1 Einleitung
Die Chalkogene (Erzbildner) sind eine sich in vielen Facetten präsentierende Gruppe von Elementen. Im Periodensystem stehen die ihr zugehörigen Elemente in der sechsten Hauptgruppe. Die Atome der Chalkogene nehmen entweder zwei Elektronen auf (wie Sauerstoff) oder geben bis zu sechs ab (wie Schwefel), um eine stabile Elektronenkonfiguration einnehmen zu können.
Schwefel kennt man schon seit einigen tausend Jahren, Sauerstoff, Selen und Tellur seit etwa 200 Jahren, Polonium seit gut 100 und Livermorium auch schon 15 Jahre. Eine ziemlich „alte“ Elementenfamilie also? Mitnichten. Die Chemie dieser Stoffe ist so vielseitig, an Anwendungen gibt es außerordentlich viele. Sauerstoff ist bei Raumtemperatur ein Gas, die anderen Elemente sind unter diesen Bedingungen alle Feststoffe. Sauerstoff und Schwefel sind reine Nichtmetalle, aber schon Selen zeigt stärker metallischen als nichtmetallischen Charakter; dieser Effekt verstärkt sich noch bei Tellur. Polonium und Livermorium sind nahezu rein metallisch. Sie finden sie alle im unten stehenden Periodensystem in der Gruppe 16 (VI A).
Die Einzeldarstellungen der insgesamt sechs Vertreter der Gruppe der Chalkogene enthalten dabei alle wichtigen Informationen über das jeweilige Element, so dass ich hier nur eine kurze Einleitung vorangestellt habe.
Elemente werden eingeteilt in Metalle (z. B. Natrium, Calcium, Eisen, Zink), Halbmetalle wie Arsen, Selen, Tellur sowie Nichtmetalle wie beispielsweise Sauerstoff, Chlor, Iod oder Neon. Die meisten Elemente können sich untereinander verbinden und bilden chemische Verbindungen; so wird z. B. aus Natrium und Chlor die chemische Verbindung Natriumchlorid, also Kochsalz).
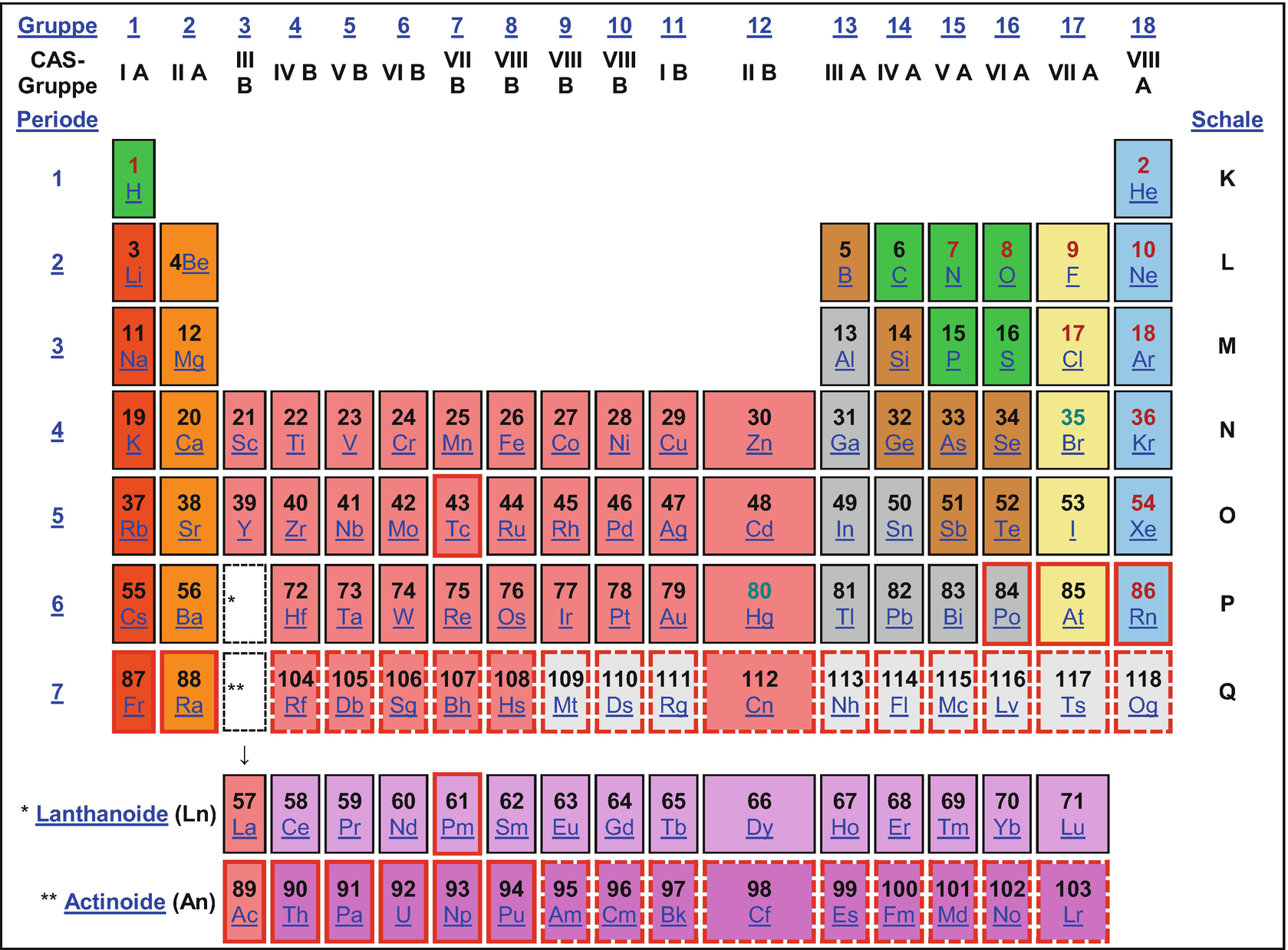
Periodensystem der Elemente
2 Vorkommen
Sauerstoff ist das häufigste Element in der Erdhülle und ist in der Luft zu gut einem Fünftel des Gesamtvolumens enthalten. Sauerstoff, aber vor allem Schwefel, Selen und Tellur kommen in der Natur meist in Form von Erzen und Mineralien vor, wenngleich auch mit sehr unterschiedlicher Häufigkeit. Oxide und Sulfide sind weit verbreitet. Oxide sind beispielsweise Kohlendioxid in der Erdatmosphäre und Quarz (Siliciumdioxid), der den Hauptbestandteil der Erdkruste bildet. Zu den Sulfiden gehören u. a. die Mineralien Bleiglanz, Zinnober, Pyrit, Zinksulfid und Kupferkies. Wesentlich seltener kommen Selenide und Telluride vor. Darüber hinaus gibt es viele weitere Verbindungen wie z. B. Sulfite, Sulfate und Selenate.
Sauerstoff, Schwefel, Selen und Tellur kommen in der Natur auch in elementarer Form vor.
3 Herstellung
Gasförmiger Sauerstoff wird durch fraktionierte Destillation flüssiger Luft gewonnen. Auch für die Herstellung des Schwefels sind die Lagerstätten des Elements die wichtigste Quelle. Selen und Tellur sind Bestandteile des Anodenschlamms der Produktion hochreinen Nickels und Kupfers und können durch dessen Aufarbeitung gewonnen werden. Polonium ist in winzigsten Spuren in Pechblende enthalten; man gewinnt es bevorzugt aber in Kernreaktoren durch Beschuss von Bismutatomen mit Neutronen.
4 Eigenschaften
4.1 Physikalische Eigenschaften
Wie bereits erwähnt, sind Sauerstoff und Schwefel reine Nichtmetalle, Selen und Tellur Halbmetalle und Polonium sowie Livermorium Metalle. Die physikalischen Eigenschaften sind nach steigender Atommasse abgestuft. So nehmen vom Sauerstoff zum Tellur die Dichte, Schmelz- und Siedepunkte zu. Ab Polonium nehmen die Schmelzpunkte wieder ab; dies dürfte wesentlich auf die starke radioaktive Strahlung zurückzuführen sein, die die Stabilität des Kristallgitters deutlich verringert.
Generell weicht bei den Chalkogenen, wie auch bei allen anderen Hauptgruppen, das Kopfelement (hier: Sauerstoff) in seinen Eigenschaften deutlich von allen anderen ab. Schwefel als zweites Element dieser Gruppe steht den höheren Homologen, Selen und Tellur, viel näher als Sauerstoff; auch seine Verbindungen (z. B. Wasserstoffverbindungen, Oxosäuren) sind denen des Selens und Tellurs sehr ähnlich, so dass man hier von einer homologen Reihe spricht.
4.2 Chemische Eigenschaften
Chalkogene reagieren mit Metallen meist heftig zu Oxiden, Sulfiden usw., mit Wasserstoff zu Chalkogenwasserstoffen (H2X: Wasser, Schwefelwasserstoff, Selenwasserstoff usw.). Sie gehen auch untereinander Verbindungen wie Schwefeloxide oder Selensulfid ein. Chalkogenoxide bilden, zusammengebracht mit Wasser, Säuren: z. B. Schweflige Säure und Selenige Säure (Summenformel H2XO3; X=S, Se) aus den Dioxiden, sowie Schwefelsäure, Selensäure usw. (Summenformel H2XO4; X=S, Se) aus den Trioxiden.
5 Einzeldarstellungen
Im folgenden Teil sind die Chalkogene jeweils einzeln mit ihren wichtigen Eigenschaften, Herstellungsverfahren und Anwendungen beschrieben.
5.1 Sauerstoff
In den 1770er-Jahren entdeckten Scheele (Kurzbiografie siehe „Molybdän“) und Priestley (Kurzbiografie siehe „Stickstoff“) voneinander unabhängig den Sauerstoff (Frey und Mayrhofer 1955; Priestley 1775). Hierdurch wurde die Phlogiston-Theorie abgelöst, also die Vorstellung, dass bei einem Brand ein unsichtbarer Stoff in Form von Wärme aus dem brennenden Stoff entweicht. So erhielt Scheele beim Erhitzen von Mangan-IV-oxid bzw. Kaliumpermanganat mit konzentrierter Schwefelsäure ein farbloses Gas, das die Verbrennung förderte. Priestley stellte dagegen durch Erhitzen von Quecksilberoxid Sauerstoff dar und publizierte seine Arbeit 1774, Scheele erst 1777.
Entscheidend waren Lavoisiers spätere Resultate (Kurzbiografie siehe „Wasserstoff“), die offen legten, dass bei einer Verbrennung nicht Phlogiston freigesetzt, sondern Sauerstoff gebunden wird. Wenn nach einem Verbrennungsvorgang ein Stoff schwerer war, führte er dies auf das zusätzliche Gewicht des im Zuge der Verbrennung aufgenommenen Sauerstoffs zurück. In der Anfangszeit glaubte man, Sauerstoff sei ein unverzichtbarer Bestandteil von Säuren. Davy (Kurzbiografie siehe „Natrium“) berichtigte diese Vorstellung ab 1808 dahingehend, dass vielmehr Wasserstoffatome charakteristisch für Säuren sind.
Sauerstoff (O2) ist das häufigste Element auf der Erde und kommt in der Atmosphäre, der Lithosphäre, der Hydrosphäre und der Biosphäre vor (Allègre et al. 2001). Er besitzt einen außergewöhnlich hohen Massenanteil von 50,5 % (!) an der Erdhülle. In der Luft ist elementarer Sauerstoff mit einem Massenanteil von 23,2 % enthalten, am gesamten Wasser der Erde mit durchschnittlich 89 % in gebundener und gelöster Form (Holleman et al. 2007).
Nahezu alle Minerale und damit Gesteine enthalten Sauerstoff, beispielsweise Silikate wie Feldspäte, Glimmer und Olivine, Carbonate wie Kalkstein (Calciumcarbonat) sowie Oxide [Silicium-IV-oxid (Quarz)]. Die Menge des in der Luft enthaltenen elementaren Sauerstoffs bleibt ungefähr konstant, da Sauerstoff produzierende Pflanzen diejenige Menge Sauerstoff nachliefern, die durch aerob atmende Lebewesen und andere Verbrennungsprozesse verbraucht wird. Das Allotrop Ozon (O3) ist in der Atmosphäre nur in sehr geringer Konzentration vorhanden, jedoch in der Stratosphäre von entscheidender Bedeutung für die Abschirmung des von der Sonne ausgesandten UV-Lichtes.
Im Weltall ist Sauerstoff nach Wasserstoff und Helium ebenfalls sehr häufig vertreten und immerhin das dritthäufigste Element (Davies 2003).
Man gewinnt Sauerstoff fast ausschließlich durch fraktionierte Destillation flüssiger Luft nach dem Linde-Verfahren, das später durch Claude verbessert wurde. Zudem fallen kleinere Mengen als Nebenprodukt bei der Herstellung von Wasserstoff im Zuge der Elektrolyse von Wasser an.
Man verdichtet Luft auf einen Druck von 5–6 bar, kühlt sie ab und leitet sie durch Filter, die Kohlendioxid, Spuren von Wasser und andere Gase entfernen. Die verdichtete Luft wird durch vorbeiströmende Gase aus dem Prozess auf eine Temperatur nahe ihrem Siedepunkt abgekühlt. Danach wird sie in Turbinen expandiert. Dabei kann man ein Teil der zur Kompression eingesetzten Energie wieder zurückgewinnen. Dadurch wird das Verfahren – im Gegensatz zum Linde-Verfahren, bei dem keine Energie zurückgewonnen wird – deutlich wirtschaftlicher.
Die beiden Hauptbestandteile der Luft, Stickstoff (78 Vol.-%) und Sauerstoff (21 Vol.-%) trennt man in zwei unter verschiedenem Druck stehenden Destillationskolonnen. In der ersten, der mit einem Druck von 5–6 bar gefahrenen Mitteldruckkolonne, sammelt sich der tiefer siedende Stickstoff(−196 °C) am Kopf, der höher siedende Sauerstoff (−183 °C) am Fuß der Kolonne. Die sich im unteren Teil der Kolonne sammelnde, mit Sauerstoff angereicherte flüssige Luft durchläuft dann eine weitere fraktionierte Destillation in der mit einem Druck von 0,5 bar betriebenen Niederdruckkolonne (Greenwood und Earnshaw 1988, S. 775–839). Der leichter flüchtige Stickstoff entweicht zuerst. Relativ konzentrierter flüssiger Sauerstoff bleibt zurück, der noch die höher siedenden Edelgase Krypton und Xenon enthält. Jene trennt man in einer zusätzlichen Kolonne ab.
Zur Gewinnung kleinerer Mengen an Sauerstoff für medizinische Anwendungen leitet man Luft durch Molekularsiebe, die Stickstoff und Kohlendioxid absorbieren, Sauerstoff und Argon aber durchlassen.
Vorkommen, physikalische und chemische Eigenschaften von Sauerstoff
|
Symbol: |
O |
|
|
|
Ordnungszahl: |
8 | ||
|
CAS-Nr.: |
7782-44-7 | ||
|
Aussehen: |
Farbloses Gas |
Flüssiger Sauerstoff (Hillier 2006) |
Sauerstoff in Gasentladungsröhre (pse-mendelejew 2006) |
|
Entdecker, Jahr |
Scheele (Schweden), 1771 Priestley (England), 1774 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
168O (99,76) |
Stabil |
---- | |
|
178O (0,04) |
Stabil |
---- | |
|
188O (0,2) |
Stabil |
---- | |
|
Massenanteil in der Erdhülle (ppm): |
494.000 (49,4 %!) | ||
|
Atommasse (u): |
15,999 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
3,44 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial für: O2 + 4 e− > 2 O2− (V) |
+1,23 | ||
|
Atomradius (pm): |
60 | ||
|
Van der Waals-Radius (berechnet, pm): |
152 | ||
|
Kovalenter Radius (pm): |
66 | ||
|
Elektronenkonfiguration: |
[He] 2s22p4 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
1314 ♦ 3388 ♦ 5300 | ||
|
Magnetische Volumensuszeptibilität: |
1,9 · 10−6 | ||
|
Magnetismus: |
Paramagnetisch | ||
|
Kristallsystem: |
<−249,3 °C: monoklin; −249,3 bis −229,35 °C: rhomboedrisch; −229,35 bis −218,75 °C: kubisch | ||
|
Dichte (kg/m3, bei 273,15 K) |
1,429 | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
17,36 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit ([W/(m · K)]): |
0,0266 | ||
|
Spezifische Wärme ([J/(mol · K)]): |
29,38 | ||
|
Schmelzpunkt (°C ♦ K): |
−218,3 ♦ 54,8 | ||
|
Schmelzwärme (kJ/mol): |
0,222 | ||
|
Siedepunkt (°C ♦ K): |
−183 ♦ 90,15 | ||
|
Verdampfungswärme (kJ/mol): |
5,58 | ||
|
Tripelpunkt (°C ■ kPa): |
−218,79 ■ 0,1463 | ||
|
Kritischer Punkt (°C ■ MPa): |
−118,57 °C ■ 5,043 | ||


Sauerstoff ist in reinem Wasser nur wenig löslich (bei 0 °C ca. 14 mg/L unter seinem Partialdruck aus der Luft). In der Gasentladungsröhre (Druck: 5–10 mbar, Spannung: 1,8 kV, Stromstärke: 18 mA, Frequenz: 35 kHz) leuchtet Sauerstoff blau.
Im Grundzustand zeigt das Sauerstoffmolekül die Spins beider Valenzelektronen parallel, also gleichsinnig, angeordnet. Dieser Triplett-Sauerstoff (Termsymbol 3Σg) stellt den energieärmsten Zustand dar. Die beiden π∗-Orbitale sind nur halb mit jeweils einem ungepaarten Elektron besetzt, was den diradikalischen Charakter und den Paramagnetismus des Sauerstoff-Moleküls bedingt.
In den zwei angeregten Zuständen des Sauerstoffs sind die Spins der Elektronen antiparallel, also gegensinnig, ausgerichtet (Singulett-Sauerstoff), die sich hinsichtlich der Anordnung der beiden ungepaarten Elektronen unterscheiden [beide Elektronen in einem π∗-Orbital (Termsymbol: 1Δg) oder in beiden π∗-Orbitalen (Termsymbol: 1Σg)]. Letzterer ist energiereicher und diamagnetisch; er wandelt sich schnell in den 1Δg -Zustand um. Dieser hat wegen seines Bahnmomentes einen Paramagnetismus, der dem des Triplett-Sauerstoffs vergleichbar ist (Hasegawa 2008; Shinkarenko und Aleskovskiji 1981; Lechtken 1974).
Singulett-Sauerstoff kann man fotochemisch aus Triplett-Sauerstoff erzeugen sowie aus chemischem Weg aus anderen Sauerstoffverbindungen. Der Einsatz von Licht scheidet aber wegen nicht erfüllter quantenmechanischer Auswahlregeln aus, da nur durch eine simultane Bestrahlung mit Photonen und Kollision zweier Sauerstoffmoleküle Singulett-Sauerstoff gebildet wird. Dieser noch am ehesten in der flüssigen Phase ablaufenden Vorgang führt zur blauen Farbe des flüssigen Sauerstoffs. Chemisch kann man den stark oxidierend wirkenden Singulett-Sauerstoff aus Peroxiden gewinnen. Im Gegensatz zu normalem Sauerstoff reagiert er mit 1,3-Dienen in einer [4 + 2]-Cycloaddition zu Peroxiden.
Ein Allotrop des Sauerstoffs ist Ozon (O3), dessen Molekül drei Sauerstoffatome enthält. Es ist ein farbloses bis bläuliches, in hoher Konzentration tiefblaues Gas (Ahrens et al. 2016) von typischem Geruch. In der Luft befindliche Ozonmoleküle zerfallen unter Normalbedingungen innerhalb weniger Tage zu Sauerstoff. Ozon schmilzt bzw. siedet bei Temperaturen von −192,5 °C bzw. −111,9 °C und hat bei 0 °C eine Dichte von 2,154 kg/m3. Es ist ein starkes und giftiges Oxidationsmittel, das bei Menschen und Tieren zu Reizungen der Atemwege und der Augen führt, jedoch schützt die in der Stratosphäre enthaltene Ozonschicht die Lebewesen auf der Erde vor der ultravioletten Strahlung der Sonne.
Die Konzentration des Ozons in der Stratosphäre beträgt bis zu einige ppm. In Städten ist seine Konzentration niedriger als in Gebieten mit reinerer Luft. Das in den Abgasen von Verbrennungsmotoren enthaltene Stickstoffmonoxid trägt mit zum Abbau des Ozons bei.
Ozon kann man im Labor durch Reaktion von konzentrierter Schwefelsäure mit Kaliumpermanganat darstellen. Dabei bildet sich zuerst Mangan-VII-oxid, das bei Raumtemperatur zu Mangan-IV-oxid und ozonreichem Sauerstoff zerfällt. Eine weitere Herstellmöglichkeit ist die Elektrolyse einer etwa 20 %igen Schwefelsäure an Edelmetallanoden, an denen bei hohen Stromdichten Ozonkonzentrationen von bis zu 5 % im als Hauptprodukt entstehenden Sauerstoff erzielt werden können. Eine geeignete Kühlung trägt mit anderen möglichen Maßnahmen dazu bei, dass noch höhere Ausbeuten erreichbar sind. Ebenfalls im Labor kann man mittels elektrischer Entladungen oder durch Bestrahlung mit Ultraviolettlicht Ozon erzeugen (Brauer 1963, S. 337).
Da sich Ozon sehr leicht zersetzt, kann man es nicht in Druckflaschen abfüllen und verkaufen, sondern es muss an der Stelle seines Verbrauchs erzeugt werden. In der Großtechnik leitet man ein hauptsächlich aus Sauerstoff bestehendes Trägergas durch einen mit elektrischen Entladungen gespeisten Ozongenerator.
In reinem Sauerstoff sind dann Ozongehalte von 6–13 % möglich. Die Herstellung eines kg Ozon erfordert etwa 7–14 kWh Strom und 1,8 m3/h Kühlwasser.

Ozon setzt man in der Wasseraufbereitung zur Entkeimung und zum Abbau störender Stoffe ein. Bei der oxidierenden Gaswäsche wird Ozon als Oxidationsmittel verwendet. Sogar in einigen modernen Waschmaschinen arbeitet ein Ozongenerator zur Desinfektion der Wäsche. Zur Entfernung von Gerüchen aus dem Innenraum gebrauchter Fahrzeuge verwendet man es ebenfalls (Baumbach 1992, S. 385).
Sauerstoff reagiert mit fast allen Elementen, außer mit Helium, Neon und Argon. Sauerstoff ist stark elektronegativ und tritt daher fast immer mit der Oxidationszahl −2 auf, mit Ausnahme von Peroxiden, in denen er mit der Oxidationsstufe −1 erscheint. Nur in seinen Verbindungen mit Fluor hat Sauerstoff positive Oxidationszahlen (+1 im Disauerstoffdifluorid O2F2; + 2 im Sauerstoffdifluorid OF2). Die ionischen Formen des Sauerstoffs sind Oxid (O2−), Peroxid- (O22−), Hyperoxid- (O2−), Ozonid (O3−) und das Dioxygenylkation (O2+).

Mit steigender Oxidationsstufe des Metallions reagieren Oxide zunehmend amphoter (Aluminium-III-oxid, Titan-IV-oxid) und schließlich sauer (Chrom-VI-oxid).
Mit Nichtmetallen bildet Sauerstoff nur kovalente Oxide. Die mit Wasserstoff gebildeten Sauerstoffverbindungen sind Wasser (H2O) und Wasserstoffperoxid (H2O2); letzteres ist instabil und wird als Oxidations- und Bleichmittel eingesetzt. Beide Verbindungen werden im Kapitel „Wasserstoff“ behandelt.
Fluorverbindungen Disauerstoffdifluorid (O2F2) wurde erstmals durch Einwirkung elektrischer Entladungen auf ein aus Sauerstoff und Fluor bestehendes Gasgemisch erzeugt (Ruff und Menzel 1933). Das Molekül der Verbindung hat die Strukturformel F-O-O-F mit einem FOO-Winkel von 109,5° sowie Bindungslängen O-O von 122 pm bzw. O-F von 158 pm. Das braune Gas kondensiert bei −57 °C zu einer kirschroten Flüssigkeit einer Dichte von 1,45 g/cm3 (am Siedepunkt), die nach weiterem Abkühlen bei einer Temperatur von −163,5 °C zu einem orangegelben Feststoff erstarrt (Bridgeman und Rothery 1999).
Schon oberhalb einer Temperatur von −95 °C zersetzt sich Disauerstoffdifluorid und ist ansonsten ein starkes Oxidations- und Fluorierungsmittel (Streng 1963). So überführt es Chlor in Chlor-I-fluorid und Chlor-III-fluorid, Schwefelwasserstoff in Schwefel-VI-fluorid sowie Verbindungen des Neptuniums bzw. Plutoniums schon bei niedrigen Temperaturen in die jeweiligen Hexafluoride (NpF6 und PuF6) (Eller et al. 1998).
Sauerstoffdifluorid (OF2) wurde 1929 erstmalig durch Elektrolyse einer Mischung von geschmolzenem Kaliumfluorid und konzentrierter Flusssäure erzeugt. Heute gewinnt man es durch Einleiten von Fluor in Natron- oder Kalilauge (Holleman et al. 2017, S. 532; Brauer 1975, S. 179). Die Verbindung ist eines der stärksten Oxidationsmittel überhaupt, das sogar Xenon zu Xenontetrafluorid (XeF4) oxidiert. Mit Schwefel-IV-oxid reagiert es zu Schwefel-VI-oxid und Fluor. Die Löslichkeit in Wasser ist gering, außerdem zersetzt sich Sauerstoffdifluorid in Wasser zu Fluorwasserstoff und Sauerstoff. Man testete die Verbindung in Treibstoffen für Raketen, allerdings verhinderte die Aggressivität des Sauerstoffdifluorids gegenüber den Materialien des Behälters und des Motors bisher eine Anwendung in größerem Maßstab.
Sauerstoffdifluorid kondensiert bei −144,8 °C zu einer orangefarbenen Flüssigkeit, die bei −223,8 °C erstarrt. Das Gas hat bei einer Temperatur von 0 °C eine Dichte von 2,42 g/L. Die Struktur des Moleküls ähnelt der des Wassermoleküls; der F-O-F-Bindungswinkel beträgt 103°, die O-F-Bindung ist jeweils 140,5 pm lang. Wird das Gas eingeatmet, sind starke und lang anhaltende Atembeschwerden die Folge.
Trisauerstoffdifluorid (O3F2) erhält man durch Reaktion von Sauerstoff mit Fluor unter Einwirkung elektrischer Entladungen bei niedrigem Druck und Temperaturen von ca. −190 °C (Brauer 1975, S. 176). Erstmals gelang Aoyama und Sakuraba die Synthese (Kirshenbaum und Von Grosse 1959).
Die tiefrote Flüssigkeit erstarrt bei −190 °C und zersetzt sich bereits oberhalb einer Temperatur von −157 °C in Sauerstoff und Disauerstoffdifluorid. Die Struktur des Moleküls ist gewinkelt und entspricht der Abfolge F‐(O)3‐F (Riedel und Janiak 2011, S. 423; Steudel 2008, S. 426). Die Verbindung ist gewöhnlich sicherer handzuhaben als Ozon und kann ohne Auftreten einer Explosion verdampft, thermisch zersetzt oder einem Funkenstrahl ausgesetzt werden. Sobald sie aber mit organischen und/oder brennbaren Materialien in Kontakt kommt, erfolgt fast immer eine Detonation. So führt schon die Zugabe nur eines Tropfens von Trisauerstoffdifluorid zu festem, wasserfreiem Ammoniak bei −183 °C zu einer schwachen Explosion (Streng 1963).
Tetrasauerstoffdifluorid (O4F2) konnte als roter, instabiler, bei −196 °C schmelzender Feststoff durch Einwirkung elektrischer Entladungen auf ein aus Sauerstoff und Fluor im Volumenverhältnis 2:1 bestehendes, in einem Pyrexkolben befindliches Gemisch bei −196 °C und niedrigem Druck erhalten werden (Gardiner und Turner 1972). Das O4F2 -Molekül steht im Gleichgewicht mit dem O2F -Radikal. Tetrasauerstoffdifluorid ist ein extrem starkes Fluorierungs- und Oxidationsmittel, so oxidiert es Ag-II zu Ag-III oder Au-III zu Au-V. Leichter oxidierbare Stoffe reagieren selbst bei sehr tiefen Temperaturen äußerst heftig, so reagiert bei −180 °C Schwefel unter Explosion zu Schwefel-VI-fluorid.
Sauerstoff wird für Zwecke der intensivmedizinischen Versorgung in weiß gekennzeichneten Flaschen abgefüllt (Bundesministerium der Justiz 2005). Die Anreicherung des Sauerstoffs im Blut ist mittels der Pulsoxymetrie oder anhand von Blutgasanalysen messbar (Andrews und Nolan 2006). Bei chronischem Sauerstoffmangel im Blut verbessert eine langfristige zusätzliche Verabreichung von Sauerstoff die Lebensqualität und auch die Überlebensdauer (Deutsche Gesellschaft für Pneumologie 1993). Die Verwendung reinen Sauerstoffs kann aber zu Problemen führen, da er das Kohlendioxid aus den Gefäßen verdrängt (Iscoe und Fisher 2005), was eine Erhöhung der Hirnaktivität zur Folge hat. Dies wird durch Zumischung von Kohlendioxid vermieden (Macey et al. 2007).
Bei der Herstellung von Roheisen und Stahl sowie bei der Raffination von Kupfer setzt man Sauerstoff oder mit diesem angereicherte Luft sowohl zum Erreichen hoher Temperaturen als auch zum Verbrennen unerwünschter Beimengungen von Kohlenstoff, Silicium, Mangan und Phosphor ein. In chemischen Prozessen wird Sauerstoff meist zur Oxidation diverser Grundstoffe verwendet, wie z. B. bei der Oxidation von Ethen zu Ethylenoxid, zur Erzeugung von Wasserstoff- und Synthesegas und bei der Produktion von Schwefel- und Salpetersäure. Weitere durch Oxidation mit Sauerstoff hergestellte wichtige Produkte sind Acetaldehyd, Essigsäure, Vinylacetat und Chlor. Zusätzliche Einsatzgebiete sind die Herstellung von Ozon, Brennstoffzellen und die Halbleiterindustrie. Flüssiger Sauerstoff dient als Treibstoff für Raketen.
Bei der Reinigung von Abwässern bewirkt zusätzlich in diese eingeleiteter Sauerstoff einen schnelleren Abbau organischer Schadstoffe, bei der Aufbereitung von Trinkwasser löst Einleiten von Ozon (O3) ebenfalls eine beschleunigte Oxidation organischer Stoffe und auch von Bakterien aus.
Sauerstoff ist ein Lebensmittelzusatzstoff (E 948) und wird als Treib- und Packgas verwendet.
Sauerstoff wird in der Natur im Wesentlichen durch Fotosynthese erzeugt. Die meisten aeroben Organismen (Säugetiere einschließlich des Menschen, die meisten Fische, Wirbellose, Pflanzen und viele Bakterien) benötigen den auf diese Weise erzeugten Sauerstoff zum Leben. Bei der Atmung wird der Sauerstoff in der Atmungskette wieder in Wasser und Kohlendioxid umgewandelt. Sauerstoff und einige seiner gelegentlich auch radikalischen Verbindungen sind sehr reaktionsfähig und können Zellmaterial angreifen. Daher besitzen viele Organismen schützende Enzyme wie Katalase und Peroxidase; sind diese nicht vorhanden, wirkt Sauerstoff toxisch. Der oxidative Stoffwechsel produziert reaktive freie Radikale, die abgefangen werden müssen, da sie sonst wichtige Funktionsmoleküle im Körper zerstören können.
Atmet der Mensch reinen Sauerstoff oder Luft höheren Sauerstoffgehaltes ein, so können nach einiger Zeit die Lungenbläschen anschwellen (Lorrain-Smith-Effekt), was unter ungünstigen Umständen auch zum Tode führen kann. Werden komprimierte Sauerstoff-Stickstoff-Gemische eingeatmet, besteht das Risiko einer Vergiftung des zentralen Nervensystems (Paul-Bert-Effekt).
5.2 Schwefel
In China und Ägypten nutzte man Schwefel schon seit etwa 5000 v. Chr. zur Desinfektion, als Arzneimittel und zum Bleichen von Textilien. Verwendet wurde Schwefel entweder aus natürlichen Quellen, oder man erzeugte ihn aus leicht zu verarbeitenden Erzen wie Pyrit (Figurowski 1981, S. 179). Auch im antiken Griechenland setzte man schon früh Schwefel in oben genannten Anwendungen ein, darüber hinaus zum Desinfizieren von Behältnissen, in denen Wein aufbewahrt wurde (Rapp 2009, S. 242). In Land- und Seekriegen verwendete man brennenden Schwefel als Brandwaffe. In China stellte man um 1000 n. Chr. explosionsfähige Mischungen aus Kaliumnitrat, Holzkohle und Schwefel her, das über einige Jahrhunderte hinweg der einzige verfügbare Explosivstoff war (Seel 1988, S. 9). Es gibt keine klaren historischen Hinweise darauf, dass der deutsche Mönch Berthold Schwarz diese später „Schwarzpulver“ genannte Mischung wieder entdeckt haben soll (Feldhaus 1910, S. 617).
Schwefel ist ein gelber, nichtmetallischer Feststoff, der in mehreren allotropen Modifikationen auftritt. Er kommt in der Lithosphäre, Hydrosphäre, Atmosphäre und Biosphäre vor, sowohl in Form von Sulfiden (Oxidationszahl −2), in Aminosäuren (Oxidationszahl −1/−2), als elementarer Schwefel (Oxidationszahl 0), als Schwefel-IV-oxid in Vulkangasen und der Atmosphäre generell (Oxidationszahl +4) und in Form von Sulfaten (Oxidationszahl +6) (Schmidt 1973). Sein Massenanteil an der Erdhülle beträgt 0,048 %, Schwefel steht damit an 15. Position in der Rangliste der häufigsten Elemente.
Elementar kommt Schwefel in großen Lagerstätten vor, oft in Gebieten mit starker tektonischer Aktivität, wie etwa im Iran, auf Sizilien, in Mexiko, auf dem Meeresboden des Golfes von Mexiko sowie des Mittelatlantischen und Ostpazifischen Rückens. Man findet ihn aber auch in vielen anderen Regionen (Polen, südliche US-Bundesstaaten). Er erscheint dann als gelber Schwefel („Schwefelblüte“).
Wesentlich häufiger tritt Schwefel in der Lithosphäre in Form von Sulfiden, Oxiden, Sulfaten, Halogeniden etc. auf. Man kennt ca. 1000 schwefelhaltige Minerale; die bekanntesten sind Pyrit und Markasit (FeS2, Schwefelgehalt ca. 53,5 %).
Für die Gewinnung des Schwefels wichtig sind fossile Brennstoffe wie Erdöl, Erdgas und Kohle. Erdgas kann hohe Konzentrationen an Schwefelwasserstoff (H2S) aufweisen. In Braunkohle beträgt der Schwefelgehalt bis zu 10 % (Adolphi und Ullrich 2000).
In der Hydrosphäre tritt Schwefel meist in Form des Sulfations auf, das sich vor allem im Meerwasser findet. In Süßwasser kommt Sulfat aus natürlichen Quellen in sehr unterschiedlicher Konzentration vor; es ist ein wichtiger Bestandteil der Wasserhärte. Manche Alpenquellen enthalten Sulfat in hoher Konzentrationen. Da Sulfat, in größeren Mengen eingenommen, Durchfall verursachen kann, beschränkt die deutsche Trinkwasserverordnung die maximal erlaubte Sulfatkonzentration in Trink- oder Mineralwässern auf 250 mg/L.
Verbrennungsprozesse schwefelhaltiger Brennstoffe und Vulkanausbrüche bewirken das Vorkommen von Schwefel in Form von Schwefel-IV-oxid (SO2) in der Troposphäre. Die gesamte jährliche Emission an SO2 beträgt 400 Mio. t (!) (Wellburn et al. 1997). Schwefel-IV-oxid wird leicht durch Pflanzenblätter absorbiert und dem Stoffwechsel zugeführt.
Außerhalb der Erde sind Schwefel oder seine Verbindungen im Sonnensystem nachgewiesen worden. Die Wolken unseres sonnennäheren Nachbarplaneten Venus enthalten große Mengen an Schwefel-IV-oxid und Schwefelsäure; erklärbar bei einer Oberflächentemperatur von ca. 400 °C. Ebenso fanden die Viking-Sonden Eisen- und Magnesiumsulfat auf unserem sonnenferneren Nachbarplaneten Mars, was auf das frühere Vorhandensein von Wasser dort hindeuten könnte. Auf dem einen ungewöhnlich starken Vulkanismus zeigenden Jupitermond Io -dessen mittlere Oberflächentemperatur bei −140 °C liegt- finden sich zahlreiche heiße Stellen an der Oberfläche; einige davon sind Seen, die aus geschmolzenem Schwefel bestehen. Forschungsergebnisse der NASA lassen vermuten, dass sich auf der Oberfläche des Jupitermondes Europa erhebliche Mengen Sulfate und Schwefelsäure befinden.
Im Weltall wurden mit Hilfe der Radioteleskopie bisher einige Schwefelverbindungen nachgewiesen, wie beispielsweise Kohlenstoffdisulfid (CS2), Carbonylsulfid (COS), Schwefelwasserstoff (H2S), Thioformaldehyd (H2CS) und Schwefel-IV-oxid (SO2) (Gottlieb et al. 1978; Sinclair et al. 1973). Astronomen haben die Hoffnung, mittels der Detektion von Schwefel-IV-oxid Vulkanismus auf Planeten außerhalb unseres Sonnensystems nachzuweisen.
Das Ende des 19. Jahrhunderts entwickelte Frasch-Verfahren erlaubt die Ausbeutung unterirdischer Lagerstätten elementaren Schwefels (Schmidt 1973). Die wichtigsten Vorkommen liegen in Kanada, in den Vereinigten Staaten von Amerika, der ehemaligen Sowjetunion und Westasien. Die Volksrepublik China ist der weltweit größte Importeur, Kanada der größte Exporteur, gefolgt von Russland und Saudi-Arabien.
Der geförderte Schwefel wird fast ausschließlich zu Schwefelsäure weiterverarbeitet. Der erste Schritt dieses Prozesses ist das Rösten sulfidischer Erze. Elementarer Schwefel wird weltweit gewonnen und gehandelt.
Physikalische Eigenschaften: Diese hängen stark von der Temperatur ab, da bei gegebener Temperatur eine Reihe allotroper Modifikationen vorliegen können. Wird Schwefel auf über 119 °C erhitzt, bildet sich zunächst eine niedrigviskose Flüssigkeit hellgelber Farbe, in der überwiegend S8-Ringmoleküle vorhanden sind.
Die Dichte festen Schwefels beträgt unter Normalbedingungen etwa 2,0 g/cm3 und seine Mohshärte rund 2,0. Meist tritt er in Form hell- bis dunkelgelber Prismen oder Pyramiden auf. Auf einer Strichtafel hinterlässt Schwefel einen weißen Strich. Größere Kristalle sind durchsichtig bis durchscheinend und zeigen einen fettigen Glanz. Pulvrige Aggregate sind jedoch undurchsichtig matt.
Die Moleküle des rhombischen Schwefels sind S8-Ringe (Cyclooctaschwefel); der Nachweis mittels Röntgenstrukturanalyse gelang 1935. Wird Schwefel zunächst erhitzt, und dann die Temperatur längere Zeit gehalten, so brechen die S8-Ringe teilweise auf, wodurch es zu einer Senkung des Schmelzpunktes auf bis zu 114,5 °C kommt. Weiteres Erhitzen über den Schmelzpunkt hinaus führt zu einem drastischen Anstieg der Viskosität infolge Bildung langer, kettenförmiger Moleküle, die ihr Maximum bei 187 °C erreicht. Bei noch höherer Temperatur zerbrechen die langen Kettenmoleküle in kleinere Einheiten, was wiederum mit einer Senkung der Viskosität der Schmelze einhergeht.
Schwefel zeigt bis zu dreißig diverse Allotrope. Die unter Normalbedingungen thermodynamisch stabilen enthalten alle S8-Molekülringe (Cyclooctaschwefel), aber es gibt Ring- und Kettenmoleküle mit unterschiedlicher Zahl an Schwefelatomen:
Im natürlich vorkommenden Schwefel liegen kronenförmig gezackte S8-Ringmoleküle (Cyclooctaschwefel) vor. Bei Raumtemperatur am stabilsten ist der orthorhombisch kristallisierende, geruch- und geschmacklose, gelbe α-Schwefel, der auch in der Natur vorkommt.
Oberhalb einer Temperatur von 95 °C liegt der monoklin kristallisierende, farblose β-Schwefel vor, der durch Erhitzen auf 100 °C und anschließendes schnelles Abkühlen auf 20 °C für einige Zeit stabilisiert werden kann. Der monoklin kristallisierende γ-Schwefel (Rosickýit) kommt natürlich im Death Valley (USA) vor, wo er durch mikrobiologische Reduktion von Sulfat gebildet wird.


Er kann in Abhängigkeit vom Herstellverfahren in vier unterschiedlichen allotropen Modifikationen auftreten (α-, β-, γ-, δ-Cycloheptaschwefel), die sämtlich temperaturempfindlich sind und bei Temperaturen oberhalb von 20 °C schnell in die thermodynamisch stabilste Form übergehen. Länger haltbar sind alle Modifikationen nur dann, wenn sie durch Trockeneis gekühlt(−78 °C) aufbewahrt werden.

So bildet Cyclononaschwefel (S9) ebenfalls vier Allotrope, von denen zwei (α- und β-Modifikation) genauer beschrieben sind.
Cyclododecaschwefel (S12) mit seiner kronenförmig gezackten Molekülstruktur ist nach Cyclooctaschwefel das thermodynamisch stabilste Molekül.
Cyclooctadecaschwefel (S18) bildet zwei konformationsisomere Ringe aus (Holleman et al. 2007, S. 549, 552).
Die Moleküle des polymeren Schwefels bestehen aus langen durch Schwefelatome gebildeten Ketten. Er kann durch Erhitzen von Schwefel auf Temperaturen oberhalb von 120 °C und darauf folgendes schnelles Abkühlen (Eiswasser, flüssiger Stickstoff) dargestellt werden.
Vorkommen, physikalische und chemische Eigenschaften von Schwefel
|
Symbol: |
S |
|
|
|
Ordnungszahl: |
16 | ||
|
CAS-Nr.: |
7704-34-9 | ||
|
Aussehen: |
Hellgelber Feststoff |
Tupan Patera, ein mit flüssigem Schwefel gefüllter Krater auf dem Jupitermond Io (NASA 2001) |
Schwefel, Pulver (Sicius 2015) |
|
Entdecker, Jahr |
China, Ägypten (5000 v. Chr.) | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
3216S (95,02) |
Stabil |
---- | |
|
3316S (0,75) |
Stabil |
---- | |
|
3416S (4,21) |
Stabil |
---- | |
|
Massenanteil in der Erdhülle (ppm): |
480 | ||
|
Atommasse (u): |
32,06 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
2,58 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial für: S + 2 e− > S2− (V) |
−0,48 | ||
|
Atomradius (pm): |
100 | ||
|
Van der Waals-Radius (berechnet, pm): |
180 | ||
|
Kovalenter Radius (pm): |
103 | ||
|
Elektronenkonfiguration: |
[Ne] 3s23p4 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
1000 ♦ 2252 ♦ 3357 | ||
|
Magnetische Volumensuszeptibilität: |
−1,3 ⋅ 10−5 | ||
|
Magnetismus: |
Diamagnetisch | ||
|
Kristallsystem: |
Orthorhombisch | ||
|
Elektrische Leitfähigkeit([A/(V · m)], bei 300 K): |
10−22 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
k. A ♦ 7,7 ♦ k. A. | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
32 ♦ k. A. | ||
|
Schallgeschwindigkeit (m/s, bei 273,15 K): |
Keine Angabe | ||
|
Dichte (g/cm3, bei 273,15 K) |
2,07 | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
15,53 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit ([W/(m · K)]): |
0,205 | ||
|
Spezifische Wärme ([J/(mol · K)]): |
22,75 | ||
|
Schmelzpunkt (°C ♦ K): |
115,21 ♦ 388,36 | ||
|
Schmelzwärme (kJ/mol): |
1,713 | ||
|
Siedepunkt (°C ♦ K): |
444,6 ♦ 717,75 | ||
|
Verdampfungswärme (kJ/mol): |
45 | ||
|
Kritischer Punkt (°C ■ MPa): |
1037 ■ 20,7 | ||

Oberhalb des Siedepunktes enthält gasförmiger Schwefel zunächst S8-Ringe, die bei noch höheren Temperaturen aufbrechen. Zunächst weist der Schwefeldampf die gleiche gelbe Farbe auf wie eine Schmelze von Cyclooctaschwefel. Bei einer Temperatur oberhalb von 550 °C zerfallen die Ringe in kleinere Moleküle wie S2–S4, die rot bis dunkelrotbraun erscheinen. Oberhalb einer Temperatur von 700 °C enthält der Dampf zumeist S2-Moleküle, bei >1800 °C liegen einzelne Schwefelatome vor.

Schwefel reagiert bei höherer Temperatur mit fast allen Metallen unter Bildung von Sulfiden, ausgenommen nur Platin, Iridium und Gold. Mit Quecksilber reagiert Schwefel sogar schon beim Verreiben bei Raumtemperatur zu Quecksilbersulfid. Schwefel reagiert auch mit den meisten Nichtmetallen direkt, nur nicht mit Tellur, Stickstoff, Iod und Edelgasen.
An Luft entzündet sich Schwefel ab einer Temperatur von etwa 250 °C und verbrennt mit blauer Flamme unter Bildung von Schwefel-IV-oxid (SO2). Schwefel wird durch Salpetersäure zu Sulfat oxidiert. In alkalischer Lösung disproportioniert Schwefel zu Sulfid und Sulfit. In sulfidischer Lösung ist Schwefel unter Bildung von Polysulfiden löslich, und mit Sulfit reagiert es zu Thiosulfat.
In seinen Verbindungen nimmt Schwefel alle Oxidationsstufen zwischen −2 (Sulfide) und +6 (Sulfate, Schwefel-VI-oxid und Schwefelsäure) ein.

Das so gebildete Produkt ist allerdings durch die Ausgangsprodukte mit Gasen wie Wasserstoff, Kohlendioxid, Stickstoff und Sauerstoff verunreinigt. Bei Verwendung von natürlichem Eisensulfid (z. B. Pyrrhotin) kann das Produkt zusätzlich auch noch mit Arsen-, Selen und Tellurwasserstoff sowie Monophosphan verunreinigt sein. Reinen Schwefelwasserstoff erhält man durch Erhitzen einer konzentrierten Lösung von Magnesiumhydrogensulfid oder aus den Elementen, ebenso aus Natriumsulfid und Phosphorsäure (Brauer 1963, S. 344).
In der Petrochemie (Raffinerien) fällt Schwefelwasserstoff in großen Mengen bei der Hydrodesulfurierung von Erdöl an. Schwefelwasserstoff ist die Hauptquelle für elementaren Schwefel, der wiederum zu über 95 % zu Schwefelsäure umgesetzt wird. Dabei wird ein Teil des Schwefelwasserstoffs zu Schwefel-IV-oxid verbrannt. Ein Teil des übrigen Schwefelwasserstoffs reagiert mit dem entstandenen Schwefel-IV-oxid unter Komproportionierung zu elementarem Schwefel.
Im klassischen Kationentrennungsgang wird Schwefelwasserstoff zum Ausfällen einer ganzen Gruppe benutzt (Schwefelwasserstoffgruppe). Durch Einleiten von H2S-Gas in schwach saure Lösungen fallen aus: As2S3, SnS2, Sb2S3, HgS, SnS, PbS, Bi2S3, CuS und bei Verdünnen mit Wasser auch CdS. Diese Kationen sind dann weiter aufzutrennen und mithilfe von Nachweisreaktionen zu identifizieren.
Wegen seiner Giftigkeit verzichtet man im Kationen-Trennungsgang zunehmend auf Schwefelwasserstoff. Stattdessen erzeugt man die benötigten Sulfid-Anionen in situ, z. B. mit Hilfe von Thioacetamid, in kleineren Mengen auch durch Erhitzen von Schwefel mit Kerzenwachs.
Schwefelwasserstoff hat die Eigenschaft, die Geruchsrezeptoren zu betäuben, weshalb man eine Erhöhung seiner Konzentration nicht mehr über den Geruch wahrnimmt. Die eigentliche Giftwirkung beruht auf einer Zerstörung des roten Blutfarbstoffes Hämoglobin und damit einer Lähmung der intrazellulären Atmung. Man vermutet, dass allgemein schwermetallhaltige, sauerstoffübertragende Enzyme infolge der Bildung schwer löslicher Sulfide inaktiviert werden. Der kleinere, nichtoxidierte Teil des Schwefelwasserstoffs kann Schäden im zentralen und evtl. auch peripheren Nervensystem hervorrufen (Reiffenstein et al. 1992).
Unter extrem hohem Druck (1,5 Mio. bar) wird die Verbindung bereits bei −70 °C supraleitend (Drozdov et al. 2015).
Disulfan (H2S2) besitzt einen campherähnlichen Geruch und zersetzt sich leicht zu Schwefelwasserstoff und elementarem Schwefel (Steudel 2003). Die Flüssigkeit siedet bei 70 °C, erstarrt bei −89,6 °C und hat die Dichte 1,334 g/cm3. Man gewinnt die Verbindung durch thermische Zersetzung von Rohsulfan (Brauer 1975, S. 364). Die gelbe luftempfindliche Flüssigkeit hydrolysiert in Wasser und Alkoholen; sie löst sich in Kohlenstoffdisulfid, Benzol und Tetrachlormethan.

Flüssiges Schwefel-IV-oxid (Onyxmet 2020)
Flüssiges Schwefel-IV-oxid ist bei Raumtemperatur schon durch Anwendung niedriger Überdrücke (ab 3,3 bar) herstellbar. Es löst zahlreiche Stoffe und hat sich daher als wertvolles aprotisch-polares Lösungsmittel etabliert.
In der Lebensmittelindustrie setzt man Schwefel-IV-oxid als Konservierungs-, Antioxidations- und Desinfektionsmittel ein, meist für Trockenfrüchte, Fruchtsäfte, Kartoffelgerichte, Marmelade und Wein. Wein- und Bierfässer desinfiziert man vor der Verwendung durch Behandlung mit gasförmigem Schwefel-IV-oxid.
Schwefel-IV-oxid zerstört Vitamin B1; ebenso zeigen Laborversuche Hinweise auf eine Zerstörung von B12-Vitaminen. In der EU ist es als Lebensmittelzusatzstoff der Nummer E 220 auch für „Bio“-Produkte zugelassen. Es dient des Weiteren zur Herstellung von Sulfuryl- und Thionylchlorid. In der Sulfochlorierung ist es Ausgangsmaterial zur Produktion von Tensiden. Ferner ist Schwefel-IV-oxid ein wichtiges Edukt zur Produktion von Schwefel-VI-oxid, um anschließend daraus konzentrierte Schwefelsäure etwa mittels des Kontaktverfahrens herzustellen.
Schwefel-IV-oxid ist ferner unverzichtbar zur Herstellung vieler Chemikalien, Medikamente und Farbstoffe sowie zum Bleichen von Papier und Textilien. Es lässt Tinte verblassen. Zudem setzt man es als Schutzgas ein, um flüssige Metallschmelzen in der Gießerei an der Oxidation zu hindern (Bünck 2014, S. 36).

Ca. 60 g Schwefel-VI-oxid in einer Ampulle (H. Hiller 2010)
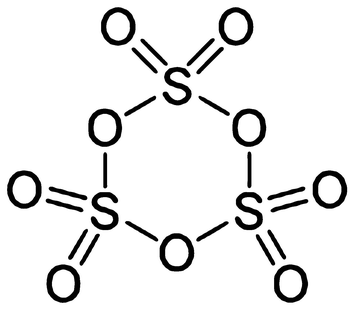
Kondensiert man Schwefel-VI-oxid allerdings bei Temperaturen oberhalb von 27 °C, wird α‐SO3 gebildet, das faserähnliche Kristalle bildet und einen Schmelzpunkt von 62,3 °C hat. Seine Molekülstruktur entspricht der eines Polymeren [[S = O)2(μ−O)]3]n, wobei jedes Ende der Polymerkette OH-Gruppen trägt. Das β‐SO3 bildet zwar ebenso faserähnliche Kristalle, hat aber ein zur zuletzt genannten Modifikation verschiedenes Molekulargewicht und schmilzt bei 32,5 °C. Nur die α‐SO3-Form ist stabil und wird aus den beiden anderen beim Stehenlassen gebildet; dieser Prozess wird durch Spuren von Wasser katalysiert.
Die relativen Dampfdrücke des festen Schwefel-VI-oxids nehmen vom α‐ über das β‐ zum γ‐SO3 hin zu. Flüssiges Schwefel-VI-oxid hat einen Dampfdruck, der demjenigen der γ‐SO3‐Form entspricht. Daher hat das Erhitzen eines Kristalls von α‐SO3 einen plötzlichen Anstiegs des Dampfdrucks zur Folge, wodurch das zu seiner Aufheizung verwendete, geschlossene Gefäß explodieren kann. Gasförmiges Schwefel-VI-oxid liegt in Form von Monomeren vor. Außerdem ist Schwefel-VI-oxid äußerst hygroskopisch, verkohlt Baumwolle, Holz und Papier schnell, wobei sich diese Materialien gelegentlich auch entzünden.
Technisch stellt man Schwefel-VI-oxid im Kontaktverfahren unter Verwendung von Vanadium-V-oxid aus Schwefel-IV-oxid und Sauerstoff bei 420 °C her. In reiner Form erhält man die Verbindung, wenn man sie aus Oleum abdestilliert. Im Labor sind kleinere Mengen durch Erhitzen einer Mischung von konzentrierter Schwefelsäure mit Phosphor-V-oxid herstellbar, wobei das bei der Reaktion gebildete Schwefel-VI-oxid abdestilliert.

Bei der Produktion von Tensiden nutzt man diese Reaktion, da man diese Ester mit Natronlauge neutralisiert und so die gewünschten Fettalkoholsulfate erhält.
Oxosäuren des Schwefels
|
Oxidationszahl des Schwefels |
Säuren des Typs H2SOn (Salze) |
Säuren des Typs H2S2On (Salze) |
|---|---|---|
|
+1 |
Thioschweflige Säure, H2S2O2 (Thiosulfite) | |
|
+2 |
Sulfoxylsäure, H2SO2 (Sulfoxylate) |
Thioschwefelsäure, H2S2O3 (Thiosulfate) |
|
+3 |
Dithionige Säure, H2S2O4 (Dithionite) | |
|
+4 |
Schweflige Säure, H2SO3 (Sulfite) |
Dischweflige Säure, H2S2O5 (Disulfite) |
|
+5 |
Dithionsäure, H2S2O6 (Dithionate) | |
|
+6 |
Schwefelsäure, H2SO4 (Sulfate) |
Dischwefelsäure, H2S2O7 (Disulfate) |
|
+6 |
Peroxoschwefelsäure, H2SO5 (Peroxosulfate) |
Peroxodischwefelsäure, H2S2O8 (Peroxodisulfate) |

Schwefelsäure 96 % (W. Oelen 2011)
Schwefelsäure gehört zu den technisch wichtigsten Chemikalien und wird vor allem in der Produktion von Düngemitteln und anderen Mineralsäuren, etwa der Salz- oder Phosphorsäure, eingesetzt.
Grundstoff zu ihrer Herstellung ist meist elementarer Schwefel, der in großen Mengen bei der Entschwefelung von Erdgas und Rohöl nach dem Claus-Prozess anfällt oder aber nach dem Frasch-Verfahren abgebaut wird. Der so gewonnene Schwefel wird zunächst zu Schwefel-IV-oxid verbrannt (Riegel und Kent 2003, S. 503). Ebenfalls große Mengen an Schwefel-IV-oxid entstehen bei der Verhüttung schwefelhaltiger Erze, etwa im Zuge der Produktion von Kupfer, Zink oder Blei aus ihren Sulfiden. Auch Pyrit (FeS2) ist eine wichtige Quelle für Schwefel-IV-oxid.
In Ländern mit geringen Vorkommen elementaren Schwefels oder sulfidischen Erzen steht nur die energieintensive, im Drehrohrofen betriebene Produktion aus Gips (Calciumsulfat) und Kohle nach dem Müller-Kühne-Verfahren zur Verfügung (Kühne 1949).
Die Produktion der Vorstufe der Schwefelsäure, des Schwefel-VI-oxids, erfolgt nur noch nach dem Kontaktverfahren mit Vanadium-V-oxid und. Alkalisulfat als Katalysator bzw. Co-Katalysator. Die Reaktionstemperatur in diesen Hordenöfen beträgt 420–620 °C (Lapina et al. 1999). Beim Doppelkontaktverfahren wäscht man vor der letzten Horde das schon im Gasgemisch enthaltene Schwefel-VI-oxid mit konzentrierter Schwefelsäure aus, wodurch die Ausbeute auf >99,8 % gesteigert wird.
Nach Bildung des Schwefel-VI-oxids setzt man dieses dann mit konzentrierter Schwefelsäure um und verdünnt jene dann entsprechend auf die gewünschte Konzentration. Vorher aber muss man zunächst nichtumgesetztes Schwefel-IV-oxid durch Zugabe von Ammoniak oder Natriumthiosulfat entfernen.
Die oft bräunliche Färbung technischer Schwefelsäure liegt am Vorhandensein organischer Verunreinigungen, die durch Entwässerung verkohlt werden. Oberhalb des Siedepunktes wasserfreier Schwefelsäure (279,6 °C) bilden sich Dämpfe, die überschüssiges Schwefel-VI-oxid enthalten, wobei das Wasser in der siedenden Schwefelsäure verbleibt. Die wasserfreie Schwefelsäure geht so in eine 98,33 %ige Schwefelsäure über, die konstant bei 338 °C siedet. Bei dieser Temperatur hat auch der Dampf einen Säuregehalt von 98,33 % (azeotropes Gemisch).
Schwefelsäure ist der Bezugspunkt für die Definition einer Supersäure. Alle Säuren, die stärker als reine Schwefelsäure sind und diese somit protonieren können, werden als Supersäuren bezeichnet.
Da das erste Proton des Schwefelsäuremoleküls viel leichter abdissoziiert als das zweite, liegt in verdünnter Schwefelsäure (c=1 mol/L) größtenteils Hydrogensulfat vor. Die zweite Dissoziation zum Sulfat erfolgt nur in viel geringerem Maße (etwa 1,3 % bei 1 mol/L) (Holleman et al. 2019, S. 511). Schwefelsäure ist im Hinblick auf die Definition von Supersäuren der Ausgangspunkt, da man alle Säuren, die in der Lage sind, Schwefelsäure zu protonieren, als Supersäuren bezeichnet.
Beim Verdünnen konzentrierter Schwefelsäure mit Wasser werden erhebliche Wärmemengen freigesetzt. Daher gibt man immer die Säure unter gutem Rühren ins Wasser und nie (!) das Wasser in die Säure, da dann die geringe Wassermenge schlagartig verdampft und mit der Säure verspritzt.

Schwefel-VI-fluorid in Ampulle (Onyxmet 2020)
Man setzt es unter Anderem als Isolator für Hochspannungsschaltungen ein, ebenso, aber in immer geringerem Maße, als Tracer zum Nachweis von Windströmungen, obwohl es Infrarotlicht stark absorbiert und somit ein hohes Treibhauspotenzial besitzt. Schon 1 t des Gases belastet nach einer Angabe des Umweltbundesamtes von 2002 die Atmosphäre in einer Größe, die 24.000 t Kohlendioxid entspricht.
Seit Ende der 1970er-Jahre setzte man Schwefel-VI-fluorid als Füllgas in Autoreifen und Schallschutz-Isolierglasscheiben ein. Bei Kraftfahrzeugen nutzte man es anstelle von Luft zur Füllung der Reifen, weil die SF6-Moleküle im Vergleich zu den Molekülen der in der Luft enthaltenen Gase (Stickstoff und Sauerstoff) größer sind und daher langsamer aus dem Reifen entweichen. Das spart dem Nutzer Zeit, da der Luftdruck der Reifen wesentlich seltener kontrolliert werden muss. Auch sollten diese gasbefüllten Reifen zu einer höheren Fahrsicherheit beitragen. Aber spätestens bei der Demontage des Reifens entweicht Schwefel-VI-fluorid ungehindert und vollständig in die Atmosphäre. Autoreifen sollen zu ca. 25 %, Schallschutzfenster zur Hälfte zu den SF6-Emissionen beigetragen haben.
Dischwefeldecafluorid (S2F10) ist eine farblose, stechend nach Schwefel-IV-oxid riechende Flüssigkeit der Dichte 2,08 g/cm3, die bei −55 °C erstarrt und schon bei 28,7 °C siedet. Es bildet sich als Nebenprodukt bei der Reaktion von Schwefel mit Fluor (Denbigh und Whytlaw-Gray 1934), die üblicherweise zum Schwefel-VI-fluorid führt oder durch lichtkatalysierten Abbau des Schwefelchloridpentafluorids (SF5Cl). Die Flüssigkeit ist durch die leicht erfolgende Bildung von SF5•-Radikalen sehr reaktionsfreudig. Bei Erhitzung zersetzt sie sich in Schwefel-VI- und Schwefel-IV-fluorid. Mit überschüssigem Chlor reagiert sie zu Schwefelchloridpentafluorid.
Dischwefeldecafluorid ist unlöslich in Wasser und ist überraschenderweise sehr stabil gegenüber Hydrolyse. Es reagiert nicht mit Wasser (Holleman et al. 1995, S. 565), auch nicht mit sauren oder alkalischen wässrigen Lösungen, sondern nur mit einer 10 %igen Lösung von Kaliumhydroxid in Methanol. Die Verbindung ist sehr giftig und noch deutlich toxischer als Phosgen. Seine Nutzung als Kampfstoff war im Zweiten Weltkrieg geplant, da es nur schwer rechtzeitig erkannt wird; diese Pläne wurden glücklicherweise nicht mehr umgesetzt.

Das farblose Schwefel-IV-fluorid kondensiert bei −40 °C zu einer Flüssigkeit, deren Dichte bei −73 °C 1,919 g/cm3 beträgt. Der Erstarrungspunkt beträgt −121 °C. Die Verbindung hydrolysiert heftig mit Wasser zu Schwefel-IV-oxid und Fluorwasserstoff (Pavone et al. 2004). Schwefel-IV-fluorid ist eine schwache Lewis-Säure und bildet Addukte mit organischen Basen wie Pyridin und Triethylamin. Man nutzt es zum Fluorieren anorganischer Oxide, Sulfide oder Carbonyle; dabei ist vor allem die selektive Umwandlung einer Ketogruppe (=C=O) zu einer Difluormethylengruppe (=CF2) interessant (Holleman et al. 1995, S. 564).
Schwefel-II-fluorid (SF2) ist aus Schwefel-II-chlorid und Kaliumfluorid bei ca. 170 °C zugänglich. Bei niedrigen Drücken (<25 mbar) funktioniert auch Quecksilber-II-fluorid (bei 150 °C) oder Silber-I-fluorid (bei 20 °C), allerdings entstehen dabei auch andere Schwefelfluoride. Eine weitere Möglichkeit der Darstellung ist die Reaktion von Fluor mit Carbonylsulfid (Holleman et al. 1995, S. 564). Das farblose Gas ist nur verdünnt und in Abwesenheit von Fluoriden kurze Zeit haltbar, zerfällt dann aber über sein Di- und Trimer schnell zu Thiothionylfluorid und Schwefel-IV-fluorid.

Das farblose, übelriechende Gas der Dichte 4,34 kg/m3 kondensiert bei −15 °C; die Flüssigkeit erstarrt bei −133 °C (Latscha und Klein 2002, S. 341; Blachnik 1998, S. 704). Bei höheren Temperaturen und Drücken geht es in das dann beständigere Thiothionylfluorid und dann durch Disproportionierung weiter in Schwefel-IV-fluorid und Schwefel über. Schon bei Raumtemperatur erfolgt bei Gegenwart von Fluorid diese Umwandlung zu Thiothionylfluorid. Mit Stickstoffdioxid reagiert jedoch nur Difluordisulfan, im Gegensatz zu Thiothionylfluorid, zu Nitrosylfluorosulfat (Holleman et al. 1995, S. 379)


Das bei −10,6 °C kondensierende und bei −164,6 °C erstarrende Thiothionylfluorid (Holleman et al. 1995, S. 379) entsteht ebenfalls aus Difluordisulfan in Gegenwart von Alkalifluoriden (Steudel 2008, S. 475). Bei höheren Temperaturen und Drücken disproportioniert es zu Schwefel-IV-fluorid und Schwefel. Fluorwasserstoff überführt es in Schwefel-IV-fluorid und Schwefelwasserstoff.
Thionylfluorid (SOF2) erhält man durch Reaktion von Fluorwasserstoff oder anderen Fluoriden mit Thionylchlorid (Brauer 1975, S. 187). Im Labormaßstab erwies sich die Umsetzung von Thionylchlorid mit Antimon-III-fluorid in Gegenwart von Antimon-V-chlorid als Katalysator am geeignetsten (Blachnik 1998, S. 712). Das farblose Gas kondensiert bei −43,7 °C; die Flüssigkeit der Dichte 1,78 g/cm3(−100 °C) erstarrt bei einer Temperatur von −110,5 °C. Die Verbindung ist gut löslich in trockenem Benzol und Diethylether (Housecroft 2005, S. 450).
Festes Thionylfluorid kristallisiert im monoklinen Gitter (Raumgruppe 14). Trockenes Thionylfluorid greift Metalle wie Magnesium, Nickel, Quecksilber und Aluminium bis zu einer Temperatur von 125 °C nicht an, Glas oberhalb von 400 °C. Kaltes Wasser hydrolysiert es nur langsam (Brauer 1975, S. 187), auch raucht es bei Kontakt mit feuchter Luft nur schwach (Smith und Muetterties 1960). Fluor überführt es in Schwefeloxidtetrafluorid (SOF4). Es ist gut löslich in Diethylether und Benzol.
Sulfurylfluorid (SO2F2) ist ein farb- und geruchloses, giftiges Gas der Dichte 3,723 kg/m3, das bei −55 °C kondensiert (Erstarrungspunkt: −135,8 °C). Man nutzt es als Insektizid zur Konservierung von Lebensmitteln (Getreide, Nüsse, Schalen- und Trockenfrüchte) und auch zur Bekämpfung von Holzschädlingen. Im Gegensatz zu anderen Mitteln zur Schädlingsbekämpfung schädigt es nicht die Ozonschicht der Erdhülle, besitzt aber ein nicht unerhebliches Treibhauspotenzial (Papadimitriou et al. 2008).
Sulfurylfluorid erhielt vom deutschen Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (BVL) eine Zulassung als Konservierungsmittel für Lebensmittel. Zunächst erging 2007 die Zulassung nur für die Anwendung bei Trockenobst, später auch für die Desinfektion leerer Mühlen. 2009 folgte die Zulassung zur Behandlung von Nüssen, Schalenfrüchten und für Getreide, die in Lagern oder unter gasdichten Planen zwischengelagert werden. Schließlich erging 2012 eine Zulassung zur Bekämpfung von Schädlingen für geschlagenes Holz, Holzpaletten und Packholz. Dabei wird angenommen, dass bei der Anwendung von Sulfurylfluorid die für die EU gültigen Höchstkonzentrationen für Fluorid von 25 mg/kg für Nüsse und 3 mg/kg für Trockenobst eingehalten werden könnten, aber das BVL weist darauf hin, dass weitere Fluoridquellen (Zahncreme) zu einer Überschreitung der maximal für Menschen tolerierbaren Gesamtdosis führen können.
- (I)

- (II)

- (III)

- (IV)

Das Molekül des Sulfurylfluorids hat eine verzerrt-tetraedrische Struktur mit dem Schwefelatom in der Tetraedermitte. Im Vergleich zu Sulfurylchlorid ist Sulfurylfluorid reaktionsträger und thermisch stabiler. In Wasser tritt bis hinauf zu Temperaturen von 150 °C keine starke Hydrolyse zu Schwefelsäure und Fluorwasserstoff ein, im basischen Milieu aber schon bei Raumtemperatur (Holleman et al. 2007, S. 590).
Schwefeloxidtetrafluorid (Thionyltetrafluorid, SOF4) entsteht bei der Reaktion von Thionylfluorid mit Brompentafluorid bei 280 °C, bei der bei 200 °C durch Stickstoffdioxid katalysierten Reaktion von Schwefel-IV-fluorid mit Sauerstoff (beide Brauer 1975, S. 187) oder bei der Umsetzung von Fluor mit Thionylfluorid (Ruff 1968). Das stechend riechende, farblose Gas kondensiert bei −48,5 °C zu einer farblosen Flüssigkeit der Dichte 1,95 g/cm3(−82 °C). Diese erstarrt bei −99,6 °C. Bei Kontakt mit Wasser erfolgt heftige Hydrolyse zu Sulfurylfluorid und Fluorwasserstoff, Natronlauge setzt es komplett zu Natriumfluorid und Natriumsulfat um.


Das farblose, ziemlich reaktive Schwefelmonochloridpentafluorid kondensiert bei einer Temperatur von −19,5 °C; die Flüssigkeit erstarrt bei −64 °C. Die Verbindung ist ein kräftiges Oxidationsmittel und ist erheblich reaktionsfähiger als Schwefel-VI-fluorid, so erfolgt die Hydrolyse viel leichter (Paquette 2013, S. 426). Wird das Gas auf Temperaturen oberhalb von 400 °C erhitzt oder mit UV-Licht bestrahlt, zersetzt es sich zu Schwefel-IV- und Schwefel-VI-fluorid. Man nutzt die Verbindung zur Einführung von SF5-Gruppen und auch in der präparativen organischen Chemie zur Darstellung von Verbindungen, deren Moleküle Kohlenstoff-Schwefel-Doppel- und Dreifachbindungen enthalten (Seppelt 1987).
Schwefel-IV-chlorid (SCl4) schmilzt zwischen −31 °C und −20 °C unter gleichzeitiger Zersetzung. Die Verbindung ist nur im festen Zustand stabil. Man stellt es durch direkte Chlorierung von Schwefel her.
Das dunkelrote, giftige Schwefel-II-chlorid (SCl2) ist eine bei 59,5 °C siedende und bei −65 °C erstarrende Flüssigkeit der Dichte 1,62 g/cm3 (15 °C). Die Verbindung ist empfindlich gegenüber Hydrolyse und hat einen chlorähnlichen Geruch. Schon ab Mitte des 19. Jahrhunderts setzte man Schwefel-II-chlorid zur Kaltvulkanisation von Kautschuk ein, im Ersten Weltkrieg war es Vorstufe der Synthese des Kampfstoffes S-Lost. Man stellt es im Labor durch Umsetzung von Dischwefeldichlorid mit Chlor her. Oder man leitet Chlor über geschmolzenen Schwefel, erhält dabei allerdings ein Gemisch aus Dischwefeldichlorid und Schwefel-II-chlorid, das dann unter Inertgas destillativ getrennt werden muss (Brauer 1963, S. 370).
Schwefel-II-chlorid ist schon bei Raumtemperatur instabil und zerfällt zu Chlor und Dischwefeldichlorid. Man kann die Verbindung durch Zusatz von Phosphorchloriden stabilisieren. Der Aufbau des Moleküls ist gewinkelt (Cl-S-Cl: 103°), die Länge der S-Cl-Bindung beträgt 201 pm.
Anwendung findet Schwefel-II-chlorid als Sulfidierungs- und Chlorierungsmittel.
Dischwefeldichlorid (S2Cl2) erhält man ebenfalls durch Chlorieren von Schwefel (Brauer 1963, S. 371). Ist Chlor im Überschuss zugegen, läuft die Reaktion zum Schwefel-II-chlorid weiter. Jenes kann aber durch Zugabe elementaren Schwefels wieder zum gewünschten Dischwefeldichlorid umgesetzt werden. Die gelborange, an der Luft rauchende Flüssigkeit der Dichte 1,69 g/cm3 (20 °C) erstarrt bei −80 °C und besitzt einen erstickenden Geruch; der Siedepunkt beträgt 138 °C (Normaldruck). Mit Wasser erfolgt heftige Zersetzung zu einem Gemisch von Schwefel und verschiedener seiner Verbindungen. Die reine Verbindung löst Schwefel in großer Menge auf; dabei bilden sich Ketten der Formel SnCl2 mit n=2–100 (Kolditz 1983, S. 469). Die Anwendung des Dischwefeldichlorids erfolgt als Sulfidierungs- und Chlorierungsmittel (wie etwa Oxalylchlorid und Chloressigsäure), außerdem setzt man es zur Synthese diverser organischer Schwefelverbindungen ein, als Zusatzstoff für Hochdruckschmiermittel, Schneidöle und Kautschuk-Vulkanisationsmitteln.


Thionylchlorid (W. Oelen 2011)
- (I)

- (II)

Thionylchlorid hat sehr viele industrielle Anwendungen. Unter anderem erzeugt man aus ihm aromatische Sulfonsäurechloride, Carbonsäurechloride sowie Alkylchloride aus aliphatischen Alkoholen.

Sulfurylchlorid (W. Oelen 2011)
Mit Wasser reagiert Sulfurylchlorid unter sehr heftiger Hydrolyse zu Schwefelsäure und Chlorwasserstoff. Ebenso setzt es sich sehr heftig mit Basen, immer noch lebhaft auch mit niederen Alkoholen um.
Man stellt aus Sulfurylchlorid aromatische Sulfonsäurechloride her; auch kann man mit seiner Hilfe aliphatische Kohlenwasserstoffe radikalisch chlorieren
Dischwefeldibromid (S2Br2) ist durch Umsetzung von Schwefel mit Brom erhältlich (Brauer 1975, S. 383). Ebenso funktioniert die Reaktion von Dischwefeldichlorid mit Bromwasserstoff. Die dunkelrote, ölige, Glas nicht benetzende Flüssigkeit erstarrt bei −46 °C zu Kristallen orthorhombischer Struktur und hat die Dichte 2,63 g/cm3. Mit Wasser tritt schnell Hydrolyse zu Bromwasserstoff, Schwefel-IV-oxid und Schwefel ein. Beim Erhitzen zersetzt sich die Verbindung, weshalb sie nur im Hochvakuum unzersetzt destillierbar ist.
Von Schwefeliodiden wurden bisher S2I2 und SI2 charakterisiert, aber nur bei tiefer Temperatur. Außerdem werden diverse Schwefeliodid-Kationen wie S2I42+ und S7I+ beschrieben (Klapotke und Passmore 1989).

Herstellung von Pentaschwefelhexanitrid (Steffen 962 2017)
Eine andere Synthesemöglichkeit besteht in der in Dichlormethan durchgeführten Umsetzung von Brom mit Tetrabutylammonium-tetraschwefelpentanitrid (Holleman et al. 2017, S. 682). Die orangen Kristalle neigen zu explosivem Zerfall, sind aber bei Raumtemperatur unter Inertgas stabil. Kontakt mit Luftsauerstoff führt zu sofortiger Verfärbung ins Schwarze. Unter Vakuum (2–10 mbar) kann man die Verbindung bei einer Temperatur um 45 °C sublimieren. Die Länge der S-N-Bindung beträgt im S4N4-Teil (Tetraschwefelnitrid) des Moleküls 161 pm, in der NSN-Brücke dagegen ca. 153 pm.


![$$ 2\, {\mathrm{S}\mathrm{Cl}}_4+2\, {\left[{\left({\mathrm{Me}}_3\mathrm{Si}\right)}_2\mathrm{N}\right]}_2\mathrm{S}\to {\mathrm{S}}_4{\mathrm{N}}_4+8\, {\mathrm{Me}}_3\mathrm{Si}\mathrm{Cl} $$](../images/454224_2_De_6_Chapter/454224_2_De_6_Chapter_TeX_Equy.png)
Tetraschwefeltetranitrid ist ebenfalls eine Käfigverbindung, schmilzt bzw. siedet bei 178 °C bzw. 185 °C und hat die Dichte 2,22 g/cm3. Alle S-N-Bindungen sind 162 pm lang, was für Mehrfachbindungen mit delokalisierten π-Elektronen spricht. Zusätzlich liegen im Käfig zwei schwache S-S-Bindungen (258 pm) vor (Janiak et al. 2012, S. 129; Steudel 2014, S. 515). Mit der Temperatur ändert Tetraschwefeltetranitrid seine Farbe von farblos (bei −196 °C), über orange (bei 25 °C) zu rot (bei 100 °C). In Wasser ist es unlöslich. Festes Tetraschwefeltetranitrid zerfällt beim Erhitzen über eine Temperatur von 130 °C oder bei Stoß in der Regel unter Explosion in Schwefel und Stickstoff. Die Hydrolyse führt zu Ammoniak, Thiosulfat und Sulfit. Starke Säuren protonieren eines der Stickstoffatome, Lewis-Säuren bilden Additionsverbindungen wie etwa S4N4 ⋅ SbCl5, (S4N4)2 ⋅ SnCl4 oder S4N4 · BCl3. Silber-II-fluorid überführt die Verbindung in tetrameres Thiazylfluorid [(NSF)4] unter Beibehaltung des molekularen Achtrings.
Reduktionen erfolgen am Stickstoffatom, das in eine Iminogruppe überführt wird. Zersetzt man die Verbindung kontrolliert thermisch, so entsteht Dischwefeldinitrid, das im weiteren Verlauf in Polythiazyl übergeht (Steudel 2014, S. 515).

- (I)
![$$ 2\, {\mathrm{S}}_7\mathrm{N}\mathrm{H}+\mathrm{Hg}{\left[\mathrm{CO}\left(\mathrm{O}\right){\mathrm{CH}}_3\right]}_2\to \mathrm{Hg}{\left({\mathrm{S}}_7\mathrm{N}\right)}_2+2\, {\mathrm{CH}}_3\mathrm{C}\left(\mathrm{O}\right)\mathrm{OH} $$](../images/454224_2_De_6_Chapter/454224_2_De_6_Chapter_TeX_IEq7.png)
- (II)

Der dunkelrote, diamagnetische Feststoff schmilzt bei 23 °C und hat eine Dichte von 1,71 g/cm3. Die Verbindung ist instabil und zersetzt sich bei 0 °C innerhalb weniger Stunden, beim Erhitzen auf 100 °C meist unter Explosion. Tetraschwefeldinitrid ist in zahlreichen organischen Lösungsmitteln löslich, aber nur wenig in Alkoholen und gar nicht in Wasser. In letzterem tritt nur eine langsame Hydrolyse ein, die in Basen deutlich stärker ist. Gegenüber verdünnten Säuren ist die Verbindung nahezu inert. Die Struktur des Moleküls entspricht einem Sechsring in seiner Halbsesselform, in der ein oberhalb der S–N=S=N–S-Ebene liegendes Schwefelatom eine Brücke über den Ring bildet (Zhu und Gimarc 1983).
Das Molekül des Dischwefeldinitrids (S2N2) ist planar, nahezu quadratisch und hat formal 6 π-Elektronen. Es ist kein Aromat, da es ein Diradikal mit vier bindenden π-Elektronen sowie zwei nichtbindenden π-Radikalelektronen ist (Holleman et al. 2017, S. 681; Alsfasser et al. 2007). Die Bindungslängen bewegen sich im Bereich von 165,1–165,9 pm, die NSN- bzw. SNS-Bindungswinkel betragen 89,9° bzw. 90,4°. Darstellen kann man die Verbindung durch Umsetzung von Tetraschwefeltetranitrid mit Silber oder durch kontrolliertes Erwärmen von Tetraschwefeltetranitrid (Brauer 1975, S. 275). Die farblose Verbindung kristallisiert monoklin (Raumgruppe 14) (Blachnik 1998, S. 706) und ist ein leicht flüchtiger Feststoff mit durchdringendem Geruch. Ist Luftfeuchtigkeit in Spuren zugegen, polymerisieren etwa zwei Drittel der Verbindung zu Polythiazyl [(SN)x], und rund ein Drittel geht in Tetraschwefeltetranitrid über. Letztere Reaktion erfolgt sofort und ausschließlich in Gegenwart von Spuren an Alkalimetallen. Mit Lewis-Säuren bildet Dischwefeldinitrid Addukte wie beispielsweise S2N2 ⋅ BCl3, S2N2 ⋅ 2AlCl3 oder S2N2 ⋅ SbCl5.
Polythiazyl [(SN)x] ist ein elektrisch leitfähiges Polymer, das bei −272,85 °C supraleitend wird (Labes et al. 1979). Erstmals stellte man es durch Erhitzen von Tetraschwefeltetranitrid im Vakuum über Silberwolle dar (Burt 1910). Es entsteht durch Polymerisation des nur in der Kälte stabilen Dischwefeldinitrids. Das langsame Erwärmen des Dischwefeldinitrids auf eine Temperatur von −10 °C ergibt zuerst ein dunkelblaues, paramagnetisches Zwischenprodukt, das innerhalb von 2–3 d in das kristalline Polythiazyl übergeht (Alsfasser et al. 2007, S. 129). Alternativ ist die Pyrolyse von Tetraschwefeltetranitrid an Silberwolle bei 300 °C und unter Vakuum möglich.
Das metallisch-golden glänzende Material ist ziemlich stabil gegenüber Sauerstoff und Wasser, zersetzt sich aber an der Luft langsam zu einem grauen Pulver (MacDiarmid et al. 2009). Bei Schlag oder plötzlichem Erhitzen auf hohe Temperaturen tritt oft Explosion ein. Das Polymermolekül stellt eine gezackte Kette dar mit einer Länge der SN-Bindungen von 159 pm bzw. 163 pm; die Bindungswinkel betragen 120° (SNS) bzw. 106° (NSN) (MacDiarmid et al. 1975). Entlang der SN-Ketten ist die Verbindung elektrisch leitfähig, aber senkrecht dazu besitzt sie die Eigenschaften eines Isolators (Archer 2001, S. 213). Die monokline Struktur geht schon bei Einwirkung mechanischer Energie (Mahlen) in die orthorhombische über (Baughman et al. 1977). Man kann Polythiazyl in LEDs, Solarzellen, Transistoren und Elektroden von Batterien einsetzen (King 2009).
Organische Verbindungen Schwefel ist zudem in vielen organischen Verbindungen, auch natürlich vorkommenden, enthalten. Beispiele hierfür sind die zwei essenziellen Aminosäuren Cystin und Cystein, Thiole (darunter auch Mercaptane, R-SH) und Thioether (R-S-R′). Manche von ihnen sind als Aromastoffe in Knoblauch oder Kaffee enthalten, aber auch im Abwehrstoff des Stinktiers (3-Methylbutanthiol) und besitzen Gerüche von kräftig-aromatisch bis zu faulem Kohl (Ethylmercaptan). Synthetische Produkte sind die oft als anionische Tenside in Waschmitteln verwendeten Natriumsulfonate (Na‐SO2‐OR). Bei der Gewinnung seltener Metalle, etwa im Zuge der Aufbereitung von Erzen oder der Flüssig-flüssig-Extraktion, setzt man u. a. Xanthogenate (Ester der Dithiokohlensäure, H2CS2O), Dithiophosphorsäureester, Mercaptane oder Alkylsulfonate ein. Nur ist die Kohlenstoff-Schwefel-Einfachbindung schwächer als die Einfachbindung zwischen zwei Kohlenstoffatomen (Pauling 1973), weswegen sie relativ leicht hydrolytisch gespalten wird.
Die Reaktionsfähigkeit und auch -richtung organischer Schwefelverbindungen unterscheidet sich teilweise stark von der der jeweiligen Sauerstoffanaloga. So stellt man Thiole, darunter auch Mercaptane, durch Reaktion von Kaliumhydrogensulfid mit Alkylhalogeniden her. Alkylierung von Thioharnstoff und nachfolgende Hydrolyse mit Natronlauge ergibt gleichfalls Thiole. Sie sind oxidativ leicht in Disulfide und deren Folgeprodukte überführbar.
Disulfid-Brücken stabilisieren die Struktur von Proteinen; eine praktische Anwendung dafür ist das Legen von Dauerwellen. Zunächst bricht man mittels Thioglycolsäure die Cystinbindungen im Keratin reduktiv auf. Danach bringt man die Haare in die gewünschte Form und oxidiert die Thiolgruppen (-SH) mit Hilfe von Wasserstoffperoxid zurück zu Disulfidbrücken, die die Struktur der Haarproteine durch Vernetzung stabilisieren.
Thioether sind durch Umsetzung von Alkalisulfid mit Alkylhalogeniden zugänglich. Mit überschüssigem Alkylhalogenid werden Trialkylsulfoniumsalze gebildet. Thioether sind leicht zu Sulfoxiden und Sulfonen oxidierbar.
Sulfoxide, deren Moleküle zwei voneinander verschiedene Alkylgruppen tragen (R-S(O)-R′), besitzen am Schwefelatom ein Chiralitätszentrum, weil das freie Elektronenpaar dabei formal den vierten Substituenten darstellt. Einige Sulfoxide, wie Dimethylsulfoxid, sind als dipolare, aber protonenfreie Lösungsmittel oft im Einsatz.
Im Bereich der Heterocyclen ist das aromatisch riechende Thiophen bekannt. Generell befinden sich unter den Aromastoffen viele schwefelhaltige Substanzen, die längst nicht nur übel riechend sind (Ohloff 1990). Der intensivste, aus natürlichen Aromen gewonnene Geruchsstoff überhaupt ist Thioterpinol (Geruchsschwellenwert >4 ppb), das aus der Pampelmuse/Grapefruit isoliert wurde (Demole et al. 1982). Nur wenig schwächer, aber immer noch äußerst intensiv duftet der Geruchsstoff der schwarzen Johannisbeere (8-Thio- p-menth-3-on, Ohloff 1990, S. 13). Ein Monoterpen, dessen Molekül einen Thiophenring enthält, bildet den Aromaträger des Hopfens. In Shiitake-Pilzen ist der Aromastoff 1,2,3,5,6-Pentathiepan (Lenthionin) enthalten, in Spargel 1,2-Dithiolan sowie in Rettich und Radieschen das 4-Methylsulfinyl-3-butenyl-isothiocyanat.
Schwefel wird zu vielen Synthesen genutzt, allen voran für die Herstellung von Schwefelsäure. Auch Farbstoffe, Insektizide, Düngemittel werden unter Verwendung von Schwefel produziert.
Schwefelsäure wird seit fast 200 Jahren mittels des Kontaktverfahrens hergestellt, bei dem im ersten Schritt durch Rösten sulfidischer Erze oder durch Verbrennen von Schwefel an der Luft Schwefel-IV-oxid (SO2) erzeugt wird, das über einen Vanadium-V-oxid-Kontakt (V2O5) geleitet wird und dabei mit dem Luftsauerstoff spontan zu Schwefel-VI-oxid (SO3) reagiert. Dessen nachfolgende Absorption in Wasser liefert Schwefelsäure. Rund 90 % des Schwefels gehen in die Produktion von Schwefelsäure.

Schwefelsäure findet weithin Einsatz zum Aufschluss von Erzen, als Elektrolyt in Autobatterien, als Katalysator bei der Alkylierung von Olefinen, der Herstellung von Phenol und Aceton nach dem Cumolhydroperoxid-Verfahren, zur Herstellung von Fluorwasserstoff, der Produktion von Caprolactam und dem Aufschluss von Zellstoff, der den Beginn der Papierherstellung darstellt. Mit Schwefel-VI-oxid werden Dodecylaromaten und Fettalkohole zu Tensiden sulfoniert (Tadros 2005).
Bei der Vulkanisation von Kautschuk, einem der Teilschritte zur Herstellung von Autoreifen, ist Schwefel unverzichtbar. Ein Zusatz von Schwefel vernetzt die polymeren Moleküle des Kautschuks durch Bildung von Sulfidbrücken miteinander und stabilisiert so das Polymer.
In der Medizin wird Schwefel nur äußerlich angewendet (Pharmacopoea Europaea 2008), wo er bakteriostatisch wirkt, meist in Form von Seifen, Salben und Gelen gegen Pilzerkrankungen. Eingenommen bewirkt er Durchfall. Schwefel kann auch bestimmte Pilze und Parasiten abtöten.
Zusätze von Schwefel erhöhen die Festigkeit von Stahl durch Bildung kleiner Einschlüsse weichen Mangansulfids, die die mechanische Verarbeitbarkeit des Stahls erleichtern. Jedoch kann aus dem Schwefel, wenn der Stahl ständig hoher Luftfeuchtigkeit ausgesetzt ist, leicht Schwefel-IV-oxid und am Ende Schwefelsäure gebildet werden, die korrosiv auf Stahl wirkt. Darauf ist bei Stahlkonstruktionen unbedingt zu achten (Küster et al. 1984).
Flüchtige, im Erdöldestillat enthaltene Schwefelverbindungen sind oft starke Gifte für Metallkatalysatoren, da sie mit diesen reagieren. Die Vergiftung führt in der Regel zur Verminderung der Aktivität eines Katalysators, zu dessen verringerter Selektivität oder sogar zur völligen Deaktivierung. In einigen Fällen setzt man teuren Katalysatoren, wie solche aus einer Platin-Rhenium-Legierung, von vornherein Schwefel zu, um des Katalysators Anfangsaktivität auf die ungünstigen Eigenschaften des Reaktionsmediums einzustellen.
Schwefel findet als leicht entzündlicher Stoff bei der Herstellung von Schwarz- oder Schießpulver und auch in Zündmitteln für Feuerwerkskörper Verwendung.
Schwefel-IV-oxid setzt man zum Desinfizieren leerer Weinfässer sowie von Trockenobst ein. Im Pflanzenschutz verwendet man schwefelhaltige Präparate gegen den an der Blattoberfläche wachsenden Mehltaupilz, der ein Schädling für Reben ist. Schwefel oxidiert dort langsam zu Schwefel-IV-oxid und hemmt so die Keimung der Pilzsporen.
Katastrophale Auswirkungen hatte das in den Abgasen alter, ohne Rauchgaswäsche arbeitenden Kohlekraftwerken der früheren DDR oder Tschechoslowakei enthaltene Schwefel-IV-oxid, da es durch Regen aus der Luft ausgewaschen wurde und in großem Umkreis die Oberfläche des Waldbodens derart ansäuerte, dass es zu einem Waldsterben großen Ausmaßes, vor allem im Böhmerwald und im Erzgebirge, kam.
Schwefel und seine Verbindungen werden als Schmierstoffadditive verwendet, z. B. in nicht wassermischbaren Kühlschmierstoffen bei Anwendung unter extremem Druck. Auch Eisensulfide (FeS, FeS2 oder Fe2S3) oder Molybdän-IV-sulfid (MoS2) werden als feste Schmierstoffe eingesetzt.
In wiederaufladbaren Akkumulatoren setzt man in japanischen Speicherkraftwerken flüssigen Schwefel als Kathode von Natrium-Schwefel-Zellen ein. Die Temperatur beträgt 300 bis 350 °C, flüssiges Natrium fungiert als Anode, und als fester Elektrolyt eine Keramik aus Natrium-β-aluminat (NaAl11O17).
-
(Weitere aktuelle Patente siehe https://worldwide.espacenet.com)
-
M. Yu und F. Zhou, Printing nanoporous ultrathin membranes for lithium-sulfur batteries (Rensselaer Polytech Institute, WO 2020112753 A1, veröffentlicht 4. Juni 2020)
-
J. R. Johnson und G. Biausque, Methods of producing 1,3-butadiene from ethylene and sulfur (Sabic Global Technologies BV, US 2020172451 A1, veröffentlicht 4. Juni 2020)
-
H. Nakatani und S. Nozato, Sulfur-carbon material composite body, positive electrode material for lithium sulfur secondary batteries, and lithium sulfur secondary battery (Sekusui Chemical Co., Ltd., US 2020176763 A1, veröffentlicht 4. Juni 2020)
-
N. H. You und B.-C. Ku, Sulfur-doped reduced graphene, manufacturing method therefor, and polyimide nanocomposite containing sulfur-doped reduced graphene oxide (Korea Institute for Science and Technology, WO 2020105926 A1, veröffentlicht 28. Mai 2020)
-
E. R. Fruchey und S. K. Berkhous, Low sulfur marine fuel compositions (Exxonmobil Research and Engineering Co., US 2020165535 A1, veröffentlicht 28. Mai 2020)
-
C. S. Ozkan und M. Ozkan, Sulfur silicon sell and methods (University of Calfornia, US 2020161648 A1, veröffentlicht 21. Mai 2020)
-
J. Sato und T. Tsuruta, Sulfur containing organo-silicon compound and resin composition (Kurakay Co., US 2020157390 A1, veröffentlicht 21. Mai 2020)
-
L. Ma und X. Ma, Novel battery systems based on lithium difluorophosphate (Tesla Motors Canada ULC, CA 3071314 A1, veröffentlicht 7. Februar 2019)
5.3 Selen
Der Name des Selens ist vom griechischen Wort σελήνη (selene, =Mond) abgeleitet. Das Element entdeckte Berzelius 1817 im Bleikammerschlamm einer Schwefelsäurefabrik. Zuerst hielt Berzelius die Substanz für Tellur [vom lateinischen Wort tellus (Erde) abgeleitet], jedoch ergaben weitere von ihm durchgeführte Versuche, dass er hier ein neues, wenngleich dem Tellur sehr ähnliches Element vorliegen hatte. Er nannte es daher Selen.
Selen und auch sein höheres Homologes Tellur kommen viel seltener in der Erdkruste vor als Schwefel. Elementares (gediegenes) Selen tritt in kleinen Mengen natürlich auf, gebunden ebenfalls selten in Selenmineralien wie Clausthalit (Bleiselenid, PbSe). Selen begleitet oft Sulfide des Kupfers, Bleis, Zinks und Eisens. Werden diese Erze geröstet, so konzentriert sich das bei Raumtemperatur feste Selen-IV-oxid in der Flugasche oder im darauf folgenden Prozess zur Herstellung von Schwefelsäure als Selenige Säure.
Selen ist essenzielles Spurenelement und Baustein der einundzwanzigsten biogenen Aminosäure Selenocystein. Somit ist es zur Gesunderhaltung des Menschen unverzichtbar, wirkt aber in größeren Mengen stark giftig. Den höchsten Gehalt an Selen unter den Nahrungsmitteln hat die Paranuss [knapp 2 ppm (Universität Düsseldorf 2013)].

2000 t Selen wurden im Jahr 2011 weltweit produziert, meist in Deutschland (650 t), Japan (630 t), Belgien (200 t) und Russland (140 t). Die auf der Erde verfügbare Reserve schätzt man auf insgesamt 93.000 t. In diesen Zahlen sind aber nicht zwei der größten Produktionsländer enthalten, China und die USA. Der Preis lag zwischen 2004 und 2010 ziemlich stabil bei US$ 30.-/lb. (= ca. € 66.-/kg), verdoppelte sich dann aber bis 2011, nicht zuletzt weil China der größte Verbraucher von Selen mit 1500–2000 t/a ist. 2010 gingen ca. 30 % des erzeugten Selens in die Metallindustrie, weitere 30 % in die Glasherstellung und je 10 % in die Landwirtschaft, die Elektronikindustrie (Halbleiter) und die Chemieindustrie (Pigmente). 2019 sackte der Preis zwischenzeitlich auf etwa US$ 20.-/kg ab, um 2022 wieder deutlich anzusteigen.
Selen tritt in verschiedenen Modifikationen auf (Brauer 1963, S. 415–418). Rotes Selen ist wie Schwefel nichtmetallisch, löst sich in Kohlenstoffdisulfid und ist ein elektrischer Nichtleiter. Seine Moleküle enthalten zu etwa einem Drittel Se8-Ringe und zu zwei Dritteln längere Ketten oder Ringe von Selenatomen. Rotes Selen geht beim Erhitzen auf Temperaturen oberhalb von 80 °C in graues, halbmetallisches Selen über.
Schwarzes amorphes Selen wandelt sich beim Erwärmen auf 60 °C zunächst in das schwarze, glasartige Selen um; weiteres Erhitzen auf Temperaturen >80 °C ergibt auch hier die Bildung grauen Selens.
Diese nichtmetallische schwarze Modifikation erhält man durch Reduktion von Selenverbindungen in der Hitze; es gibt eine glasartige und eine amorphe Modifikation. In der glasartigen Form bilden die Selenatome größere Ringe und Ketten, die oberhalb einer Temperatur von 50 °C elastisch sind; bei weiterer Erhöhung der Temperatur wird Selen zunehmend plastisch.

Von links nach rechts: Schwarzes, graues und rotes Selen (Tomihahndorf 2006)
Vorkommen, physikalische und chemische Eigenschaften von Selen
|
Symbol: |
Se |
|
|
|
Ordnungszahl: |
34 | ||
|
CAS-Nr.: |
7782-49-2 | ||
|
Aussehen: |
Rotes Pulver Grau, metallisch glänzend Schwarzer, amorpher Feststoff |
Graues und rotes Selen, W. Oelen 2011) |
Selen, Pulver (Sicius 2015) |
|
Entdecker, Jahr |
Berzelius (Schweden), 1817 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
7434Se (0,87) |
Stabil |
---- | |
|
7634Se (9,36) |
Stabil |
---- | |
|
7734Se (7,63) |
Stabil |
---- | |
|
7834Se (23,78) |
Stabil |
---- | |
|
8034Se (49,61) |
Stabil |
---- | |
|
Massenanteil in der Erdhülle (ppm): |
0,8 | ||
|
Atommasse (u): |
78,971 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
2,48 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial für: Se + 2 e− > Se2‐(V) |
−0,92 | ||
|
Atomradius (pm): |
115 | ||
|
Van der Waals-Radius (berechnet, pm): |
190 | ||
|
Kovalenter Radius (pm): |
120 | ||
|
Elektronenkonfiguration: |
[Ar] 3d104s24p4 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
941 ♦ 2045 ♦ 2974 | ||
|
Magnetische Volumensuszeptibilität: |
1,9 · 10−5 | ||
|
Magnetismus: |
Diamagnetisch | ||
|
Kristallsystem: |
Hexagonal | ||
|
Elektrische Leitfähigkeit([A/(V · m)], bei 300 K): |
10−10 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
10 ♦ 8,3 ♦ 3,7 | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
29 ♦ 736 | ||
|
Schallgeschwindigkeit (m/s, bei 293,15 K): |
3350 | ||
|
Dichte (g/cm3, bei 273,15 K) |
4,48 (rotes, 25 °C) 4,82 (graues, 25 °C) 4,28 (schwarzes, 60 °C) | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
16,42 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit ([W/(m · K)]): |
0,52 | ||
|
Spezifische Wärme ([J/(mol · K)]): |
25,36 | ||
|
Schmelzpunkt (°C ♦ K): |
221 ♦ 494,15 | ||
|
Schmelzwärme (kJ/mol): |
5,4 (grau: 6,7) | ||
|
Siedepunkt (°C ♦ K): |
685 ♦ 958,15 | ||
|
Verdampfungswärme (kJ/mol): |
95,5 | ||
|
Kritischer Punkt (°C ■ MPa): |
1493 ■ 27,2 | ||


Wird graues Selen belichtet, so verändert sich seine elektrische Leitfähigkeit. Diese wird durch Leitung von Löchern (positiv geladenen Fehlstellen von Elektronen) verursacht. Den Mechanismus hierfür interpretiert man als „Hopping“, also einem Springen der Löcher von einer Kristallfehlstelle zur nächsten (Mott 1969).
Beim Erhitzen in Luft verbrennt Selen mit blauer Flamme zu Selen-IV-oxid, SeO2. Oberhalb von 400 °C setzt es sich mit Wasserstoff zum Selenwasserstoff, H2Se, um. Mit Metallen bildet es Selenide, z. B. Natriumselenid, Na2Se, oder Eisen-II-selenid, FeSe.
Das chemische Verhalten des Selens ist dem des Schwefels ähnlich, nur ist Selen schwerer zu oxidieren. So ergibt beispielsweise die Umsetzung mit Salpetersäure Selenige Säure (H2SeO3) und nicht Selensäure (H2SeO4).
Wasserstoffverbindungen Selenwasserstoff (Monoselan, H2Se) entsteht beim Lösen salzartiger Selenide in verdünnten Säuren. Das farblose Gas ist sehr giftig und besitzt einen Geruch nach faulem Rettich. Schon kurzes Einatmen kleiner Mengen führt zu lange anhaltenden Reizungen der Schleimhäute („Selenschnupfen“). Bei Laborversuchen muss daher unbedingt unter einem Abzug gearbeitet werden. Man setzt Selenwasserstoff in der Elektronikindustrie zur Dotierung von Halbleitern mit Selen ein.
Das Molekül der Verbindung ist gewinkelt (HSeH: 91°), die Länge der Se–H-Bindung beträgt 146 pm (Holleman et al. 2007, S. 627). Selen- wirkt stärker reduzierend als Schwefelwasserstoff. Wässrige Lösungen von Selenwasserstoff werden durch den in der Luft enthaltenen Sauerstoff schnell oxidiert (Sennikov et al. 1996), wobei rotes Selen ausfällt. Reiner Selenwasserstoff entsteht beispielsweise beim Lösen von Aluminiumselenid in Wasser (Brauer 1963, S. 418).

Selen-IV-oxid (Onyxmet 2020)
Selen-IV-oxid ist hygroskopisch und löst sich gut in Wasser unter Bildung von Seleniger Säure (H2SeO3), einer mittelstarken Säure, deren Salze man Selenite bzw. Oxoselenate-IV nennt. Umgekehrt kann man Selen-IV-oxid durch Entwässern von seleniger Säure darstellen (Brauer 1963, S. 428). Da die Oxidationsstufe +4 des Selens die beständigste des Elements ist, wirkt Selen-IV-oxid deutlich schwächer reduzierend als Schwefel-IV-oxid. Man setzt Selen-IV-oxid bei der Riley-Synthese als mildes Oxidationsmittel ein. Shampoos zur gleichzeitigen Therapie starker Bildung von Schuppen können ebenfalls Selen-IV-oxid enthalten. Jedoch ist die Verbindung wie fast alle des Selens stark giftig.
- (I)

- (II)


Selenige Säure (Onyxmet 2020)
Es gibt einige, aber sehr unterschiedliche Einsatzgebiete für Selenige Säure. Man nutzt sie als Katalysator zur Synthese von 1,2-Dialdehyden (Ronzio und Waugh 1944), zur Färbung von Stahloberflächen (Angier 1936) und als Reagenz in Testkits zum Nachweis des menschlichen Drogenkonsums. Selenige Säure ist wie nahezu alle Selenverbindungen für Säugetiere und aquatische Organismen stark toxisch und kann auch zu Langzeitschäden führen.




Selensäure (Onyxmet 2020)
Selensäure verätzt Haut und Schleimhäute. Darüber hinaus ist Selensäure giftig.

Selen-IV-sulfid (Onyxmet 2020)
Selensulfide wirken fungizid und schuppenlösend; sie sind daher in Anti-Schuppen-Shampoos oft in einer Konzentration von 1 % enthalten. In höherer Konzentration (2,5 %) sind Selensulfide in apothekenpflichtigen Pasten und Suspensionen zur Behandlung des seborrhoischen Ekzems enthalten. Höhere Konzentrationen sind verschreibungspflichtig. Da Selensulfide nicht resorbiert werden, besteht bei korrekter Anwendung der Präparate keine Vergiftungsgefahr.
Halogenverbindungen Selen-VI-fluorid (Selenhexafluorid, SeF6) ist ein farbloses, giftiges Gas (Kimmerle 1960) der Dichte 8,69 kg/m3, das bei einer Temperatur von −34,7 °C kondensiert und einen abstoßenden Geruch aufweist. Es ist durch Leiten von Fluor über Selen darstellbar. Der Sublimationspunkt der festen Verbindung liegt bei Normaldruck bei −46,6 °C. Im Molekül des Selen-IV-fluorids sind die Fluoratome oktaedrisch um das Atom des Selens angeordnet. Die Verbindung ist etwas reaktionsfähiger als das nahezu inerte Schwefel-VI-fluorid. Es wird durch Wasser nur sehr langsam hydrolysiert, dabei bilden sich Fluorwasserstoff und Selensäure. Man setzt das Gas als elektrischen Isolator beispielsweise in Elektroden von Bogenlampen ein (Holleman et al. 2007, S. 628).
Auch Selen-IV-fluorid (Selentetrafluorid, SeF4) erhält man durch Überleiten von Fluor über Selen bei Einsatz stöchiometrischer Mengen beider Elemente. Ebenso entsteht die Verbindung durch Umsetzung von Selen-IV-oxid mit Schwefel-IV-fluorid, Bor-III-fluorid oder auch Fluorwasserstoff (Holleman et al. 1995, S. 620). Die farblose Flüssigkeit der Dichte 2,72 g/cm3 erstarrt bei −9,5 °C und siedet bei 106 °C. Mit Wasser erfolgt heftige Hydrolyse, und Glas wird von Selen-IV-fluorid angegriffen (Perry 2011, S. 360). Alkalifluoride lösen sich in Selen-IV-fluorid unter Bildung von Pentafluoroseleniten auf (Brauer 1975, S. 195). Mischbar ist Selen-IV-fluorid mit Iod-V-fluorid, Schwefelsäure, Ethanol und Diethylether.

Selen-IV-chlorid (Onyxmet 2020)
Das bei Raumtemperatur unbeständige Selen-II-chlorid (SeCl2) entsteht durch Umsetzung grauen Selens mit Sulfurylchlorid. Die Verbindung löst sich mit rotbrauner Farbe in Ether und Tetrahydrofuran (Maaninen et al. 1999). Nur Addukte mit einigen organischen Thioverbindungen sind halbwegs charakterisiert (Jolleys et al. 2013). In Lösung disproportioniert Selen-II-chlorid zu Selen-IV-chlorid und Diselendichlorid (Se2Cl2).


Die Verbindung löst sich in Chloroform, Tetrachlorkohlenstoff und Kohlenstoffdisulfid, hat einen stechenden Geruch und kristallisiert im festen Zustand monoklin (Raumgruppe 14) (Blachnik et al. 1998, S. 720).
Selenoxiddichlorid (SeOCl2) erhält man aus Selen-IV-oxid und Chlorwasserstoff in stark schwefelsaurer Lösung. Alternativ kann Selen-IV-oxid auch mit Thionylchlorid bei 50 °C zu Selenoxiddichlorid umgesetzt werden (Brauer 1975, S. 423). Die gelbe Flüssigkeit erstarrt bzw. siedet bei 8,5 °C bzw. 176 °C, hat die Dichte 2,42 g/cm3 und reagiert empfindlich gegenüber Wasser und bereits feuchter Luft, an der sie unter Freisetzung von Chlorwasserstoff stark raucht. Die Hydrolyse ergibt Chlorwasserstoff und Selenige Säure. Die Verbindung ist unbegrenzt mischbar mit Trichlormethan, Tetrachlorkohlenstoff, Kohlenstoffdisulfid, Benzol und Toluol. In wenigen Fällen nutzt man Selenoxiddichlorid als Lösungsmittel.

Selen-IV-bromid (Onyxmet 2020)

Die dunkelrote, ölige Flüssigkeit der Dichte 3,6 g/cm3 erstarrt bei −49 °C, siedet unter teilweiser Zersetzung bei 225 °C, ist empfindlich gegenüber Hydrolyse und besitzt einen unangenehmen Geruch. Beim Stehen an der Luft spaltet sie schnell Brom ab; außerdem bildet sich in ihr ein Niederschlag von Selen. In Wasser sinkt die Verbindung zuerst in Form öliger Tropfen zu Boden und zersetzt sich dann langsam zu Selen, Selen-IV-oxid und Bromwasserstoff (Lamoureux und Milne 1989). Im festen Zustand ist die orthorhombische α-Form am stabilsten Raumgruppe 41 (Kniep et al. 1983; Katryniok et al. 1980); es existiert aber bei −80 °C auch eine metastabile β-Form mit monokliner Kristallstruktur (Goerdeler et al. 2014).
Pnictogenverbindungen Tetraselentetranitrid (Se4N4) ist wie das Schwefelanalogon (S4N4) ein oranger Feststoff, ist noch zersetzlicher als jenes und explosiv. Man kann es etwa aus Selen-IV-chlorid und Bis(trimethylsilylamino)selen darstellen (Silvari et al. 1933). Meist erhält man dabei die α-Form, die in Form einer wenig symmetrischen Käfigverbindung vorliegt. Eine hierzu etwas abgewandelte Synthese geht von Selen-IV-oxid und N-Trimethylsilyl-P-trimethylphosphanoimin aus (Dehnicke et al. 1994). Im letzteren Fall erhält man rotbraune Kristallnadeln der β-Form nach dem Umkristallisieren aus Acetonitril.
Umsetzung von Selen mit Grignard-Verbindungen (R-Mg-Hal) ergibt nach Hydrolyse der zunächst entstehenden Grignard-Addukte (R-Se-Mg-Hal) zu Selenolen (R-Se-H), die die Selenanaloga zu Alkanolen und Mercaptanen sind (Rheinboldt 1955). Die niederen Selenole haben einen widerwärtigen Geruch und sind giftig.
Selen ist für nahezu alle Lebensformen essenziell. Daher gibt man Selenverbindungen Nahrungsergänzungsmitteln zu. Nichtmetallisches Selen setzt man zum Rotfärben von Glas und -da Rot die Komplementärfarbe zu Grün ist- zum Entfärben grünen Glases ein.
In Belichtungstrommeln für Fotokopierer und Laserdrucker findet es ebenso Verwendung wie in Tonern für die Schwarzweißphotographie, um den Kontrast zu erhöhen, und in analogen Belichtungsmessern für die Fotografie. Mit Hilfe von Zinkselenid (ZnSe) ist die Herstellung stark reflektierender Oberflächen möglich; Licht lässt Selen nur im infraroten Wellenlängenbereich durch, was es zur Produktion von Fokuslinsen von Kohlendioxidlasern geeignet macht.
Kupferlegierungen beigefügt, erleichtert es deren mechanische Bearbeitbarkeit. Selen-IV-oxid setzt man auch zur Oberflächenbehandlung von Aluminium und Messing ein, um einen dunkleren Farbton zu erzielen. Bei der elektrolytischen Gewinnung von Mangan werden relativ große Mengen an Selen-IV-oxid zugegeben (2 kg SeO2 pro t erzeugtes Mangan), da so der Energieverbrauch bei der Elektrolyse gesenkt werden kann.
Bei der Herstellung von Halbleitern wird Selen ebenfalls eingesetzt, früher auch oft im Selen-Gleichrichter und in der Selenzelle, wobei es in letzteren Anwendungen aber schon meist durch Silicium ersetzt wurde. Zusammen mit Kupfer und Indium ist es in der lichtaktiven Beschichtung von CIGS-Solarzellen enthalten.
Selendisulfid ist ein hochwirksamer Inhaltsstoff von Anti-Schuppen-Haarshampoos.
Selen bindet im Körper Schwermetalle als Selenide; seine Verbindungen haben darüber hinaus starke antioxidative Wirkung. Daher werden die Körperzellen wirksam vor freien Radikalen geschützt. Daher ist Selen essenziell für Menschen und Tiere. In Futtermitteln für Nutztiere ist es daher oft als Natriumselenit und -selenat enthalten.
Viele Menschen leiden unter Selenmangel. Dabei gibt es viele Lebensmittel, die gute Selenlieferanten sind, wie z. B. Paranüsse, Kokosnüsse und ihre Produkte, Hirse, Sonnenblumenkerne, Vollkorngetreide, Hülsenfrüchte, Knoblauch, Steinpilze, Eigelb, Sesam, Rind- und Hühnerfleisch und viele Fischarten (Russell 2003; Ekmekcioglu 2000).
Ausgesprochene Zeichen einer Mangelversorgung kommen nur in Ländern mit generell starker Unterversorgung vor. Dazu gehören Erkrankungen des Herzmuskels in jugendlichem Alter (Keshan-Krankheit/China), Degeneration der Gelenkknorpel (Kaschin-Beck-Krankheit/Nordostsibirien, Mongolei und Nordchina), Schädigung des Nervensystems (Epidermische Neuropathie, Kuba) und Erkrankung und Rückbildung von Muskeln (Rees et al. 2013; Behne et al. 1990; Arthur et al. 1993; Schweizer et al. 2004).
Als Zusatzstoff für Nahrungs- und Futtermittel setzt man Selenomethionin ein, das durch Hefen (Saccharomyces cerevisiae (Sel-Plex®, Lalmin®) in einem aus Melasse und Natriumselenit bestehenden Substrat erzeugt wird.
In höherer Konzentration wirkt Selen aber schnell toxisch; die Spanne der „verträglichen“ Konzentration ist sehr schmal. Zudem ist die Toxizität von Selen abhängig von der chemischen Bindungsform. Selen ist Bestandteil der Aminosäure Selenocystein, die ein wichtiger Baustein des Enzyms Glutathionperoxidase ist. Jenes bewirkt den Abbau oxidativer Zellgifte wie Wasserstoffperoxid zu Wasser. Generell dienen Selenverbindungen wegen deren Reaktivität mit Sauerstoff und anderen aggressiven freien Radikalen als deren Fänger.
Die meisten selenhaltigen Eiweiße (Selenoproteine) enthalten das als 21. Aminosäure klassifizierte Selenocystein und kommen in dieser Form nur in tierischen Organismen, Bakterien und Archaeen vor. Der Gehalt des Selens im Erdboden entscheidet, ob Selen in Pflanzen -meist unspezifisch- angereichert wird.
Die Wirkung des Selens auf den menschlichen Körper ist bereits lange Gegenstand zahlreicher Untersuchungen (Stranges et al. 2007; Bleys et al. 2007; BioMed Central 2010). Der hemmende Einfluss von Selen auf die Entwicklung mancher Krankheiten wie Diabetes mellitus ist nicht eindeutig belegt, wie sich durch Vergleich der jeweiligen Studien ergibt (Bleys et al. 2007; BioMed Central 2010).
-
(Weitere aktuelle Patente siehe https://worldwide.espacenet.com)
-
T. Chen und Z. Lin, DNA immunoadsorbent using nano-selenium as adsorbent carrier, preparation method therefor, and application thereof (University of Jinan, WO 2020093989 A1, veröffentlicht 14. Mai 2020)
-
M. Cai und R. Gu, PEA oligopeptide selenium, preparation method therefor, and application thereof (China National Research Institute of Food and Fermentation Industries Co., Ltd., WO 2020073486 A1, veröffentlicht 16. April 2020)
-
K. Wiktorska und L. Mielczarek, Conjugates of selenium and isothiocyanate nanoparticles for use in the treatment of tumors (Narodowy Instytut Lekow w Warszawie; Uniwersytet Warszawski, WO 2020046153 A1, veröffentlicht 5. März 2020)
-
M.-L. Tan und S.-C. Li, Method for recycling copper-indium-gallium-selenium waste (Hanergy New Material Technology Co., Ltd., TW 202000931 A, veröffentlicht 1. Januar 2020)
-
P. L. Figueroa und K. Juárez López, Klebsiella pneumoniae strain for the biotransformation of Chrome, iron, selenium, and uranium metals (Universidad Nacional Autónoma de México, MX 2018005790 A; veröffentlicht 11. November 2019)
5.4 Tellur
Tellur wurde 1782 von dem österreichischen Chemiker und Mineralogen Franz Joseph Müller von Reichenstein bei Untersuchungen von Gold-Erzen aus der Grube Mariahilf beim heute rumänischen Sibiu (dt. Hermannstadt) entdeckt. Er fand, dass das neu erhaltene Metall im Unterschied zu Antimon und Bismut kaum eine Reaktion mit Schwefelsäure zeigte. Wie wir heute wissen, enthielt das damals erstmals dargestellte Tellur noch weitere Tellurmineralien. Er sandte 1783 zwar noch Proben zum schwedischen Chemiker Bergman; jener verstarb aber ein Jahr später, ohne zuvor die Ergebnisse seiner Untersuchungen zurückgeschickt zu haben, worauf Müller von Reichenstein die Untersuchungen beendete.
1797 schickte jener weitere Proben des Minerals an den preußischen Chemiker Klaproth, der im Wesentlichen die Befunde Müller von Reichensteins bestätigte und jenem die Entdeckung des neuen Elements zuschrieb. Klaproth benannte dieses aber selbst mit dem Namen Tellur (lat. tellus: „Erde“).
Der österreichische Naturforscher Franz Joseph Müller von Reichenstein [* 1. Juni 1740 Hermannstadt, Siebenbürgen (oder 4. Oktober 1742 Poysdorf, Österreich); † 12. Oktober 1825 Wien] studierte in Wien Philosophie, Mineralogie und Bergbau und wurde 1768 zum niederungarischen Markscheider ernannt. 1770 wurde er Bergwerksdirektor im Banat, ging 1775 nach Schwaz (Tirol) zur dortigen Mine als deren Direktor und kehrte schließlich 1778 als Thesaurariatsrat nach Siebenbürgen zurück. 1788 erhob ihn Kaiser Joseph in den erbländischen Ritterstand. Müller von Reichenstein wurde 1798 Wirklicher Hofrat in Siebenbürgen und 1802 zur Hofstelle nach Wien berufen. Er erhielt er das Ritterkreuz des St. Stephan-Ordens und 1820 seine Erhebung in den Freiherrnstand. Unter anderem entdeckte er 1782 das Element Tellur, nahezu gleichzeitig mit Paul Kitaibel (Benda 1975; von Gümbel 1885; Szabadváry 1997; Wurzbach 1868).
Tellur ist ein sehr seltenes Element; sein Anteil an der Erdhülle beträgt nur etwa 0,01 ppm (g/t). Vereinzelt kommt es mit Gold, gelegentlich auch mit Silber, Kupfer, Blei, Wismut und den Platinmetallen vergesellschaftet in elementarer Form (gediegen) in der Natur vor.
Gediegenes Tellur kann Selen enthalten. Trotz seiner Seltenheit bildet Tellur viele eigene Minerale und tritt wegen der Größe seines Atoms nicht als Ersatz für Schwefel- oder Selenatome in deren Mineralien auf; letztere findet man aber öfters als Ersatz für Telluratome in Tellurmineralien.
Mineralien mit Tellurid- (Te2−) bzw. Ditellurid- (Te22−) Anionen sind Goldtellurid (!), daneben kommen Edelmetall-, Blei- und Bismuttelluride vor. Dagegen sind Mineralien mit Te4+-Ionen seltener, wie bereits das wichtigste Oxid des Tellurs, das Tellur-IV-oxid (TeO2) in seinen zwei Modifikationen Tellurit (orthorhombisch) und Paratellurit (tetragonal). Weitere Mineralien, die Tellur in der Oxidationszahl +4 enthalten, sind Tellurite der Formeln [TeO3]2− oder [TeO4]4−.
Von Mineralen mit Tellur in der Oxidationsstufe +6 gibt es rund zwanzig, meist vom Kupfer oder Blei, die [TeO6]6−-Anionen enthalten. Es existieren auch gemischte Tellurminerale, darunter das Calciumorthotellurat (Carlfriesit, CaTe3O8) mit einem Te4+ : Te6+-Verhältnis von 2:1 (Williams und Gaines 1975; Effenberger et al. 1978).
- a.
Telluride: Hessit (Ag2Te) und Altait (PbTe),
- b.
Ditelluride: Calaverit (AuTe2) und Sylvanit [(Au, Ag) Te2]
- c.
Telluridsulfide: Nagyágit [AuPb(Pb, Sb,Bi) Te2–3S6] und Tetradymit (Bi2Te2S)
- d.
Tellurite [Tellurit, TeO2 und Zemannit [Mg6,5ZnFe[TeO3]3 ⋅ 4,5 H2O]


Das bei der Reaktion jeweils gebildete Natriumtellurit (Na2TeO3) löst man dann in Wasser; durch Zugabe von Schwefelsäure fällt man das sehr schwer in Wasser lösliche Tellur-IV-oxid aus. Dieses wird durch Umsetzung mit Schwefel-IV-oxid zu elementarem, amorphen Tellur reduziert. Zur Gewinnung hochreinen Tellurs wendet man das Zonenschmelzverfahren an.
Vor einigen Jahren wurden durchschnittlich etwa 120 t Tellur jährlich hergestellt. Die wichtigen Produzentenländer sind mit jeweils rund 50 t/a die USA und Japan, sowie mit ca. jeweils 13 t/a Peru und Kanada. Möglicherweise wird Tellur auch noch in einigen Ländern Westeuropas erzeugt; dafür liegen aber keine Zahlen vor (British Geological Survey 2011).
- a.
Te-II, monoklin, im Druckbereich von 4 bis 6,6 GPa.
- b.
Te-III orthorhombisch, im Druckbereich von 6,6 bis 10,6 Pa stabil.
- c.
Te-IV, trigonal, im Druckbereich von 10,6 bis 27 GPa, und
- d.
Te-V, kubisch-raumzentriert, im Druckbereich oberhalb von 27 GPa.

Vorkommen, physikalische und chemische Eigenschaften von Tellur
|
Symbol: |
Te |
|
|
|
Ordnungszahl: |
52 | ||
|
CAS-Nr.: |
13494-80-9 | ||
|
Aussehen: |
Silberweiß, metallisch glänzend |
Tellur, Scheibe ø 3,5 cm (N. N.) |
Tellur, Pulver (Sicius 2015) |
|
Entdecker, Jahr |
Von Müller-Reichenstein (Österreich), 1783a, b Klaproth (Preußen), 1797 | ||
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit (a) |
Zerfallsart, -produkt | |
|
12252Te (2,60) |
Stabil |
---- | |
|
12452Te (4,82) |
Stabil |
---- | |
|
12552Te (7,14) |
Stabil |
---- | |
|
12652Te (18,95) |
Stabil |
---- | |
|
12852Te (31,69) |
7,2 ⋅ 1024 |
β−β−>12854Xe | |
|
13052Te (33,80) |
7,9 ⋅ 1020 |
β−β−>13054Xe | |
|
Massenanteil in der Erdhülle (ppm): |
0,01 | ||
|
Atommasse (u): |
127,60 | ||
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
2,1 ♦ K. A. ♦ K. A. | ||
|
Normalpotenzial für: Te + 2 e− > Te2−(V) |
−1,14 | ||
|
Atomradius (pm): |
140 | ||
|
Van der Waals-Radius (berechnet, pm): |
206 | ||
|
Kovalenter Radius (pm): |
138 | ||
|
Elektronenkonfiguration: |
[Kr] 4d105s25p4 | ||
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
869 ♦ 1790 ♦ 2698 | ||
|
Magnetische Volumensuszeptibilität: |
−2,4 ⋅ 10−5 | ||
|
Magnetismus: |
Diamagnetisch | ||
|
Kristallsystem: |
Trigonal | ||
|
Elektrische Leitfähigkeit([A/(V · m)], bei 300 K): |
10.000 | ||
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
43 ♦ 65 ♦ 16 | ||
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
57 ♦ 180–270 | ||
|
Schallgeschwindigkeit (m/s, bei 293,15 K): |
2610 | ||
|
Dichte (g/cm3, bei 273,15 K) |
6,24 | ||
|
Molares Volumen (m3/mol, im festen Zustand): |
20,46 ⋅ 10−6 | ||
|
Wärmeleitfähigkeit ([W/(m · K)]): |
3,0 | ||
|
Spezifische Wärme ([J/(mol · K)]): |
25,73 | ||
|
Schmelzpunkt (°C ♦ K): |
449,51 ♦ 722,66 | ||
|
Schmelzwärme (kJ/mol): |
17,5 | ||
|
Siedepunkt (°C ♦ K): |
990 ♦ 1263 | ||
|
Verdampfungswärme (kJ/mol): |
114 | ||


Chemische Eigenschaften Tellur wird durch Wasser nicht angegriffen, ebenso kaum durch Salz- und Schwefelsäure und in Laugen. Salpetersäure löst es dagegen oxidativ unter Bildung von Tellurit auf. Geschmolzenes Tellur seinerseits greift Kupfer, Stahl und auch Edelstahl an.
An Luft verbrennt es mit blaugrüner Flamme zu Tellur-IV-oxid (TeO2). Mit Halogenen reagiert es heftig zu Halogeniden; diese sind stabiler als die des Schwefels und Selens. So existieren auch thermodynamisch stabile Iodide, darunter Telluriodid (TeI). Zink und andere unedle Metalle reagieren mit pulverförmigem Tellur beim Erhitzen heftig zu den jeweiligen Telluriden.
Die häufigsten Oxidationsstufen des Tellurs sind −2 (Tellurid) und +4 (in den Tetrahalogeniden, in Tellur-IV-oxid und Telluriten).
Wasserstoffverbindungen Tellurwasserstoff (H2Te) entsteht bei der elektrolytischen Reduktion von Tellur an der Kathode in 50 %iger Schwefelsäure oder durch Lösen salzartiger Telluride in Säuren, wie etwa bei der Umsetzung von Aluminiumtellurid mit Salzsäure (Brauer 1963, S. 439). Das knoblauchartig riechende Gas der Dichte 5,76 kg/m3 kondensiert bei −1 °C (Erstarrungspunkt der Flüssigkeit: −46 °C), ist leicht in Wasser löslich und wird durch Luftsauerstoff schnell zersetzt. In Wasser bildet sich die mittelstarke, unbeständige Tellurwasserstoffsäure (pKs1: 2,64; pKs2: 8,80) (Holleman et al. 2007, S. 627).

Tellur-IV-oxid (Onyxmet 2020)

Der tetragonal kristallisierende, farblose Paratellurit (Raumgruppe 92) der Dichte 6,02 g/cm3 schmilzt bei 732,6 °C zu einer roten Flüssigkeit, die bei 1245 °C in ein gelbes Gas übergeht. Mit Wasser bildet Tellur-IV-oxid die fast amphotere Tellurige Säure (H2TeO3), die sich mit starken Säuren sogar zu Tellur-IV-Salzen umsetzt. Basen dagegen lösen es zu Hydrogentellurat-IV (HTeO3−) oder Tellurat-IV (TeO32−).
Aus Tellur-IV-oxid kann man Gläser eines sehr hohen Brechungsindex herstellen; ebenso akustooptische Modulatoren. Die Verbindung ist giftig.
Das gelbe, trigonal-rhomboedrisch kristallisierende Tellur-VI-oxid (TeO3) hat eine Dichte von 5,07 g/cm3 und schmilzt bei einer Temperatur von 400 °C unter Zersetzung zu Tellur-IV-oxid und Sauerstoff. Die Verbindung ist das Anhydrid der Ortho-Tellursäure (H6TeO6). Tellur-VI-oxid kommt in Form zweier Modifikationen vor, der gelborangen amorphen α-Form und der grauen, trigonal kristallisierenden, deutlich reaktionsträgeren β-Form (Raumgruppe 167; Kjekshus et al. 2000). Beide Modifikationen erhält man durch unterschiedlich verlaufende Entwässerung von Orthotellursäure. Dehydratisiert man jene bei ca. 310 °C, entsteht die α-Form. Die β-Form bildet sich beim Erhitzen von Orthotellursäure im geschlossenen Rohr bei 320 °C bei Gegenwart katalytischer Mengen konzentrierter Schwefelsäure. Tellur-VI-oxid ist weder in Wasser noch in Säuren und heißen Laugen löslich (Brauer 1963, S. 450).

Orthotellursäure (Onyxmet 2020)
Kristallstrukturen von Tellurhexafluorid (Ben Mills 2009)

Die geschmolzene Verbindung ist elektrisch leitfähig. Festes Tellur-IV-fluorid liegt als Polymer mit orthorhombischer Mikrostruktur vor (Edwards und Hewaidy 1968); es greift Glas, Kupfer, Nickel, Gold, Quecksilber an. Man verwendet die Verbindung als Fluorierungsmittel.

Tellur-IV-chlorid, idealisierte Struktur (links) und als Feststoff (Onyxmet 2020)
- (I)

- (II)


Der gelbe bis orange Feststoff der Dichte 4,31 g/cm3 schmilzt erst bei einer Temperatur von 380 °C; die Schmelze siedet bei 420 °C unter Zersetzung. Zusatz von Wasser bewirkt schnelle Hydrolyse zu Bromwasserstoff und Tellur-IV-oxid. Die Verbindung löst sich in Ether und reiner Essigsäure. Die Kristallstruktur des festen Tellur-IV-bromids entspricht der des Chloranalogons, also einem Kuban-Typ (Holleman et al. 1995, S. 632).


Tellur-IV-iodid (Onyxmet 2020)
Pnictogenverbindungen Der Versuch, binäre und gut charakterisierbare Tellurnitride darzustellen, scheiterte trotz vieler Anstrengungen bisher. Noch unbestätigt ist die Existenz des Tetratellurtetranitrids, das das Homologe der entsprechenden Selen- und Schwefelverbindung wäre. Allerdings ist schon lange bekannt (Strecker und Ebert 1925), dass Ammoniak mit Tellur-IV-chlorid reagiert, also in analoger Weise zum Selen-IV- oder Schwefel-IV-chlorid (Laitinen et al. 1998). Die Umsetzung von Tellur-IV-chlorid mit in Tetrahydrofuran (THF) gelöstem Tris(trimethylsilyl)amin ergibt zumindest ein mit Hilfe zusätzlicher Liganden stabilisiertes Tellurnitrid der Formel [Te6N8(TeCl2)4(THF)4] (Massa et al. 1998).
Organische Verbindungen Telluralkyle sind sehr instabil. Bekannt sind die reinen Tellurorganyle der Form R2Te, R2Te2, R4Te und R6Te (R jeweils Alkyl-, Aryl-). Außerdem kennt man noch Diorganotellurdihalogenide R2TeX2 (R = Alkyl-, Aryl-; X = F, Cl, Br, I) und Triorganotellurhalogenide R3TeX (R = Alkyl-, Aryl-; X = F, Cl, Br, I).



Tellur ist nur aufwändig herzustellen und daher teuer. Es wird daher meist durch andere Elemente bzw. Verbindungen ersetzt, wo dies möglich ist. Man verwendet es z. B. als Zusatz (<1 %) für Stahl, Gusseisen, Kupfer- und Blei-Legierungen sowie in rostfreien Edelstählen, weil so die Beständigkeit gegenüber Korrosion und die mechanische Bearbeitbarkeit verbessert werden. In seiner Funktion als Halbleiter findet es lediglich im Cadmiumtellurid Verwendung, das man in Fotodioden und Solarmodulen einbaut.
Bismuttellurid (Bi2Te3) setzt man z. B. in Radionuklidbatterien zur Stromerzeugung oder in Peltier-Elementen zur Kühlung ein. Bestandteile optischer Speicherplatten (CD-RW, DVD, Phase Change Random Access Memory) sind unter anderem auch Kombinationen aus Germanium- und Antimontelluriden. Gläser aus Tellur-IV-oxid besitzen einen besonders hohen Brechungsindex und werden anstelle von Quarz (SiO2) in Lichtwellenleitern eingesetzt.
Kleine Mengen an Tellur verwendet man zur Vulkanisierung von Gummi, in Sprengkapseln und zum Färben von Glas und Keramik. Selten, weil teuer, ist der Einsatz von Verbindungen des Tellurs zur Erzeugung einer grasgrünen Farbe bei Feuerwerken.
Tellur ist giftig und wird daher meist mit dem Gefahrensymbol „T“ gekennzeichnet. Tellur ist aber sehr schlecht in Wasser und körpereigenen Säuren löslich, weswegen das Institut für Arbeitsschutz der Deutschen Gesetzlichen Unfallversicherung Tellur auf gesundheitsschädlich (Xn) herabstufte. Viele Hersteller verwenden aber nach wie vor das alte Gefahrensymbol „T“ in Verbindung mit dem R-Satz 25 („Giftig beim Verschlucken“).
Tellur besitzt jedoch nicht die Giftigkeit des Selens. Wird Tellur vom menschlichen Körper aufgenommen, wird dort durch Reduktion Dimethyltellur (Me2Te) gebildet, das für Blut, Leber, Herz und Nieren giftig ist und stark nach Knoblauch riecht. Tellurpulver wird neben den Symbolen Xn bzw. T zusätzlich mit den R-Sätzen 36/37/38 sowie S-Sätzen 26 und 37 gekennzeichnet. Die Maximale Arbeitsplatz-Konzentration (MAK) ist auf den geringen Wert von 0,1 mg/m3 festgelegt worden.
-
M. Yamamoto, Method for producing organic tellurium compound and method of producing vinyl polymer (Otsuka Chemical Co., Ltd., WO 2020116144 A1, veröffentlicht 11. Juni 2020)
-
J. Bradley und A. Knights, Tellurium-oxide coated silicon nitride optical waveguides and methods of fabrication thereof (University of McMaster, CA 3025925 A1, veröffentlicht 30. Mai 2020)
-
P. Fantini und M. Bernasconi, Transition metal doped germanium-antimony-tellurium (GST) memory device components and composition (Micron Technology Inc., WO 2020041047 A1, veröffentlicht 27. Februar 2020)
-
G. Mestl und K. Wanninger, Syntheis of a MOVNBTE catalyst having a reduced niobium and tellurium content and higher activity for the oxidative dehydrogenation of ethane (Clariant Produkte Deutschland, US 2020061583 A1, veröffentlicht 27. Februar 2020)
-
F. Carroll und K. W. Hang, Thick film pastes containing lead-tellurium-lithium oxides, and their use in the manufacture of semiconductor devices (Dupont Electronics Inc., US 2020013910 A1, veröffentlicht 9. Januar 2020)
5.5 Polonium
Marie und Pierre Curie (Kurzbiografie siehe „Thorium“) entdeckten 1898 das später Polonium genannte Element (Curie und Curie 1898; Pfützner 1999; Adloff 2003; Kabzinska 1998) bei Untersuchungen der Pechblende. Diese war nach der Entfernung von Uran und Thorium erstaunlicherweise noch stärker radioaktiv als zuvor; also mussten in ihr noch weitere radioaktive Elemente enthalten sein. Neben Polonium isolierten die Curies einige Monate später auch noch Radium (Curie et al. 1898). Unabhängig davon und drei Jahre später isolierte der deutsche Chemiker Marckwald 3 mg Polonium und glaubte, es sei ein neues Element. Erst 1905 wurde nachgewiesen, dass es sich dabei ebenfalls um Polonium handelte (Neufeldt 2012).
Während des Zweiten Weltkriegs produzierten die Vereinigten Staaten unter dem Namen des Manhattan-Projekts Polonium, das im Zünder der Atombombe verwendet wurde. Die US-amerikanische Atomenergiebehörde und die Verantwortlichen des Manhattan-Projekts führten zwischen 1943 und 1947 auch Versuche an fünf lebenden Personen aus, weil man den Weg des Poloniums im menschlichen Stoffwechsel untersuchen wollte. Die Versuchspersonen erhielten täglich eine Dosis zwischen 400 und 800 kBq) an Polonium (Moss und Eckhardt 1995).
P. und M. Curie arbeiteten 1898 mit Pechblende und führten die starke radioaktive Strahlung einiger Fraktionen dieses Rohproduktes auf die Existenz eines bis dahin unbekannten Elements zurück (Neufeldt 2012), das sie, ohne es zuvor entdeckt oder isoliert zu haben, Polonium nannten. Vier Jahre später gelang W. Marckwald die erstmalige Darstellung kleinster Mengen des Elements (Marckwald 1907). Die in 1000 t Uranpechblende enthaltene Menge an Polonium schätzt man auf 0,07 bis 30 mg (Holleman et al. 2007, S. 617).
Isotope des Poloniums entstehen als Zwischenprodukte der Thorium- und Uranzerfallsreihen. Bei letztgenannter fällt das Isotop 21084Po an, so dass es aus Pechblende gewonnen werden kann. Bei der Aufarbeitung reichert es sich zunächst zusammen mit Bismut an, von dem es durch fraktionierte Fällung der Sulfide (PoS ist schwerer wasserlöslich als Bi2S3) getrennt werden kann.

Beide Metalle können durch Destillation separiert werden (Siedepunkt von Polonium: 962 °C; Siedepunkt von Bismut: 1564 °C) (U. S. Department of Energy 2015). Alternativ extrahiert man das Polonium aus geschmolzenem Wismut unter Luftausschluss und bei Temperaturen von 400 °C mit Hilfe geschmolzenen Natriumhydroxids (Siemens Jr et al. 1977). Die Jahresproduktion beträgt ca. 100 g.
Polonium ist ein silberweiß glänzendes Metall, das als einziges Metall kubisch-primitiv kristallisiert. Die chemischen Eigenschaften sind in mancher Hinsicht sehr ähnlich zu denen seiner linken Nachbarn im Periodensystem, Bismut, Blei und Thallium. Bezüglich seines Redoxpotenzials für die Reaktion M2+ + 2 e− > M ist es etwa mit Kupfer vergleichbar; bezüglich höherer Oxidationsstufen stehen seine jeweiligen Redoxpotenziale sogar in der Nähe derjenigen von Edelmetallen.
Polonium löst sich, begünstigt durch Luftzutritt, in starken Mineralsäuren unter Bildung des rosaroten Po2+-Kations, das aber in wässriger Lösung langsam in das gelbe Po4+-Ion übergeht. Die starke von Polonium ausgesandte α-Strahlung reagiert mit Wassermolekülen unter Bildung oxidierender Verbindungen bzw. Radikale, die für die Entstehung der Po4+-Ionen verantwortlich sind (Holleman et al. 2007, S. 620).
Vorkommen, physikalische und chemische Eigenschaften von Polonium
|
Symbol: |
Po |
|
|
Ordnungszahl: |
84 | |
|
CAS-Nr.: |
7440-08-6 | |
|
Aussehen: |
Silbrig glänzend |
Polonium auf Edelstahl (Lapp 1965) |
|
Entdecker, Jahr |
Curie, Marie (Polen) und Curie, Pierre (Frankreich), 1898 Marckwald (Deutsches Reich), 1902 | |
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit |
Zerfallsart, -produkt |
|
20884Po (synthetisch) |
2,898 a |
α>20482Pb, ε>20883Bi |
|
20984Po (synthetisch) |
103 a |
α>20582Pb, ε>20983Bi |
|
21084Po (99,998) |
138,376 d |
α>20682Pb |
|
21184Po (5 ⋅ 10−10) |
0,516 s |
α>20782Pb |
|
21284Po (2 ⋅ 10−12) |
304 ns |
α>20882Pb |
|
Massenanteil in der Erdhülle (ppm): |
2,1 ⋅ 10‐11 | |
|
Atommasse (u): |
209,98 | |
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
2,0 ♦ K. A. ♦ K. A. | |
|
Normalpotenzial für: Po2+ + 2 e− > Po (V) |
0,37 | |
|
Atomradius (pm): |
190 | |
|
Van der Waals-Radius (berechnet, pm): |
197 | |
|
Kovalenter Radius (pm): |
140 | |
|
Ionenradius (Po6+, pm) |
67 | |
|
Elektronenkonfiguration: |
[Xe] 4f145d106s26p4 | |
|
Ionisierungsenergie (kJ/mol), erste: |
812 | |
|
Magnetische Volumensuszeptibilität: |
Keine Angabe | |
|
Magnetismus: |
Diamagnetisch | |
|
Kristallsystem: |
Kubisch (α-Po), >309 K rhomboedrisch (β-Po) | |
|
Elektrische Leitfähigkeit([A/(V · m)], bei 300 K): |
2,5 ⋅ 106 | |
|
Elastizitäts- ♦ Kompressions- ♦ Schermodul (GPa): |
26 ♦ 26,3 ♦ k. A. (α-Po) | |
|
Vickers-Härte ♦ Brinell-Härte (MPa): |
Keine Angabe | |
|
Schallgeschwindigkeit (m/s, bei 293,15 K): |
Keine Angabe | |
|
Dichte (g/cm3, bei 273,15 K) |
9,20 | |
|
Molares Volumen (m3/mol, im festen Zustand): |
22,97 ⋅ 10−6 | |
|
Wärmeleitfähigkeit ([W/(m · K)]): |
20 | |
|
Spezifische Wärme ([J/(mol · K)]): |
26,4 | |
|
Schmelzpunkt (°C ♦ K): |
254 ♦ 527 | |
|
Schmelzwärme (kJ/mol): |
13 | |
|
Siedepunkt (°C ♦ K): |
962 ♦ 1235 | |
|
Verdampfungswärme (kJ/mol): |
102,9 | |

Wasserstoffverbindungen und Polonide Poloniumwasserstoff (Monopolan, H2Po) lässt sich nicht wie Schwefelwasserstoff aus den Elementen gewinnen. Stattdessen wird zur Herstellung elementares Polonium mit Magnesium in Gegenwart verdünnter Salzsäure reduziert. Dabei entstehen aber nur Spuren von Poloniumwasserstoff.
Poloniumwasserstoff ist wie die leichteren analogen Verbindungen Selenwasserstoff und Tellurwasserstoff eine endotherme Verbindung und zerfällt unter Wärmeabgabe in die Elemente, ist also sehr instabil. Die dabei abgegebene Wärmemenge ist mit >100 kJ/mol die größte aller Chalkogen-Wasserstoffverbindungen. Gegenüber auch in Spuren vorkommendem Sauerstoff ist die Verbindung sehr empfindlich und wird sofort zersetzt. Bei Raumtemperatur ist Poloniumwasserstoff eine Flüssigkeit, die bei 36,1 °C siedet und bei −35,3 °C erstarrt (Weigel 1959). Die Verbindung bildet, wie auch die anderen Chalkogen-Wasserstoff-Verbindungen, zwei Reihen von Salzen, die Polonide und Hydrogenpolonide. Von letzteren sind aber keine Vertreter bekannt. Bleipolonid kommt dagegen immer in Poloniumproben vor, da beim α-Zerfall von Polonium Blei entsteht (Holleman et al. 2007, S. 626).


Die Verbindung kristallisiert wie auch das homologe Natriumtellurid im Antifluorittyp, einem kubischen System der Raumgruppe Fm3m (Raumgruppe 225) (Emeléus und Sharpe 1963, S. 209).
Magnesiumpolonid (MgPo) ist ein grauer Feststoff der Dichte 6,7 g/cm3, der durch Zusammenschmelzen der Elemente im Temperaturbereich von 300 °C bis 400 °C dargestellt werden kann (Eméleus und Sharpe 1963, S. 210). Er kristallisiert im hexagonalen Kristallsystem.
Bleipolonid (PbPo) stellt man durch Einleiten von Poloniumdampf in flüssiges Blei im Vakuum dar (Hagen 2009, S. 161). Der graue Feststoff der Dichte 9,64 g/cm3 schmilzt oberhalb einer Temperatur von 550 °C unter Zersetzung (Miura et al. 2007) und kristallisiert analog zu Bleitellurid in der kubischen Natriumchlorid-Struktur (Raumgruppe 225) (Dalven 1973).
Chalkogenverbindungen Polonium-IV-oxid (PoO2) stellt man durch Verbrennen von Polonium im Sauerstoffstrom bei <250 °C her (Steinmeyer und Kershner 1971). Die Verbindung existiert in Form zweier Modifikationen. Die gelbe ist bei 20 °C stabil, kristallisiert in einer fluoritähnlichen Struktur (Raumgruppe 225) und schmilzt bei 500 °C. Die rote Modifikation kristallisiert tetragonal und ist nur bei erhöhter Temperatur beständig. Unter reduziertem Druck zersetzt sich die Verbindung oberhalb einer Temperatur von 500 °C in die Elemente.
Polonium-II-oxid (PoO) entsteht z. B. durch spontanen Zerfall von Polonium-II-sulfit (Emeléus und Sharpe 1963, S. 213). Zugabe von Alkalihydroxid zu Polonium-II-oxid gewinnt man Poloniumhydroxid, das im Alkalischen leicht zu Po4+ oxidiert.
Polonium-VI-oxid (PoO3) ist bisher nur in Form von Spuren nachgewiesen worden (Bagnall 1962, S. 197). Über eine eventuelle Darstellungsmöglichkeit besteht keine Klarheit. Eine Voraussage ist, dass man die Verbindung durch Erhitzen von Polonium-IV-oxid mit Chrom-VI-oxid in Luft erzeugen könne. Das Wissen über die chemischen Eigenschaften von Poloniumverbindungen ist auch deshalb bislang nicht sehr umfangreich, weil die meisten Versuche mit dem relativ leicht zugänglichen, aber stark radioaktiven Isotop 21084Po gemacht wurden. Eine Hoffnung zum besseren Nachweis des Po-VI liegt also darin, dass man entsprechende Versuche mit den längerlebigen Isotopen 20884Po oder 20984Po durchführen kann. Es gibt Annahmen, die besagen, dass Po-VI unter Umständen durch Komplexbildung stabilisiert wird, so etwa in den Anionen (PoF8)2‐ oder (PoO6)6− (Thayer 2005).
Polonium-II-sulfid (PoS) erhält man durch Fällung von in Säure gelöstem Polonium mit Schwefelwasserstoff; es ist ein schwarzer, schwer wasserlöslicher Feststoff.
Halogenverbindungen Polonium-VI-fluorid (PoF6) ist immer noch eine hypothetische Verbindung, da ihre Synthese aus Fluor und 21084Po schon 1945, jedoch erfolglos, versucht wurde. Möglicherweise war der 1960 durchgeführte, analoge Versuch der Synthese unter Einsatz des stabileren Isotops 20884Po erfolgreicher, da in ihr ein flüchtiges Poloniumfluorid erzeugt wurde (Seppelt 2015), aber auch hier konnte das entstandene Produkt nicht vollständig charakterisiert werden, da es durch die starke radioaktive Strahlung zerstört wurde (Weinstock und Chernick 1960).
Polonium-IV-chlorid (PoCl4) ist ein hellgelber, hygroskopischer Feststoff, der sich oberhalb einer Temperatur von 200 °C in Polonium-II-chlorid und Chlor zersetzt (Holleman et al. 2001, S. 594). Unter Chlorgas wird der Schmelzpunkt von 300 °C erreicht. Die Schmelze siedet bei 390 °C und ist dann scharlachrot; der dann gebildete Dampf ist purpurrot. Die Verbindung stellt man durch Umsetzung von Polonium-IV-oxid mit Chlorwasserstoff, Phosphor-V-chlorid oder Thionylchloriddampf her, ferner sind auch das Lösen metallischen Poloniums in konzentrierter Salzsäure bei Luftzutritt, das Verbrennen von Polonium im Chlorstrom oder das Erhitzen von Polonium-IV-oxid mit Tetrachlorkohlenstoff im Autoklaven bei ca. 200 °C möglich. Die letztgenannten zwei Darstellungsmethoden liefern das kristallwasserfreie Salz.

Kristallstruktur von Polonium-II-chlorid (Ben Mills 2009)
Die Darstellung erfolgt durch Chlorieren von Polonium oder durch Dehalogenierung von Polonium-IV-chlorid durch dessen Erhitzen auf 300 °C (Holleman et al. 2001, S. 594). Ebenfalls ist die Reduktion von Polonium-IV-chlorid durch Schwefel-IV-oxid, Schwefelwasserstoff oder Kohlenmonoxid möglich (Bagnall et al. 1955). Polonium-II-chlorid hat die Dichte 6,5 g/cm3, schmilzt bei 355 °C und löst sich in verdünnter Salzsäure unter Bildung einer rosafarbenen Lösung, die leicht zu Po-IV autoxidiert. Ebenso überführen Wasserstoffperoxid oder Chlorwasser Po-II schnell in Po-IV.
Polonium-II-bromid (PoBr2) ist bei Raumtemperatur ein purpurbrauner kristalliner Feststoff (Holleman et al. 2001, S. 594). Es zersetzt sich am Schmelzpunkt (270 °C), der nur unter Stickstoffatmosphäre erreicht wird. Die Verbindung erhält man durch thermische Entbromierung von Polonium-IV-bromid (PoBr4) im Vakuum bei 200 °C oder durch dessen Reduktion bei niedrigerer Temperatur mit Schwefelwasserstoff. In bromwasserstoffsaurer Lösung bildet die Verbindung purpurrote Lösungen. Ammoniak reduziert Polonium-II-bromid zum Metall (Bagnall 1962).
Polonium ist Bestandteil transportabler Neutronenquellen. Einige industriell genutzte Ionisatoren enthalten 21084Po, z. B. in Anlagen, die zum Aufrollen von Papier, Textilien ect. dienen, oder aber zum Aufheben elektrostatischer Ladungen auf optischen Linsen.
Ein g 21084Po entwickelt durch seinen radioaktiven Zerfall 140 W Wärme, die ausreicht, festes Polonium zum Schmelzen zu bringen (Petrjanow-Sokolow 1977). Früher setzte man Polonium in kurzlebigen Radionuklidbatterien ein. Die auf Hiroshima und Nagasaki abgeworfenen Atombomben enthielten aus Polonium und Beryllium bestehende Starter für die Kernspaltungsreaktion.
Polonium entsteht durch α-Zerfall von Radonkernen, die ihrerseits durch Abbau höherer radioaktiver Nuklide (Radium, Uran) im Erdreich gebildet werden. So können selbst die großvolumigen Radonatome durch Risse im Fundament oder durch Leitungen in Häuser eindringen, wo sich das Gas wegen seiner hohen Dichte immer am Boden konzentriert. Einatmen von auch nur spurenweise in der Luft enthaltenem Radon erhöht das Risiko, an Lungenkrebs zu erkranken; daher sollte man Kellerräume oft lüften. Man schätzt, dass das Einatmen von Radon nach Rauchen die zweithäufigste Ursache für die Entstehung von Lungenkarzinomen ist. Gründliche Abhilfe schafft nur ein komplettes Versiegeln des Untergrundes.
Die eigentliche Ursache für die Krebserkrankung ist aber nicht Radon, sondern die Inhalation der sich schnell aus Radon bildenden, ebenfalls kurzlebigen Isotope, die sich im Gegensatz zu Radon in der Lunge anreichern. Unter den Zerfallsprodukten befinden sich auch die Poloniumisotope 21084Po, 21284Po, 21484Po, 21684Po und 21884Po, die eine starke Wirkung auf Körpergewebe entfalten, weil sie α-Teilchen aussenden.
Die tödliche (letale) Dosis des Isotops 21084Po liegt bei 1,7 ng/kg (!) Körpergewicht, weshalb es eines der stärksten Gifte überhaupt ist. Die toxische Wirkung beruht dabei fast nur auf radiologischer, nicht auf chemischer Wirkung. Auch jene wäre aber sehr stark, da sich Polonium ähnlich wie seine linken Nachbarn im Periodensystem, Quecksilber, Thallium oder Blei, verhält und schwefelhaltige Enzyme/Aminosäuren unwirksam macht. Poloniumionen verteilen sich gleichmäßig im gesamten Körper und werden nur allmählich wieder ausgeschieden. Da Poloniumisotope bei ihrem Zerfall kaum γ-Strahlung erzeugen, sind diese im Körper schwer nachzuweisen.
5.6 Livermorium

Das Target von 24896Cm, das zur Synthese von Isotopen des Livermoriums benutzt wurde
- (I)

- (II)

Reproduktionsversuche einiger Labors im Folgejahr scheiterten allerdings, sogar das Team in Berkeley war dazu nicht in der Lage. Im Juni 2002 veröffentlichte der Leiter des Labors in Berkeley, dass die Versuche auf Basis der von Ninov im Jahr 2000 vorgegebenen Daten beruhten (Dalton 2002).
- (I)

- (II)

- (III)

Die Eigenschaften des am Ende gebildeten Isotops des Fleroviums waren mit denen desjenigen Fleroviumisotops identisch, das im Juni 1999 dargestellt wurde. Dieses hielt man erst für 288114Fl, was später zu 289114Fl berichtigt wurde (Oganessian et al. 2004). In einem zweiten Versuch, der im Frühjahr 2001 durchgeführt wurde, kam es zur Beobachtung zweier weiterer Atome des Livermoriums. Das als Zerfallsprodukt entstehende Isotop des Fleroviums war, wie später erhaltene Ergebnisse bestätigten, vermutlich 289m114Fl, was wiederum ein Isotop 293m116Lv als Mutterisotop bedingt hätte. Im Frühjahr 2005 wiederholte das Team in Dubna den Versuch erneut und erhielt dabei acht Atome des Livermoriums. Das dabei erzeugte Isotop wurde zu 293116Lv bestätigt. Ein weiterer Versuch wurde mit dem Curiumisotop 24596Cm durchgeführt, und dabei wurden folgerichtig die Isotope 291116Lv und 290116Lv erhalten (Barber et al. 2011).
Im Mai 2009 bestätigte eine Kooperation des LLNL in Berkeley und der deutschen GSI die Bildung der Fleroviumisotope 286114Fl bis 289114Fl, die unmittelbare Töchter der oben genannten Livermoriumisotope sind. Die im Jahr 2011 von der IUPAC vorgenommene Bewertung erkannte die zwischen 2004 und 2006 durchgeführten Experimente als Identifizierung des Livermoriums an (Hofmann et al. 2012).
Unabhängig voneinander bestätigten das GSI 2012 sowie das RIKEN 2014 und 2016 die Synthese des Livermoriums (Morita et al. 2014). Im vom RIKEN 2016 durchgeführten Versuch entdeckte man wahrscheinlich ein Atom des Isotops 294116Lv, das α-Zerfall zu 290114Fl und 286112Cn erlitt; letzteres konnte man nachweisen (Kaji et al. 2014). Die IUPAC empfahl 1979 zunächst den Namen Ununhexium als Platzhalter (Chatt 1979; Koppenol 2002), aber meist nannte man es „Element 116“ oder „116“. Den Namen Livermorium verlieh die IUPAC am 23. Mai 2012 (Loss und Corish 2015), wobei der Name das Lawrence Livermore National Laboratory anerkennt, das in der Stadt Livermore, CA gelegen ist.
Vorkommen, physikalische und chemische Eigenschaften von Livermorium
|
Symbol: |
Lv | |
|
Ordnungszahl: |
116 | |
|
CAS-Nr.: |
54100-71-9 | |
|
Aussehen: |
Unbekannt, wahrscheinlich metallisch | |
|
Entdecker, Jahr |
Vereinigtes Institut für Kernforschung (Russland) und Lawrence Livermore National Laboratory (USA), 2010 GSI Helmholtzzentrum für Schwerionenforschung (Deutschland), 2014 | |
|
Wichtige Isotope [natürliches Vorkommen (%)] |
Halbwertszeit |
Zerfallsart, -produkt |
|
292116Lv (synthetisch) |
18 ms |
α>288114Fl |
|
293116Lv (synthetisch) |
53 ms |
α>289114Fl |
|
Massenanteil in der Erdhülle (ppm): |
----- | |
|
Atommasse (u): |
(293) | |
|
Elektronegativität (Pauling ♦ Allred&Rochow ♦ Mulliken) |
Keine Angabe | |
|
Atomradius (pm): |
183* | |
|
Van der Waals-Radius (berechnet, pm): |
Keine Angabe | |
|
Kovalenter Radius (pm): |
162–166* | |
|
Elektronenkonfiguration: |
[Rn] 5f146d107s27p4 | |
|
Ionisierungsenergie (kJ/mol), erste ♦ zweite ♦ dritte: |
724* ♦ 1332* ♦ 2846 * | |
|
Magnetische Volumensuszeptibilität: |
Keine Angabe | |
|
Magnetismus: |
Diamagnetisch | |
|
Kristallsystem: |
Keine Angabe | |
|
Schallgeschwindigkeit (m/s, bei 273,15 K): |
Keine Angabe | |
|
Dichte (g/cm3, bei 273,15 K) |
12,9* | |
|
Molares Volumen (m3/mol, im festen Zustand): |
22,7 ⋅ 10−6∗ | |
|
Wärmeleitfähigkeit ([W/(m · K)]): |
Keine Angabe | |
|
Spezifische Wärme ([J/(mol · K)]): |
Keine Angabe | |
|
Schmelzpunkt (°C ♦ K): |
364–507 ♦ 637–780* | |
|
Schmelzwärme (kJ/mol): |
7,6* | |
|
Siedepunkt (°C ♦ K): |
762–862 ♦ 1035–1135* | |
|
Verdampfungswärme (kJ/mol): |
42* | |
Die Chemie des Livermoriums ähnelt vermutlich der des Poloniums. Die Stabilität höherer Oxidationszahlen nimmt in der Gruppe vom Schwefel über Selen, Tellur und Polonium stetig ab. Der beständigste Oxidationszustand des Livermorium ist wohl wegen der Anwesenheit zweier nahezu „inerter“ Elektronenpaare +2; die Stufe +4 wurde als wesentlich instabiler berechnet und sollte höchstens in Verbindung mit stark elektronegativen Elementen wie Fluor vorkommen, etwa im hypothetischen Livermorium-IV-fluorid (LvF4). Auch die bei den niederen Chalkogenen stets beobachtete Stufe −2 wird wegen der Destabilisierung der 7p3/2-Orbitale besonders unbeständig sein und daher kaum existieren, weshalb man beim Livermorium von einer praktisch ausschließlichen Kationenchemie ausgeht. Das Element sollte sich also fast durchgehend wie ein typisches Metall unter Bildung von Lv2+-Kationen verhalten (Fricke 1975; Thayer 2010).
So ist das hypothetische Livermoran (LvH2) wahrscheinlich mehr ein Hydrid als ein Wasserstofflivermorid, wäre aber eine immer noch mehrheitlich kovalente Verbindung (Nash und Crockett 2006). Die Lv-H-Bindung wäre wohl länger als üblicherweise zu erwarten und der H-Lv-H-Bindungswinkel größer als der in den anderen Chalkogenwasserstoffmolekülen, was laut Berechnungen darauf schließen lässt, dass sogar unbesetzte, relative „energiearme“ 8s-Orbitale an den Bindungen des vermutlich extrem instabilen Moleküls beteiligt sind. Dieses „supervalente Hybridisierung“ genannte Phänomen kommt aber auch in den nicht-relativistischen Regionen des Periodensystems vor (Greenwood und Earnshaw 1997b, S. 117). Bei den höheren Dihalogeniden des Livermoriums nimmt man eine lineare Struktur der Moleküle an, bei den leichteren eine gebogene (Van Wüllen und Langermann 2007).
Da der obengenannte Herstellprozess durch Beschießen eines Curium-Ziels mit schweren Calciumatomen immer noch der einzige Weg zur Herstellung des Elements ist, beruhen die in diesem Kapitel gemachten Aussagen auf gemessenen und vorhergesagten Eigenschaften weniger Atome, die bisher erzeugt werden konnten. Wegen der sehr kurzen Halbwertszeiten aller Isotope des Livermoriums wird die Sammlung weiterer Daten auch in Zukunft schwierig sein.