Biochemistry
PROTEIN STRUCTURE AND FUNCTION
Amino Acid Structure
Amino acids are the building blocks of proteins. Amino acids are composed of an α-amino group (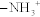 ) and an α-carboxyl group (-COOH), with different characteristic side chains. Twenty amino acids are naturally incorporated into polypeptides (Fig. 2-1). Nine of these amino acids are “essential,” cannot be synthesized by humans, and must be obtained in the diet. These essential amino acids include phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, leucine, and lysine. Additionally, three amino acids are essential in children: arginine, tyrosine, and cysteine. In adults, the remaining 11 amino acids can be synthesized.
) and an α-carboxyl group (-COOH), with different characteristic side chains. Twenty amino acids are naturally incorporated into polypeptides (Fig. 2-1). Nine of these amino acids are “essential,” cannot be synthesized by humans, and must be obtained in the diet. These essential amino acids include phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, leucine, and lysine. Additionally, three amino acids are essential in children: arginine, tyrosine, and cysteine. In adults, the remaining 11 amino acids can be synthesized.
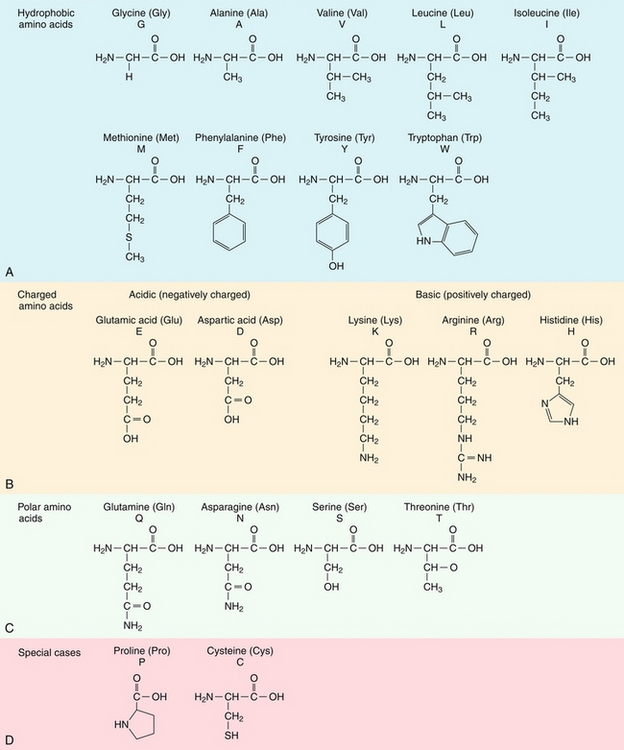
The nature of the amino acid side chain is critical to how proteins interact with their environment. Acidic, basic, and uncharged polar side chains tend to be found on the exterior of soluble proteins and on the interior of proteins found within membranes. Isoleucine, valine, and leucine are branched-chain amino acids that are increased in maple syrup urine disease.
 Alanine is an important substrate for gluconeogenesis.
Alanine is an important substrate for gluconeogenesis.
Methionine is a precursor of homocysteine, a product associated with atherosclerosis. Increased levels of homocysteine are seen in patients with classic homocystinuria, with a deficiency in cystathionine β-synthase. These patients have early onset of vascular disease.
Phenylalanine accumulates in phenylketonuria (PKU). Tryptophan is a precursor of serotonin, niacin, and melatonin. Arginine and histidine stimulate growth hormone and insulin. Arginine is a precursor of nitric oxide.
Glutamine is the most common amino acid and is an important nitrogen donor in the synthesis of purines and pyrimidines.
Cysteine forms disulfide bonds and is sensitive to oxidation state.
Proline is different from other amino acids because its side chain forms a five-membered ring. This unique side chain means that proline is often found in collagen and is used to interrupt α-helices in globular proteins.
The pKa (-log of the acid dissociation constant, Ka) is a measure of the strength of an acid in solution. The pKa of amino acid side chains gives insight into the pH characteristics of proteins and stability. Histidine is unique in that its side group, imidazole, has a pKa of 6. This means that histidine has a positive charge at pH 7; and at physiologic pH, small shifts in pH change the charge on histidine, and the side group acts as a buffer. Aspartic acid and glutamic acid (acidic amino acids) have a negative charge at pH 7; albumin is a strong binding protein for positively charged molecules in part because of its high content of these acidic amino acids.
The isoelectric point (pI) is the pH value at which an amino acid, or any other molecule, has a net zero electrical charge. Amino acids are zwitterions, with positive and negative charges. When pH is greater than pI, the net charge on the molecule is negative. When pH is less than pI, the net charge on the molecule is positive. At physiologic pH, lysine, arginine, and histidine have a positive charge, whereas aspartate and glutamate have a negative charge. Proteins can be separated based on their pI using isoelectric focusing on a polyacrylamide gel, which separates proteins using a pH gradient. This is the first step in two-dimensional gel electrophoresis.
Protein Structure
The primary structure of a protein is the linear sequence of amino acids (Fig. 2-2). Peptide bonds are formed between the α-carboxyl group and the α-amino group, creating a covalent amide linkage.
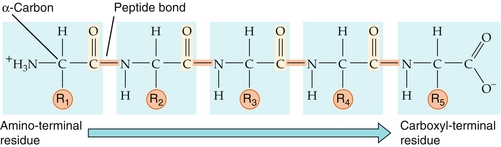
Figure 2-2 Protein primary structure. By convention, the amino-terminal (N-terminal) residue is depicted at the far left of the protein sequence, and the carboxyl-terminal (C-terminal) residue is at the far right. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
The secondary structure of proteins involves the arrangements of the amino acids located near each other in the amino acid sequence (Fig. 2-3). Important secondary structure components include α-helices and β-sheets.
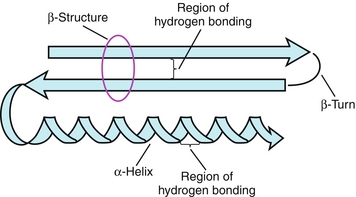
Figure 2-3 Protein secondary structure components. (From Pelley JW. Elsevier’s Integrated Biochemistry. Philadelphia: Elsevier; 2007.)
α-Helices are the most common kind of polypeptide helix. Extensive hydrogen bonding occurs between the peptide bonding of carbonyl oxygen and amide hydrogen. Each α-helical turn contains 3.6 amino acids. Therefore, amino acids that are spaced 3 to 4 amino acids apart in the primary structure are quite close together in an α-helix. Proline disrupts α-helices because its five-membered ring structure forms kinks in the chain.
β-Sheets, or β-pleated sheets, occur when two or more polypeptide chains known as β-strands are arranged in parallel or antiparallel to each other. The hydrogen bonds form between the polypeptide backbones of separate polypeptide chains and are known as interchain bonds. Globular proteins tend to have β-sheets with a right-handed curl or twist, which form their core. Silk is a packed β-sheet.
Proteins that bind to DNA also have important secondary structure motifs. For example, the DNA-binding zinc finger motif is found in many transcription factors.
The tertiary structure of proteins includes the three-dimensional shape of the folded protein chain. Several different kinds of interactions stabilize tertiary structure.
Disulfide bonds are covalent links formed from the sulfhydryl group (-SH) of two cysteine residues. Disulfide bonds are important in proteins that are secreted from cells, such as immunoglobulins.
Ionic interactions include negatively charged side groups interacting with positively charged groups.
Hydrophobic interactions force amino acids with nonpolar side chains into the interior of polypeptide molecules, where they are able to associate with other hydrophobic residues. Amino acids with polar side chains tend to be located on the surface of a protein and contact the polar solvent to be most energetically favorable.
Hydrogen bonds can form between amino acid side chains that have oxygen- or nitrogen-bound hydrogen, such as serine and threonine. This can help to improve the solubility of proteins in aqueous environments.
The quaternary structure is the arrangement of polypeptide subunits in a protein structure that has more than one polypeptide chain. These subunits are usually held together by noncovalent interactions.
Proteins fold in a very short time period (milliseconds to microseconds), whereas the translation of protein from messenger RNA (mRNA) progresses at a much slower rate (5 to 20 amino acids per second). Chaperone proteins such as the heat shock proteins are critical in keeping some proteins properly folded. Proteins can be denatured by heat, solvents, strong acids or bases, and detergents. Some proteins can also misfold, and deposition of these misfolded proteins is associated with several diseases.
Amyloidoses are diseases in which an altered protein accumulates. In some amyloidoses, insoluble, fibrillar proteins aggregate in a form that resembles β-sheets. In Alzheimer disease, plaques form that contain amyloid β (Aβ), which is hypothesized to be neurotoxic. These Aβ peptides aggregate, forming amyloid in brain tissue and in blood vessels. Another protein that is deposited in Alzheimer disease is neurofibrillary tangles, made of an abnormal, hyperphosphorylated form of the tau (τ) protein.
Prion diseases are caused by the prion protein (PrP), an infectious protein that causes normal protein to change structure and form insoluble aggregates of fibrils. Prion diseases include transmissible spongiform encephalopathies (TSEs) such as Creutzfeldt-Jakob disease in humans, scrapie in sheep, and bovine spongiform encephalopathy (BSE) in cattle (mad cow disease). It has been shown that many α-helices present in noninfectious PrP are replaced by β-sheets in the infectious form; this makes the protein highly resistant to proteolytic degradation.
Heinz bodies are formed when red blood cells undergo oxidative stress, and hemoglobin is denatured to form aggregates on the membrane of red blood cells (RBCs). This occurs in conditions such as glucose-6-phosphate dehydrogenase (G6PD) deficiency.
OXYGEN-BINDING PROTEINS: HEMOGLOBIN AND MYOGLOBIN
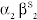 .
.Hemoglobin A, the most common hemoglobin in adults, is composed of two α-globin subunits and two β-globin subunits (α2β2). Each globin protein subunit is composed of an α- or β-protein chain plus a heme group. Heme, in turn, is composed of an Fe2 + held in a protoporphyrin IX ring. Hemoglobin has two primary forms. Hemoglobin that is desaturated with oxygen is deoxyhemoglobin (T, tense form), which has a low oxygen affinity and little available movement. Hemoglobin that is saturated with oxygen is oxyhemoglobin (R, relaxed form), which has a high oxygen affinity and more available movement.
A single hemoglobin molecule can bind up to four oxygen molecules. The four globin subunits work cooperatively in hemoglobin, in which the binding of oxygen to one subunit of the tetramer increases the affinity of the other subunits for oxygen. The first oxygen binds with low affinity, but this leads to a transition from T to R form. The second through fourth oxygen molecules bind with increasing affinity, which leads to a sigmoidal oxygen-binding curve for hemoglobin (Fig. 2-4).
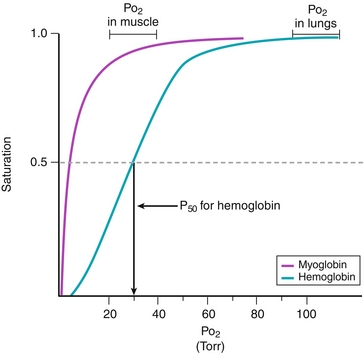
Figure 2-4 Comparison of oxygen-binding curves for hemoglobin and myoglobin. The P50 is the Po2 at half saturation. Having a lower P50 means having a greater affinity for oxygen. (From Pelley JW. Elsevier’s Integrated Biochemistry. Philadelphia: Elsevier; 2007.)
Deoxyhemoglobin (T form) preferentially binds hydrogen ion (H+), 2,3-bisphosphoglycerate (2,3-BPG), and CO2. This leads to stabilization of the T state, decreased affinity for oxygen, and a rightward shift in the oxygen saturation curve. Exercise and increase in temperature can also cause a rightward shift. Conversely, a leftward shift in the oxygen saturation curve occurs in the presence of a decrease in CO2, alkalosis (high pH, low hydrogen ion concentration), and decrease in 2,3-BPG.
Myoglobin is a single polypeptide chain with a single heme group. Myoglobin is present in heart and skeletal muscle and acts as an oxygen carrier and location for storage of oxygen. Myoglobin can only bind to one oxygen molecule; therefore, the binding curve is not sigmoidal because the binding of O2 is not cooperative. Instead, the myoglobin curve is hyperbolic (see Fig. 2-4).
Carbon monoxide (CO) binds to hemoglobin and forms carboxyhemoglobin, which has a high affinity for CO and displaces O2. This leads to stabilization of the R state, a leftward shift of the oxygen saturation curve, and an oxygen saturation curve for hemoglobin that resembles the curve for myoglobin.
Figure 2-4 compares the oxygen-binding curves for hemoglobin and myoglobin. Myoglobin has a lower P50 than hemoglobin, meaning that it has a greater affinity for oxygen. This ensures that oxygen is bound to myoglobin in all cases except hypoxia.
Hemoglobinopathies (Fig. 2-5) are a group of genetic disorders caused by abnormal hemoglobin structure or insufficient synthesis of normal hemoglobin.
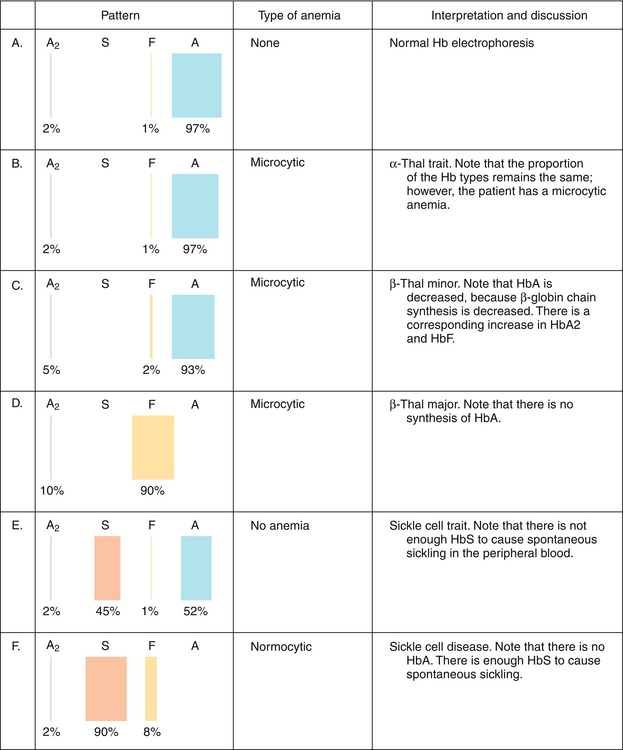
Figure 2-5 Hemoglobinopathies. Hb, hemoglobin. (From Goljan EF, Sloka KI. Rapid Review Laboratory Testing in Clinical Medicine. Philadelphia: Elsevier; 2007.)
Sickle cell disease (HbS disease) is a homozygous recessive genetic disorder that results from production of hemoglobin with an altered amino acid sequence, caused by a single point mutation in the β-globin gene. The mutation is a glutamate-to-valine mutation at position 6 in the β-globin chain. During electrophoresis at a basic pH, HbS migrates more slowly toward the anode (positive electrode) than does HbA, owing to the absence of the negatively charged glutamate residues in the two β-globin chains, which makes HbS less negative. The mutant β-globin chain is designated βS, and the resulting hemoglobin, 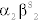 , is HbS. The α-globin chains are normal. An infant does not begin to show symptoms of the disease until sufficient HbF has been replaced by HbS so that sickling can occur. This amino acid substitution also forms a protrusion on the β-globin that polymerizes HbS in the deoxygenated state into fibers. This polymerization creates sickled erythrocytes, which occlude blood flow in the capillaries. Microinfarcts then cause tissue anoxia and result in severe pain. Sickling is increased by anything that increases the proportion of HbS in the deoxygenated state, such as decreased O2 tension, increased Pco2, decreased pH, dehydration, and increased concentration of 2,3-BPG in erythrocytes. Hydroxyurea is used to treat sickle cell disease because it increases circulating levels of HbF, which decreases RBC sickling. Sickle cell disease is tested for at birth to allow prophylactic antibiotic therapy to begin soon after because these children are at risk for sepsis.
, is HbS. The α-globin chains are normal. An infant does not begin to show symptoms of the disease until sufficient HbF has been replaced by HbS so that sickling can occur. This amino acid substitution also forms a protrusion on the β-globin that polymerizes HbS in the deoxygenated state into fibers. This polymerization creates sickled erythrocytes, which occlude blood flow in the capillaries. Microinfarcts then cause tissue anoxia and result in severe pain. Sickling is increased by anything that increases the proportion of HbS in the deoxygenated state, such as decreased O2 tension, increased Pco2, decreased pH, dehydration, and increased concentration of 2,3-BPG in erythrocytes. Hydroxyurea is used to treat sickle cell disease because it increases circulating levels of HbF, which decreases RBC sickling. Sickle cell disease is tested for at birth to allow prophylactic antibiotic therapy to begin soon after because these children are at risk for sepsis.
Hemoglobin C disease (HbC disease) results from production of hemoglobin with an altered amino acid sequence, in which a lysine is substituted for a glutamate at position 6 in the β-globin gene. The substitution of a positively charged amino acid for a negatively charged amino acid causes HbC to move more slowly toward an anode than HbA or HbS does. Homozygous patients have a mild, chronic hemolytic anemia.
Hemoglobin SC disease occurs when some β-globin genes have the sickle cell mutation, while others have the mutation found in HbC disease. These patients are compound heterozygotes, or doubly heterozygous, because both of their β-globin genes are abnormal but are different from each other. Affected individuals have fewer vasoocclusive events than those with sickle cell disease, but their course is more serious than those with HbC disease.
Thalassemias are caused by decreased production of normal hemoglobin due to defective synthesis of either the α- or the β-globin chain. In β-thalassemia, synthesis of β-globin chains is decreased or absent, but the α-globin chain synthesis is normal. Other forms of hemoglobin may be present in elevated amounts, including HbF (α2γ2) in β-thalassemia and hemoglobin Bart (γ4) in α-thalassemia. Because there are only two copies of the β-globin genes, individuals either have β-thalassemia trait (minor) or β-thalassemia major (Cooley anemia). Because β-globin is not expressed until late in gestation, symptoms of β-thalassemia appear only after birth. α-Thalassemias occur when α-globin chains are decreased or absent. Every cell contains four copies of the α-globin genes, so there are several possible levels of deficiency. If one copy of the α-globin is absent or defective, the person is a silent carrier; two defective genes lead to α-thalassemia trait; three defective genes lead to hemoglobin H (HbH) disease. If all four α-globin genes are defective, hydrops fetalis and fetal death result because α-globin chains are required for the synthesis of HbF.
Fibrous Proteins
Collagen is the most abundant protein in humans and is found primarily in connective tissue and muscle. Collagen is composed of a triple helix of three α-chains held together by hydrogen bonds. There are more than 20 types of collagen; collagen I is the most common. Collagen has a large amount of proline and glycine. Proline helps in the formation of the α-chain, and glycine is found in every third amino acid. The sequence is –Gly–X–Y–, where X is often proline and Y is often hydroxyproline or hydroxylysine.
The biosynthesis of collagen (Fig. 2-6) occurs as follows:
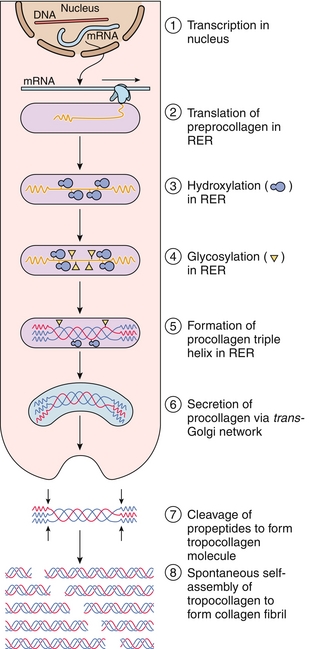
Figure 2-6 Collagen synthesis and assembly. RER, rough endoplasmic reticulum. (From Gartner LP, Hiatt JL: Color Textbook of Histology. 3rd ed. Philadelphia: Saunders; 2007:77.)
1. It begins with transcription in the nucleus of a fibroblast or related cell.
2. mRNA is translated into preprocollagen on the rough endoplasmic reticulum (RER), and these peptide chains are directed into the lumen of the RER and become pro-α-chains.
3. Proline and lysine residues are next hydroxylated by prolyl hydroxylase and lysyl hydroxylase.
4. Some hydroxylysine residues are glycosylated with glucose and galactose.
5. Pro-α-chains form procollagen, which has a central triple helix with N- and C-terminal propeptide extensions; these prevent premature assembly of collagen within the endoplasmic reticulum.
6. Procollagen is transported to the Golgi apparatus, where it is released into the extracellular space.
7. After release of procollagen, peptidases remove the terminal propeptides.
8. Collagen fibrils then spontaneously assemble into tropocollagen. The collagen fibers are cross-linked by lysyl oxidase, which oxidatively deaminates lysyl and hydroxylysyl residues in collagen, forming covalent cross-linked, mature collagen fibers.
The hydroxylation reaction requires both oxygen and the reducing agent vitamin C (ascorbic acid) for the hydroxylating enzymes prolyl hydroxylase and lysyl hydroxylase to function. Vitamin C deficiency leads to a lack of prolyl and lysyl hydroxylation, making collagen fibers unable to be cross-linked, which decreases the tensile strength of the assembled collagen fiber. This is called scurvy. Because of the weak collagen structure, patients often have bruises, corkscrew hairs, and perifollicular hemorrhage due to capillary fragility. Copper is also a cofactor for lysyl oxidase.
Ehlers-Danlos syndrome (EDS) is a connective tissue disorder caused by defects in collagen synthesis. EDS arises from lysyl hydroxylase deficiency, procollagen peptidase deficiency, or mutations in collagen amino acid sequences, most importantly collagen type III. Stretchy skin and loose joints are seen in patients with EDS.
Osteogenesis imperfecta (OI), also known as “brittle bone disease,” is a genetic disorder caused by defects in connective tissue, usually type I collagen, such as decreased production of collagen α-chains, leading to bones that are prone to bending and fracture. OI is an autosomal dominant disorder but arises sporadically in one-third of cases. Type I OI is the most common form of OI and is known as osteogenesis imperfecta tarda. Patients with type I OI have bones that fracture easily, early hearing loss, and a blue-gray tint to the sclera due to thinned scleral tissue. The blue tint of the sclera is secondary to defective type I collagen, which allows visualization of underlying choroidal veins.
Elastin is a connective tissue protein composed of elastin and glycoprotein microfibrils that are found primarily in the lungs, arterial walls, and elastic ligaments. Elastin is synthesized from tropoelastin, a precursor protein. After secretion from the cell, tropoelastin deposits onto fibrillin. Mutations in fibrillin cause Marfan syndrome. In the alveoli, elastin is broken down by elastase from activated neutrophils. α1-Antitrypsin, an enzyme produced in the liver, usually blocks elastase and protects the lungs. However, genetic defects in α1-Antitrypsin can lead to pulmonary emphysema at a young age due to increased breakdown of lung connective tissue.
Enzymes
Enzymes are protein catalysts. They have active sites, which permit substrate binding. Stabilization of the transition state leads to decreased activation energy, leading to increased rates of substrate to product, but no change in reaction equilibrium. Enzymes are described mathematically using Michaelis-Menton kinetics (Fig. 2-7):
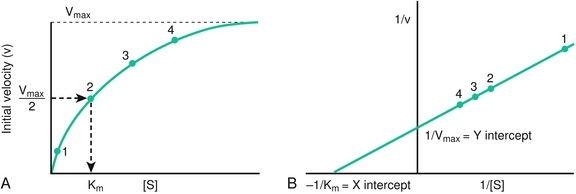
Figure 2-7 Michaelis-Menton enzyme kinetics. A, Substrate concentration [S] versus initial velocity (V) for a reaction with Michaelis-Menton kinetics with a constant enzyme concentration. B, Lineweaver-Burk double reciprocal plot. Km and Vmax are determined from the intersection of the resulting straight line with the horizontal and vertical axes, respectively. (From Pelley JW, Goljan EF. Rapid Review Biochemistry. 3rd ed. Philadelphia: Elsevier; 2010.)
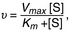
where Km is the substrate concentration at which the reaction rate is 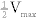 , v is the velocity of the reaction, and [S] is the substrate concentration. A higher Km means a lower affinity of the substrate for the enzyme.
, v is the velocity of the reaction, and [S] is the substrate concentration. A higher Km means a lower affinity of the substrate for the enzyme.
When [S] > > than Km, the rate of the reaction is independent of [S]. This is known as zero-order kinetics.
When [S] = Km, then the initial velocity (v) = Vmax/2
When [S] < Km, the reaction rate is proportional to [S]. This is first-order kinetics.
Vmax is the maximal reaction velocity. This occurs when the enzyme is saturated with substrate.
A Lineweaver-Burk plot is a double reciprocal plot of 1/v versus 1/[S]. This produces a straight line. The Y-intercept is 1/Vmax, and the X-intercept is − 1/Km.
Enzyme kinetics are affected by inhibitors, which may be classified in several ways (Fig. 2-8).
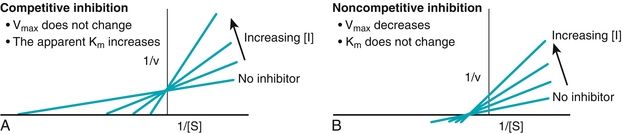
Figure 2-8 Lineweaver-Burk plots of competitive (A) and noncompetitive (B) enzyme inhibition. (From Pelley JW, Goljan EF. Rapid Review Biochemistry. 3rd ed. Philadelphia: Elsevier; 2010.)
Competitive inhibitors: Km is increased, Vmax is unchanged (there is always competition between two lines so that they cross on graph; solute concentration changes, but velocity remains the same). Plots intersect on the vertical axis (Vmax is the same). Competitive inhibitors include methanol and ethylene glycol (antifreeze), which compete with ethanol for binding to alcohol dehydrogenase. Giving a patient ethanol reduces methanol toxicity by competing for the enzyme active site. For competitive inhibitors, high substrate concentration can reverse competitive inhibition because the enzyme is saturated with substrate.
Noncompetitive inhibitors: Km is unchanged, Vmax is decreased. Plots intersect on the horizontal axis (Km is the same). Noncompetitive inhibitors bind at a site distant from the active site and form unreactive complexes with the enzyme. Increased substrate amount does not change the level of inhibition. Physostigmine, a cholinesterase inhibitor, is a noncompetitive inhibitor.
A third kind of enzyme inhibitor is an irreversible inhibitor, which permanently inactivates enzymes. Examples include heavy metals, aspirin (an irreversible inhibitor of cyclooxygenase), fluorouracil, and organophosphates. The effect of irreversible inhibitors is only overcome by synthesis of new enzymes.
Enzymes may be useful in serum as diagnostic markers (Table 2-1).
Table 2-1
Serum Enzyme Markers Used for Diagnosis
| Serum Enzyme | Diagnostic Use |
| Alanine aminotransferase (ALT) | Viral hepatitis (ALT > AST) |
| Aspartate aminotransferase (AST) | Alcoholic hepatitis (AST > ALT) |
| Alkaline phosphatase | Osteoblastic bone disease Obstructive liver disease |
| Amylase | Acute pancreatitis Mumps (parotitis) |
| Creatine kinase (CK) | Myocardial infarction (CK-MB) Duchenne muscular dystrophy (CK-MM) |
| γ-Glutamyltransferase (GGT) | Obstructive liver disease, increased in alcoholic patients |
| Lactate dehydrogenase (LDH, type I) | Myocardial infarction |
| Lipase | Acute pancreatitis (more specific than amylase) |
NITROGEN METABOLISM
Disposal of Amino Acid Nitrogen
Free amino acids are produced by degradation of dietary protein, synthesis of nonessential amino acids, and degradation of body protein. Nitrogen is removed from amino acids because amino acids cannot directly take part in energy metabolism. Amino groups are removed from amino acids by two sequential reactions:
Transaminases, such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST), transfer amino groups to α-ketoglutarate, producing an α-keto acid and glutamate. The α-keto acid can enter the citric acid cycle. Aminotransferases require pyridoxal phosphate for function (a derivative of vitamin B6). Transaminases are intracellular enzymes found primarily in hepatic tissue; thus, elevated serum levels of transaminases can be diagnostic of liver damage.
Next, glutamate dehydrogenase oxidatively deaminates glutamate to α-ketoglutarate and free ammonia (NH3). The NH3 can be stored and transported to the liver as glutamine or as alanine (as part of the glucose-alanine cycle).
Aspartate and ammonia then enter the urea cycle, which is the body’s primary method for disposing of amino groups from amino acids. The nitrogen of aspartate, CO2, and NH3 are incorporated into urea.
The urea cycle (Fig. 2-9) is a five-step metabolic pathway that takes place within the liver. It removes nitrogen waste from the amino groups of amino acids that occurs during protein turnover. The rate-limiting step for the urea cycle is carbamoyl phosphate synthetase I, which is activated by N-acetylglutamate, which is synthesized from acetyl coenzyme A (CoA). Two molecules of NH3 and one of CO2 are converted into urea. Urea is then transported in the blood to the kidneys for excretion in urine. Urea levels in patients with kidney failure are elevated. Urease produced by bacteria in the gut creates a significant amount of ammonia, which can lead to hyperammonemia. Neomycin orally administered can reduce the number of urease-producing bacteria.
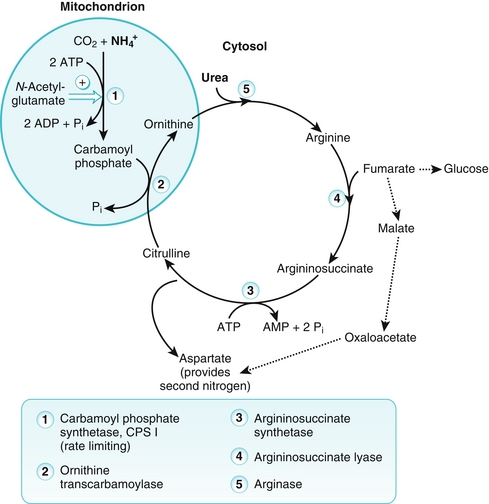
Figure 2-9 Urea cycle. This pathway occurs in the liver and disposes of toxic ammonia (NH3). ADP, adenosine diphosphate; AMP, adenosine monophosphate; ATP, adenosine triphosphate. (From Pelley JW, Goljan EF. Rapid Review Biochemistry. 3rd ed. Philadelphia: Elsevier; 2010.)
Hyperammonemia occurs when there are genetic defects of the urea cycle, or liver disease. Ammonia has a toxic effect on the central nervous system (CNS), causing tremors, cerebral edema, and blurring of vision. Urea cycle disorders (Table 2-2) are rare and, with the exception of X-linked ornithine transcarbamylase deficiency (which is the most common hereditary hyperammonemia), are inherited as autosomal recessive traits. Deficiencies of enzymes in the urea cycle cause intolerance to protein from the accumulation of ammonia in the body (hyperammonemia). These increased ammonia levels are toxic to the CNS and can lead to coma and death. These hereditary hyperammonemia disorders include those listed in Table 2-2.

In each of these disorders, urea is unable to be synthesized, which leads to hyperammonemia during the first weeks following birth. Mental retardation is common. Treatment of these disorders includes protein limitation in the diet and administration of compounds that bind covalently to amino acids so that they can then be excreted in the urine. For example, phenylbutyrate, a prodrug that is metabolized to phenylacetate, combines with glutamine to form phenylacetylglutamine, which can be excreted in the urine. This assists in clearance of nitrogen from the blood.
Amino Acid Synthesis and Degradation
Although essential amino acids must be obtained from the diet, nonessential amino acids can be synthesized by several different pathways.
Aspartate, alanine, and glutamate are synthesized from transamination of α-keto acids. Aspartate is derived from oxaloacetate, glutamate is from α-ketoglutarate, and alanine is from pyruvate.
Glutamine and asparagine are synthesized by amidation. Glutamine synthetase forms glutamine from glutamate. This reaction also helps to reduce ammonia levels. Asparagine synthetase forms asparagine from aspartate.
Serine is synthesized from the glycolysis intermediate 3-phosphoglycerate. Glycine, in turn, can be synthesized from serine.
Proline is synthesized from glutamate.
Arginine is synthesized from citrulline, an intermediate in the urea cycle.
Two amino acids can be synthesized from essential amino acids. Cysteine is synthesized from homocysteine and serine; homocysteine is derived from methionine. Tyrosine is synthesized from phenylalanine by phenylalanine hydroxylase. This reaction requires tetrahydrobiopterin (BH4). Because tyrosine and cysteine are formed from essential amino acids, tyrosine and cysteine are only nonessential in the presence of adequate dietary intake of methionine and phenylalanine.
When amino acids are catabolized, the α-amino group is removed and enters the urea cycle for excretion (see previous section, “Disposal of Amino Acid Nitrogen”), while the carbon skeleton is metabolized. Amino acids are classified as glucogenic, ketogenic, or both based on which intermediates are produced during catabolism.
Glucogenic amino acids yield pyruvate or one of the intermediates of the tricarboxylic acid (TCA) cycle (oxaloacetate, α-ketoglutarate, succinyl CoA, or fumarate) when they are catabolized. This yields lipids and energy in addition to glucose. Most amino acids are exclusively glucogenic.
Ketogenic amino acids yield acetoacetate or one of its precursors, such as acetyl CoA or acetoacetyl CoA, providing lipids and energy. Leucine and lysine, the two exclusively ketogenic amino acids, cannot therefore produce glucose or glycogen in the liver or glycogen in muscle.
Four amino acids are both glucogenic and ketogenic. These include isoleucine, phenylalanine, tryptophan, and tyrosine.
Figure 2-10 depicts the location at which each amino acid is integrated into the TCA cycle.
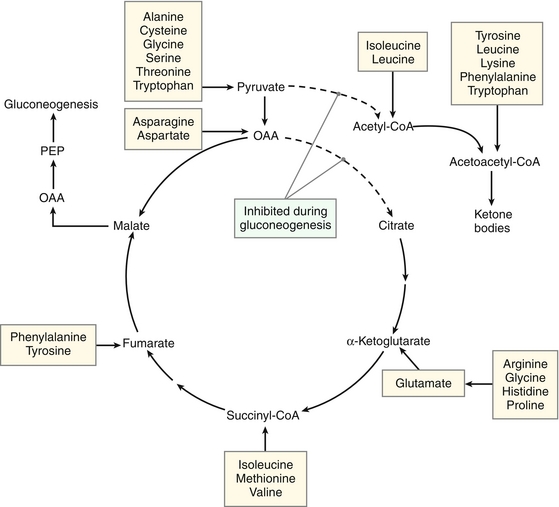
Figure 2-10 Metabolic intermediates from amino acid degradation. CoA, coenzyme A; OAA, oxaloacetic acid; PEP, phosphoenolpyruvate. (From Pelley JW. Elsevier’s Integrated Biochemistry. Philadelphia: Elsevier; 2007.)
Several important disorders arise in patients with deficiencies in the enzymes of amino acid synthesis and degradation pathways, including maple syrup urine disease, albinism, and PKU (Fig. 2-11).
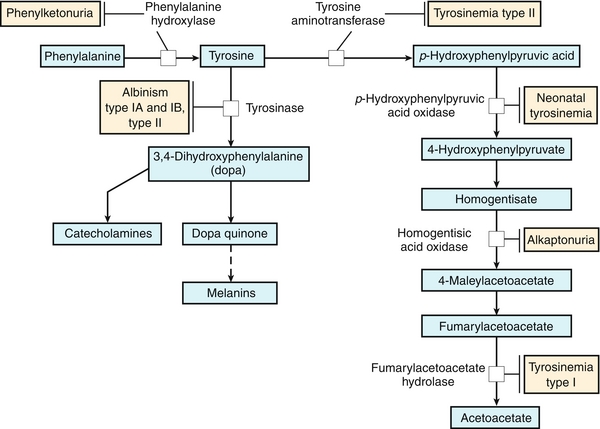
Figure 2-11 Disorders of amino acid metabolism. (From Adkison LR, Brown MD. Elsevier’s Integrated Genetics. Philadelphia: Elsevier; 2007.)
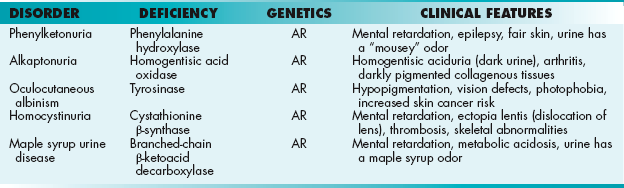
PKU is a disease of amino acid metabolism that is common but can be effectively treated through changes in diet. PKU is caused by a deficiency of phenylalanine hydroxylase. Patients are characterized by a deficiency of tyrosine and elevated phenylalanine. High levels of phenylalanine lead to elevated levels of phenylalanine metabolites such as phenyllactate, phenylacetate, and phenylpyruvate, which give urine a characteristic “musty” or “mousey” odor. Phenylpyruvate in the urine was the phenylketone that PKU was named after. Clinically, patients with PKU have mental retardation, seizures, tremor, microcephaly, and failure to thrive. Symptoms of mental retardation start by 1 year of age. The first step in the pigment melanin formation is hydroxylation of tyrosine by tyrosinase, which is competitively inhibited by the high levels of phenylalanine in PKU. Because of this, patients with PKU also often have hypopigmentation (“fair hair, blue eyes, light skin color”). Early screening and diagnosis are critical because PKU can be treated by diet. However, newborns with PKU may have normal phenylalanine levels at birth owing to maternal transfer; therefore, screening tests are typically done at least 1 to 2 days after birth.
Albinism is a group of conditions in which there is a deficiency in melanin production due to a defect in tyrosine metabolism, causing decreased pigmentation of eyes, hair, and skin. The most severe kind of albinism is called complete albinism and is caused by a complete lack of tyrosinase activity.
Alkaptonuria is a disorder due to the deficiency of homogentisic acid oxidase, an enzyme in the pathway that degrades tyrosine. This leads to accumulation of homogentisic acid, causing homogentisic aciduria, in which the patient’s urine has an elevated level of homogentisic acid. When allowed to oxidize/stand, the urine is oxidized to a dark pigment. Patients with alkaptonuria also exhibit arthritis of large joints and black pigmentation of cartilage and collagen. Treatment includes diets low in phenylalanine and tyrosine. Although this is not a life-threatening disorder, arthritis may be severe.
Maple syrup urine disease is an autosomal recessive disorder in which there is a deficiency of branched-chain α-keto acid dehydrogenase. The inability to oxidatively decarboxylate the branched-chain amino acids leucine, valine, and isoleucine leads to the buildup of α-ketoisocaproic acid, α-ketoisovaleric acid, and α-keto-β-methyl-valeric acid, respectively. This leads to a buildup of branched-chain α-keto acids in the urine, causing a sweet odor. They also accumulate in the blood, leading to toxic effects on the brain. Typically, symptoms present within a few days of birth and include vomiting, severe metabolic acidosis, and a maple syrup odor to the urine. Treatment is with formula with reduced levels of leucine, valine, and isoleucine. However, branched-chain amino acids are important in growth, so low levels are present in the formula.
Homocystinuria is a group of autosomal recessive disorders caused by abnormal homocysteine metabolism. Patients have high levels of homocysteine and methionine in the urine but a low level of cysteine. One important enzyme involved in homocysteine metabolism, cystathionine β-synthase, is mutated in a common form of homocystinuria. Without cystathionine β-synthetase to convert homocysteine to cystathionine, patients have ectopia lentis (lens displacement), osteoporosis, and mental retardation, in addition to early vascular disease. Some patients can improve with treatment with pyridoxine (vitamin B6) because this is a coenzyme of cystathionine β-synthetase. Treatment of homocystinuria includes reduced intake of methionine and administration of vitamin supplements.
Other disorders can also arise from genetic mutations in enzymes important in the amino acid metabolism pathways. These include tyrosinemia type I, methylmalonyl CoA mutase deficiency, histidinemia, and cystathioninuria.
A summary of several important hereditary disorders is included in Table 2-3.
Amino Acid Derivatives
In addition to their role in proteins, amino acids are the precursors of many nitrogen-containing molecules such as pyrimidines, purines, heme, neurotransmitters (glycine and glutamate), and other small molecules.
Figure 2-12 depicts the synthetic pathway of the catecholamines dopamine, norepinephrine, and epinephrine from tyrosine. This synthetic pathway occurs primarily in the CNS, peripheral ganglia, and the adrenal medulla. First, tyrosine is hydroxylated by tyrosine hydroxylase into dopa, a reaction that requires tetrahydrobiopterin (BH4). Second, dopa is decarboxylated, forming dopamine. Dopamine levels are reduced in Parkinson disease; one treatment for Parkinson disease is l-dopa, the precursor to dopamine. Norepinephrine is formed by hydroxylation of dopamine, and epinephrine is then formed by methylation.

The degradation of catecholamines is shown in Figure 2-13. Two enzymes are critical in catecholamine degradation: monoamine oxidase (MAO) and catechol-O-methyltransferase (COMT). Homovanillic acid and vanillylmandelic acid are excreted in the urine. The catecholamine breakdown pathway is important in two different clinical scenarios.
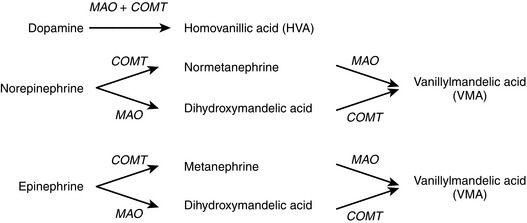
Figure 2-13 Degradation of catecholamines with major degradation products. COMT, catechol-O-methyltransferase; MAO, monoamine oxidase.
MAO inhibitors (MAOIs) are a class of medications used to treat depression. MAOIs can inhibit the breakdown of amines in the diet, which can cause hypertensive crisis if an individual taking an MAOI also consumes foods containing tyramine, such as cheese.
Neuroendocrine tumors of the adrenal medulla are known as pheochromocytomas. The tumorous chromaffin cells secrete large amounts of catecholamines, leading to elevated heart rate, elevated blood pressure, and sweating. Diagnosis of pheochromocytoma includes measuring plasma for catecholamines and metanephrines or urine for vanillylmandelic acid (VMA). Neuroblastomas may also produce elevated levels of catecholamines and are tested for in a similar manner.
The synthesis and breakdown of serotonin, also called 5-hydroxytryptamine (5-HTP), is important in the balance of serotonin in the body (Fig. 2-14). Tryptophan is hydroxylated to form 5-HTP, which is then decarboxylated to form serotonin. Serotonin is degraded by MAO. The presence of high levels of 5-hydroxyindole acetic acid in the urine is diagnostic of cancers that produce large amounts of serotonin.

Several other important nitrogen-containing molecules synthesized from amino acids.
Heme Synthesis and Metabolism
Heme is composed of a tetrapyrrole ring (four linked pyrrole rings) of protoporphyrin IX, with one coordinating ferrous (Fe2 +) iron ion in the left. Heme is found not only in hemoglobin and myoglobin but also in cytochromes and catalase. Heme is synthesized (Fig. 2-15) primarily in the liver (especially cytochrome P-450, which is a heme protein) and the bone marrow. The first reaction and the last three reactions occur in the mitochondria, whereas the rest of the reactions occur in the cytosol. Because reactions occur in the mitochondria, mature RBCs cannot synthesize heme because of their lack of mitochondria. The steps in heme synthesis include the following:
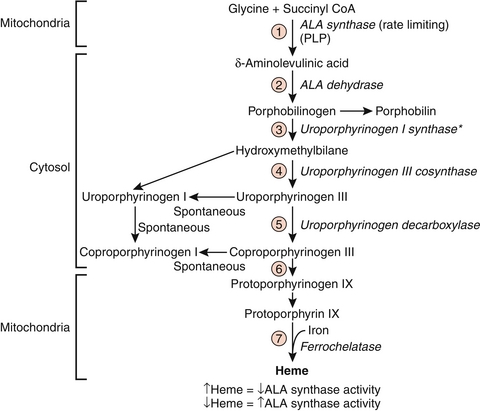
Figure 2-15 Heme synthesis pathway. ALA, δ-aminolevulinic acid. (From Pelley JW, Goljan EF. Rapid Review Biochemistry. 2nd ed. St. Louis: Mosby; 2007, Fig. 8-7.)
First, δ-aminolevulinic acid (ALA) is formed from glycine and succinyl CoA, in a reaction catalyzed by ALA synthase and requiring pyridoxal phosphate as a coenzyme. This is the rate-limiting step of heme synthesis. Hepatic ALA synthase is inhibited by hemin, a protein that forms from heme when porphyrin production is higher than globin production. However, erythropoietic heme synthesis is instead controlled by erythropoietin and iron levels.
When drugs that are metabolized by the cytochrome P-450 system are metabolized, there is increased production of cytochrome P-450 enzymes and a decrease in the heme concentration in the liver. This causes upregulation of ALA synthase production and activity.
In the second step of heme synthesis, porphobilinogen is produced by ALA dehydrase, an enzyme that is inhibited by lead. Another enzyme, ferrochelatase, is also inhibited by lead. The anemia and elevation in ALA commonly seen in lead poisoning are explained by this fact.
A group of disorders known as porphyrias are due to problems with synthesis of heme (Table 2-4). This causes accumulation of heme precursors. (The term porphyria means “purple pigment,” referring to the purple color of the urine of some patients.) There are two kinds of porphyria: deficiencies in liver heme synthesis, called hepatic porphyrias; and deficiencies in bone marrow heme synthesis, called erythropoietic porphyrias. In some of these porphyrias, there is photosensitivity caused by accumulation of tetrapyrrole intermediates, which form superoxide free radicals that destroy cellular components.
Table 2-4
Inborn Errors of Porphyrin Metabolism

AD, autosomal dominant; AR, autosomal recessive; CNS, central nervous system.
Porphyria cutanea tarda is the most common porphyria. It is caused by deficiency in uroporphyrinogen decarboxylase. Photosensitivity is caused by porphyrin accumulation. Urine has a characteristic red-brown color.
Hepatic porphyrias (acute intermittent porphyria, hereditary coproporphyria, and porphyria variegate) cause acute abdominal pain, psychiatric symptoms, and cardiovascular problems. Drugs that induce cytochrome P-450 synthesis can precipitate attacks of hepatic porphyria because increased cytochrome P-450 synthesis reduces heme, increasing ALA synthase synthesis and thus increasing the accumulation of heme precursors.
Erythropoietic porphyrias (congenital erythropoietic porphyria and erythropoietic protoporphyria) present with photosensitivity, blisters, and urine that changes to a red-brown color.
Heme degradation occurs in the spleen by erythrocytes and in the liver by hepatocytes. Within macrophages, hemoglobin and cytochromes are digested into heme, which is converted into biliverdin, a green pigment, by heme oxygenase. Biliverdin is reduced, forming bilirubin, a yellow pigment. Because bilirubin is relatively insoluble in plasma, bilirubin is conjugated to albumin for transport in the blood to the liver. Upon arrival in hepatocytes in the liver, bilirubin becomes bilirubin glucuronide (conjugated bilirubin) using the enzyme bilirubin glucuronyltransferase, an enzyme that is deficient in many patients with Crigler-Najjar and Gilbert syndromes. The conjugated bilirubin is secreted into bile and transported to the intestine, where it is oxidized into stercobilin and excreted (brown pigment in feces), is converted into urobilin and excreted in urine (yellow pigment in urine), or enters the enterohepatic urobilinogen cycle.
NUCLEIC ACID STRUCTURE AND FUNCTION
Nucleic Acid Structure
Nucleic acids include DNA (deoxyribonucleic acid) and RNA (ribonucleic acid). Nucleic acids are assembled from nucleotides, which are composed of a 5-carbon sugar, a nitrogenous base, and one to three phosphate groups. The sugar can be ribose (RNA) or deoxyribose (DNA). The nitrogen-containing base may be a purine (adenine or guanine) or a pyrimidine (cytosine, thymidine, or uracil). DNA contains thymine, whereas RNA contains uracil. DNA has several structural forms; the B form is active DNA, whereas the Z form is inactive.
Purine Metabolism
Purine nucleotides are synthesized de novo from several compounds, including CO2, glycine, glutamine, aspartate, and N10-formyl tetrahydrofolate. The synthesis of purines is an 11-step process (Fig. 2-16A). The first step is synthesis of PRPP (5-phosphoribosyl-1-pyrophosphate) from adenosine triphosphate (ATP) and ribose-5-phosphate, a reaction catalyzed by PRPP synthetase. The second step is the regulated step of purine synthesis, using the enzyme glutamine phosphoribosyl pyrophosphate aminotransferase. The next nine steps lead to the synthesis of IMP (inosine 5′-monophosphate). Two of these steps require N10-formyl tetrahydrofolate. Several drugs target the purine synthesis pathway by inhibiting the formation of tetrahydrofolate (THF). Inhibition of purine synthesis leads to a lack of DNA synthesis, resulting in inhibition of cell growth and division (Fig. 2-16B).
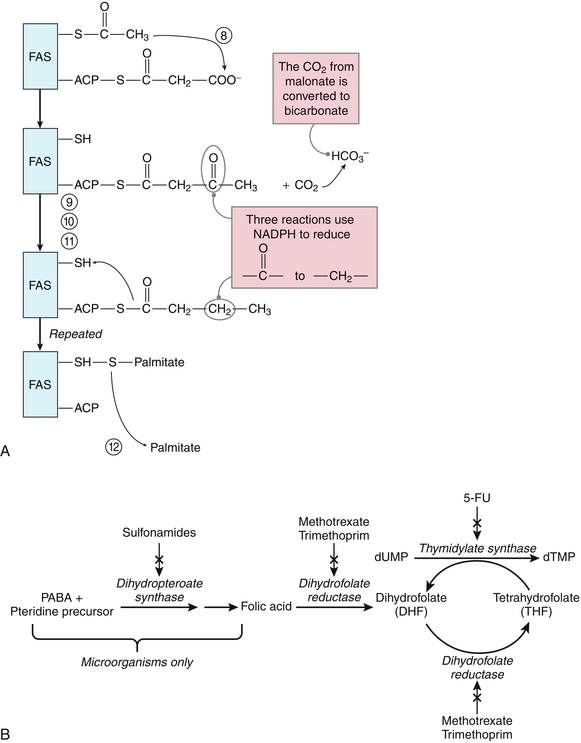
Figure 2-16 A, Simplification of the purine synthesis pathway. B, Folate synthesis and inhibitors. 5-FU, 5-fluorouracil; dTMP, thymidine-5′-phosphate; dUMP, deoxyuridine monophosphate; PABA, paraaminobenzoic acid. (A, from Pelley JW, Goljan EF. Rapid Review Biochemistry. 3rd ed. Philadelphia: Elsevier; 2010.)
The next steps in purine synthesis involve formation of adenosine monophosphate (AMP) or guanosine monophosphate (GMP) from IMP. Mycophenolic acid, an immunosuppressive drug used to prevent organ transplant rejection, is an inhibitor of IMP dehydrogenase, the first reaction to convert IMP to GMP. This reduces the proliferation of T and B cells, resulting in mycophenolic acid’s immunosuppressive effects. The final step in production of purines is synthesis of nucleoside diphosphates from nucleoside monophosphates using nucleoside monophosphate kinases.
Sulfonamides are analogs of paraaminobenzoic acid (PABA) and competitively inhibit the synthesis of folic acid by bacteria. This inhibits the synthesis of THF, which slows the purine synthesis pathway in microorganisms, leading to antibacterial effects. Humans do not synthesize folic acid, but instead obtain it from the diet, so sulfonamides affect only bacterial purine synthesis.
Methotrexate and trimethoprim inhibit dihydrofolate reductase. The inhibition of dihydrofolate reductase inhibits growth of cancer cells (in the case of methotrexate) and bacterial growth (for trimethoprim, a specific inhibitor of bacterial dihydrofolate reductase) because these drugs inhibit synthesis of THF and slow the purine synthesis pathway. However, methotrexate has toxicity for all dividing cells, including cells in the bone marrow, skin, immune system, and gastrointestinal tract, leading to numerous drug side effects.
Another pathway to obtain purines is the purine salvage pathway, in which purines from the diet or cellular breakdown can be recycled. Hypoxanthine and guanine are converted to IMP and GMP, respectively, by the enzyme hypoxanthine guanine phosphoribosyltransferase (HGPRT). Lesch-Nyhan syndrome is an X-linked recessive disorder in which there is a deficiency of HGPRT. These patients are unable to salvage guanine or hypoxanthine using the purine salvage pathway; this leads to increased PRPP and an increase in de novo purine synthesis. This then leads to increased purine turnover and hyperuricemia. Patients with Lesch-Nyhan syndrome present with neurologic features such as self-mutilation, spasticity, and cognitive defects. Uric acid crystals found in their diaper may be due to elevated uric acid in the urine. Although there is no cure, treatment with allopurinol can decrease the hyperuricemia, but it does not alter the neurologic symptoms.
Purines are degraded into uric acid, which is excreted in the urine. Xanthine oxidase converts hypoxanthine to xanthine, and then converts xanthine to uric acid. Two diseases are closely associated with degradation of purines.
Gout occurs when there are high levels of uric acid in the blood (hyperuricemia), which can be caused by overproduction or underexcretion of uric acid. Monosodium urate crystals deposit in the joints, leading to inflammatory arthritis. Most patients with gout underexcrete uric acid. These patients are treated with uricosuric drugs, such as probenecid or sulfinpyrazone, which help to increase the amount of uric acid that is excreted. Patients with gout that overproduce uric acid are treated with allopurinol, which is an inhibitor of xanthine oxidase. This leads to an increase in hypoxanthine and xanthine, which are more soluble than uric acid and do not form crystal deposits. For both kinds of patients, acute gout attacks are treated with anti-inflammatory drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs) and colchicine. Colchicine stops the polymerization of microtubules, which inhibits migration of neutrophils into the inflamed area.
Adenosine deaminase (ADA) deficiency can lead to a form of SCID.
Pyrimidine Synthesis
The first step in pyrimidine synthesis is the formation of carbamoyl phosphate (CAP) by carbamoyl phosphate synthetase II. This is the regulated step of pyrimidine synthesis. After several more steps, uridine 5′-monophosphate (UMP) is synthesized. UMP can then be converted into the pyrimidines thymidine monophosphate (TMP, also known as thymidylate), UTP, and cytidine triphosphate (CTP). Deficiency of UMP synthase (which is a bifunctional enzyme composed of both orotidine phosphate decarboxylase and orotate phosphoribosyltransferase) causes orotic aciduria. Patients have severe anemia, poor growth, and orotate excreted in the urine. Treatment involves feeding synthetic uridine to supply the pyrimidine nucleotides that are needed for DNA and RNA synthesis.
DNA Replication
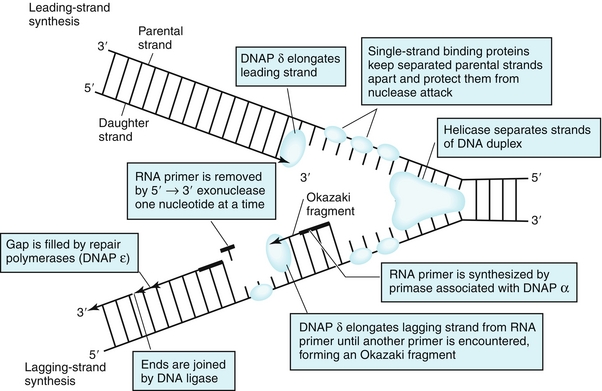
DNA replication begins at the replication fork, where helicases help to separate the strands of DNA (Fig. 2-17). Topoisomerases act to remove supercoiled structures that are formed by this process. Primase adds an RNA primer for DNA synthesis. DNA polymerases (DNAP) synthesize DNA in the 5′ → 3′ direction. Two daughter strands are formed: one leading strand, synthesized continuously in the 5′ → 3′ direction toward the replication fork; and one lagging strand, synthesized discontinuously in the 5′ →3′ direction away from the replication fork using short DNA fragments called Okazaki fragments.
DNA Repair
DNA may need repair due to environmental damage, such as ultraviolet (UV) light or chemicals, mistakes in DNA synthesis, or spontaneous loss of bases. Cells have three principal mechanisms to counteract single-stranded DNA damage.
In DNA mismatch repair (MMR), mismatches in daughter DNA strands compared with the parent DNA are removed by an exonuclease and correctly filled in by DNA polymerase. Hereditary nonpolyposis colorectal cancer (HNPCC) is caused by defects in mismatch repair, most commonly in theMSH2 gene, and leads to an increased risk for colon cancer.
Nucleotide excision repair (NER) is used to correct errors created by UV radiation damage. UV light can cause DNA damage by covalent joining of two thymines that are next to each other, creating a thymine dimer. In NER, UV-specific endonucleases cut one side of the double helix, and then DNA polymerases synthesize a new DNA strand. DNA polymerase has an exonuclease activity, which excises the damaged strand from 5′ to 3′. Finally, DNA ligase joins the ends together. Xeroderma pigmentosum is an autosomal recessive disorder that results from defective nucleotide excision repair due to a mutant UV-specific endonuclease. This leads to high rates of skin cancer after exposure to UV light, including melanoma, squamous cell carcinoma, and basal cell carcinoma.
In base excision repair (BER), altered bases are recognized by DNA glycosylases and cleaved from the DNA backbone, and several other enzymes assist in repair of the site.
In addition, ataxia-telangiectasia, Fanconi syndrome, and Bloom syndrome are also associated with defects in DNA repair.
CARBOHYDRATE STRUCTURE AND METABOLISM
Carbohydrate Structure
Glucose oxidation provides much of the energy needed by cells in the fed state.
Monosaccharides are classified as either aldoses (aldehydes) or ketoses (ketones). Although most sugars can exist as either D or L form optical isomers, most human sugars are D form. The general formula for monosaccharides is (CH2O)x, with the number of carbons being key. Triose sugars have 3 carbons; tetrose have 4 carbons; pentose/furanose have 5 carbons (ribose, fructose, deoxyribose); hexose have 6 carbons (glucose, galactose, fructose).
Monosaccharides can link together through condensation reactions to form disaccharides and oligosaccharides. The bond linking sugars is called a glycosidic bond, which can be either α or β.
Polysaccharides can be linear, such as amylose, or branched, such as glycogen. Starch is the primary glucose storage molecule in plants. Starch can be broken down by humans using amylase. Starch has two components: amylose (linear, α-1,4 linkages) and amylopectin (branched, α-1,4 linkages and α-1,6 linkages). Glycogen is the main way that animals store glucose. The linkages are α-glycosidic (branched, α-1,4 and α-1,6). Each glycogen has one reducing end and many nonreducing ends. Glycogen is produced by the liver and muscle from excess glucose.
Cellulose has β-1,4 linkages and is an insoluble fiber. Humans do not have enzymes that can break it down. Instead, human digestive enzymes cleave α-glycosidic bonds, such as in starch.
Glycolysis
Glycolysis involves the oxidation of glucose and occurs in the cytosol of all cells. Glycolysis can be either aerobic or anaerobic.
In aerobic glycolysis (with oxygen), glucose is oxidized to pyruvate. The pyruvate and NADH created in aerobic glycolysis can be used by the citric acid cycle and the mitochondrial electron transport system to generate 36 to 38 ATP molecules by oxidative phosphorylation.
In anaerobic glycolysis (without oxygen), glucose is oxidized to lactate. Lactate dehydrogenase converts pyruvate to lactate, forming 2 ATP molecules per glucose molecule and reoxidizing NADH to NAD+. Anaerobic glycolysis occurs in anoxic tissues, red blood cells, and skeletal muscle during intense exercise.
There are nine steps in glycolysis. Three regulated enzymes catalyze irreversible reactions in glycolysis: hexokinase, phosphofructokinase, and pyruvate kinase (PK). The first step in glycolysis (Step 1, Fig. 2-18) is phosphorylation of glucose into glucose-6-phosphate by either hexokinase or glucokinase. Phosphorylation traps glucose inside cells because phosphate is negatively charged and charged compounds cannot easily cross cell membranes, and there is no specific transporter for glucose-6-phosphate.
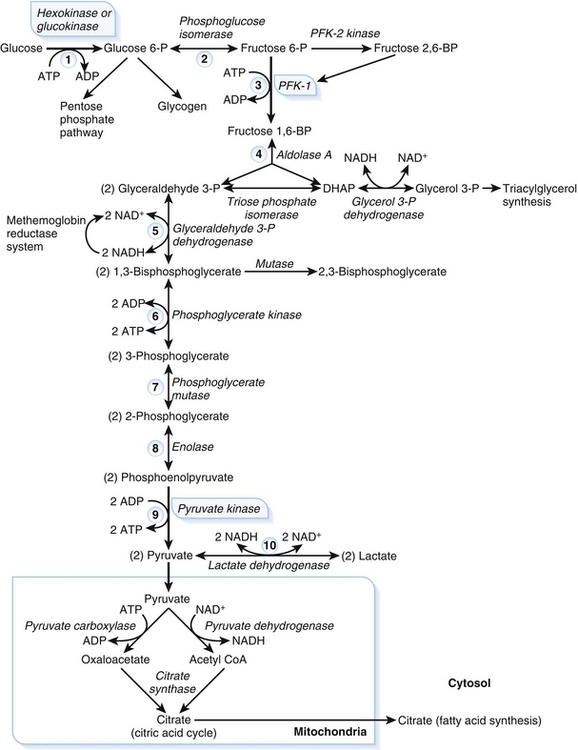
Figure 2-18 Glycolysis pathway. ADP, adenosine diphosphate; ATP, adenosine triphosphate; CoA, coenzyme A; DHAP, dihydroxyacetone phosphate; PFK, phosphofructokinase. (From Pelley JW, Goljan EF. Rapid Review Biochemistry. 3rd ed. Philadelphia: Elsevier; 2010.)
Hexokinase is found in the cytosol of most tissues. Hexokinase has a low Km (high affinity for glucose), ensuring that hexokinase is saturated at normal blood glucose concentrations. Hexokinase is not specific for glucose and can catalyze the phosphorylation of many different hexoses. Hexokinase is also inhibited by glucose-6-phosphate, which prevents too much glycolysis occurring and cells accumulating too much glucose.
Glucokinase is present in the liver and beta cells of the pancreas. In contrast to hexokinase, glucokinase has a high Km (low affinity for glucose), meaning that glucokinase is not saturated at normal blood glucose concentrations, but instead is able to act as a glucose sensor for the liver and beta cells to maintain glucose homeostasis. Glucokinase is highly specific for glucose. Glucokinase is inhibited by fructose-6-phosphate, which prevents glucose from being phosphorylated faster than it is metabolized.
The rate-limiting step of glycolysis is the phosphorylation of fructose-6-phosphate by phosphofructokinase-1 (PFK-1) (Step 3, see Fig. 2-18). Fructose 2,6-bisphosphate strongly activates PFK-1. Fructose 2,6-bisphosphate is created by phosphofructokinase-2 (PFK-2), a bifunctional enzyme that can either produce fructose 2,6-bisphosphate using kinase activity or produce fructose-6-phosphate using phosphatase activity. After a meal, insulin levels increase and glucagon levels decrease; this causes an increase in fructose 2,6-bisphosphate, activating PFK-1 and increasing the rate of glycolysis when glucose is plentiful. In contrast, in the fasting state, there is a decrease in fructose 2,6-bisphosphate, leading to a decrease in the rate of glycolysis.
The final irreversible reaction of glycolysis is formation of pyruvate from phosphoenolpyruvate, which is catalyzed by pyruvate kinase (Step 9, see Fig. 2-18). A mutation in pyruvate kinase causes hemolytic anemia. This is because the ATP generated by anaerobic glycolysis in red blood cells is necessary for red blood cell function. When pyruvate kinase is mutated, the red blood cells die prematurely and are lysed, resulting in hemolytic anemia.
Gluconeogenesis
Gluconeogenesis, the process by which glucose is synthesized, occurs primarily in the liver and kidneys. The carbon sources for the synthesis of glucose is from small precursors, including pyruvate, lactate, glycerol, and glucogenic amino acids. The glucose released from gluconeogenesis is released into the bloodstream. Gluconeogenesis is composed of the reversible reactions of glycolysis. To bypass the irreversible reactions of glycolysis, several reactions specific to gluconeogenesis must occur: (1) conversion of pyruvate to phosphoenolpyruvate (PEP), which bypasses pyruvate kinase; (2) conversion of fructose 1,6-bisphosphate to fructose-6-phosphate, which bypasses phosphofructokinase; and (3) conversion of glucose-6-phosphate to glucose, which bypasses hexokinase.
To convert pyruvate to PEP, pyruvate is first carboxylated to oxaloacetate by pyruvate carboxylase. In a second reaction, oxaloacetate is converted to PEP by PEP-carboxykinase (PEP-CK).
Fructose 1,6-bisphosphate is converted to fructose-6-phosphate by fructose-1,6-bisphosphatase. Fructose 1,6-bisphosphatase is inhibited by fructose-2,6-bisphosphate, a molecule that activates PFK-1 in glycolysis. This ensures that both pathways are not active at the same time.
Glucose-6-phosphate is converted to glucose by glucose-6-phosphatase. This enzyme is also essential for the last steps of glycogenolysis, and a deficiency of glucose-6-phosphatase leads to type 1a glycogen storage disease (Von Gierke disease). There is hypoglycemia in this disorder because the patients are unable to produce glucose by glycogenolysis or gluconeogenesis.
Glycogen Synthesis and Degradation
Glycogen, a polysaccharide, is the way that the body stores glucose in a form that can be rapidly mobilized to maintain glucose homeostasis and to serve as fuel for muscles. Glycogen is stored primarily in liver and muscle tissue, but small amounts are present in most cells.
Glycogenesis is the production of glycogen and occurs in the cytosol. The substrate for glycogenesis is uridine diphosphate (UDP) glucose. Glycogen synthase adds to the nonreducing ends of chains in α(1→4) linkages. Branches with α(1→6) linkages are created by a branching enzyme, amylo-α(1→4)→(1→6)-transglucosidase. Glycogenesis is stimulated by insulin and inhibited by glucagon and epinephrine.
Glycogenolysis is the breakdown of glycogen and also occurs in the cytosol. First, glycogen phosphorylase cleaves α(1→4) glycosidic bonds between individual glucosyl residues’ nonreducing ends, forming glucose-1-phosphate. Next, glucose-1-phosphate is converted to glucose-6-phosphate by phosphoglucomutase. Glucose-6-phosphatase can then convert the glucose-6-phosphate into glucose. The debranching enzyme releases free glucose from α(1 → 6) bonds at the branch points. Glucagon stimulates glycogenolysis, while insulin inhibits glycogenolysis.
Glycogen Storage Diseases
Several inherited deficiencies of glycogen metabolism lead to glycogen storage diseases (Table 2-5).

Metabolism of Sugars
Lactase metabolizes lactose to glucose and galactose. Galactokinase converts galactose to galactose-1-phosphate. In several reactions, galactose-1-phosphate becomes glucose-1-phosphate. Lactase deficiency leads to milk intolerance and causes bloating and diarrhea after ingestion of lactose-containing products. Galactokinase deficiency causes a mild galactosemia, and cataracts can form early. Galactose-1-phosphate uridyl transferase deficiency causes severe galactosemia, resulting in failure to grow, lowered mental capacity, and possibly early death.
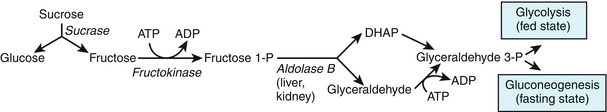
Sucrase converts sucrose to glucose and fructose (Fig. 2-19). Note that hexokinase can convert fructose to fructose-6-phosphate by phosphorylation in the liver and kidney. Fructokinase deficiency leads to essential fructosuria, which is a benign disorder. However, fructose-1-phosphate aldolase deficiency leads to hereditary fructose intolerance. This causes severe hypoglycemia after ingesting fructose or sucrose.
Pentose Phosphate Pathway
The pentose phosphate pathway can either be used for glycolysis, or for oxidation of glucose. The oxidative processes, which are irreversible, generate NADPH needed for pathways such as synthesis of fatty acids and cholesterol. The nonoxidative processes, which are reversible, rearrange sugars so that they can enter glycolytic pathways. Additionally, ribose-5-phosphate can be formed by the pentose phosphate pathways and used for synthesis of nucleotides. G6PD is an enzyme of the pentose phosphate pathway that provides a key regulatory role. Patients with G6PD deficiency, an X-linked recessive disorder, are unable to effectively produce NADPH through the pentose phosphate pathway. This prevents cells from maintaining reduced glutathione, which results in increased oxidative stress on cells, particularly erythrocytes, leading to hemolytic anemia. Some patients with G6PD deficiency only develop anemia when they experience oxidative stress, such as by taking oxidant drugs (sulfamethoxazole, primaquine), eating fava beans, or infection.
TCA Cycle
The tricarboxylic acid (TCA) cycle (Fig. 2-20), also known as the citric acid cycle or the Krebs cycle, occurs in the mitochondria. Red blood cells do not have mitochondria. The pyruvate produced from glycolysis becomes acetyl coenzyme A (acetyl CoA) through the enzyme pyruvate dehydrogenase. Fatty acids can also yield acetyl CoA by beta oxidation. The TCA cycle begins when oxaloacetate condenses with acetyl CoA to form citrate. For one pass through the TCA cycle, oxaloacetate is regenerated, two moles of CO2 are released, 1 guanosine triphosphate (GTP) is produced, and 11 molecules of ATP are produced by oxidative phosphorylation.
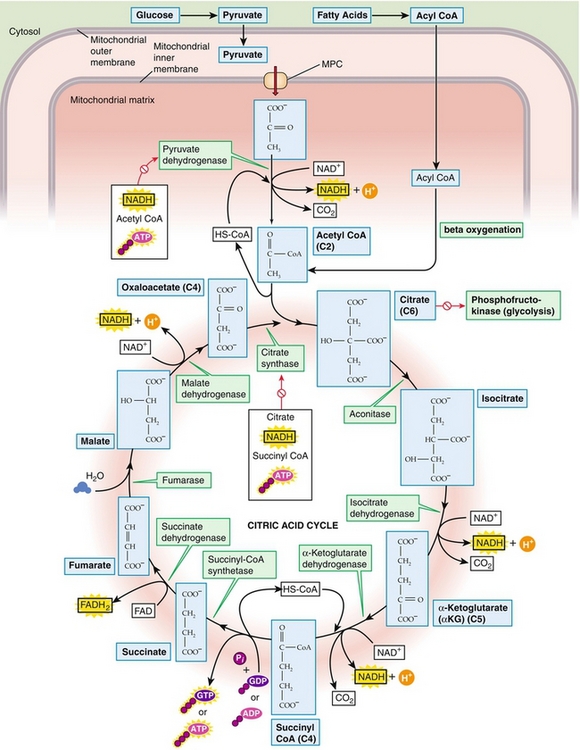
Electron Transport and Oxidative Phosphorylation
The chemiosmotic hypothesis describes the coupling of the electron transport chain to the synthesis of ATP through flow of electrons. As electrons are pumped through the complexes of the electron transport system, hydrogen ions are pumped into the intermembrane space of mitochondria. This forms a proton-motive force from a pH and electrical potential gradient across the mitochondrial membrane. Hydrogen ions passing through ATP synthase down their concentration gradient drive the formation of ATP through ATP synthase.
Uncoupling agents carry hydrogen ions across the inner mitochondrial membrane without transport through ATP synthase. This uncouples flow of electrons and ATP synthesis. This leads to energy dissipated as heat instead of synthesis of ATP. Uncoupling proteins play an important role in animals that hibernate, where stored energy generates heat. In newborn mammals, brown fat has thermogenin, which is an uncoupling protein. This allows energy to dissipate as heat. However, in humans, uncoupling agents such as dinitrophenol (taken in the past for weight reduction) are poisonous.
Several compounds inhibit flow of electrons through the electron transport chain, including cyanide, carbon monoxide, hydrogen sulfide, and amobarbital.
LIPID STRUCTURE AND METABOLISM
Lipid Structure
Fatty acids are the building blocks of lipids (Fig. 2-21). Fatty acids are oxidized in the fasting state to provide energy to cells. Two fatty acids must be supplied in the diet: linoleic and linolenic acid. Fatty acids are made up of an unbranched hydrocarbon chain with a terminal carboxyl group. Most fatty acids have an even number of carbon atoms and 16 to 20 total carbons.
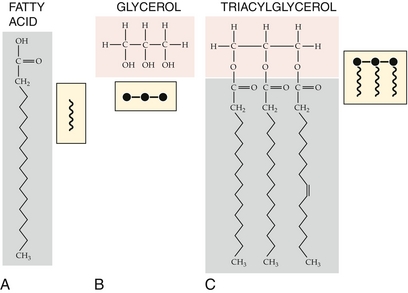
Figure 2-21 Structure of triacylglycerol, 2-monoacylglycerol, and fatty acids. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
Short-chain fatty acids have 2 to 4 carbons, and medium-chain fatty acids have 6 to 10 carbons. They can be directly absorbed in the small intestine. They can also diffuse into the mitochondrial matrix and be oxidized directly.
Long-chain fatty acids have 12 or more carbons. They are in triacylglycerols (fat). To move from the cytosol into the mitochondria, they require the carnitine shuttle.
Unsaturated fatty acids have at least one double bond, most commonly cis (rather than trans) configuration. Trans fatty acids are formed during the production of hydrogenated vegetable oils and have been associated with increase in atherosclerosis.
Triacylglycerols are formed by esterification of fatty acids with glycerol. These have 9 kcal/g and are stored in adipose tissues.
Steroids are a kind of lipid with a four-membered ring structure and a hydroxyl or keto group on the third carbon. There are five major groups of steroids:
Cholesterol (27 carbons): the most abundant steroid in humans, important in cellular membrane fluidity, precursor of steroid hormones, skin-derived vitamin D, and bile acids
Bile acids (24 carbons): include cholic acid
Progesterone and adrenocortical steroids (21 C)
Estrogens (18 carbons): derived from aromatization of androgens
Cholesterol synthesis is regulated by 3-hydroxy 3-methylglutaryl (HMG) CoA reductase, and a key intermediate in cholesterol synthesis is HMG CoA.
Phospholipids are created from phosphatidic acid (diacylglycerol + phosphate group on C3) and are a major component of cell membranes. The higher the melting point of the membrane phospholipid, the less fluid the lipid membrane. Phospholipids are cleaved by phospholipases. Phospholipase A1 and A2 remove fatty acyl groups. Phospholipase A2 in cell membranes is activated by cytosolic Ca2 +, which causes damage to cell membranes when tissues undergo hypoxia. Phospholipase A2 is inactivated by corticosteroids, reducing the release of arachidonic acid. Phospholipase C frees its component compounds, diacylglycerol and inositol triphosphate, which are important in intracellular signaling. Phospholipase D makes phosphatidic acid from phospholipids.
Lung surfactant is important in decreasing the surface tension in alveoli. It is particularly rich in phosphatidylcholines. Insufficient production of lung surfactant by premature infants leads to respiratory distress syndrome, which is characterized by poor gas exchange and partial lung collapse.
Sphingolipids and Sphingolipid Storage Diseases
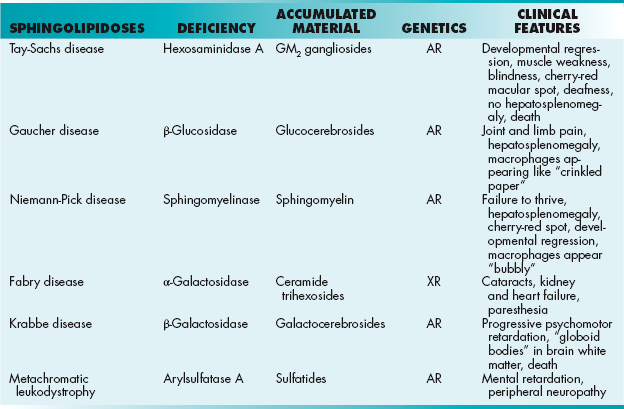
Sphingolipids are derived from ceramide, a molecule that is formed by coupling a fatty acid and sphingosine (sphingosine + fatty acids = ceramide). Sphingolipids are found in the white matter of the CNS. Sphingolipids are essential components of membranes throughout the body and are particularly abundant in nervous tissue. Lysosomal enzymes degrade sphingolipids to sphingosine using several hydrolytic reactions. Sphingolipidoses are a group of hereditary lysosomal enzyme deficiency diseases in which one of these hydrolytic enzymes in the degradative pathway is deficient (Table 2-6). Deficiencies of sphingolipid-degrading enzymes in lysosomes (lysosomes contain hydrolytic enzymes) lead to the accumulation of the substrate in lysosomes and thus to lysosomal storage diseases. In most of these diseases, neurologic deterioration and early death occur. Also note that Fabry disease is X-linked recessive rather than autosomal recessive.
Eicosanoids
Eicosanoids (icosanoids) are important short-range (autocrine/paracrine) signaling molecules that are formed by oxidation of 20-carbon essential fatty acids, including eicosapentaenoic acid (an omega-3 fatty acid) and arachidonic acid (an omega-6 fatty acid). There are four subtypes of eicosanoids: leukotrienes (LTs) and three types of prostanoids—prostaglandins (PG), prostacyclins (PGI), and thromboxanes (TX).
LTs are noncyclic. They are synthesized by hydroxylation of arachidonic acid by lipoxygenase. Leukotriene B4 (LTB4) is an important chemotactic agent for neutrophils, and also increases neutrophil adhesion. LTC4, LTD4, and LTE4 are known as slow-reacting substance of anaphylaxis and increase bronchoconstriction, vasoconstriction, and vascular permeability. LT inhibitors are used for treatment of asthma and include zileuton, an inhibitor of lipoxygenase, and zafirlukast and montelukast, which act to block receptors for leukotrienes on cells.
PGs are created when cyclooxygenase acts on arachidonic acid. Prostaglandin H2 (PGH2) is the first stable prostaglandin produced in this pathway. PGs produce inflammation, inhibit or stimulate muscle contraction, and promote vasodilation or vasoconstriction depending on the vascular bed. PGE2 interacts with several different prostaglandin receptors, which are G-protein-coupled receptors, and leads to vasodilation, inflammation, and an increase in gastric mucus secretion. PGE2 is known as dinoprostone and is used in labor to prepare the cervix for induction of labor. PGF2α stimulates uterine contractions and also increases vasoconstriction. Analogs of PGF2α include dinoprost, latanoprost, bimatoprost, and travoprost. They are used in medicine to induce labor and as abortifacients. Aspirin inhibits cyclooxygenase.
Prostacyclin (PGI2) is an effective vasodilator and bronchodilator and inhibits platelet activation. Synthetic prostacyclin analogs, such as iloprost and cisaprost, are used as vasodilators in severe Raynaud disease, in pulmonary hypertension, and in primary pulmonary hypertension. As a drug, PGI2 is known as epoprostenol. PGI2 is produced in endothelial cells from PGH2. Prostacyclin is in cardiovascular homeostasis with thromboxane A2 (TXA2).
TXA2 is produced in platelets from PGH2 by thromboxane synthase. TXA2 promotes contraction of arterioles and aggregation of platelets. Dipyramidole inhibits thromboxane synthase. Aspirin and other NSAIDs acetylate and inhibit cyclooxygenase, leading to reduced synthesis of prostaglandins (anti-inflammatory effect) and reduced synthesis of TXA2 (antithrombotic effect due to reduced platelet activation).
By inhibiting phospholipase A2, corticosteroids inhibit the production of all the eicosanoids.
Fatty Acid Oxidation and Synthesis
Fatty acids are oxidized to CO2 and H2O in the mitochondrial matrix. Long-chain fatty acids must be shuttled into the mitochondrial matrix by the carnitine transport system because they cannot cross the mitochondrial inner membrane alone. Medium-chain fatty acids are able to pass directly through the mitochondrial membrane. The oxidation of fatty acids occurs by the β-oxidation system of the mitochondria, where each cycle produces 17 ATP molecules using the electron transport system and citric acid cycle.
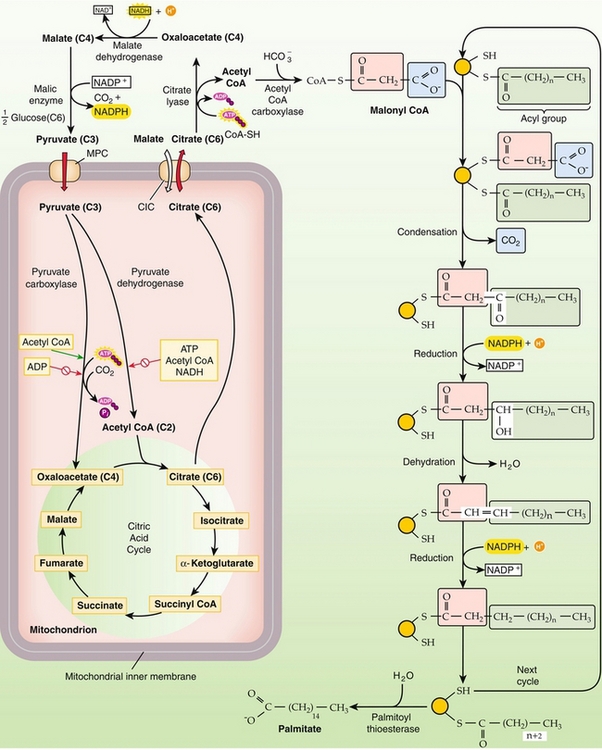
Fatty acids are synthesized from acetyl CoA and malonyl CoA by fatty acid synthase (Fig. 2-22). Seven reaction cycles yield palmitate and fatty acid synthase. Palmitate acts as the precursor to other fatty acids. Longer fatty acids are synthesized by chain-lengthening systems, and unsaturated fatty acids are synthesized by a desaturating system. However, the desaturating enzymes are only able to desaturate double bonds greater than 10 carbons from the C terminus. Therefore, linoleic acid and linolenic acid are essential fatty acids.
INTEGRATION OF METABOLISM
Vitamins
Vitamins are classified as water soluble, excreted in the urine and rarely reach toxic levels, or fat soluble, functioning as hormones, cofactors, and antioxidants (Fig. 2-23). Because fat-soluble vitamins are transported in chylomicrons and stored in the liver and adipose tissue, toxicity can occur.
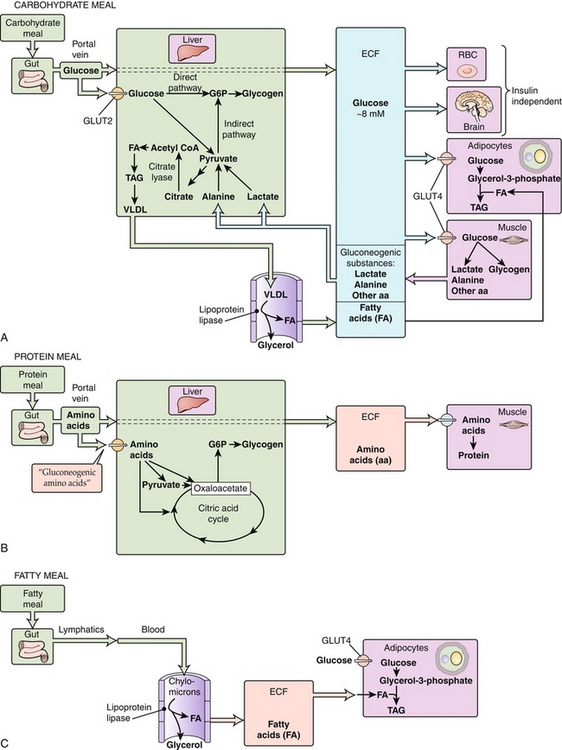
Figure 2-23 Energy utilization and storage following meals. aa, amino acid; CoA, coenzyme A; ECF, extracellular fluid; RBC, red blood cell; TAG, triacyglycerol; VLDL, very-low-density lipoprotein. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
The water-soluble vitamins include thiamine, riboflavin, niacin, pyridoxine, folic acid, vitamin B12, and vitamin C.
Thiamine (vitamin B1) is a water-soluble vitamin that acts as a coenzyme in the decarboxylation of pyruvate and α-ketoglutarate for the enzymes transketolase, pyruvate dehydrogenase, and α-ketoglutarate dehydrogenase. These pathways are important in cellular energy metabolism and production of ATP and are particularly important in the nervous system. Because of its role as a cofactor for transketolase, thiamine deficiency can be diagnosed by either low transketolase activity levels or by increased erythrocyte transketolase activity on addition of thiamine. Two disorders are caused by thiamine deficiency.
 Beriberi is a disorder classically caused by malnutrition due to a diet consisting primarily of polished rice, which is thiamine deficient. Beriberi can occur in two forms: dry and wet.
Beriberi is a disorder classically caused by malnutrition due to a diet consisting primarily of polished rice, which is thiamine deficient. Beriberi can occur in two forms: dry and wet.
Dry beriberi affects the peripheral nervous system, causing wasting, difficulty walking, and paralysis.
Wet beriberi affects cardiac tissue, leading to congestive heart failure with marked edema.
Wernicke-Korsakoff syndrome is seen primarily in chronic alcoholic patients and is due to dietary deficiency or reduced absorption of thiamine. Wernicke-Korsakoff syndrome is associated with atrophy of the mammillary bodies. Wernicke encephalopathy results from severe, acute deficiency of thiamine, whereas untreated Wernicke encephalopathy and chronic thiamine deficiency lead to Korsakoff syndrome, coma, and possibly death. Wernicke encephalopathy is characterized by ataxia, ophthalmoplegia, and confusion. Korsakoff psychosis is characterized by anterograde and retrograde amnesia, confabulation, and hallucinations. Glucose administration to malnourished patients should be preceded by thiamine treatment to avoid the risk for precipitating Wernicke encephalopathy. This is because glucose metabolism involves the action of pyruvate dehydrogenase, an enzyme that requires thiamine.
Riboflavin (vitamin B2) is active as the central component of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), cofactors required for many key steps in energy metabolism. Riboflavin is rarely deficient alone but may accompany other vitamin deficiencies. Symptoms of riboflavin deficiency, known as ariboflavinosis, include dermatitis, glossitis, and fissures at the corner of the mouth.
Niacin (vitamin B3, nicotinic acid) is active as nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+). A severe deficiency in niacin causes the disease pellagra, which is characterized by dermatitis, diarrhea, and dementia (the three Ds). If untreated, it can lead to death (the fourth D). Excessive use leads to flushing. Niacin can increase the levels of high-density lipoprotein (HDL) cholesterol but is not frequently used.
Pyridoxine (vitamin B6, pyridoxamine) is a cofactor in the metabolism of amino acids, heme, and neurotransmitters. Isoniazid (first-line treatment for tuberculosis) interferes with the action of pyridoxine, so supplementation may be necessary to prevent complications of isoniazid treatment such as peripheral neuropathy.
Biotin (vitamin B7) is a cofactor for carboxylase enzymes. Deficiency is rare and may be caused by eating raw egg whites, which bind biotin.
Folic acid (vitamin B9) is essential to the synthesis and repair of nucleic acids as its derivative tetrahydrofolate. Deficiency is caused by malnutrition (especially among alcoholic people) or increased utilization during pregnancy. Folic acid deficiency leads to macrocytic anemia, hypersegmented neutrophils, and neural tube defects. All women should take folic acid before conception and during pregnancy. Medications such as phenytoin, methotrexate, 5-fluorouracil, sulfonamides, and trimethoprim also cause folate deficiency and should be avoided in pregnancy.
Vitamin B12 (cobalamin) is important in DNA and fatty acid synthesis. Like folic acid, deficiency leads to macrocytic anemia and hypersegmented neutrophils. Additionally neurologic deficits occur such as peripheral neuropathy and dementia (cobalamin deficiency is one of the few reversible causes of dementia). Cobalamin is absorbed in the terminal ileum when bound to intrinsic factor (produced by gastric parietal cells). In pernicious anemia, autoimmune destruction of gastric parietal causes vitamin B12 deficiency. Other causes of deficiency include surgical resection of the stomach/ileum, inflammation of the ileum (Crohn disease), the “fish tapeworm” Diphyllobothrium latum, or a vegan diet (because cobalamin is found only in animal products).
Vitamin C (ascorbic acid), as discussed earlier, is a cofactor necessary for the cross-linking of collagen fibers. Deficiency leads to scurvy. Additionally, vitamin C reduces Fe3 + (from dietary plants) to Fe2 + (the absorbable form). Vitamin C deficiency can therefore cause iron deficiency anemia. Vitamin C is found in vegetables and fruits.
The four fat-soluble vitamins are A, D, E, and K.
Vitamin A (retinal, β-carotene) is present within the retina’s rods and cones in the form of retinal. Retinal responds to light by changing shape. It is the first step in a cellular cascade that converts electromagnetic energy (light) into vision. Vitamin A, in its retinoic acid form, has an entirely different function as a growth factor. It plays an important role in developing the anterior-posterior axis during embryogenesis by interacting with Hox genes (which is why isotretinoin [Accutane] is teratogenic). Vitamin A also plays a role in the differentiation of cells (which is why it can be used to treat acne vulgaris and promyelocytic leukemia). Deficiency, usually due to malnutrition, causes night blindness. Toxicity causes neurologic symptoms (headache, blurry vision, confusion). When you think of vitamin A, think of the color yellow: it is found in butter, yellow vegetables, and egg yolks. Excessive consumption of β-carotene, a vitamin A precursor, can turn your skin yellow and look like jaundice–but it preferentially affects the hands and soles and never affects the eyes (no scleral icterus, unlike jaundice).
Vitamin D, when in its active form [1,25(OH)-vitamin D, calcitriol], increases intestinal absorption and renal reabsorption of calcium and phosphate. It also stimulates bone remodeling. The net effect of vitamin D is increasing serum calcium and phosphate levels through the kidneys, gut, and bone. Vitamin D is obtained in the diet or synthesized in the skin through sunlight. This inactive vitamin D then goes to the liver and kidney to become activated as calcitriol. Vitamin D deficiency can be caused by low dietary intake or low sunlight exposure. Renal dysfunction can also lead to a lack of activated calcitriol. Deficiency results in defective bone mineralization. This presents as osteomalacia in adults and rickets in children.
Vitamin E is an antioxidant that protects cell membranes from free radical damage. Deficiency is rare but can lead to a hemolytic anemia due to RBC membrane fragility.
Vitamin K is a cofactor for an enzyme that carboxylates glutamate residues. This is required for the proper synthesis of clotting factors II, VII, IX, X. It is also important for anticoagulant protein C and protein S. Vitamin K is found in leafy green vegetables and is also synthesized by intestinal flora. It must be activated, however, by the hepatic enzyme epoxide reductase. Warfarin, an anticoagulant, functions by inhibiting epoxide reductase and thus inhibiting the formation of clotting factors. Vitamin K deficiency is most commonly characterized by excessive bleeding. It is most commonly seen in people taking warfarin or in cirrhotic patients who lack functional hepatic epoxide reductase. Neonates are supplemented with vitamin K to prevent hemorrhagic disease of the newborn because milk is low in vitamin K and they have no intestinal flora at birth. Patients on broad-spectrum antibiotics reduce their intestinal flora and may also be deficient.
Trace Elements
Iodine is an important component of thyroid hormone. It is naturally found in seafood. Table salt is now fortified with iodine to prevent deficiency. Clinically, iodine deficiency causes hypothyroidism and goiters.
Zinc is second only to iron as the most abundant metal found in the human body. It plays an important role in many enzymes, particularly as zinc finger motifs in proteins that bind to DNA and collagenases that aid in wound remodeling. It also plays a role in spermatogenesis and is found in high concentrations in the prostate and in semen. Deficiency leads to poor wound healing, hypogonadism, and anosmia.
Copper is a cofactor in many enzymes. Notably, these enzymes are involved in the electron transport chain, iron absorption and transport, neurotransmitter synthesis, and collagen cross-linking (lysyl oxidase). Deficiency is rare but leads to iron deficiency anemia and poor wound healing. Wilson disease is an autosomal recessive disorder characterized by excessive total body copper. It is caused by a mutation in Wilson disease protein, an ATPase responsible for transporting copper and forming ceruloplasmin (the transport protein for copper). The inability to excrete copper leads to an increase in free serum copper. Total serum copper may be high, low, or normal, given the relative lack of ceruloplasmin to transport copper. Copper accumulates in the liver, causing cirrhosis, and in the basal ganglia, causing parkinsonism. Excess copper in the cornea is visible as Kayser-Fleischer rings. Diagnosis is based on a decrease in serum ceruloplasmin, an elevation in urinary copper, and copper deposits on liver biopsy.
Fluoride is important in the mineralization of teeth. Adequate consumption prevents dental caries. Excessive fluoride intake can stain teeth white. Fluoride is added to the drinking water in most U.S. communities.
GENETICS
Patterns of Inheritance
Pedigree Analysis
Being able to read a pedigree is critically important to understanding genetic disorders. First, the traditional symbols in pedigrees must be covered (Fig. 2-24). Females are depicted as circles, whereas males are shown as squares. If the sex is unknown, it is typically a diamond (such as in the case of an early fetus). Affected individuals have shaded circles (females) or squares (males), whereas unaffected individuals have unshaded shapes. Sometimes carriers are denoted with half-shaded shapes or dots, but often they are left blank as well. Reading a pedigree will allow you to deduce whether a condition is likely to be autosomal dominant, autosomal recessive, X-linked dominant, or X-linked recessive, or have mitochondrial inheritance. This distinction is important to understanding the risk for disease in future offspring.

Basic Mendelian Inheritance
Genes are found on autosomes and on sex chromosomes. Each individual has two copies of each gene for all autosomes. For sex chromosomes, males only have one X chromosome (because they are XY) and therefore only possess one copy of any gene that lies on the X chromosome. Females are XX, and although they technically have two copies of each gene, because of a process called lyonization (X-inactivation), only one of the X chromosomes is active (in fact, if, because of a genetic defect, a female has more than two X chromosomes, all but one will be inactivated).
There are variations in the genes people have, called alleles. The alleles that an individual has define the individual’s genotype. The expression of those alleles defines someone’s phenotype. Dominant alleles are typically denoted by capital letters (e.g., H), whereas recessive alleles are denoted by lowercase letters (e.g., h). Someone with both alleles as dominant alleles (HH) would be called homozygous dominant; a person with one dominant and one recessive allele (Hh) would be called heterozygous; and a person with two recessive alleles (hh) would be called homozygous recessive. A condition that only requires one defective gene to cause the condition (display the phenotype) is called dominant (if the genes are on autosomes, it is called autosomal dominant; if the genes are on the X chromosome, it is called X-linked dominant). A condition that requires two defective genes to display the condition is referred to as recessive.
Parents will give one of each of their chromosomes (with their alleles) to their children; therefore, one allele for each gene will come from the father, one from the mother. To show which combinations of alleles in a child are possible, Punnett squares are drawn (Fig. 2-25). Punnett deduced the probability of the offspring getting certain combinations of alleles. In a Punnett square, the alleles of each parent are drawn at the end of a 2 × 2 table. Then, in each cell of the 2 × 2 table, write in the corresponding allele for each parent to see what combinations of alleles can occur.
Figure 2-25 Punnett squares demonstrating (A) the output when one parent who is homozygous dominant and one parent who is homozygous recessive (all offspring are heterozygotes), and (B) the output when both parents are heterozygotes (25% of offspring are homozygous dominant, 50% are heterozygous, and 25% are homozygous recessive). (From Jorde LB, Carey JC, Bamshad MJ. Medical Genetics. 4th ed. Philadelphia: Elsevier; 2009.)
In Figure 2-25A, it can be seen that when one parent is homozygous dominant (HH) and the other is homozygous recessive (hh), one parent will always give an H, and the other will always give an h, leading to only Hh (heterozygous) offspring. In Figure 2-25B, it can be seen that both parents are heterozygotes. This leads to a diverse combination in which 25% off the offspring will be homozygous dominant (HH) if both parents give an H and 25% of the offspring will be homozygous recessive (hh) if both parents give an h. The other 50% of the time, the offspring will be heterozygous (Hh) when one parent gives an H and the other gives an h (or vice versa). Practice drawing Punnett squares to familiarize yourself with the concept.
Autosomal Dominant
Autosomal dominant means that for two alleles (which again are alternative forms of the same gene), only one must be present for the disease to manifest itself. For instance, Marfan syndrome is autosomal dominant. This is a syndrome due to a defect in the gene that codes a protein in connective tissue called fibrillin. This connective tissue defect leads to individuals who are tall, have long limbs and fingers, and are predisposed to aortic dissections, eye lens dislocations, and other conditions. Fibrillin is on chromosome 15 (an autosome, as it is not a sex chromosome); each individual has two copies of the gene that codes for fibrillin. Since Marfan syndrome is autosomal dominant, only one gene must be abnormal for an individual to have Marfan syndrome. Figure 2-26 depicts what the pedigree for an autosomal dominant condition for Marfan syndrome might look like.
Figure 2-26 A, Pedigree depicting an autosomal dominant condition. Note that all the affected individuals are shaded; they possess only one “A” (are all heterozygous by genotype) but display the features of the disease (phenotype). Therefore, because only one allele had to be present for the disease to manifest, this is autosomal dominant. B, Punnett square showing one affected parent and one affected parent: half of their children will be affected. (From Jorde LB, Carey JC, Bamshad MJ. Medical Genetics. 4th ed. Philadelphia: Elsevier; 2009.)
To further familiarize yourself with Punnett squares and with pedigrees, refer again to Figure 2-26. It can be seen that the father has Marfan syndrome (shaded square) and the mother does not (blank circle). If a Punnett square were to be drawn between the two (see Fig. 2-26B), it would be seen that there is a 50% chance of a child having Marfan syndrome. In this case, one of the two children (50%) did have Marfan syndrome. The unaffected daughter (aa) went on to have children with an unaffected man, and therefore none of the children could possibly have Marfan syndrome. The affected son had children with an unaffected woman and subsequently still had a 50% risk for having children with Marfan syndrome.
Because only one allele must be present to display the disease, and having the allele causes expression of the disease, this brings a few “rules” for autosomal dominant inheritance. Note that if a new mutation occurred in one of the genes, these rules do not apply (e.g., if one of the father’s sperm had a new mutation for the fibrillin gene and subsequently caused a child with Marfan syndrome).
The affected offspring must have had one affected parent (unlike recessive).
Unaffected individuals cannot have children with the disease (unlike recessive).
Males and females have an equal likelihood of getting and passing on the disease (unlike X-linked).
As a general rule, patients with autosomal dominant conditions live to reproductive age. Otherwise, these conditions would be rapidly eliminated from the population (no carrier state). Consider Huntington disease, autosomal dominant polycystic kidney disease, and Marfan syndrome as examples.
Autosomal Recessive
Autosomal recessive diseases are those in which two mutated alleles must be present for the disease to manifest itself (Fig. 2-27). Therefore, if a child has an autosomal recessive disease but neither parent displays the disease, the parents must have both been heterozygotes (Hh). Refer to Figure 2-25B, which shows that when both parents are heterozygotes, they have a 25% chance of producing a homozygous recessive offspring (hh). An example of an autosomal recessive disease is cystic fibrosis (see Fig. 2-27 for details; see also Chapter 17). In cystic fibrosis, there is an abnormal chloride channel (called the cystic fibrosis transmembrane conductance regulator, or CFTR); loss of function of this channel leads to thickened mucus production in the lungs (predisposing to infections) and thickened pancreatic secretions (leading to pancreatitis and type 1 diabetes mellitus). Having one functionalCFTR gene allows for normal function. Therefore, because both genes must have mutant alleles, cystic fibrosis is homozygous recessive.
Figure 2-27 Autosomal recessive disease. A, Pedigree showing affected children (shaded) at the bottom. The dots in this case indicate carriers. The two carriers who produced the affected children are both heterozygotes. The likelihood of them both being heterozygotes was increased because of consanguinity; both individuals were close relatives. B, Punnett square showing two heterozygotes with a one in four chance of having an affected child. (From Jorde LB, Carey JC, Bamshad MJ. Medical Genetics. 4th ed. Philadelphia: Elsevier; 2009.)
There are also “rules” for autosomal recessive inheritance.
Suspect autosomal recessive inheritance when a child has a genetic disease but has two unaffected parents (who then must be heterozygotes themselves).
If both parents are heterozygotes (carriers), only 25% of their children will be affected, on average.
As with autosomal dominant inheritance, males and females are equally likely to be affected (unlike X-linked).
Consanguinity (mating with close relatives) increases the likelihood of recessive diseases to manifest because the likelihood of close relatives being heterozygotes for a disease is higher than the likelihood of two unrelated individuals both being heterozygotes for a particular disease.
X-Linked Recessive
Males are XY and females are XX, so the likelihood of expression of disease differs in X-linked diseases (unlike autosomal diseases, which do not favor one sex or the other because males and females have the same autosomal chromosomes). Because males only have one X chromosome, an X-linked recessive disease will cause the male to have the phenotype of the disease with just one mutant allele. Females, on the other hand, have two X chromosomes, and therefore having a single mutant allele will not necessarily cause disease (because they have another X chromosome with a functional allele). This is complicated somewhat by X-inactivation, whereby the female cells inactivate one of the two X chromosomes in each cell. Therefore, in females with one abnormal allele on an X chromosome, about 50% of the body’s cells will have inactivated the chromosome with the abnormal allele, but 50% will have inactivated the other. Usually, this does not lead to significant disease.
The classic X-linked recessive disease is hemophilia (both hemophilia A and hemophilia B are X-linked recessive). Hemophilia A causes a deficiency in factor VIII, and hemophilia B causes a deficiency in factor IX; both of these deficiencies in clotting factors lead to deep tissue bleeding, joint bleeding, and other bleeding-related symptoms. Another common X-linked disease is Duchenne muscular dystrophy and its milder subtype, Becker muscular dystrophy. Both of these cause skeletal muscle degeneration, eventually leading to paralysis and death (with Duchenne muscular dystrophy nearly inevitably causing inability to walk by age 12 years; Becker muscular dystrophy patients may be able to ambulate for significantly longer).
In Figure 2-28A, it is clear that only males are affected. This is a striking feature of X-linked recessive diseases. In Figure 2-28B, the mating between a normal XY father and a carrier XX mother is shown (X1 is the normal allele, X2 is the abnormal allele). Because the female offspring are XX, one X must be from the father, and one must be from the mother. In this case in which the mother is the carrier, 50% of the daughters then will also be carriers. Male offspring must receive the Y from the father because the mother cannot provide a Y chromosome. Therefore, an interesting finding in X-linked recessive disease is that a male with the disease cannot give the disease to his son (because by definition the male offspring X chromosome must have come from the mother and not the father).
Figure 2-28 X-linked recessive disease. A, Pedigree showing that only males are affected; there are numerous female carriers. B, Punnett square showing mating between an unaffected male (XY) and a carrier female (XX). X1 is the normal allele, X2 is the abnormal allele. (From Jorde LB, Carey JC, Bamshad MJ. Medical Genetics. 4th ed. Philadelphia: Elsevier; 2009.)
Knowing what we know now about X-linked recessive inheritance, we can form the “rules”:
Affected males cannot give the disease to their sons (because they have to give their Y chromosome to make a son); the only way that a son of an affected father can have the disease is if the mother is an asymptomatic carrier.
Affected males will always give their X chromosome (with their mutated allele) to their daughters (because if they gave their Y chromosome, it would be a son). Therefore, all daughters of affected males are carriers.
Heterozygous women (X*X) will give their mutant allele away half the time. Therefore, 50% of the daughters will be carriers, 50% of the sons will have the disease.
X-Linked Dominant
This is incredibly rare. Because it is dominant, there cannot be any carriers. However, the defining feature that separates X-linked dominant conditions from autosomal dominant conditions is that affected males cannot transmit the disease to other males. This is because if a male is affected, he is affected because he has an abnormal allele on his X chromosome. However, to have male offspring, he has to give his Y chromosome. Suspect X-linked dominant only if the transmission appears dominant but there are numerous affected males who have normal male offspring and affected female offspring. Fragile X syndrome and Rett syndrome are examples of X-linked dominant conditions.
Mitochondrial Inheritance
Mitochondrial inheritance is easy to detect if you remember to think of this as a possible mode of transmission. All mitochondria in your entire body initially came from your mother’s egg. The sperm do not provide mitochondria. Therefore, with mitochondrial diseases, affected females will have 100% of their offspring affected, but affected males will have 0% of their offspring affected (Fig. 2-29). Affected males do not give their mitochondria to their children, so it is impossible for males to transmit the condition to their offspring. Important conditions that are inherited through mitochondria are Leber hereditary optic neuropathy, mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS), and myoclonic epilepsy and ragged red fibers (MERRF).
Figure 2-29 Mitochondrial inheritance. It can be seen that affected females pass their mitochondrial disease onto 100% of their offspring. However, males do not contribute mitochondria to their offspring (because it is only present in the egg), so none of the affected males will have affected offspring. (From Jorde LB, Carey JC, Bamshad MJ. Medical Genetics. 4th ed. Philadelphia: Elsevier; 2009.)
CHROMOSOMAL ABNORMALITIES
Nondisjunction
Meiosis is the type of cell division that occurs to produce gametes (sperm and eggs). It is incredibly complex and therefore is prone to error. One type of error that can occur is nondisjunction. There are two opportunities for nondisjunction during meiosis: during the first round of meiosis (meiosis I), or during the second round of meiosis (meiosis II). Figure 2-30 depicts a simplified illustration of this complex concept with only one pair of chromosomes. In the top image, nondisjunction has occurred during meiosis I. This means that a pair of homologous chromosomes has failed to separate. Imagine for the sake of discussion that this pair of homologous chromosomes was a pair of chromosome 21 and that this was meiosis for a female. This would lead to four eggs: Two eggs would have an additional 21st chromosome and would produce a child with trisomy 21 (Down syndrome) if fertilized by a normal sperm. The other two eggs would not have a 21st chromosome and would produce a child with monosomy 21 if fertilized by a normal sperm.
Figure 2-30 Examples of nondisjunction in meiosis I (top) and meiosis II (bottom). (From Adkison LR. Elsevier’s Integrated Review Genetics. 2nd ed. Philadelphia: Elsevier; 2011.)
Essentially all cases of monosomy are lethal in utero and lead to spontaneous abortion, except for Turner syndrome (monosomy X). Most autosomal trisomy cases are also lethal in utero, with the exception of trisomy 21 (Down syndrome), trisomy 13 (Patau syndrome), and trisomy 18 (Edwards syndrome). To remember which is which, think trisomy 18 for Edwards syndrome—at age 18 you can participate in the elections. These will be briefly discussed later; there are also abnormalities of increased numbers of sex chromosomes, which are typically not lethal.
Nondisjunction can also occur during meiosis II. Because the first division has occurred appropriately, the outcome is different. The first division produced two cells. Nondisjunction in meiosis II would only affect one of the two cells, so the unaffected cell would be expected to divide normally. Therefore, of the four gametes, two would be normal. The other gamete, which had nondisjunction in meiosis II, would produce two abnormal gametes: one with an extra chromosome (e.g., chromosome 21 causing trisomy 21), and one with a missing chromosome (e.g., causing monosomy of that chromosome).
In summary, nondisjunction in meiosis I leads to four abnormal gametes—two with an extra chromosome, two with a missing chromosome. Nondisjunction in meiosis II leads to only two abnormal gametes because the other normal gametocyte produced during meiosis I will divide normally. This leads to one gamete with an additional chromosome, one with a missing chromosome, and two normal gametes.
Autosomal Trisomies
As stated previously, there are only three autosomal trisomies that are not lethal in utero: Down syndrome (trisomy 21), Patau syndrome (trisomy 13), and Edwards syndrome (trisomy 18).
Down syndrome is the most common trisomy and also the only trisomy that allows for survival into adulthood. This is typically associated with cognitive delays and a set of distinguishing physical characteristics, including a single palmar crease, protruding tongue, hypertelorism (increased interorbital distance), and epicanthic skin folds on the inner corner of the eyes. Unfortunately, those with Down syndrome are at increased risk for acute lymphoblastic leukemia, Alzheimer disease, and congenital heart disease (specifically endocardial cushion defects). Note that in addition to nondisjunction, patients with Down syndrome can also have an extra 21st chromosome if one of the 21st chromosomes attaches abnormally to another chromosome. This is called translocation and can either be asymptomatic in the mother (e.g., mother has two 21st chromosomes, but one of them is attached to another chromosome, leading to the potential for trisomy 21 in the offspring) or can occur during rearrangement in meiosis.
Both Patau and Edwards (Fig. 2-31) syndromes are associated with extremely high mortality by 1 year of age. They both have much in common: the risk for congenital heart defects is high, both have severe cognitive impairment, and they include numerous congenital defects. The features that set them apart are that patients with trisomy 13 (Patau syndrome) have cleft lip and palate and polydactyly, and patients with trisomy 18 (Edwards syndrome) have clenched hands with overlapping fingers and rocker-bottom feet.
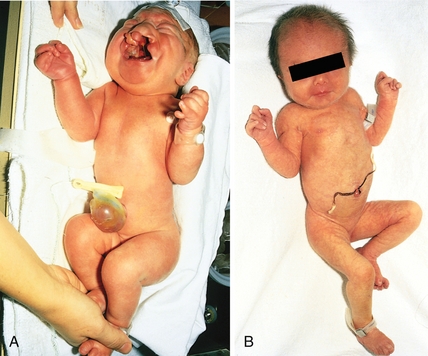
Figure 2-31 A, Trisomy 13 (Patau syndrome) with characteristic cleft lip and palate as well as polydactyly. This baby also has an omphalocele. B, Trisomy 18 (Edwards syndrome) with characteristic clenched fists with overlapping fingers. (From Adkison LR. Elsevier’s Integrated Review Genetics. 2nd ed. Philadelphia: Elsevier; 2011.)
Sex Chromosome Aneuploidy
It has been discussed how, through nondisjunction (or translocation), an extra (or one fewer) autosomal chromosome can occur. However, the same process can also occur in sex chromosomes. This leads to a few specific conditions, the most important of which are Klinefelter syndrome and Turner syndrome.
Klinefelter syndrome occurs when instead of having 46,XY (normal male) or 46,XX (normal female), the individual is 47,XXY. Therefore, the person has an extra sex chromosome (47 total chromosomes instead of 46). Patients with Klinefelter syndrome are phenotypically male but have an inactivated X chromosome in each of their cells, just as females would. This can be seen microscopically as a Barr body (the remnant of the inactivated chromosome). Those with Klinefelter syndrome typically are tall, have long arms and legs, and have small testes. They are typically sterile.
Turner syndrome is a condition in which instead of being 46,XX (normal female), the patients are 45,Xo. This means they only have a single X chromosome. They typically are short and have ovarian dysgenesis (“streak” gonads, which are nonfunctional, causing sterility and lack of estrogen and sexual infantilism). They have a characteristic physical appearance that includes a broad shieldlike chest, lymphedema of the hands and feet, and a webbed neck (Fig. 2-32). They are also at risk for coarctation of the aorta and a bicuspid aortic valve.