CHAPTER 9
ALZHEIMER’S DISEASE
Introduction
In 1905, German psychiatrist Alois Alzheimer performed an autopsy on a woman who had suffered an early cognitive decline. He discovered that about one-third of the woman’s cortical brain cells had deteriorated, leaving a plaque (an extracellular protein buildup) and tangled cell fragments. Similar findings had been reported earlier by other researchers, but Alzheimer provided a much more detailed and accurate description of the diseased brain.152 At the time, Alzheimer was working at the laboratory of Emil Kraepelin, the renowned discoverer of manic depression (now known as bipolar disorder) and the leading psychiatrist of his time. In 1910, Kraepelin published a Handbook of Psychiatry153 in which he referred to this form of dementia as Alzheimer’s disease in honor of his friend and colleague.
More than 100 years have passed, and Alzheimer’s disease (AD) is still poorly understood and generally regarded as incurable. AD is a progressive, fatal brain disease affecting about 4.5 million Americans. The first signs are reduced sense of smell and inability to retain short-term memories. As the relentless destruction of brain cells proceeds, the ability to function declines drastically. Aricept and other medications can enable the deteriorating brain to perform better for 4-12 months, but they fail to stop the rate of cell destruction. The average time between diagnosis and death is about seven years. Many AD patients attempt to disguise their symptoms in the early stages. In 2007, we evaluated an old friend who called me by name and carried on a brief conversation that didn’t betray his AD condition. His wife had reminded him of my name a few seconds before our meeting, but testing revealed that he thought Harry Truman was still president. AD is a disease feared by most senior citizens for good reason. About one-third of the population is in some stage of AD by the time they reach the age of 85. The latter stages are especially difficult as the patient no longer recognizes loved ones and may experience depression, anxiety, irritability, and declining physical health.
Stages of Alzheimer’s Disease
1. Early warning signs: Many patients exhibit subtle symptoms of AD several years prior to diagnosis. The most common indications are loss of interest in events and activities along with a decline in mental sharpness. This condition is often called mild cognitive impairment.
2. Mild Alzheimer’s disease: A formal diagnosis of AD usually is made when there is a striking loss of recent memories, while older memories are still retained. Another diagnostic symptom is a shrinking vocabulary and loss of communication skills. The popular mini-mental state examination (MMSE) test154 is helpful in establishing a diagnosis; it is based on a patient’s ability to perform simple tasks such as counting backward from 100 by threes or copying a drawing of a pentagon. A more decisive test is the computerized CANTAB Test155 (Cambridge Neuropsychological Test Automated Battery). In this stage of the disease, most persons still enjoy life and can accomplish basic tasks but require supervision for activities requiring complex thought or logic.
3. Moderate Alzheimer’s disease: The deterioration of neurons eventually spreads throughout the brain and the patient loses the ability to perform many common activities of daily living. Memory continues to deteriorate, and the patient may no longer recognize his own grandchildren or recent friends. Reading and writing skills gradually disappear, and irritability, wandering, falling, and physical aggression symptoms can be very challenging for the family. About one-third of AD victims develop delusions and urinary incontinence. Most families are forced to move their loved one to a long-term care facility when they can no longer cope with the worsening symptoms. While heartbreaking for the family, this step frequently does not seem to bother the patient.
4. Advanced Alzheimer’s disease: During this final stage, the patient becomes completely dependent on caregivers. As the disease progresses, many develop a wooden expression, lose the ability to speak, and fail to respond to visitors. In the final stages, they become bedridden, totally incontinent, and unable to feed themselves. Death usually results from an infection, respiratory problems, etc. and not directly from AD itself.156
The Role of Genetics
Although the cause of AD is still unknown, it is clear that genetics or epigenetics play an important role. There are two types of AD and both have genetic links. Familial AD157 typically develops between the ages of 40 and 55 and represents about 5% of cases. This condition is caused by mutations to genes that produce presenilin 1, presenilin 2, or amyloid precursor protein (APP). The late-onset form of AD represents about 95% of cases, usually developing after age 70. In 1993, researchers at Duke University158 reported that an apolipoprotein E (ApoE) abnormality represents a powerful risk factor in late-onset AD. ApoE is a protein containing 299 amino acids that can exist in three isoforms: E2, E3, and E4. Isoform E3 is identical to that of E2 except that a cysteine in the amino acid chain is replaced by arginine. In isoform E4, two cysteines are replaced by arginine. The risk of developing AD is lowest in ApoE2, intermediate in ApoE3, and highest in ApoE4. Persons born with an E4 allele from both parents are between 10 and 30 times more likely to develop AD by age 75 when compared with persons without an E4 allele. However, many persons with both E4 alleles live to a ripe old age without developing memory loss or other symptoms of dementia. Moreover, 40% of AD victims do not have the E4 gene. Genetic testing can determine a person’s ApoE type, but it has not become popular due to the perception that the disease is incurable. The bottom line is that ApoE4 represents an AD risk factor but does not doom a person to acquiring the disease. In a positive development, several risk factors have been identified that may enable a person to reduce the likelihood of acquiring AD.
Risk Factors
The following factors increase the likelihood that a person will experience Alzheimer’s disease.
 Age: Life expectancy was only 45-50 years in the early 1900s, and few persons lived long enough to develop AD. For many years, the connection between AD and advanced age went unnoticed. In 1960, British researchers159 discovered that most cases of senility were actually AD, with clear evidence of amyloid plaques and tangles within brain cells. We now know that more than 90% of AD cases are diagnosed after age 70. The likelihood of this disease increases sharply with age, and about one-third of persons reaching age 85 are in some stage of the disorder. Surprisingly, there is evidence that the risk of AD declines after age 90.
Age: Life expectancy was only 45-50 years in the early 1900s, and few persons lived long enough to develop AD. For many years, the connection between AD and advanced age went unnoticed. In 1960, British researchers159 discovered that most cases of senility were actually AD, with clear evidence of amyloid plaques and tangles within brain cells. We now know that more than 90% of AD cases are diagnosed after age 70. The likelihood of this disease increases sharply with age, and about one-third of persons reaching age 85 are in some stage of the disorder. Surprisingly, there is evidence that the risk of AD declines after age 90.
Head injury: Studies160 have found a strong association between head injuries and AD. This factor is most evident in persons who have had multiple blows to the head, including boxers and football players. In 1993, the British Medical Association161 reported evidence that boxers have an increased risk for AD, and some acquire the disorder before age 40. An example is provided by three boxers in the Quarry family who all developed serious dementia, including Jerry Quarry, who once fought for the world heavyweight championship, His family has established a foundation for AD research.
Education level: In 2008, Washington University scientists162 reported that persons with higher education levels had a lower AD risk when compared with the rest of the population. This was based on positron emission tomography (PET) scans that directly measured the amount of plaque in brains of living persons. The data indicated that persons who didn’t complete grade school were four times more likely to acquire AD than college graduates. A recent study163 has indicated that higher education delays the onset of AD but that the disease progresses more rapidly once AD has begun in these persons.
Mental activity: Three studies164 have indicated that mentally stimulating activities such as playing bridge, doing puzzles, or learning a new language reduce the risk of AD. This suggests that sitting on the couch and watching TV too regularly may be dangerous to your health.
Physical activity: Persons engaging in regular physical activity165 were found to have reduced AD risk. However, it’s possible this is due to better cardiovascular health or reduced obesity rather that a direct result of physical exercise.
Vascular factors: The first brain areas damaged in AD are very close to blood vessels. In addition, there is published evidence166 that a variety of cardiovascular problems increase AD risk. Examples include strokes, hypertension, hypotension, atrial fibrillation, atherosclerosis, and elevated serum homocysteine.
Alcohol use: An interesting finding is that persons who drink moderate amounts of alcohol are better protected against AD than those who never consume alcohol. In the Copenhagen Heart Study,167 red wine was found to be especially effective. One or two glasses of red wine daily have been recommended for persons with no history of alcoholism. There is also evidence that high alcohol consumption (including red wine) increases the likelihood of AD.
Physical illness: Persons with a history of diabetes or other autoimmune diseases appear to have a higher risk of AD. The strongest data168 are for type 2 diabetes and insulin resistance.
Toxic metals: There is some evidence169 that aluminum, mercury, and other toxic metals are associated with AD. Mercury in particular has become a highly controversial issue. On one hand, there is published data showing a positive association between mercury exposure and the risk of AD, and this has led many persons to have their dental amalgams removed. In addition, mercury exposure causes increased oxidative stress and this factor is elevated in AD. Dental amalgams result in a significant vapor pressure of mercury, and these vapors can be directly absorbed by lung capillaries. On the other hand, other studies have reported no correlation between the number of amalgam fillings and AD, and the American Dental Association has aggressively denied any relationship between amalgams and AD. Another source of mercury for many seniors is an annual flu shot.
Poor nutrition: Inadequate nutrition is a common problem in elderly populations, and low levels of zinc and vitamins A, C, and E result in increased oxidative stress. Several studies170 indicate that oxidative stress plays an important role in the development of AD. In addition, elevated serum homocysteine is a known AD risk factor171 that can result from deficiencies of folic acid and vitamin B-12. A high-quality diet may represent an important protective factor against AD.
Alzheimer’s Disease Theories
1. Cholinergic theory: The cholinergic theory172 was prominent in the 1980s and early 1990s and continues to be the basis for most of today’s AD medications. This theory states that AD begins with the depletion of acetylcholine activity in the brain. Acetylcholine is a major neurotransmitter important for memory processes and is seriously depleted in AD brains. This has been attributed to a shortage of enzymes (choline acetyltransferase and acetylcholine esterase) necessary for production and regulation of acetylcholine. For years, these low enzyme levels were believed to cause physical problems with at-risk neurons and trigger the cell death process. However, it was eventually learned that the low enzyme levels are not present during early stages of AD but appear later. This is strong evidence173 that the cholinergic theory of AD onset is incorrect and that the low acetylcholine and enzyme levels are an aftermath and not a cause of the disease. Aricept and other acetylcholine-enhancing medications are still widely used since they can provide temporary memory improvement for the dying patient. The bottom line is that these medications provide 4-12 months of improved brain function, but they do little or nothing to stop the relentless destruction of the brain.
2. Amyloid plaque hypothesis: The theory with the largest following is the amyloid hypothesis174-175 that assigns a central role to beta-amyloid (Aβ), a small protein that is the main constituent of the plaques found in AD brains. Researchers have learned that Aβ is formed when APP is cut into small sections by enzymes called secretases. The factors that cause the disintegration of APP into Aβ fragments are a subject of active research. The Aβ plaques tend to clump together like a ball of spaghetti and form a mass outside brain cells. Advocates of the amyloid theory believe production and aggregation of Aβ to be the key event in the brain cell destruction process.
The amyloid theory has led to several experimental treatment approaches. An early strategy for removing plaques involved immunizing AD patients with Aβ peptides to produce antibodies that can clear Aβ from the brain. An early vaccine176 was successful in reducing plaques and improving mental functioning in mice. However, this therapy failed in human testing due to serious side effects. In a 2009 study, a different vaccine that had removed Aβ plaques in mice was given to 80 volunteer patients. The vaccine effectively reduced or completely eliminated the plaques in these persons. However, the researchers were disappointed to discover that removing the Aβ plaques had no measurable impact on the progressive destruction of brain cells and the course of the disease.177 This finding suggests that Aβ plaques may be a result and not a cause of AD. A second amyloid-targeting strategy involves inhibition of the secretase enzymes that cut APP into fragments and produce Aβ. Several pharmaceutical companies are developing secretase inhibitors aimed at prevention of AD.
3. The tau hypothesis: The second most popular AD theory178 assigns a primary role to tau, a protein that normally helps organize and stabilize a cell’s internal skeleton. Tiny brain axons receive structural support from microtubules, tiny structures that resemble soda straws packed together. The microtubules also provide a conduit for vesicle and enzyme transport within the cell. Tau and other proteins assist in keeping the delicate tubules intact and in the proper place. However in AD patients, tau proteins become chemically modified and clump together, resulting in microtubule disintegration and disabling tangles. The net result is loss of nutrient transport and death of the brain cell. Figure 9-1 illustrates plaques and tangles observed in AD brain cells. Although no therapies targeting tau have reached clinical trials, many experts are convinced that understanding tau will reveal crucial clues about AD’s devastating effects on nerve cells and lead to effective therapies.
Figure 9-1.
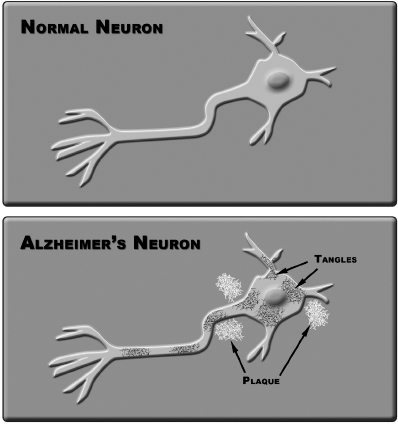
4. Inflammation theory: In 2004, the Scripps Research Institute proposed a theory179 that chronic inflammation is central to the development of AD. This theory was supported by mouse studies that found inflammation triggered a suspected AD mechanism and caused cognitive decline. The body’s response to inflammation involves chemical changes, especially protein modifications. The Scripps theory suggests that amyloid proteins are modified during inflammation, causing them to misfold and accumulate into the characteristic plaques found in AD brains. Scientists have launched several clinical trials to investigate whether anti-inflammatory drugs actually reduce AD risk. Other research groups are trying to identify molecular mechanisms that could explain protective effects of these drugs. Testing of primates has failed to confirm the findings of the Scripps mouse studies. Some experts believe anti-inflammatory therapies may have more application for AD prevention rather than addressing actual AD disease states.
5. Oxidative stress theories: There is considerable evidence180-181 for the presence of excessive oxidative stress (free radicals) in AD brains. Oxidative free radicals perform several useful functions in the body, such as killing bacteria and burning glucose to produce energy. However, a chronic excess of free radicals can lead to death of brain cells. Sources of free radicals include physical injury, bacteria, viruses, inflammation, heavy metals, and nuclear radiation. The body has a supply of antioxidant molecules that have the job of keeping free radicals from reaching concentrations lethal to brain cells. A healthy brain requires (a) a competent blood-brain barrier to reduce influx of toxics, and (b) enough antioxidant protection within the brain to cope with the number of free radicals present. If the free radicals win the war, then neurodegeneration (death of brain cells) will occur.
Several antioxidant therapies have been the subject of formal studies, including vitamin E, vitamin E plus selenium, coenzyme Q10, glutathione, and lipoic acid. There is evidence182 that some of these therapies increase the life span of AD patients but none has shown the ability to stop the advancing death of brain cells.
6. Metal metabolism theories: A number of research studies have implicated trace metal imbalances in the development of AD. Australian doctor Ashley Bush, MD, and colleagues183 reported that copper overloads cause increased Aβ in the brain. Copper and iron are major sources of free radicals in the human brain, and elevated copper levels have been found in Aβ. Metallothionein and Cu/Zn SOD protect against copper free radicals, but both are depleted in AD brains. This theory suggests that excess copper prevents the natural removal of Aβ from AD brains. Interestingly, two 2009 studies184-185 have reported a protective role for copper in AD. It’s possible that either deficiency or excess of copper can be harmful to the brain and that homeostatic regulation of copper levels is essential.
Advanced Photon Source Measurements
In 2005, I organized a research study measuring metal concentrations in brain tissues from AD and age-matched control subjects. My collaborators were Barry Lai and Stefan Vogt of Argonne National Laboratory and Walter Lukiw of the Louisiana State University (LSU) Health Sciences Center. The tissues were 10-micron thick sections processed under highly stringent conditions at the LSU Neuroscience Center Brain Tissue Bank. The chemical assays were made at Argonne’s Advanced Photon Source, using high-brilliance photon beams focused to 0.4 micron diameter. Samples were raster-scanned, yielding tens of thousands of individual measurements for zinc, copper, phosphorus, sulfur, chlorine, iron, aluminum, and calcium. Large calcium-rich circular areas were observed in the AD samples and very small calcium-rich zones in the controls. In both cases, the calcium concentrations were 15 times higher in the calcium-rich areas when compared with adjacent tissues.186 Another interesting finding was the presence of very elevated Cu/Zn ratios in parts of the AD samples but not in the controls.
The Case for Metallothionein
Two separate autopsy studies187-188 have reported severe deficiency of MT protein levels in deceased AD patients. MT proteins have several protective functions in the brain including the following:
Preventing toxic metals from passing the blood-brain barrier
Regulation of copper levels
Powerful antioxidant action against free radicals
The protective properties of MT depend on ample amounts of glutathione and selenium, and I often refer to these antioxidants as the Three Musketeers. Gene expression of MT is zinc dependent, and most AD patients are depleted in zinc. MT proteins are far more powerful than selenium, coenzyme Q10, vitamins C and E, and other antioxidants that have been used in experimental AD therapies. In 2007, I received a US patent for an MT-Promotion formulation147 for the treatment of autism, and a patent is pending for use of the same formulation for the treatment of AD. This formulation is comprised of 22 biochemical factors known to enhance the gene expression and functioning of MT. The treatment is a two-step process: normalization of plasma zinc levels followed by MT-Promotion therapy. A reliable caretaker is essential for treatment compliance since the AD patients may forget to take the capsules or take them repeatedly the same day. This treatment has been provided to more than 100 AD patients, resulting in many reports of improvement.
The first case involved an elderly Minnesota woman who had been diagnosed with AD and was a resident in a health care facility in the year 2000. She no longer remembered her grandchildren and was reported to have lost her sparkle and enthusiasm for life. After two months of MT-Promotion therapy, the family reported a partial return of memory and recognition of her grandchildren. They added that her irritability and malaise had disappeared and she wanted to go shopping. Her mental functioning remained fairly stable for the last seven years of her life.
Encouraged by this case history, we provided MT-Promotion therapy to increasing numbers of AD patients. In 2007, researcher Aditi Gulibani and I reviewed the progress of the first 60 AD patients who received MT-Promotion therapy. We learned that approximately 70% of the families reported a partial return of memory followed by stable mental functioning that continued for several years. In collaboration with Dr. John Crayton, CANTAB system testing was performed before and after treatment for dozens of patients, and in most cases, the data confirmed the reports of improved memory and stable cognitive function. Several of the improved patients were eventually told that they had been misdiagnosed, apparently since they hadn’t deteriorated and died. Diagnosis of AD is preliminary prior to autopsy, so we cannot be certain that all of these patients actually had AD. It’s also possible that some of the nonresponders had Lewy body disease or vascular dementia and not AD. In addition, the absence of a blinded control study means that the MT-Promotion approach must be regarded as unproven. Still, the positive reports from the first 100 persons treated with the MT-Promotion protocol suggests this approach is promising and worthy of additional study.