verwendete als Erster Platindraht in der anorganischen Analyse und entdeckte die Cobaltperle. Er entwickelte zusammen mit Scheele ein Verfahren, Phosphor aus gemahlenen Knochen zu erzeugen. Außerdem isolierte Gahn 1774 metallisches Mangan durch Reduktion von Mangan-IV-oxid (Braunstein); im gleichen Jahr stellte er erstmals Bariumoxid durch Glühen von Schwerspat dar.
Gahn verbesserte wesentlich die Effektivität der Erzverhüttung im Bergwerk Falun und gewann aus dem Grubenwasser unter anderem Eisen- und Kupfersulfat. Seit 1793 war er Mitglied der Königlich Schwedischen Akademie der Wissenschaften und von 1808 an auswärtiges Mitglied der Bayerischen Akademie der Wissenschaften (Pötsch et al. 1989, S. 161; Hofberg et al. 1906, S. 378).
Der österreichische Arzt und Apotheker Ignatius Gottfried Kaim (* 1746; † 1778) war wohl noch vor Gahn der Erste, der metallisches Mangan entdeckte, denn er beschrieb sein Verfahren der Reduktion von Braunstein mit pulverförmiger Aktivkohle und das so hergestellte, brüchige, blauweiß glänzende Metall in seiner 1770 in Wien veröffentlichten Dissertation „De metallis dubiis“ („Über zweifelhafte Metalle“). Diese Arbeit fand aber wenig Beachtung in der damaligen wissenschaftlichen Öffentlichkeit, auch weil die Analyse des von ihm dargestellten Mangans unvollständig war. Allgemein galt daher Gahn als Entdecker des Mangans, obwohl dies wahrscheinlich ungerechtfertigt ist (Rumpf 1980, S. 36–42; Weeks 1956, S. 169).

Rhodochrosit (rot) und Manganit (schwarz) (Lawinsky 2010)
Mangan kommt größtenteils in drei verschiedenen Erzformen vor. Zum einen sind dies Rhodochrosit-Braunit-Erze, die in Brasilien sowie West- und Zentralafrika gefunden werden.
Eine weitere wichtige Quelle für Mangan sind eisen- und silikathaltige Sedimente, die unter anderem in Brasilien und Südafrika riesige Lagerstätten bilden. Eine wichtige Verbindung des Mangans ist dabei Braunstein (Mangan-IV-oxid, MnO2). Schließlich kommt Mangan in Schiefergesteinen vor, die rund um das Schwarze Meer und auch in Teilen Westafrikas zu finden sind (Corathers und Machamer 2006).
Die mit Abstand bedeutendsten und auch intensiv geförderten Vorkommen lagern unter der südafrikanischen Kalahari-Wüste; andere wichtige Förderländer sind China, Australien, Brasilien, Indien, Gabun und die Ukraine. 2014 belief sich die weltweit geförderte Menge auf 18 Mio. t Erz bei geschätzten Reserven in Höhe von 570 Mio. t (Corathers 2015).
Die Erze müssen im Allgemeinen einen Mindestgehalt von 35 % Mangan aufweisen, damit ein Abbau wirtschaftlich möglich ist. Für die verschiedenen, später geplanten Anwendungen ist eine Vorklassifizierung der Erze erforderlich. Soll das erzeugte Mangan als Legierungsbestandteil eingesetzt werden, so sollte das Erz zwischen 38 und 55 % Mangan enthalten. Ist die Verwendung des Metalls dagegen -in Form von Braunstein- in Alkali-Mangan-Batterien vorgesehen, so muss der Mangananteil bei mindestens 44 % liegen, bei zugleich geringem Anteil anderer Schwermetalle. Noch höheren Anforderungen müssen diejenigen Erze genügen, die man zur Herstellung reinen Mangans sowie dessen Verbindungen verwendet.
Auf dem Boden der Tiefsee kommt Mangan in Mengenanteilen bis zu 50 % in knollenartigen, bis zu 20 cm dicken, aus Oxiden von Schwermetallen bestehenden Agglomerationen vor. Diese enthalten auch die im Periodensystem benachbarten Nebengruppenelemente Kobalt, Nickel und Kupfer in Form ihrer Oxide. Bisher ergab sich aber kein wirtschaftlicher Nutzen aus einer etwaigen Förderung, da die für Mangan auf dem Weltmarkt erzielbaren Preise nicht ausreichen, die Kosten einer Produktion zu decken (Wellbeloved et al. 2005).
Wahrscheinlich gelang dem Österreicher Kaim 1770 die erstmalige Darstellung des Elements in unreiner Form durch Umsetzung von Mangan-IV-oxid mit Kohle, bevor Scheele und Gahn vier Jahre später mittels derselben Reaktion reineres Mangan darstellten (Rancke-Madsen 1975). Der Name des Mangans ist der antiken Bezeichnung für Braunstein („manganesia nigra“) entlehnt (Liebig et al. 1851).
1856 erkannte man, dass die Produktivität des damals zur Gewinnung von Stahl herangezogenen Bessemer-Verfahrens durch Zusatz von Mangan erheblich gesteigert werden kann (Corathers und Machamer 2006).
Die Mehrzahl der technischen Anwendungen erfordern kein reines Mangan; daher beschränkt man sich oft auf die Produktion einer Eisen-Mangan-Legierung mit einem Gehalt von 78 % Mangan („Ferromangan“). Jenes stellt man durch Reduktion eines Gemisches aus Mangan- und Eisenoxiden mit Koks im elektrischen Ofen her. Ähnlich verläuft die Produktion weiterer Manganlegierungen, wie z. B. Silicomangan, zu dessen Herstellung Quarzsand der im Ofen befindlichen Mischung zugesetzt wird.
Es ist aber nicht möglich, reines Mangan auf diesem Weg herzustellen, da dann neben dem Metall auch dessen stabile Carbide (beispielsweise Mn7C3) entstehen. Erst bei Temperaturen über 1600 °C zersetzen sich diese wieder unter Freisetzung reinen Mangans, das unter diesen Bedingungen schon sehr flüchtig ist, sodass dieses Verfahren kein wirtschaftlich gangbares ist. Bevorzugt elektrolysiert man daher Lösungen von Mangan-II-sulfat (MnSO4) bei Spannungen von 5 bis 7 V an Elektroden aus Edelstahl. An der Kathode schlägt sich reines Mangan nieder und an der Anode Sauerstoff, der die in wässriger Lösung befindlichen Mn2+-Ionen zu Mangan-IV-oxid (Braunstein, MnO2) oxidiert.

Vorkommen, physikalische und chemische Eigenschaften von Mangan
|
Symbol: |
Mn |
|
|
|
Ordnungszahl: |
25 | ||
|
CAS-Nr.: |
7439-96-5 | ||
|
Aussehen: |
Grauweiß glänzend |
Mangan (Tomihahndorf 2006) |
Mangan, Stücke (Sicius 2016) |
|
Entdecker, Jahr |
Kaim (Österreich), 1770 Gahn (Schweden), 1774 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
5325Mn (Spuren) |
3,74 ⋅ 106 |
ε >5324Cr | |
|
5525Mn (100) |
Stabil |
---- | |
|
Massenanteil in der Erdhülle (ppm): |
850 | ||
|
Atommasse (u): |
54,938 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
1,55 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial: Mn2+ + 2 e− > Mn (V) |
−1,18 | ||
|
Atomradius (berechnet) (pm): |
140 (161) | ||
|
Van der Waals-Radius (pm): |
Keine Angabe | ||
|
Kovalenter Radius (pm): |
139 (low spin), 161 (high spin) | ||
|
Ionenradius (Mn2+, pm) |
80 | ||
|
Elektronenkonfiguration: |
[Ar] 3d54s2 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte ♦ vierte ♦ fünfte ♦ sechste ♦ siebte: |
717 ♦ 1509 ♦ 3248 ♦ 4940 ♦ 6990 ♦ 9220 ♦ 11500 | ||
|
Magnetische Volumensuszeptibilität: |
9 ⋅ 10−4 | ||
|
Magnetismus: |
Paramagnetisch | ||
|
Kristallsystem: |
Kubisch-verzerrt | ||
|
Elektrische Leitfähigkeit( [A/(V ∙ m)], bei 300 K): |
6,94 ⋅ 105 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
198 ♦ 120 ♦ 79,5 (α-Mn) | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
Keine Angabe ♦ 196 (α-Mn) | ||
|
Mohs-Härte |
6,0 | ||
|
Schallgeschwindigkeit (longitudinal, m/s, bei 293,15 K): |
5150 | ||
|
Dichte (g/cm3, bei 293,15 K) |
7,43 | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
7,35 · 10−6 | ||
|
Wärmeleitfähigkeit [W/(m ∙ K)]: |
7,8 | ||
|
Spezifische Wärme [J/(mol ∙ K)]: |
23,35 | ||
|
Schmelzpunkt (°C ♦ K): |
1246 ♦ 1519 | ||
|
Schmelzwärme (kJ/mol) |
13,2 | ||
|
Siedepunkt (°C ♦ K): |
2100 ♦ 2373 | ||
|
Verdampfungswärme (kJ/mol): |
225 | ||


Zwischen Temperaturen von 727 und 1095 °C liegt β-Mangan mit ebenfalls kubisch-verzerrter Struktur vor, deren Elementarzelle 20 Atome enthält, und in der jedes Manganatom von 12 bis 14 Manganatomen umgeben ist (Shoemaker et al. 1978). β-Mangan wird bei tiefen Temperaturen nicht antiferromagnetisch (Kasper et al. 1956).
Oberhalb einer Temperatur von 1095 °C liegt das in der kubisch-flächenzentrierten Struktur kristallisierende γ-Mangan vor (Kupfer-Typ), die bei weiterem Erhitzen oberhalb von 1133 °C in die kubisch-innenzentrierte Struktur (δ-Mangan, Wolfram-Typ) übergeht (Schubert 1974).
Das Isotop 5525Mn ist das einzige stabile des Elements. Damit ist Mangan ein Reinelement.
Chemische Eigenschaften: Mangan ist nicht nur hinsichtlich seines Normalpotenzials, sondern vorrangig wegen seiner Unfähigkeit, eine schützende Passivschicht auf seiner Oberfläche auszubilden, nach Scandium das reaktionsfähigste Metall der ersten Periode der Übergangsmetalle (3d-Elemente). Es reagiert, vor allem bei erhöhter Temperatur, teils heftig mit vielen Nichtmetallen. Fein verteiltes Mangan ist an der Luft pyrophor und verbrennt schnell zu Mangan-II,III-oxid (Mn3O4). Nur mit Stickstoff reagiert das Element erst bei Temperaturen von über 1200 °C zu Mangannitrid (Mn3N2), Hydride bildet es nicht (Hartwig 2006; Holleman et al. 2007, S. 1608).
Mangan reagiert schon mit verdünnten Mineralsäuren unter heftiger Entwicklung von Wasserstoff, selbst durch Wasser wird es bei Raumtemperatur langsam angegriffen. Konzentrierte Salpeter- und Schwefelsäure reduziert es in stürmischer Reaktion bis hin zu Stickoxiden oder Schwefel-IV-oxid. Das Endprodukt der stark exergonischen Reaktionen (stark negative freie Bildungsenthalpie) ist jeweils das energetisch begünstigte Mn2+-Ion, da jenes eine halb gefüllte d-Konfiguration aufweist. In wässriger Lösung liegt der rosafarbene [Mn(H2O)6]2+ -Komplex vor. Sofern die entsprechenden Salze in Wasser löslich sind und sich nicht dabei zersetzen, lösen sich Mn3+-Ionen in Wasser mit roter Farbe. „Mn4+“ in Form von Braunstein ist braunschwarz; in wässriger Lösung existiert diese Spezies aber nicht. Verbindungen des Mangans in der Oxidationsstufe +5 (Hypomanganat, MnO43−) sind blau, die der Oxidationsstufe +6 (Manganat, MnO42−) grün und die der Oxidationsstufe +7 (Permanganat, MnO4−) violett.
Generell kann Mangan in allen Oxidationsstufen zwischen −3 und +7 auftreten. Am beständigsten sind diejenigen Verbindungen, die Mangan in den Oxidationsstufen +2, +3 und +4 enthalten.

Mangan-VII-oxid (Oelen 2006)


Kaliumpermanganat (Mangl 2007)


Mangan-IV-oxid (Benjah-bmm27 2007)



Mangan-IV-oxid ist das Kathodenmaterial in Alkali-Mangan-Batterien und zersetzt sich bei der Entladung der Batterie zu Manganoxidhydroxid und Mangan-II-hydroxid.


Mangan-III-oxid (Onyxmet 2018)
Mangan-II,III-oxid (Mn3O4) gehört zur Klasse der Spinelle, eine Klasse gemischter Oxide der allgemeinen Formel AB2X4 (A: Metall in der Oxidationsstufe +2; B: Metall mit der Oxidationsstufe +3; X: Chalkogen in der Oxidationsstufe −2). Es kommt in der Natur in Form des Minerals Hausmannit vor. Die Verbindung entsteht bei der Pyrolyse von Mangandioxid (MnO2) und auch bei der Verbrennung organischer Verbindungen des Mangans. Mangan-II,III-oxid kristallisiert in einer durch den Jahn-Teller-Effekt tetragonal verzerrten Spinellstruktur (Holleman et al. 2007, S. 1614). Man setzt die Verbindung als Futtermittelzusatzstoff sowie zur Herstellung von Halbleitern und magnetischen Materialien ein.

a Mangan-II-oxid (Onyxmet 2018). b Mangan-II-oxid Sputtertarget (QS Advanced Materials 2018)


β-Mangan-II-sulfid (Onyxmet 2018)

Durch Umsetzung wässriger Lösungen von Mangan-II-salzen mit Natronlauge entstehen Ausfällungen rosafarbenen Mangan-II-hydroxids [Mn(OH)2], das vor allem im alkalischen Milieu schon durch Luftsauerstoff leicht zu basischen Mangan-III, IV-oxiden oxidiert wird.
Mangan-II-selenid (MnSe) ist wie viele andere Chalkogenide der Übergangsmetalle technisch interessant als Halbleiter. Es bildet graue Kristalle oder ein graues Pulver, der Schmelzpunkt liegt bei 1460 °C, seine Dichte bei 5,59 g/cm3. Man erhält die Verbindung durch Erhitzen einer pulverförmigen Mischung stöchiometrischer Zusammensetzung der Elemente auf 500–600 °C bei Ausschluss von Luft.
Nanoröhrchen aus kristallinem Mangan-II-selenid konnten 2013 in solvothermischer Reaktion unter Einsatz von Polyvinylpyrrolidon als Schutzmatrix hergestellt werden. Die Ergebnisse spektroskopischer Untersuchungen zeigten, dass die Nanoröhrchen entlang der [2 0 0]-Achse wachsen. Auch hat das Volumenverhältnis von N,N -Dimethylformamid zu entionisiertem Wasser bedeutenden Einfluss auf den Habitus der Kristalle. Die Verbindung erwies sich als geeignet, Mikrowellen zu absorbieren (Zhang et al. 2013).

Mangan-II-tellurid Sputtertarget (QS Advanced Materials 2018)
- (I)
Mn + 2 F2 → MnF4
- (II)
MnF2 + 2 TbF4 → MnF4 + 2 TbF3
- (I)
2 MnI2 + 13 F2 → 2 MnF3 + 4 IF5
- (II)
2 Mn + 3 F2 → 2 MnF3

Mangan-II-fluorid Sputtertarget (QS Advanced Materials 2018)
- (I)
MnCO3 + 2 HF → MnF2 + CO2 ↑ + H2O
- (II)
Mn + F2 → MnF2
- (I)
Mn + 2 HCl → MnCl2 + H2↑
- (II)
MnCO3 + 2 HCl → MnCl2 + CO2 ↑ + H2O
- (III)
MnO2 + 4 HCl → MnCl2 + 2 H2O + Cl2↑

Mangan-II-chlorid-Tetrahydrat (Walkerma 2005)
- (I)
Mn + Br2 → MnBr2
- (II)
MnO2 + 4 HBr → MnBr2 + Br2 + 2 H2O

Mangan-II-bromid (Onyxmet 2018)
Das ebenfalls rosafarbene Mangan-II-iodid (MnI2) stellt man aus Mangan-II-oxid oder -carbonat und Iodwasserstoff her (Schuman 2007, S. 352). Die in wasserfreiem Zustand trigonal, in Form des Tetrahydrats monoklin kristallisierende Verbindung (Moore et al. 1985) ist ziemlich oxidationsempfindlich, färbt sich an der Luft unter Freisetzung von Iod braun und löst sich leicht in Wasser unter hydrolytischer Zersetzung. Die Verbindung der Dichte 5 g/cm3 schmilzt in wasserfreiem Zustand bei einer Temperatur von 701 °C.

Tetramanganmononitrid, Mn4N (Onyxmet 2018)
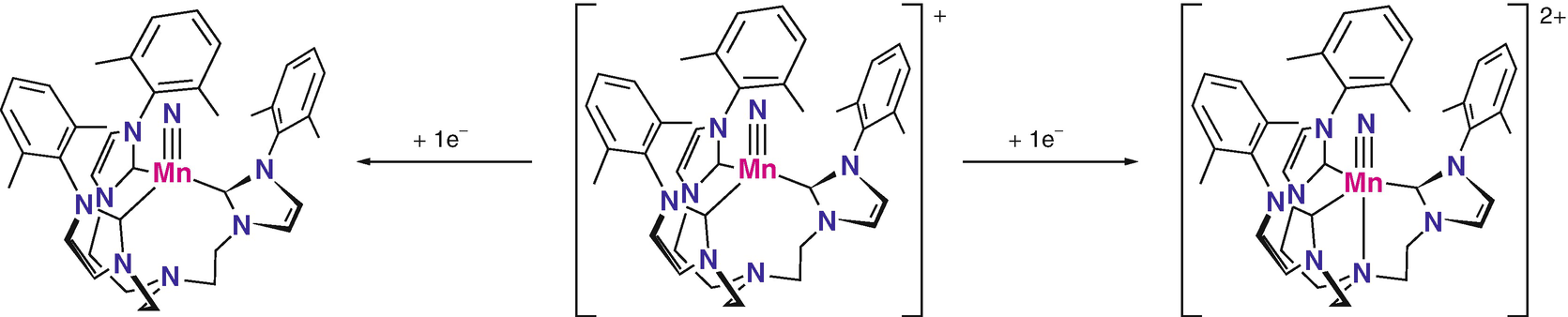
Mangan-Tris[2-(3-xylylimidazol-2-yliden)ethyl]amin-Komplexe (Kropp et al. 2012)
Mangan-II-phosphid (Mn3P2) ist ein schwarzes Pulver der Dichte 5,9 g/cm3, das bei 1095 °C schmilzt. Man setzt es als Halbleiter und als Zusatz zu Keramiken ein. Das gleichfalls feste und kristalline Mangan-III-arsenid (MnAs) wird ebenfalls in der elektronischen Industrie als Halbleiter und in optischen Anwendungen eingesetzt. Sehr interessant ist der jüngst getestete mögliche Einsatz in Sensoren mit hohem magnetischen Widerstand; diese sind wesentlich leistungsfähiger aufgrund größerer Speicherplatzdichten (Humphries 2014).


Trimanganmonocarbid, Mn3C (Onyxmet 2018)
Durch Reduktion einer aus Fe2O3 und Mn2O3 bestehenden Mischung mit Kohlenstoff in einer Stickstoffatmosphäre werden in Abhängigkeit von der Menge zugesetzten Mangans ein cohenitischer Fe3C-, ein Fe3 −xMnxC- und ein Fe7C3- Phasentyp erhalten. Stickstoff ist an der Reduktion vor allem manganreicher Mischungen beteiligt; ein relativ reines Mangancarbonitrid (Mn2C0,6N0,21) mit hexagonaler Kristallstruktur ist durch Erhitzen einer Mischung von Mangan-III-oxid und Kohlenstoff unter Stickstoff zugänglich (Zhang und Schleich 1994).
Höhere Mangansilicide wie MnSi1,73 sind erfolgversprechend als p-Halbleiter, die in thermoelektrischen Materialien im Temperaturbereich von 100–400 °C eingesetzt werden. Zur Größe von Nanoteilchen gemahlenes Mangansilicid mit Zusätzen von Ytterbium wurde durch Funkenplasmasintern hergestellt. Die Aufklärung der Kristallstruktur erfolgte mittels Röntgendiffraktometrie und Scan-Elektronenmikroskopie (Saleemi et al. 2015). Nanoröhrchen von Pentamangantrisilicid (Mn5Si3) stellten Lu et al. aus einer pulverförmigen Mischung von Mangan-III-oxid, Silicium und Magnesium im Autoklaven her (2018). Die aus der Röntgendiffraktometrie erhaltene Struktur ist hexagonal.
Das im orthorhombischen Eisenborid-Gitter kristallisierende Manganmonoborid (MnB) ist ferromagnetisch mit einer Curie-Temperatur von 273 °C (546 K) und der hohen Magnetisierung von bis zu 155,5 emu/g. Die asymptotische Vickers-Härte ist mit 15,7 GPa deutlich höher als diejenige traditionell verwendeter Ferromagnetika (Cui et al. 2017).

Mangan-II-nitrat (Onyxmet 2018)

Kaliummanganat-V (Maxim Startsev 2018)
- (I)
MnO4− + SO32− + H2O → MnO43− + SO42− + 2 H+
- (II)
2 MnO42− + H2O2 + 2 OH− → 2 MnO43− + O2 + 2 H2O
- (III)
2 MnO2 + 3 OH− → MnO43− + MnOOH + H2O

Kaliummanganat-VI (Sanja Ardent 2017)
- (I)
2 MnO2 + 4 KOH + O2 → 2 K2MnO4 + 2 H2O
- (II)
2KOH + KNO3 + MnO2 → K2MnO4 + H2O + KNO2


Galliummanganarsenid [(Ga,Mn)As] ist ein magnetischer Halbleiter und eng mit dem weithin eingesetzten Galliumarsenid verwandt. Im Unterschied zu II-VI-Halbleitern ist es nicht para-, aber ferromagnetisch (Furdyna 1988). Sein magnetisches Verhalten ist hysteretisch, die Manganionen wirken zudem als Akzeptor. Daher ist die Verbindung ein p-Halbleiter und gibt über Ladungsträger spin-polarisierte Ströme weiter, was die meisten anderen ferromagnetischen Halbleiter nicht können (Ohno et al. 1992; Pinto et al. 2005). Daher ist Galliummanganarsenid ein spintronisches Material.
Man stellt die Substanz durch Molekularstrahl-Epitaxie her, bei dem Kristallstrukturen in der Präzision atomarer Schichten geändert bzw. erzeugt werden können. Hier substituieren Mangan- die Galliumionen, nur diffundieren erste nur schwer in das Galliumarsenid hinein. Um trotzdem einen ausreichenden Ferromagnetismus erhalten zu können, erhitzte man das als Substrat dienende Galliumarsenid in ersten Versuchen auf ca. 600 °C. Nachteilig war, dass die sich die Manganionen nur wenig in das Substrat integrierten, sondern sich vielmehr an der Oberfläche des Kristalls mit Arsenatomen verbanden (De Simone und Wood Jr. 1982). Dieses Problem löste man durch Senken der Temperatur des Substrates auf etwa 250 °C, und es konnten in Analogie zu bereits früher hergestellten Einkristallen von Indiummanganarsenid (Munekata et al. 1989) Kristalle des Galliumanganarsenids guter Qualität hergestellt werden (Ohno et al. 1996; Ohno 1998).
In den meisten Komplexen tritt Mangan in der Oxidationsstufe +2 auf. Die oktaedrisch koordinierten sind meist paramagnetisch und rosa gefärbt. Niedrigere Oxidationszahlen findet man im Dimangandecacarbonyl [Mn2(CO)10; 0] oder im Mn(NO)3CO [-3 (!)]. Als Resonanzmittel für die Magnetresonanzspektroskopie der Leber dient das anderen Produkten überlegene Mangafodipir (Bellin 2006).
Dimangandecacarbonyl [Mn2(CO)10] erhält man durch Carbonylierung von Mangan-II-Salzen (Brauer 1981, S. 1634) bei Kohlenmonoxiddrücken von ca. 300 bar in Gegenwart von Triethylaluminium, so etwa bei Verwendung von Mangan-II-acetat.
Das goldgelbe, lichtempfindliche Dimangandecacarbonyl schmilzt bei 152 °C und ist relativ beständig gegenüber Luftsauerstoff. Es ist unlöslich in Wasser, aber löslich in fast allen organischen Lösungsmitteln. Im Vakuum sublimiert es schon merklich bei Raumtemperatur. Dimangandecacarbonyl ist Ausgangsmaterial zur Herstellung von Mangancarbonyl-Verbindungen und wird darüber hinaus als Katalysator und Antiklopfmittel verwendet.

Reines Mangan wird technisch kaum genutzt. Nahezu die gesamte abgebaute Menge an Mangan (130.000 t/a) wird dagegen mit Stahl zu Ferromangan legiert, bindet darin Sauerstoff und Schwefel und erhöht gleichzeitig die Härte des Stahls. Es verhindert durch Bindung des Schwefels beispielsweise die Bildung des leicht schmelzenden Eisensulfides. Dadurch, dass es dem Stahl Sauerstoff entzieht, erhöht es dessen Aufnahmevermögen für Stickstoff, was die Resistenz des Stahls gegenüber Korrosion erhöht.
Ähnliche Effekte zeigt es in Legierungen mit Kupfer und Aluminium, wo es ebenfalls die Festigkeit, Korrosionsbeständigkeit und Verformbarkeit verbessert. Eine besondere Wirkung besitzt eine in elektrischen Messgeräten eingesetzte, aus 83 % Kupfer, 12 % Mangan und 5 % Nickel bestehende Legierung, deren elektrischer Widerstand nur wenig von der Temperatur abhängig ist.
In LEDs findet man manganhaltige Aktivatoren, so emittiert BaMgAl10O17 : Eu2+, Mn2+ grünes, Mg14Ge5O24 : Mn4+ rotes Licht (S. Shionoya et al. 2006). Mangan-IV-oxid geht als Kathode in Alkali-Mangan-Batterien und wurde schon in der Steinzeit als Pigment in Höhlenmalereien nachgewiesen (Chalmin et al. 2003, 2006). Auch die Römer nutzten bereits Manganverbindungen in der Färbung von Glas (Sayre und Smith 1961; McCray 1998). Mangan-IV-oxid diente lange Zeit im Weldon-Verfahren zur Oxidation von Salzsäure (Chlorwasserstoff) zu Chlor.
Mangan ist für alle Lebewesen essenziell und Komponente diverser Enzyme, in deren Molekülen es wegen seiner zahlreichen Oxidationsstufen in unterschiedlichen Funktionen auftreten kann. Diese findet man in der Fotosynthesereaktion (Yano et al. 2006) ebenso wie bei anaeroben Vorgängen (Madigan und Martinko 2009).
Enzyme, die Superoxid abbauen (Superoxiddismutasen; Alscher 2002; Law et al. 1998) oder Sauerstoff in bestimmte organische Moleküle einbauen können (Dioxygenasen), enthalten als wirksames Prinzip redoxaktive Manganionen. Manganperoxidase ist als eines der wenigen bekannten Enzyme zum Abbau von Lignin in der Lage. An vielen anderen enzymatisch katalysierten Reaktionen ist Mangan gleichfalls beteiligt (Arginasen, Hydrolasen, Kinasen, Decarboxylasen, Transferasen, Katalasen und Ribonukleotidreduktasen).
Der Mensch nimmt Mangan über den Dünndarm auf und speichert es meist in den Nieren, der Leber und der Bauchspeicheldrüse. Die Zellsubstanz enthält es in Mitochondrien, Lysosomen und im Zellkern, oft an Eiweißmoleküle gebunden (Takeda 2003). Wegen der dennoch niedrigen vom Menschen benötigten Menge (1 mg/d) ist ein Mangel an Mangan selten, der sich bei Tieren beispielsweise in Deformationen des Skeletts, Nervenschäden und Wachstumsstörungen äußert. Nahrungsmittel mit hohem Gehalt an Mangan sind schwarzer Tee, Weizenkeime, Haselnüsse, Haferflocken, Sojabohnen, Leinsamen, Heidel- und Aroniabeeren sowie Roggenvollkornbrot (Ekmekcioglu und Marktl 2006).
Das Inhalieren manganhaltigen Staubs kann Schäden der Lunge verursachen, die sich zuerst in Form von Husten, Bronchitis oder Lungenentzündung äußern. Konzentrationen an Mangan, die den MAK-Wert dauerhaft überschreiten (0,02 mg/m3 für sehr feinen bzw. 0,2 mg/m3 für generell inhalierbaren Staub), können das zentrale Nervensystem schädigen, was in Bewegungsstörungen oder starkem Zittern zum Ausdruck kommt (Santamaria und Sulsky 2010). Durch Aufnahme von Mangan hervorgerufene Erkrankungen sind als Berufskrankheit (1105) anerkannt.

-
Y.-M. Ha und Y.-D. Jung, High manganese steel for low temperature, having excellent surface quality, and manufacturing method therefor (Posco Ltd., WO 2019078538 A1, veröffentlicht 25. April 2019)
-
H. Miura und K. Suetsugu, Electrolytic manganese dioxide and method for its production, and its application (Tosoh Corp., US 2019119124 A1, veröffentlicht 25. April 2019)
-
S. Ferrasse und F. C. Alford, Copper manganese sputtering target (Honeywell International Ltd., WO 2019075204 A1, veröffentlicht 18. April 2019)
-
G. G. Yadav und X. Wei, Process for making manganese dioxide and its polymorphs reversible (Research Foundation of the City University of New York, WO 2019023546 A2, veröffentlicht 31. Januar 2019)
-
C.-V. Muntean und M. E. Stoia, Process preparing manganese ferrite (Universitatea Politehnika din Timişoara, RO 133044 A2, veröffentlicht 30. Januar 2019)
-
T. Yuzawa und K. Inokuchi, Production method for positive electrode material and manganese dry cell using same (Panasonic IP Manufacturing Co., Ltd., WO 2019017055 A1, veröffentlicht 24. Januar 2019)
-
H.-J. Kim und H.-S. Hwang, High-manganese hot-dip aluminum coated steel sheet having excellent coating adhesion (Posco, US 2019010597 A1, veröffentlicht 10. Januar 2019)
-
B. D. Briggs und L. A. Clevenger, Hybrid electric scheme for varying liner thickness and manganese concentration (IBM, US 2019013278 A1, veröffentlicht 10. Januar 2019)
-
M. Kasaaian, Sulfide recycling in manganese production (privat, US 2019003065, veröffentlicht 3. Januar 2019)
-
D. Antonelli, Synthesis and hydrogen storage properties of novel manganese hydrides (USW Commerical Services Ltd., US 2018375136, veröffentlicht 27. Dezember 2018)
-
W.-L. Wu und Y.-C. Lee, Manganese-doped red fluoride phosphor, light emitting device, and backlight module (Lextar Electronics Corp., US 2018366614 A1, veröffentlicht 20. Dezember 2018)
-
W. Chen und Y. Cui, Ultrastable rechargeable manganese battery with solid-liquid reactions (University Leland Stanford Junior, WO 2018222609 A1, veröffentlicht 6. Dezember 2018)
-
Z. J. McAfee und J. A. Calderone III, Aviation gasoline containing branched aromatics with a manganese octane enhancer (Afton Chemical Corp., MX 2017015555 A, veröffentlicht 9. November 2018)
5.2 Technetium
Technetium kommt auf der Erde in extrem geringen Spuren natürlich vor, ist aber in größeren Mengen nur auf künstlichem Wege herstellbar. Alle Isotope des Elements sind radioaktiv, womit Technetium neben Promethium (Ordnungszahl 61) das einzige Element ist, das eine geringere Ordnungszahl als Bismut besitzt und dessen Isotope dennoch sämtlich radioaktiv sind.
Über lange Zeit bestand in dem von Mendelejev konzipierten Periodensystem der Elemente eine Lücke zwischen Molybdän (Ordnungszahl 42) und Ruthenium (Ordnungszahl 44). Mendelejev sagte einige der Eigenschaften dieses damals noch unbekannten Elementes voraus, ohne dessen Entdeckung noch zu erleben. Bereits seit Beginn des 19. Jahrhunderts erhoben verschiedene Forscher Ansprüche auf eine angebliche Entdeckung des Elements. Es begann 1828 mit dem von Osann beschriebenen Polinium, das sich später als verunreinigtes Iridium herausstellte (Kenna 1962). 1847 bezeichnete Rose das vermeintlich entdeckte, später Technetium genannte Element als Pelopium (De Jonge und Pauwels 1996).
Die erste ausschließlich auf die Entdeckung des noch unbekannten Elementes mit der Ordnungszahl 43 gerichtete Suche endete 1877 mit der Benennung Davyum (Kern 1877), das aber später als Legierung von Rhodium und Iridium identifiziert wurde. Wiederum später reklamierte Ogawa die Entdeckung des Nipponiums für sich (Yoshihara 2004), das sich später als Rhenium herausstellte (Kenna 1962).
1925 glaubten Noddack und Tacke (Kurzbiografie s. „Neptunium“), die im gleichen Jahr noch das Rhenium entdeckten, auch das Element 43 identifiziert zu haben und nannten es Masurium. Sie beschossen das Mineral Columbit mit Elektronenstrahlen und schlossen aus dem Röntgenspektrum auf das Vorliegen des neuen Elements. Die Versuche konnten jedoch weder von anderen Arbeitsgruppen reproduziert werden, noch gelang Noddack und Tacke die Darstellung des reinen Elements. Daher fand die Entdeckung zunächst keine Anerkennung (Weeks 1933). 65 Jahre später, im Jahre 1998, simulierten Armstrong (National Institute of Standards and Technology) und Curtis (Los Alamos National Laboratory) die Arbeiten von Noddack und Tacke mittels moderner Methoden und kamen überraschenderweise zu ähnlichen Ergebnissen, weshalb das Recht auf die erstmalige Entdeckung des Technetiums wieder offen ist (Zingales 2005).

Der deutsche Chemiker Walter Noddack (* 17. August 1893 Berlin; † 7. Dezember 1960 Bamberg) entdeckte 1925 zusammen mit seiner späteren Frau Ida Tacke und Otto Berg das Element Rhenium. Die gleichzeitige Entdeckung des Elements mit der Ordnungszahl 43 veröffentlichten sie ebenfalls und nannten das neue Element Masurium, nur erschienen der wissenschaftlichen Öffentlichkeit die von ihnen vorgelegten Nachweise als nicht ausreichend. Das neue Element erklärte man daher erst 1937 als sicher nachgewiesen und nannte es Technetium.
Noddack und seine Frau erhielten 1931 die Liebig-Denkmünze der Gesellschaft Deutscher Chemiker. 1935 lehrte Walter Noddack als Ordentlicher Professor für Physikalische Chemie an der Universität Freiburg Chemie und von 1941 bis 1945 an der von den Nationalsozialisten in Reichsuniversität umbenannte Hochschule in Straßburg. 1947 wechselte er als Professor zur Philosophisch-Technischen Hochschule Bamberg, wurde dann 1957 Honorarprofessor an der Universität Erlangen und Leiter des Staatlichen Forschungsinstituts für Geochemie in Bamberg (Engel 1999, S. 307; Klee 2005, S. 438).
Der italienische Physiker Emilio Gino Segrè (* 1. Februar 1905 Tivoli; † 22. April 1989 Lafayette, CA) führte 1937 den ersten eindeutigen Nachweis des Elements Technetium. Er studierte zunächst in Rom Ingenieurswissenschaften, wechselte 1927 zur Physik und promovierte 1928 bei Enrico Fermi. Nach zwei Forschungsjahren in Deutschland und den Niederlanden war er bis 1936 Assistenzprofessor für Physik an der Universität von Rom und sollte gemäß einem Vertrag bis 1938 die Physiklabors der Universität Palermo leiten. Schon 1937 beschoss er zusammen mit Lawrence in Berkeley Molybdän mit Deuteronen; die hiernach vom Molybdän emittierten Teilchen wiesen auf ein im Versuch erzeugtes radioaktives Element hin, das er Technetium nannte.
1938 untersagte die Regierung Mussolini Juden die weitere Bekleidung universitärer Stellen, sodass Segrè in den USA blieb. Zuerst war er bei Lawrence und der University of California angestellt. In Berkeley arbeitete Segrè mit seinen Kollegen an der Entdeckung des Elements Astat und des Plutoniumisotops 23994Pu, dann zwischen 1943 und 1946 am Manhattan-Projekt. Zwischen 1946 und 1972 lehrte er Physik an der Universität Berkeley, kehrte 1974 nach Italien zurück, wo er in Rom einen Lehrstuhl für Kernphysik erhielt. 1959 erhielten Segrè und Chamberlain den Physik-Nobelpreis für ihre Entdeckung des Antiprotons (Segrè 1995).
Der österreichisch-ungarische Physiker Josef Mattauch (* 21. November 1895 Mährisch-Ostrau (Moravska Ostrava); † 10. August 1976 Klosterneuburg) trat 1941 die Nachfolge von Lise Meitner in der physikalischen Abteilung des Berliner Kaiser-Wilhelm-Instituts für Chemie an. Zwischen 1944 und 1949 wurde das Institut ins württembergische Tailfingen ausgelagert und dann unter dem Namen Max-Planck-Institut für Chemie in Mainz wieder aufgebaut, dessen Direktor er wurde. Schwerpunkt seiner Arbeit war die Untersuchung der Häufigkeit von Isotopen mit Hilfe der Massenspektrografie; 1934 stellte er die Mattauchsche Isobarenregel auf (Mattauch 1934; Hintenberger 1990, S. 388). Er votierte 1957 mit siebzehn anderen Kernphysikern gegen die Ausrüstung der Bundeswehr mit Atomwaffen.
Schon 1952 gelang der spektroskopische Nachweis, dass zumindest in einigen Roten Riesen wegen der in ihnen herrschenden extrem hohen Temperaturen und Drücke Isotope des Technetiums vorkommen (Paul und Merrill 1952). Da das Alter dieser Sterne schon mehrere Mrd. a beträgt, das längstlebige Isotop des Technetiums aber nur eine Halbwertszeit von ca. 4 Mio. a besitzt, galt dies als erster klarer Beweis, das Isotope derartig schwerer Elemente im Inneren solcher Sterne gebildet werden können (Moore 1951). Die Masse und Energie unserer Sonne reicht hingegen nicht zur Bildung solcher Isotope aus, denn bestenfalls lässt sich das Vorhandensein von Isotopen des Eisens (Ordnungszahl 26) im Sonnenspektrum nachweisen.

Jedes Jahr entstehen in Kernreaktoren weltweit mehrere t des Metalls durch Spaltung des Uranisotops 23592U , weswegen abgebrannte Brennstäbe einen Anteil an Technetium von rund 6 % (!) aufweisen. Infolgedessen dürfte sich die bislang auf künstlichem Weg hergestellte Menge des Metalls auf -extrapoliert- rund 100 t belaufen und übertrifft damit die natürlich vorkommende um Größenordnungen.
Bei der Wiederaufarbeitung dieser abgebrannten Kernbrennstäbe löst man das Technetium in wässrigem Medium auf und oxidiert es zu Pertechnetat (TcO4−). Nach Abwarten einer gewissen Abklingzeit kann man es dann von Salzen anderer in der Lösung enthaltenen Elemente (Uran, Neptunium, Plutonium) abtrennen. Nach Umwandlung zu Ammoniumpertechnetat (NH4TcO4) oder auch -hexachlorotechnetat-IV [(NH4)2TcCl6] lässt man diese Verbindungen dann bei erhöhter Temperatur mit Wasserstoff reagieren, worauf sich metallisches Technetium bildet. Ein zweiter Darstellungsweg ist die Elektrolyse einer schwefelsauren, wasserstoffperoxidhaltigen Lösung von Ammoniumpertechnetat.
Es wird jedoch weit mehr an Technetium produziert, als genutzt werden kann. Daher muss die größte Menge des im Reaktor hergestellten Materials endgelagert werden. Erschwerend wirkt die lange Halbwertszeit der erzeugten Isotope des Technetiums. Zurzeit wird noch die Lagerung in Salzstöcken favorisiert, jedoch bestehen Bedenken wegen einer eventuell zu leicht möglichen Auswaschung durch Grundwasser.
Soll Technetium in der Medizin eingesetzt werden, so beschießt man Molybdän mit Neutronen. Das so erhaltene Isotop 9942Mo erleidet β−-Zerfall zum Isotop 99m43Tc, das unter Aussendung der therapeutisch eingesetzten γ-Strahlen in das Isotop 9943Tc übergeht. Bei diesem Prozess entsteht nach Auflösen des Metalls schließlich Pertechnetat (TcO4−) in Konzentrationen von 10 bis 1000 nmol/L. Nach dessen Konzentration und Aufarbeitung entstehen Verbindungen, die man mit Wasserstoffgas zu metallischem Technetium reduziert.
Vorkommen, physikalische und chemische Eigenschaften von Technetium
|
Symbol: |
Tc |
|
|
|
Ordnungszahl: |
43 | ||
|
CAS-Nr.: |
7440-26-8 | ||
|
Aussehen: |
Silbergrau glänzend |
Technetium, (http://www.webqc.org 2016) |
Mit 9943Tc beschichtete Goldfolie (Onyxmet 2018) |
|
Entdecker, Jahr |
Segrè und Perrier (Italien), 1937 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit |
Zerfallsart, -produkt | |
|
9643Tc (synthetisch) |
4,28 d |
ε >9642Mo | |
|
9743Tc (synthetisch) |
2,6 ∙ 10 6 a |
ε >9742Mo | |
|
9843Tc (synthetisch) |
4,2 ∙ 10 6 a |
β−>9844Ru | |
|
9943Tc (synthetisch) |
211.100 a |
β−>9944Ru | |
|
Massenanteil in der Erdhülle (ppm): |
1,2 ⋅ 10−19 | ||
|
Atommasse (u): |
98,906 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
1,9 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial: TcO2 + 4 e− + 4 H+ → Tc + 2 H2O (V) |
0,272 | ||
|
Atomradius (berechnet) (pm): |
135 (185) | ||
|
Van der Waals-Radius (pm): |
Keine Angabe | ||
|
Kovalenter Radius (pm): |
147 | ||
|
Ionenradius (Tc2+/Tc4+, pm) |
95/72 | ||
|
Elektronenkonfiguration: |
[Kr] 4d55s2 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
702 ♦ 1472 ♦ 2850 | ||
|
Magnetische Volumensuszeptibilität: |
3,9 ⋅ 10−4 | ||
|
Magnetismus: |
Paramagnetisch | ||
|
Kristallsystem: |
Hexagonal | ||
|
Elektrische Leitfähigkeit ([A/(V ∙ m)], bei 300 K): |
4,54 ⋅ 106 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
407 ♦ 297 ♦ 123 | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
Keine Angabe | ||
|
Mohs-Härte |
5,5 | ||
|
Schallgeschwindigkeit (longitudinal, m/s, bei 293,15 K): |
16.200 | ||
|
Dichte (g/cm3, bei 293,15 K) |
11,5 | ||
|
Molares Volumen (m3/mol , im festen Zustand): |
8,63 · 10−6 | ||
|
Wärmeleitfähigkeit [W/( ∙ K)]: |
51 | ||
|
Spezifische Wärme [J/(mol ∙ K)]: |
24,27 | ||
|
Schmelzpunkt ( °C ♦ K): |
2157 ♦ 2430 | ||
|
Schmelzwärme (kJ/mol) |
23 | ||
|
Siedepunkt (°C ♦ K): |
4265 ♦ 4538 | ||
|
Verdampfungswärme (kJ/mol): |
550 | ||


Bei Raumtemperatur ist Technetium schwach paramagnetisch. Unterhalb einer Temperatur von −265,45 °C wird das hochreine Metall supraleitend. Im supraleitenden Zustand wird die Durchlässigkeit für Magnetfelder sehr hoch und wird nur noch von der des Niobs übertroffen.
Alle bisher bekannten 34 Isotope des Elements sind radioaktiv. Die längstlebigen sind 9843Tc,9743Tc bzw. 9943Tc mit Halbwertszeiten von 4,2 Mio., 2,6 Mio. bzw. 211.100 a. 9943Tc ist ein weicher β−-Strahler. Ist die Massenzahl der Isotope des Technetiums <98, so fangen sie Elektronen ein (ε) und gehen in Molybdänisotope gleicher Massenzahl über. Die Isotope mit Massenzahlen 99 und ≥101 erleiden dagegen β−-Zerfall zu den jeweiligen Isotopen des Rutheniums. Nur für das Isotop 10043Tc sind beide Zerfallswege möglich.
Warum ist Technetium als Element relativ niedriger Massenzahl instabil? Dies wird erklärt durch die von Mattauch aufgestellten Thesen (Mattauch’sche Isobarenregel). Atomkerne sind nur dann stabil, wenn sich die Zahl der Neutronen und Protonen miteinander im Gleichgewicht befindet. Für Technetium liegt dieser Bereich möglicherweise beständiger Isotope bei Massenzahlen zwischen 95 und 101. Nur sind auch diese Isotope sämtlich radioaktiv.
Kerne einer konstanten Zahl von Nukleonen (Protonen plus Neutronen), aber verschiedener Protonen- (Kernladungs- oder Massen)zahl heißen Isobare, wie beispielsweise 9942Mo,9943Tc und 9944Ru. Die Kernbindungsenergie der Atomkerne dieser Isobare ist parabelförmig abhängig von der Zahl der Protonen, wobei der Scheitelpunkt der Parabel die Kernbindungsenergie innerhalb des stabilsten Atomkerns wiedergibt (Mattauch 1934).
Bei ungerader Nukleonenzahl liegen die Energiewerte aller Kerne ebenfalls auf einer Parabel und nur das stabilste existiert auch, also das, dessen Kernbindungsenergie in der Nähe des Parabelmaximums liegt. Für den in Betracht kommenden Bereich der Nuklide von 95 bis 101 sind dies aber nur Isobare der zu Technetium benachbarten Elemente Molybdän und Ruthenium, sodass alle Technetiumisotope ungerader Nukleonenzahl schon einmal instabil, d. h. radioaktiv, sind.
Ist die Nukleonenzahl dagegen gerade, können Isobare mehrerer Elemente stabil sein. Dafür existieren zwei unterschiedliche Energiekurven, eine für gg-Kerne (jeweils gerade Zahl an Protonen und Neutronen) und eine für die energiereicheren, also instabileren uu-Kerne (jeweils ungerade Zahl an Protonen und Neutronen). Das Vorhandensein von gg-Kernen ist bevorzugt, und nur wenn es keinen gg-Kern gleicher Nukleonenzahl gibt, existiert der entsprechende uu-Kern. Die Atomkerne des Technetiums gerader Massenzahl (9643Tc,9843Tc und 10043Tc) sind alle uu-Kerne und könnten nur dann beständig sein, falls es keine stabilen gg-Kerne gleicher Nukleonenzahl gäbe. Da aber jedes der Isotope 9642Mo,9842Mo,10042Mo,9644Ru,9844Ru und 10044Ru stabil ist, entfällt auch diese Möglichkeit, dass Technetium stabile Isotope besitzen könnte.
Chemische Eigenschaften: Technetium ähnelt in seinen chemischen Eigenschaften weit mehr dem Rhenium als dem Mangan. Ist die beständigste Oxidationsstufe des Mangans +2 (Mn2+), so liegt Technetium in seinen Verbindungen oft auch in anderen Oxidationsstufen vor (+4, +5 und +7). Auch +1, +3, 0 und sogar −1 sind beschrieben, während gerade die bei Mangan verbreitete Oxidationszahl +2 nur selten auftritt (Rimshaw und Hampel 1968).
Pulverförmiges Technetium ist leicht brennbar und reagiert auch heftig mit reaktiven Nichtmetallen. Auch das kompakte Metall läuft an feuchter Luft infolge Korrosion langsam an. Hingegen ist Technetium überraschend beständig gegenüber Säuren wie Salz- oder Flusssäure, nur in konzentrierter Schwefel- oder Salpetersäure löst es sich.
Chalkogenverbindungen Technetium-VII-oxid (Tc2O7) kann man durch Verbrennen von Technetium im Sauerstoffstrom bei 450–500 °C erhalten (Brauer 1981, S. 1598). Die nadelförmigen, gelben Kristalle orthorhombischer Struktur haben die Dichte 3,5 g/cm3, schmelzen bei 120 °C (die Flüssigkeit siedet bei 311 °C) und sind stark wasseranziehend. In Wasser lösen sie sich leicht unter Bildung von in höheren Konzentrationen pinkfarbenen Lösungen von Pertechnetiumsäure (HTcO4) (Krebs 1969; Herrell et al. 1977). Diese gehört zu den starken Säuren; ihr Pertechnetatanion wirkt im Gegensatz zu Permanganat aber nur noch leicht oxidierend.
- (I)
NH4TcO4 → 2 TcO2 + 4 H2O + N2
- (II)
Tc2O7 + 3 H2 → 2 TcO2 + 3 H2O


Mit den höheren Chalkogenen bildet Technetium analog Technetium-IV-selenid (TcSe2) und –tellurid (TcTe2) (Schwochau 2000).
Halogenverbindungen Technetium-VI-fluorid (TcF6) ist das höchste Fluorid des Technetiums und wird durch Erhitzen von Technetium im Fluorstrom bei Temperaturen um 400 °C erhalten (Selig et al. 1961). Der goldgelbe, oberhalb einer Temperatur von −4,5 °C kubisch, darunter orthorhombisch kristallisierende Feststoff (Siegel und Northrop 1966) der Dichte 3,6 g/cm3 (Drews et al. 2006) schmilzt bei einer Temperatur von 37 °C; die Flüssigkeit siedet bei 55 °C (Selig und Malm 1962; Osborne et al. 1978). Im Molekül der Verbindung ist ein Technetium- von sechs Fluoratomen oktaedrisch umgeben (Claassen et al. 1962, 1970).
Technetium-VI-fluorid ist ein starkes Fluorierungsmittel und unzersetzt nur in wenigen Medien, wie etwa Iod-V-fluorid, löslich. Durch Iod wird es sofort zu Technetium-V-fluorid reduziert (Binenboym und Selig 1976). Mit Alkalichloriden setzt es sich zu Alkalihexafluorotechnetat-V (TcF6−) um (Edwards et al. 1963; Hugill und Peacock 1966). Mit Natronlauge erleidet Technetium-VI-fluorid Hydrolyse und Reduktion zu schwarzem, aus der Lösung ausfallenden Technetium-IV-oxid (TcO2). In Flusssäure gelöstes Hydraziniumfluorid reduziert das in der Oxidationsstufe +6 vorliegende Element zu Tc-V bzw. Tc-IV, dies unter Bildung von Hydraziniumhexafluorotechnetat-V bzw. -IV (Frlec et al. 1967).
- (I)
Tc + 2 Cl2 → TcCl4
- (II)
Tc2O7 + 7 CCl4 → 2 TcCl4 + 7 COCl2 + 3 Cl2
Die Technetiumbromide TcBr4 und TcBr3 konnte man durch Umsetzung metallischen Technetiums mit Brom bei ca. 400 °C erhalten. Danach durchgeführte Versuche zur Bestimmung der Kristallstruktur mittels Röntgendifraktometrie zeigten, dass TcBr3 im orthorhombischen Gitter kristallisiert. In diesem liegen unendliche Ketten von flächenseitig aneinander grenzenden TcBr6-Oktaedern vor, dies mit abwechselnd langen und kurzen Abständen der Technetiumatome untereinander. Auch das rotbraune TcBr4 kristallisiert orthorhombisch; im Gitter befinden sich ebenfalls unendliche Ketten mit TcBr6-Octaedern, die jedoch lediglich kantenverknüpft sind. Anzeichen für Tc-Tc-Bindungen liegen hier dagegen nicht vor. Technetium-III-bromid ist isomorph mit Ruthenium- bzw. Molybdän-III-bromid (RuBr3, MoBr3), wogegen TcBr4 mit Platin- und Osmium-IV-bromid (PtBr4, OsBr4) isomorph ist (Poineau et al. 2009).
Es gibt zahlreiche weitere Halogenide und Oxidhalogenide, über die aber nur wenige Daten zu finden sind, wie beispielsweise die Technetium-VII-trioxidhalogenide (TcO3F, TcO3Cl, TcO3Br und TcO3l), die Technetium-VI-oxidtetrahalogenide (TcOF4, TcOCl4) und die Technetium-V-oxidtrihalogenide (TcOF3, TcOCl3 und TcOBr3). Alle diese Verbindungen sind meist sehr hydrolyseempfindlich und werden daher durch Wasser schnell zersetzt.
Pnictogenverbindungen Reines Technetiummononitrid (TcN) weist eine Dichte von 12,12 g/cm3 auf und kristallisiert im kubischen Kochsalzgitter. Die Zusammensetzung intermediärer Phasen untersuchte man während der Thermolyse von (NH4)TcCl6 und (NH4)2TcBr6 innerhalb eines Temperaturbereiches von 380–800 °C unter Argon. Bei 380 °C erhält man stickstoffuntersättigtes Nitrid TcN0,75 mit kubisch-flächenzentrierter Struktur. Mit steigender Reaktionstemperatur erfolgt eine lineare Änderung der Gitterparameter (Vinogradov et al. 1978).
Die strukturell bedingten Eigenschaften der Technetiumphosphide Tc3P und TcP4 wie Härte, Spektren und Leitfähigkeit studierten Feng et al. im weiten Druckbereich bis zu 0.40 GPa. So steigt beispielsweise die Härte von TcP4 mit dem Druck (2017).
Sonstige Verbindungen Technetium zeigt wie andere höhere Metalle der Nebengruppen kaum noch eine „metalltypische“ Kationenchemie und bildet dagegen eher kovalente Verbindungen mit Molekülstruktur oder auch Cluster. Selbst im Molekül des Hydridotechnetat-VII-Komplexes [(TcH9)2−] ist ein Technetiumion trigonal-prismatisch von insgesamt neun Wasserstoffanionen umgeben(!).
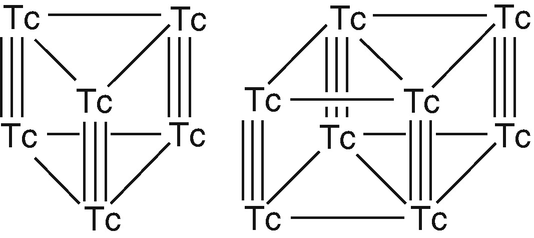
Technetiumcluster Tc6 und Tc8 (Aglarech 2005)
In Abb. 19 sind die beiden wichtigsten Technetium-Cluster abgebildet, der Tc6- und der Tc8-Cluster. In beiden sind je zwei Technetiumatome durch eine Dreifachbindung miteinander verbunden. Die Chemie dieser Cluster ist schon seit Längerem Gegenstand ausführlicher Untersuchungen (Kryutchkov 1996).
Ein Technetiumcarbid der ungefähren Zusammensetzung Tc6C wird gebildet, wenn Technetiummetall zusammen mit Kohle (Anteil ca. 16 %) erhitzt wird; die Entstehung des Carbids erkennt man dann am abrupten Wechsel der Kristallstruktur des Gitters der Technetiumatome von hexagonal nach kubisch.
Eine 2016 vorbereitete Veröffentlichung über ein angebliches Technetiummonocarbid (TcC) musste zurück gezogen werden (Jensen). Berechnungen zeigten, dass die Forscher stattdessen eine bis dahin unbekannte Phase metallischen Technetiums untersucht hatten. Wie an der Universität Stony Brook, NY, von Oganov et al. durchgeführte Rechnungen zeigten, ist die Bildung von Technetiumcarbiden aufgrund der eindeutig bevorzugten intermetallischen Bindung der Technetiumatome im Gegensatz beispielsweise zum Zirconium- oder Tantalcarbid energetisch viel ungünstiger (Wang et al. 2016).
Dagegen existieren Technetiummonoborid und -diborid (TcB und TcB2), deren sämtliche relevanten Phasen im Druckbereich 0–100 GPa hinsichtlich ihrer Elastizität untersucht wurden. Die Existenz einer neuen orthorhombisch kristallisierenden Phase von TcB wird vorhergesagt. Weiterhin wurden die Phasen Tc3B und Tc7B3 beobachtet (Yang 2014).
Ditechnetiumdecacarbonyl [Tc2(CO)10] ist aus Kohlenmonoxid und Technetium-VII-oxid unter Druck zugänglich und ein weißer Feststoff (Hileman et al. 1961). Neuerdings wurden zahlreiche Derivate des Carbonyls hergestellt (Sidorenko 2010). Im Molekül des Carbonyls sind zwei durch eine schwache Einfachbindung miteinander verbundene Technetiumatome von jeweils fünf, in oktaedrischer Symmetrie angeordneten Kohlenmonoxidmolekülen umgeben (Bailey und Dahl 1965; Wallach 1962). Die zu Technetium verwandten Elemente der siebten Nebengruppe, Mangan und Rhenium, bilden analoge Carbonyle.
Die Reaktion des oktaedrischen Technetium-I-Komplexes [Tc(NO)(Cp)Cl(PPh3)] mit Trimethylsilylpropin und Silberhexafluorophosphat [AgPF6] in einem aus Ethanol und Dichlormethan bestehenden Lösemittel liefert das dimere [{Tc(NO)(Cp)(PPh3)}2Cl]+, mit Alkinen und Phosphinen reagiert der chirale Komplex [Tc(NO)Cl(Cp)(PPh3)] unter anderem zu einem Fischer-Carben [Tc(NO)(Cp)(PPh3){C(OR)C2H4PPh3}]2+, das als Salz mit dem PF6−-Anion isoliert wird (Abram et al. 2021).
Der größte Teil des nur in geringen Mengen gehandelten Technetiums dient als Radiotherapeutikum (Schwochau 1994). Das mit Abstand wichtigste Isotop des Technetiums, das für diese Zwecke genutzt wird, ist das sehr kurzlebige 99m43Tc (Halbwertszeit: 6 h). Man gewinnt es durch Beschuss von 9842Mo mit Neutronen in speziellen Reaktoren (Dilworth und Parrott 1998), von denen auf der ganzen Welt aber nur maximal fünf im Einsatz sind. Die Überalterung dieser Reaktoren und damit sich häufende Stillstände lassen Engpässe bei der Versorgung mit diesem Isotop befürchten.
Seine kurze Halbwertszeit, die sehr weiche γ-Strahlung und die Fähigkeit, sich an viele im menschlichen Körper vorhandene Moleküle anzulagern, prädestinieren 99m43Tc als Tracer für die Szintigrafie. Dazu koppelt man das in Lösung vorliegende Technetium an Eiweiße oder Antikörper und injiziert sie in den Blutkreislauf. Das Technetium lagert sich dabei auch an Tumorzellen an und macht sie nicht nur „sichtbar“, sondern kann sie teils auch bekämpfen. Die meisten inneren Organe des Menschen sind so erfassbar.
Der größte Teil des 99m43Tc wird schnell wieder aus dem Körper ausgeschieden, ein kleiner verbleibt im Körper und zerfällt unter Aussendung der medizinisch genutzten γ-Strahlung zu 9943Tc, das eine lange Halbwertszeit von 212.000 a besitzt, ein weicher β--Strahler ist und daher auch zu diesem Zweck verwendet wird.
Überraschenderweise erwiesen sich Ammonium- oder Kaliumpertechnetat als äußerst wirksames Rostschutzmittel für Stahl. Selbst unter drastischen Bedingungen (auf bis zu 250 °C erhitzter Wasserdampf) wird Stahl nach vorheriger Behandlung mit einer Lösung von 55 mg/L Kaliumpertechnetat (KTcO4) in belüftetem entionisiertem (!) Wasser nicht oxidiert.
Technetium besitzt nach bisher vorliegenden Resultaten nur eine geringe chemische Toxizität, alle Isotope des Elementes sind aber radioaktiv und müssen gemäß ihrer Strahlungsintensität in Schutzbehältern aufbewahrt werden. Für das Isotop 99m43Tc gilt zur Abschirmung der freigesetzten weichen Röntgenstrahlung ein Sicherheitsabstand von 30 cm als ausreichend. Einatmen staubförmigen Metalls muss unbedingt vermieden werden, da dieses in den Lungen abgelagert wird und über die Zeit Krebs verursachen kann.
-
P. Schaffer und F. Benard, Process of cyclotron production of technetium-99m, system, and device (Triumf, JP 2018095967 A, veröffentlicht 21. Juni 2018)
-
W. Yang und Y. Liu, Method for preparing tricarbonyl technetium-99m intermediate (Institute of High Energy Physics CAS, WO 2018107526 A1, veröffentlicht 21. Juni 2018)
-
H. Ebara und Y. Honda, Technetium production device, technetium production method and radioactive medicine production method (Nihon Mediphysics Co., Ltd., JP 2018091708 A, veröffentlicht 14. Juni 2018)
-
E. S. Stasyuk und E. A. Nesterov, Method of obtaining the technetium-99m complex with octreotide for diagnosing of neuroendocrine tumors (Federalnoe Gosudarstvennoe Avtomnoe Obrazovatelnoe Uchrezhdenie Vysshego Obrazovaniya Natsionalnyj, RU 2655392 C1, veröffentlicht 28. Mai 2018)
-
H. Hu und L. Sun, High-sensitivity spectrophotometric method for measuring rhenium or technetium in solution and water environment (University of Fuzhou, CN 107907493 A, veröffentlicht 13. April 2018)
-
S. Rogov und E. A. Nesterov, Method of manufacturing chromatographic generator technetium-99m from molybdenum-98 irradiated by neutrons (Federalnoe Gosudarstvennoe Avtomnoe Obrazovatelnoe Uchrezhdenie Vysshego Obrazovaniya Natsionalnyj, RU 2642485 C1, veröffentlicht 25. Januar 2018)
-
B. Guerin und R. Ouellet, Processes for obtaining technetium-99m and/or molybdenum(VI) oxide (Socpra Sciences Santé et Humaines S.E.C., CA 2958749 A1, veröffentlicht 23. August 2017)
-
G. Yang und J. Hu, Method for extracting technetium from molybdenum solution adopting polyamide resin (Atom High Tech Co. Ltd., CN 106967882 A, veröfentlicht 21. Juli 2017)
-
R. C. Moore und M. D. Tucker, Apatite sequestration of technetium (Sandia Corp., US 9443627 B1, veröffentlicht 13. September 2016)
5.3 Rhenium
1925 entdeckten Noddack, Tacke und Berg Rhenium (Kurzbiografie Ida Tacke siehe „Neptunium“; Kurzbiografie Walter Noddack siehe „Technetium“). Durch Aufarbeiten des Minerals Columbit erhielten sie schließlich eine wässrige, äußerst niedrig konzentrierte Lösung von Perrhenat und wiesen das Vorliegen von Rhenium in dieser Lösung mittels Röntgenspektroskopie nach (Tacke 1925). Gleichzeitig reklamierten Noddack und Tacke auch die Entdeckung des Technetiums für sich, indem sie behaupteten, sehr geringe Mengen dieses Elements entdeckt zu haben, ohne dieses aber darstellen zu können. In Fortsetzung ihrer Arbeit isolierten Noddack und Tacke 1928 1 g Rhenium aus 660 kg Molybdänerz (Noddack und Noddack 1929).
Der deutsche Chemiker Otto Berg (* 23. November 1873 Berlin; † 1939) war an der Entdeckung des Elements Rhenium in Zusammenarbeit mit Walter Noddack und Ida Tacke beteiligt, ebenso an den Arbeiten zur nicht offiziell anerkannten Entdeckung des Elements Masurium (heute Technetium). Er studierte von 1894 bis 1898 Chemie in Berlin, Heidelberg und Freiburg im Breisgau, arbeitete dann von 1902–1911 als Privatdozent in Greifswald und wechselte danach zur Firma Siemens & Halske in Berlin-Charlottenburg.
Er war an der Identifikation der zwei neuen Elemente mittels Röntgenspektroskopie maßgeblich beteiligt. 1925 gelang dem Dreierteam die Entdeckung von Masurium und Rhenium. Rhenium wiesen sie in Röntgenspektren eindeutig nach, und sie konnten es aus Erzen in wägbarer Menge isolieren. Auch im Fall des Technetiums gilt für ca. 30 von rund 1000 Spektren der Nachweis des Elements als gesichert und in vielen weiteren als wahrscheinlich. Trotzdem erkannte man ihnen offiziell die Entdeckung des Elements nicht zu, weil sie es nicht isolieren konnten und die an der Grenze der damals möglichen Nachweisbarkeit liegenden Spektrallinien nicht reproduzierbar waren (Tilgner 2000).
Rhenium ist eines der seltensten nicht-radioaktiven Elemente und ist in der Erdkruste nur mit einem Anteil von 0,7 ppb (!) vertreten. Elementar kommt es nicht vor, sondern nur chemisch gebunden in einigen wenigen Erzen. Es tritt oft als Begleiter des Molybdäns in dessen Erzen wie etwa Molybdänglanz (Molybdän-IV-sulfid, MoS2) auf, der Rhenium in Anteilen bis zu 0,2 % enthalten kann (Greenwood und Earnshaw 1988). Columbit (Fe,Mn)[NbO3], Gadolinit Y2FeBe[O| SiO4]2 und Alvit (ZrSiO4) können ebenfalls nennenswerte Mengen an Rhenium aufweisen. Die wichtigsten Vorkommen, wenn man angesichts der Seltenheit des Elementes überhaupt davon sprechen kann, befinden sich in den USA, in Kanada, Polen, Chile und Usbekistan. Das einzige bislang aufgefundene reine Rheniummineral ist der Rhenit (Rhenium-IV-sulfid, ReS2); gefunden wurde er im Fernen Osten Russlands auf einer Kurileninsel (Korzhinsky et al. 1994).
- (I)
Re2O7 + H2O + 2 NH3 → 2 NH4ReO4
- (II)
2 NH4ReO4 + 4 H2 → 2 Re + N2 + 8 H2O
Vorkommen, physikalische und chemische Eigenschaften von Rhenium
|
Symbol: |
Re |
| |
|
Ordnungszahl: |
75 | ||
|
CAS-Nr.: |
7440-15-5 | ||
|
Aussehen: |
Grauweiß glänzend |
Rhenium, Barren 25 g, 99,99 % (Onyxmet 2019) | |
|
Entdecker, Jahr |
Noddack, Tacke und Berg (Deutschland), 1925 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
18575Re (37,4) |
Stabil |
---- | |
|
18775Re (62,6) |
Stabil |
---- | |
|
Massenanteil in der Erdhülle (ppm): |
0,001 | ||
|
Atommasse (u): |
186,207 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
1,9 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial: ReO2 + 4 H+ + 4 e− → Re + 2 H2O (V) |
−0,276 | ||
|
Atomradius (berechnet) (pm): |
135 (188) | ||
|
Van der Waals-Radius (pm): |
Keine Angabe | ||
|
Kovalenter Radius (pm): |
159 | ||
|
Ionenradius (Re6+/Re7+, pm) |
61/60 | ||
|
Elektronenkonfiguration: |
[Xe] 4f145d56s2 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte ♦ vierte: |
760 ♦ 1260 ♦ 2510♦ 3640 | ||
|
Magnetische Volumensuszeptibilität: |
9,6 ⋅ 10−5 | ||
|
Magnetismus: |
Paramagnetisch | ||
|
Kristallsystem: |
Hexagonal | ||
|
Elektrische Leitfähigkeit ([A/(V ∙ m)], bei 300 K): |
5,56 ⋅ 106 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
463 ♦ 370 ♦ 178 | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
2450 ♦ 1320 | ||
|
Mohs-Härte |
7,0 | ||
|
Schallgeschwindigkeit (longitudinal, m/s, bei 293,15 K): |
4700 | ||
|
Dichte (g/cm3, bei 293,15 K) |
21,03 | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
8,86 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit [W/(m ∙ K)]: |
48 | ||
|
Spezifische Wärme [J/(mol ∙ K)]: |
25,48 | ||
|
Schmelzpunkt (°C ♦ K): |
3186 ♦ 3459 | ||
|
Schmelzwärme (kJ/mol) |
33 | ||
|
Siedepunkt (°C ♦ K): |
5630 ♦ 5903 | ||
|
Verdampfungswärme (kJ/mol): |
707 | ||

Darüber hinaus hat Rhenium mit 3186 °C nach Wolfram und Kohlenstoff den dritthöchsten Schmelzpunkt aller Elemente und mit 5596 °C deren höchsten Siedepunkt. Unterhalb einer Temperatur von −271,45 °C wird es supraleitend. Rhenium ist gut schmied- und schweißbar und bleibt dies auch nach Rekristallisation (Gebhardt et al. 1972).
34 Isotope und weitere 20 Kernisomere des Rheniums sind bekannt, von denen ausschließlich die Isotope 18575Re und 18775Re natürlich vorkommen. Ersteres hat einen Anteil von 37,4 % an der natürlichen Isotopenverteilung und ist stabil, das andere und damit häufigere (Anteil: 62,6 %) ist schwach radioaktiv und erleidet β−-Zerfall mit einer Halbwertszeit von 4,12 ⋅ 1010 a zu 18776Os. Daher rührt die spezifische Radioaktivität natürlich vorkommenden Rheniums mit 1020 Bq/g.
Von den künstlich erzeugten Isotopen setzt man 18675Re und 18875Re als Tracer ein, das vorwiegend unter Emission von β−-Strahlung zerfallende Isotop 18675Re auch zur Therapie bei der Radiosynoviorthese (Farahati et al. 1997). Dafür verwendet man 18875Re bevorzugt als Radiopharmakon zur Bekämpfung von Tumoren.

Ähnlich wie bei Mangan und Technetium kennt man Verbindungen in allen Oxidationsstufen von −3 bis +7, wobei, im Gegensatz zu Mangan, diejenigen mit höheren Oxidationszahlen auch die stabileren sind.
Chalkogenverbindungen Rhenium-VII-oxid (Re2O7) ist das stabilste Oxid des Rheniums, das nicht nur ein Zwischenprodukt bei der Produktion von Rhenium ist, sondern auch Ausgangsmaterial für die Synthese organischer Verbindungen des Elements (Herrmann et al. 2007). Das Rösten rheniumhaltiger Manganerze liefert unter anderem das flüchtige Rhenium-VII-oxid (Schmelzpunkt 220 °C, Siedepunkt 363 °C), das aus dem Flugstaub ausgewaschen wird. Aus der so entstehenden wässrigen Lösung der -im Gegensatz zur „Permangansäure“- stabilen, relativ starken Perrheniumsäure fällt man Rhenium durch Zugabe von Ammoniumsalzen in Form farblosen Ammoniumperrhenats und reduziert jenes mit Wasserstoff bei hoher Temperatur zum Element. Das gelbe, kristalline Rhenium-VII-oxid hat trotz eines beträchtlichen Anteils an Sauerstoff die hohe Dichte von 6 g/cm3 und kristallisiert orthorhombisch (Krebs et al. 1969).
Rhenium-VII-oxid ist sehr hygroskopisch, naturgemäß sehr gut löslich in Wasser und wird durch Wasserstoff bei Temperaturen um 300 °C zu Rhenium-IV-oxid (ReO2) reduziert (Brauer 1981, S. 1616). Es dient als Katalysator bei der Oxidation von Alkanen zu Carbonsäuren (Kirillova et al. 2007) und bei der Metathese von Olefinen (Onaka und Oikawa 2002).
- (I)
Re2O7 + CO → 2 ReO3 + CO2
- (II)
3 Re2O7 + Re → 7 ReO3
- (I)
Re + 2 ReO3 → 3 ReO2
- (II)
2 NH4ReO4 → 2 ReO2 + N2 + 4 H2O

- (I)
Re + 2 S → ReS2
- (II)
Re2S7 → 2 ReS2 + 3 S
Rhenium-IV-selenid (ReSe2) kristallisiert wie andere Übergangsmetalldichalkogenide in einem Schichtengitter, in dem nur die jeweiligen Ionen einer Schicht relativ stark aneinander gebunden sind, nicht aber die Ionen benachbarter Lagen. Jene sind nur durch schwache Van der Waals-Kräfte miteinander verbunden, was sich darin äußert, dass dünne Schichten leicht von den Oberflächen des kompakten Materials abgelöst werden können.

a Wachstum des Gitters von Rhenium-IV-selenid bei der Reaktion von Rhenium-VI-oxid mit Selendampf (Jiang et al. 2018). b Ansicht des ReSe2-Gitters von oben (links) und von der Seite (rechts) (Hart 2017)

Das Gitter des Rhenium-IV-selenids hat eine sehr niedrige trikline Symmetrie, die auch beim Übergang vom kompakten zum extrem dünnen Material nicht wechselt (Dumcenco et al. 2008; Jiang et al. 2018).

Rhenium-IV-tellurid (Onyxmet 2018)
Halogenverbindungen Rhenium reagiert mit Halogenen meist zu Hexahalogeniden (ReX6). Fluor und Chlor ergeben mit Rhenium bei einer Temperatur um 120 °C direkt blassgelbes Rhenium-VI-fluorid (ReF6) (Brauer 1975, S. 271) bzw. bei 600 °C grünes, sehr leicht zu Rhenium-V-chlorid zerfallendes Rhenium-VI-chlorid (ReCl6).

- (I)
6 ReFe7 + Re → 7 ReF6
- (II)
Re + 3 F2 → ReF6

Rhenium-V-chlorid (Onyxmet 2018)
- (I)
2 ReCl5 + SbCl3 → 2 ReCl4 + SbCl5
- (II)
ReCl5 + ReCl3 → 2 ReCl4
- (III)
2 ReCl5 + C2Cl4 → 2 ReCl4 + C2Cl6
Dagegen stellt γ-Rhenium-IV-chlorid ein braunes Pulver monokliner Kristallstruktur dar, das ebenfalls nur an trockener Luft beständig ist. Auch ist es unlöslich in Benzol und Tetrachlorkohlenstoff, in Aceton löst es sich aber unzersetzt mit grüner Farbe. In wasserfreien Lösungsmitteln kann man Rhenium-IV-oxid mit Thionylchlorid zur α-Modifikation des Rhenium-IV-chlorids umsetzen, die aber immer nur in unreinem Zustand anfällt.




- (I)
HReO4 + 3 HI + 2 C2H5OH → ReI3 + 4 H2O + 2CH3C(O)H
- (II)
ReCl3 + BI3 → ReI3 + BCl3↑
Pnictogenverbindungen Aus Rheniumnitrid (ReNx) bestehende Filme konnten auf (100)-Si-Substraten mit der durch pulsierendes Laserlicht unterstützten Methode der Abscheidung aus der Gasphase (PLD-Methode) erzeugt werden. Ein Stab aus hochreinem Rhenium setzte man einer aus molekularem Stickstoff bestehenden Atmosphäre aus. Die unter anderem mittels Auger-Elektronen- und Röntgen-Spektroskopie charakterisierten Filme sind sehr gute elektrische Leiter, wenn das Verhältnis N/Re<1,3 ist. Der elektrische Widerstand von ReNx-Filmen für 0,2<x<1,3 beträgt nur 5 % desjenigen von aus reinem Rhenium bestehenden Filmen, nur für x>1,3 steigt der Widerstand sprunghaft an (Soto et al. 2007).
Weitere Untersuchungen zeigten, dass die Härte der Rheniumnitride, von denen bis jetzt Re3N, Re2N, Re3N2, ReN3 und ReN4 charakterisiert sind, mit steigendem Gehalt an Stickstoff zunimmt (Cui et al. 2014). Die hexagonal kristallisierenden Nitride Re3N und Re2N haben sehr hohe Kompressionshärten von >400 GPa und liegen damit auf einer Stufe mit den härtesten Übergangsmetallcarbiden und -nitriden. Besser für technische Anwendungen geeignet ist Re3N, weil man es bei moderaten Drücken (13–16 GPa) und Temperaturen (1300–2100 °C) aus den Elementen darstellen kann (Friedrich et al. 2010).
Rheniumphosphid (ReP) ist ein Halbleiter und kann in Hochenergieanwendungen und Laserdioden eingesetzt werden.
Sonstige Verbindungen Die Verwendung von Rheniumcarbiden in keramischen Heizelementen ist patentiert (Tatematsu et al. 1989). Dirheniummonocarbid (Re2C) konnte im Mikromaßstab unter Normalbedingungen aus einer feinpulvrigen Mischung der Elemente nach elfstündigem Mahlen erzeugt werden. Das in polyedrischen Agglomeraten kristallisierende Produkt erwies sich als bis zu Temperaturen von 800 °C stabil und besaß eine spezifische Oberfläche von 2 m2/g (Granados-Fitcha et al. 2016).
Auch Re2C ist, ähnlich wie Re2N, mit einer Härte (Bulk-Modul) von 400 GPa sehr inkompressibel. Üblicherweise und im Gegensatz zu der oben beschriebenen, nur für den Kleinstmaßstab anwendbaren Methode stellt man Rheniumcarbide üblicherweise unter Anwendung hoher Drücke und Temperaturen her (bis zu 10 GPa für Re2C und 20–30 GPa für Re2N). Hierzu verwendete man eine Mischung aus Rhenium und selbst erzeugtem, polymerem Kohlenstoffnitrid und erhielt bei Drücken unter 10 GPa nur Re2C (Yasui et al. 2015).
Rheniumdisilicid (ReSi2−x) bildet schwarze Kristalle vom Schmelzpunkt 2000 °C. Die Zusammensetzung des Materials kann je nach Synthesebedingungen wechseln. So stellten Ivanenko et al. halbleitende Einkristalle von ReSi1,75 durch Reaktion einer Mischung aus Rhenium- und Siliciumpulver im entsprechenden stöchiometrischen Verhältnis her und reinigten das Produkt durch Zonenschmelzen. Die Verbindung soll im Kristallgitter eine große Anisotropie der effektiven Massen und Beweglichkeit der Silicid-Fehlstellen besitzen (2002).
Inui untersuchte die Strukturen sowie die thermoelektrischen Eigenschaften einiger Rheniumsilicide. Die schon oben erwähnte binäre Verbindung ReSi1,75 kristallisiert mit monokliner Struktur. Die Dichte und Anordnung der Silicid-Fehlstellen kann durch Legieren mit einem dritten Metall bzw. Halbmetall beeinflusst werden (2005).

Rheniumdiborid (Kaner und Levine 2008)
Auch Rheniumdiborid ist mit einem Kompressionsmodul von 360 GPa äußerst hart und steht darin Diamant (442 GPa) kaum nach. Die Ursache für die außerordentlich hohe Härte von Rheniumdiborid, -nitriden und -carbiden ist, dass in diesen Gittern sehr viele kurze kovalente Bindungen (Zhou et al. 2007) vorliegen, und Atome des Rheniums mit einer Valenzelektronendichte von 476/nm3 (zum Vergleich: Osmium mit 572/nm3, Diamant mit 705/nm3) die diesbezüglich nahezu höchsten Werte aller Atome von Übergangsmetallen besitzen (Cumberland et al. 2007). Eine Beimengung der kleinen Bor-, Stickstoff- und Kohlenstoffatome weitet das Kristallgitter des Rheniums nur um wenige Prozent auf.

Rhenium-I-tricarbonylkomplexe zeigten ersten Versuchen zufolge eine gute Wirkung bei der Behandlung von Tumoren, da sie nicht die Nachteile von Platinkomplexen zu haben schienen, die bislang für diesen Zweck eingesetzt worden waren. Die infrage kommenden Rheniumkomplexe befinden sich aber alle noch im Stadium vorklinischer Studien. Dabei werden zahlreiche Wirkungsmechanismen untersucht wie Binden des Wirkstoffs an Nukleinsäuren, Phytotoxizität, Wirkung auf Mitochondrien, Inhibierung von Enzymen oder die Regulierung des oxidativen Stress. Je nach Ligand ändert sich das lipophile Verhalten, das Aufnahmevermögen durch die betreffende Zelle, die Luminiszenz und die Toxizität gegenüber der Zelle, kurz das pharmakologische und toxikologische Profil (Colléry et al. 2019).
Neuartige, heteroleptische Rhenium-I-Komplexe auf Basis des Liganden tfb-dmpda ([(N,N-(4,4,4-Trifluorobut-1-en-3-on)-dimethylpropylendiamin], als „L“ abgekürzt im Folgetext), nämlich [fac-Re(I)(CO)3(L)] sind gut geeignete Ausgangsmaterialien zur Herstellung polykristalliner Filme aus Rheniumnitrid (ReN) bei der um 600 °C durchgeführten Abscheidung aus der Dampfphase. Das bevorzugte Wachstum des Films läuft entlang der ⟨100⟩-Richtung. Diese Rheniumkomplexe sind ziemlich stabil, da sie ein stares molekulares Netzwerk ausbilden (Mathur et al. 2019).
Meist setzt man Rhenium in Form seiner Legierungen ein. Etwa zwei Drittel der gesamten Produktionsmenge gehen in nickelhaltige Legierungen, die sehr widerstandsfähig gegenüber Ermüdungsbrüchen sind. Derartige Eigenschaften benötigt man für Turbinenschaufeln, die in Flugzeugmotoren verbaut werden (Heckl 2011).
Da Rhenium weniger anfällig für Vergiftung durch Ablagerungen von Kohlenstoff als Platin ist, finden Katalysatoren auf Grundlage von Rhenium etwa bei der Produktion von klopffestem Benzin Verwendung, weshalb diese Reaktion unter relativ milden und somit energiesparenden Bedingungen gefahren werden kann. Auch zur Synthese aromatischer Kohlenwasserstoffe dienen Platin-Rhenium-Katalysatoren; und aus den gleichen Bestandteilen werden auch Thermoelemente zur Messung im Hochtemperaturbereich gefertigt (Breuer 2000).
Rhenium ist spektroskopisch nachweisbar, da das Element die Flamme des Brenners fahlgrün färbt [Spektrallinien bei 346 und 488,9 nm (Hurd 1936)]. Über die schwer wasserlöslichen Alkaliperrhenate ist es gravimetrisch zu bestimmen (Lexikon der Chemie 1998).
Von Rhenium kennt man aktuell weder biologische Funktionen noch toxische Wirkungen.
-
V. P. Polyakov und N. Yu. Popov, Method of processing spent catalysts containing noble metals and rhenium (Obshchestvo Ogranichennoj Otvetstvennostyu Legkie Metally, RU 2678627 C1, veröffentlicht 30. Januar 2019)
-
J. Pogorzałek und P. Rozański, Method for producing steel with rhenium (Instytut Metalurgii Zeleza Im Stanislawa Staszica, PL 422261 A1, veröffentlicht 28. Januar 2019)
-
Z. Zhang und W. Huang, Method for growing rhenium disulfide nanometer sheets on silicon substrate of non-oxidation layer (Zhaoqing South China Normal University, Optoelectronics Industry Research Institute, CN 108689432 A, veröffentlicht 23. Oktober 2018)
-
T. Tsurumi, Method for recovering high-purity rhenium sulfide (Sumitomo Metal Refining, JP 2018162479 A, veröffentlicht 18. Oktober 2018)
-
W. Cao und X. Huang, Cutting tables including rhenium-containing structures, and related cutting elements, earth-boring tools, and methods (Baker Hughes A Ge. Co., Ltd., WO 2018151965 A1, veröffentlicht 23. August 2018)
-
Y. Kon und B. Katryniok, Use of rhenium-containing supported heterogenous catalysts for the direct deoxy-dehydratation of glycerol to allyl alcohol (Centre National de Recherche Scientifique; Lille Ecole Centrale, US 2018207618 A1, veröffentlicht 26. Juli 2018)
-
T. Wilkes und F. Cichocki Jr., Tungsten-rhenium alloys for curved surgical needle applications (Ethicon Inc., AU 2018204738 A1, veröffentlicht 19. Juli 2018)
-
J. Hämälainen und M. Ritala, Atomic layer deposition of rhenium-containing thin film (ASM IP Holding BV, JP 2018095961 A, veröffentlicht 21. Juni 2018)
5.4 Bohrium

Der russische Kernphysiker Juri Zolakowitsch Oganessian (* 14. April 1933 Rostow am Don) war von 1989 bis 1996 Direktor des Flerow-Labors für Kernreaktionen (FLNR) des Vereinigten Instituts für Kernforschung (JINR) in Dubna. Von 1951 bis 1956 studierte er Kernphysik am Moskauer Institut für Physik-Ingenieurswesen und arbeitete ab 1958 am Moskauer Kurtschatow-Institut, wo er im Labor für Kernreaktionen tätig war. 1962 promovierte er zum Kandidat der Wissenschaften und habilitierte 1970. Zusammen mit Flerov war er ab 1965 maßgeblich an den Arbeiten beteiligt, die zur Entdeckung kurzlebiger Elemente mit Ordnungszahlen von 102 an aufwärts (Rutherfordium, Dubnium, Seaborgium, Bohrium, Nihonium, Flerovium und Livermorium) führten. Oganessian gilt als einer der führenden Wissenschaftler der Schwerionenforschung. Er synthetisierte und charakterisierte nicht nur schwere Nuklide, sondern konstruierte Ionenbeschleuniger und erstellte Verfahren zum Nachweis ionischer Strahlung.
Oganessian entwickelte neue Ideen zur Herstellung der Elemente 102 bis 118, an deren Entdeckung er Anteil hatte. Er arbeitete intensiv mit anderen führenden Instituten zusammen (LBNL in Berkeley, CERN in Genf und GSI in Darmstadt). 2006 wies sein Team Kerne des Elements der Ordnungszahl 118 nach. 2016 wurde deshalb für dieses Element von den an den Arbeiten beteiligten Gruppen der Name Oganesson (Og) der IUPAC vorgeschlagen. Am 30. November 2016 bestätigte diesen die IUPAC offiziell, womit Oganessian nach Seaborg erst der zweite Mensch ist, nach dem zu Lebzeiten ein Element benannt wurde.
Der deutsche Physiker Gottfried Münzenberg (* 17. März 1940 Nordhausen) war im Rahmen seiner Arbeit an der Gesellschaft für Schwerionenforschung (GSI) in Darmstadt wesentlich an der Synthese der Elemente 107 (Bohrium, Bh), 108 (Hassium, Hs), 109 (Meitnerium, Mt), 110 (Darmstadtium, Ds), 111 (Roentgenium, Rg) und 112 (Copernicium, Cn) beteiligt. Er studierte Physik in Gießen und Innsbruck, promovierte 1971 in Gießen (Münzenberg 1971) und begann 1976 seine Tätigkeit bei der GSI in der Abteilung Kernchemie. Münzenberg entwickelte die Anwendung der kalten Fusion auf die Synthese schwerer Elemente und leitete ab 1984 das GSI-Projekt des Fragmentseparators, mit dessen Hilfe man die Wechselwirkung von Materie mit schweren Ionen untersuchen sowie ionische Strahlen zum Zweck der Erforschung der Kernstruktur ableiten konnte. Er erhielt eine Professur in Physik an der Johannes-Gutenberg-Universität in Mainz. 1983 erkannte man ihm den Röntgen-Preis der Stadt Gießen zu, 1996 zusammen mit Sigurd Hofmann den Otto-Hahn-Preis der Stadt Frankfurt am Main (Münzenberg und Schädel 1996) und 2000 zusammen mit Peter Armbruster den Lise-Meitner-Preis (Bertulani et al. 2001).
Der deutsche Kernphysiker Peter Armbruster (* 25. Juli 1931 Dachau) arbeitete bei der GSI; er war maßgeblich an der Entdeckung der Elemente Hassium, Meitnerium, Darmstadtium, Roentgenium und Copernicium beteiligt. Er studierte Physik in Stuttgart und München. 1961 promovierte er in München (Armbruster 1961) und arbeitete von 1965 bis 1970 am Forschungszentrum Jülich unter anderem über Kernspaltung sowie der Wechselwirkung schwerer Ionen mit Materie. 1971 wechselte er zur GSI und war dort bis 1996 leitender Wissenschaftler, seit 1968 Professor an der Universität Köln und seit 1984 auch an der Universität Darmstadt (Armbruster 1982, 1989, 1999; Armbruster et al. 1983). Er erhielt 1988 den gemeinsam vom Londoner Institute of Physics und von der Deutschen Physikalischen Gesellschaft verliehenen Max-Born-Preis und 1997 die Stern-Gerlach-Medaille der Deutschen Physikalischen Gesellschaft. Im gleichen Jahr wurde er von der American Chemical Society als einer der wenigen Nicht-Amerikaner mit dem Nuclear Chemistry Award geehrt. Zusammen mit Gottfried Münzenberg erhielt er 2000 den Lise-Meitner-Preis.
Physikalische Eigenschaften: Bohrium kommt nicht in der Natur vor; alle seine Isotope sind radioaktiv und müssen künstlich erzeugt werden. Bisher kennt man Isotope des Bohriums mit Massenzahlen zwischen 260 und 272 sowie 274, die von diversen Arbeitsgruppen weltweit erzeugt und auch nachgewiesen wurden (Nelson et al. 2008; Gan et al. 2004; Wilk et al. 2000). Die längstlebigen sind wahrscheinlich 270107Bh bzw. 274107Bh mit Halbwertszeiten von 61 bzw. 54 s. Die anderen Isotope weisen Halbwertszeiten im Bereich von 25 s bis hinab zu wenigen ms auf. Der mit Abstand vorherrschende Zerfallsweg ist der α-Zerfall. Für das noch unbekannte Isotope 278107Bh sagt man aufgrund von Berechnungen eine deutlich längere Halbwertszeit (11,5 min) voraus, jedoch auch die Möglichkeit der spontanen Kernspaltung.
Die leichten Isotope produziert man durch kalte Fusion, die schweren durch Beschuss von Isotopen der Actinoide. Längerlebige Isotope des Bohriums gewinnt man besser aus den beim Zerfall von Nukliden des Moscoviums und Nihoniums entstehenden Produkten, wie beispielsweise 272107Bh,271107Bh und 270107Bh (Moody 2014).
Vorkommen, physikalische und chemische Eigenschaften von Bohrium
|
Symbol: |
Bh | ||
|
Ordnungszahl: |
107 | ||
|
CAS-Nr.: |
54037-14-8 | ||
|
Aussehen: |
---- | ||
|
Entdecker, Jahr |
Oganessian et al. (Sowjetunion), 1976 Armbruster, Münzenberg et al. (Deutschland), 1981 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit |
Zerfallsart, -produkt | |
|
267107Bh (synthetisch) |
17 s |
α >263105Db | |
|
270107Bh (synthetisch) |
61 s |
α >266105Db | |
|
274107Bh (synthetisch) |
54 s |
α >270105Db | |
|
Massenanteil in der Erdhülle (ppm): |
----- | ||
|
Atommasse (u): |
(270) | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
Keine Angabe. | ||
|
Atomradius (berechnet) (pm): |
128 * | ||
|
Van der Waals-Radius (pm): |
Keine Angabe | ||
|
Kovalenter Radius (pm): |
Keine Angabe | ||
|
Elektronenkonfiguration: |
[Rn] 5f146d57s2 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
743 ♦ 1689 ♦ 2567 * | ||
|
Magnetische Volumensuszeptibilität: |
Keine Angabe | ||
|
Magnetismus: |
Keine Angabe | ||
|
Kristallsystem: |
Hexagonal-dichtest * | ||
|
Elektrische Leitfähigkeit ([A/(V ∙ m)], bei 300 K): |
Keine Angabe | ||
|
Dichte (g/cm3, bei 293,15 K) |
37,1 * | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
7,28 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit [W/(m ∙ K)]: |
Keine Angabe | ||
|
Spezifische Wärme [J/(mol ∙ K)]: |
Keine Angabe | ||
|
Schmelzpunkt (°C ♦ K): |
Keine Angabe | ||
|
Schmelzwärme (kJ/mol) |
Keine Angabe | ||
|
Siedepunkt (°C ♦ K): |
Keine Angabe | ||
|
Verdampfungswärme (kJ/mol): |
Keine Angabe | ||
Chemische Eigenschaften: Bohrium ist das schwerste Element der Mangangruppe. Verbindungen, in denen Bohrium die Oxidationszahl +7 einnimmt, werden als stabil vorhergesagt, ebenso aber, dass es in Analogie zu Rhenium und Technetium auch solche Verbindungen bildet, in denen es mit der Oxidationszahl +3 oder +4 erscheint (Gäggeler et al. 2000; Malmbeck et al. 2000); von letzterer erwartet man ebenfalls stabile Verbindungen. Bohrium sollte ebenso wie seine niedrigeren Homologen ein flüchtiges Heptoxid [Bohrium-VII-oxid (Bh2O7)] bilden, das sich mit Wasser zur Perbohriumsäure (HBhO4) umsetzt. Die Existenz von Oxidhalogeniden (z. B. BhO3Cl) wird ebenfalls erwartet.
Die Moleküle dieser Oxidchloride sollten in der Reihe TcO3Cl > ReO3Cl > BhO3Cl zunehmend große Dipolmomente und somit abnehmende Flüchtigkeit besitzen, was die Messung der Adsorptionsenthalpien bestätigte (Hoffman et al. 2006).
