Hematology and Oncology
Hematology is the study of the physiology and pathology of blood and blood-forming organs. The pluripotent hematopoietic stem cell (HSC) is the precursor to all cells in blood and also gives rise to cells of the lymphoid system. As a result, lymph nodes and lymph tissue are included in the study of hematology. The organs associated with hematology are usually defined by the sites in which pathology can arise; these include the bone marrow, lymph nodes, and intravascular compartment. The intravascular compartment is where blood cells circulate and includes the endothelial cell lining of blood vessels as well as the proteins in the blood plasma.
ANATOMY
Blood
Blood is a specialized connective tissue composed of red blood cells (RBCs), white blood cells (WBCs), and platelets suspended in plasma.
Blood Cells
Blood cells include three major types: (1) erythrocytes (RBCs), (2) leukocytes (WBCs); and (3) platelets. About 45% of the total blood volume is made up of blood cells.
Plasma
The straw-colored liquid that suspends the blood cells in whole blood. Blood plasma is obtained by centrifugation of whole blood and is the acellular liquid that remains after the blood cells are removed. Plasma contains clotting factors (unlike serum) and other components, such as dissolved proteins and electrolytes. Blood plasma makes up the other 55% of total blood volume. Plasma is serum plus clotting factors.
Serum
This is blood plasma without clotting factors. As noted, the HSC is the precursor to cells in the blood and lymphoid system. The HSC has the ability to differentiate into one of two cells, the common myeloid progenitor (CMP) cell or the common lymphoid progenitor (CLP) cell (Fig. 11-1). The myeloid system originates from CMP cells which can give rise to RBCs, WBCs, and platelets. The cells of myeloid lineage arise in the bone marrow. CLP cells generate both T and B lymphocytes through a process called lymphopoiesis.
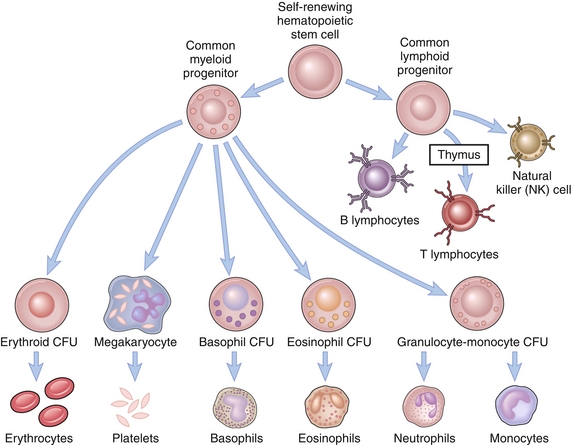
Figure 11-1 Erythropoiesis and the differentiation of the self-renewing hematopoietic stem cell (HSC) into a common myeloid progenitor cell, which can make erythrocytes, platelets, basophils, eosinophils, neutrophils, and monocytes, or a common lymphoid progenitor cell, which can make B and T lymphocytes and natural killer cells. CFU, colony-forming unit. (From Abbas AK. Cellular and Molecular Immunology Updated Edition. 6th ed. New York: Elsevier; 2009.)
The Myeloid Lineage
Erythrocytes (RBCs) are specialized biconcave anucleate cells that carry oxygen to and carbon dioxide from tissues via the hemoglobin protein. To maximize gas exchange (O2 and CO2), RBCs have a large surface area–to–volume ratio. Also, in an effort to increase their oxygen-carrying capacity, as RBCs mature, they lose most of their cell contents (including their nucleus and other organelles, such as ribosomes and mitochondria) and replace them with hemoglobin. As a result of the loss of their mitochondria, they can only use glucose as a source of energy and obtain adenosine triphosphate (ATP) mostly via glycolysis (< 10% of ATP comes from hexose monophosphate shunt). As the RBC ages, it loses its energy-producing capabilities, causing its cell membrane to stiffen. These “stiff” old RBCs are at increased risk of getting caught up in the splenic circulation and being removed by splenic macrophages. The RBC life span is about 120 days.
Key Definitions
 Erythrocytosis or polycythemia is an increase in the number of RBCs.
Erythrocytosis or polycythemia is an increase in the number of RBCs.
Leukocytes (WBCs) are the blood cells that help the body fight infections or mount inflammatory responses. The normal count ranges from 4000 to 10,000 cells/μL. Leukocytes can be divided into five subtypes, referred to as the WBC differential (in order, from highest percentage to lowest): neutrophils, lymphocytes, monocytes, eosinophils, basophils (see mnemonic in the accompanying box).
Neutrophils
Also referred to as polymorphonuclear neutrophils (PMNs) or segmented neutrophils because they contain a nucleus with multiple segments (see Fig. 11-1). Neutrophils account for 50% to 80% of the total WBC count. These cells are an essential part of the innate immune system. Neutrophils are typically the first responders in the acute phase of inflammation and are the predominant cells in pus. They act to phagocytize (engulf) and digest bacteria, cellular debris, and dead tissue. In the circulation, half are marginated (or adherent to the endothelial cells of blood vessels) and the other half are in the peripheral circulation. They exit the circulation via chemotactic stimuli in a process called diapedesis. Neutrophils can be divided into three morphologic groups:
Nonsegmented (band) neutrophils are immature neutrophils that can be seen in acute infections or inflammation. The bone marrow is sending out everything it has to fight an infection, even if the cells are not fully mature.
Segmented mature or morphologically appropriate neutrophils with three to five nuclear lobes.
Hypersegmented neutrophils usually have more than five lobes and are associated with vitamin B12 or folate deficiencies.
Monocytes are large mononuclear cells that are easily recognizable by their kidney-shaped nucleus and account for 2% to 10% of all WBCs. Monocytes become macrophages when they leave the bloodstream and enter into tissue. They form part of the mononuclear phagocyte system and act to clean up circulating debris, microorganisms, senescent RBCs, and damaged cells. Macrophages survive in tissues for up to 80 days and are named by their tissue or origin (e.g., Kupffer cells in the liver, alveolar macrophages in lungs, oligodendrocytes or glial cells in brain). Monocytes and macrophages function as antigen-presenting cells by phagocytosing pathogens and displaying proteins from them on their cell surface (via major histocompatibility complex [MHC] class II) for recognition by lymphocytes.
Eosinophils are cells with bilobed nuclei and have prominent eosinophilic (reddish-orange) granules (see Fig. 11-1). They account for 1% to 6% of WBCs. They contain proteins, such as major basic protein (MBP), in their granules that are released (degranulated) in response to immune stimulus by foreign proteins. MBP is important in defending against helminthic and protozoan infections. Eosinophils are increased in invasive parasitic infections, allergic processes, and neoplasms.
Basophils also have bilobed nuclei but have granules that stain blue (basic stain; basophilic granules). They account for less than 1% of WBCs. They are most commonly involved in inflammatory reactions that cause allergic symptoms. They express IgE receptors that release histamine, heparin, prostaglandins, leukotrienes, and other vasoactive amines when stimulated. Basophils become mast cells when they exit the circulation and enter tissues. Mast cells are involved in type I hypersensitivity reactions and mediate allergic reactions via the release of histamine. Basophil levels are often elevated in patients with myeloproliferative disorders such as chronic myelogenous leukemia.
Platelets or thrombocytes are anucleate cell fragments that play a central role in hemostasis (blood coagulation). They are derived from the cytoplasm of megakaryocytes in the bone marrow. The life span of platelets is about 7 to 10 days, with the first 2 days of life spent in the spleen. This becomes important in congestive or inflammatory disorders that can lead to the entrapment of platelets in an enlarged spleen. The normal platelet count ranges from 150,000 to 400,000/mm3.
The Lymphoid Lineage
Lymphocytes are also leukocytes (as are all white blood cells), but these originate from CLP cells in the bone marrow and are therefore of lymphoid, rather than myeloid, lineage. They are small round cells with a small amount of cytoplasm and densely staining nuclei (Fig. 11-2). They normally account for 25% to 33% of WBCs. There are four types of lymphocytes: T cells, B cells, plasma cells, and natural killer (NK) cells.
Figure 11-2 A lymphocyte, showing the characteristic large densely-staining nucleus. (From Stevens A, Lowe JS. Human Histology. 3rd ed. Philadelphia: Elsevier; 2004.)
T lymphocytes are primarily responsible for mediating the cellular immune response. They are made in the bone marrow and later migrate to the thymus, where they begin to mature. Once they mature, they leave the thymus and reside in the lymphoid tissues. T cells can be distinguished from other lymphocytes by the presence of a T cell receptor. They differentiate into helper T cells, cytotoxic T cells, and suppressor T cells.
Helper T cells or CD4 T cells help other WBCs complete their immunologic tasks (e.g., maturation of B cells into plasma cells, activation of macrophages or cytotoxic T cells). They express CD4 and recognize their targets by binding MHC II. MHC II is expressed on the surface of APCs (see Chapter 6 pg.3 for discussion of MHC).
Cytotoxic T cells or CD8 T cells help eliminate tumor cells and cells infected by viruses. They express CD8 and recognize their targets by binding MHC I. MHC I is expressed on the surface of almost every cell in the body except anucleate cells such as platelets and red blood cells.
B lymphocytes are part of the humoral immune system. Their primary functions are to create antibodies against antigens, act as APCs and to develop into memory cells for future immune response. They are made in the bone marrow, partly mature in the bone marrow, and later migrate to lymphoid tissues. Large quantities of B cells reside in the follicles of lymph nodes and white pulp of the spleen. The terminal differentiation of a B cell happens in the peripheral lymph tissues, such as lymph nodes, when they encounter antigens. Once they encounter an antigen, B cells differentiate into plasma cells that can produce antibodies.
NK cells are a part of the innate immune system and are a type of cytotoxic lymphocyte. They play a large role in the destruction of cells infected by viruses and neoplasms. They work by releasing proteins (perforin and granzyme) that induce programmed cell death (apoptosis).
PHYSIOLOGY
Blood Groups: ABO and Rh Systems
Blood grouping is based on the following: (1) the presence of surface antigen(s) on RBCs; (2) the presence of plasma antibodies (Ab) against antigen(s) that are not present on host RBCs; and (3) the presence or absence of Rh factor.
The ABO system is an excellent example of codominant expression of alleles of a gene (Table 11-1), meaning that the contributions of both alleles are visible in the phenotype. If a person has inherited the IA and IB alleles they will have the AB blood group phenotype and produce both A-type and B-type antigens on the RBC cell surface. Your body always produces antibodies (Ab) against antigens not present on your RBC cell surface.
Table 11-1
Phenotype (Expressed) and Genotype (Genetic Makeup) for Red Blood Cell Surface Proteins
| Phenotype | Genotype |
| A | AA or AO |
| B | BB or BO |
| AB | AB |
| O | OO |
AB Group
Produces A and B antigen on RBC surface and no Abs. These patients can receive blood transfusion from all blood types because no Abs against RBC cell surface antigens are produced (universal recipient).
O Group
Produces no antigens on RBC surface and anti-A and anti-B Abs. These patients can donate blood to all blood types because their RBCs lack antigen on the cell surface (universal donor).
The Rh system is based on the presence or absence of the five most important antigens—D, C, c, E and e—on the RBC cell surface. However, the most clinically important antigen is the D antigen (it is the most immunogenic). Strictly speaking, when a person is said to be Rh-positive, that person produces the D antigen. Unlike the ABO system, Rh antibodies are only made if an Rh-negative person is exposed to Rh-positive blood. At baseline, an Rh-negative person does not produce the D antigen on their RBC cell surface and does not produce anti-Rh Abs. Rh-negative individuals can become sensitized (i.e., produce anti-Rh Abs) during a blood transfusion from Rh-positive blood.
For example, an Rh-negative mother is carrying an Rh-positive fetus and significant hemorrhage occurs during pregnancy, or the mother is exposed to Rh-positive blood during birth. In this situation, Rh immunoglobulin (RhoGAM) is given to mothers at about 28 weeks of pregnancy and within 72 hours after delivery. These anti-D antibodies bind all Rh antigens and prevent the mother from being exposed to them, thus preventing sensitization in the mother (development of anti-Rh IgG Abs).
Hemostasis: The Coagulation Cascade
Hemostasis is the cessation of blood flow through a blood vessel or body tissue and is the physiologic end point following injury to a vessel wall or tissue. Normally, the endothelial cells of intact vessels prevent hemostasis by continually secreting anticoagulants and inhibitors of platelet aggregation. When injury to the vessel wall occurs, the endothelial cells cease to produce these inhibitors and instead release procoagulation factors (von Willebrand factor and tissue thromboplastin) that initiate hemostasis. Normally, hemostasis and thrombosis result from a careful interplay between protein and cell components. The first component (protein) involves a group of proteins that specialize in coagulation (clot formation), fibrinolysis (clot dissolution), and anticoagulation (regulation of clot formation). The three protein components work to balance each other and localize hemostasis to the site of injury. The second component (cellular) involves platelets, endothelial cells (cells lining the blood vessel wall), neutrophils, and monocytes.
The initial part of hemostasis involves the following: (1) the formation of a platelet plug, followed by (2) blood coagulation and (3) growth of fibrous tissue into the clot to repair the site of injury. A platelet plug (Fig. 11-3) begins to form on vessel injury and acts as a temporizing measure to repair injury to blood vessels. This process happens in three steps—adhesion, aggregation, and platelet swelling.
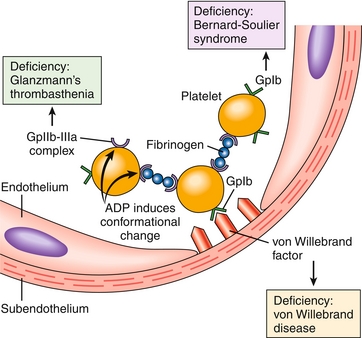
Figure 11-3 Platelet adhesion and aggregation. Von Willebrand factor is the bridge that allows the platelet glycoprotein Ib (GpIb) receptor to adhere to the subendothelium of the damaged blood vessel. To allow platelets to bind to other platelets, fibrinogen is the bridging molecule and uses the platelet GpIIb-IIIa receptor. Also shown are deficiency states causing improper platelet adhesion and/or aggregation. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009.)
Adhesion
This occurs when injury to the blood vessel wall exposes subendothelial collagen. Von Willebrand factor (vWF) is released and binds to the glycoprotein Ib (GP Ib) receptor on the platelet surface. At the site of injury, vWF is released from the endothelium and from the platelet, helping to form additional links between the platelets and exposed collagen fibrils. vWF also circulates in the plasma bound to factor VIII, acting as a stabilizer for factor VIII. Therefore, vWF deficiency also causes decreased factor VIII levels.
Aggregation
This involves a complex balance between factors that favor aggregation versus those that prevent aggregation. Undamaged endothelium secretes inhibiting factors (e.g., prostacyclin) that prevent aggregation. Increased turbulence at a site of damaged endothelium promotes aggregation. Activated platelets release adenosine diphosphate (ADP), thromboxane A2 (TXA2), vWF, calcium, serotonin, and other factors, which in turn help activate additional platelets and promote aggregation. ADP interacts with the ADP receptor on the platelet surface and initiates a cascade that promotes the insertion of glycoprotein IIb/IIIa (GP IIb/IIIa) receptors on the platelet surface. The GP IIb/IIIa receptor, a calcium-dependent receptor, on the platelet surface binds fibrinogen and helps cross-link adjacent platelets, an important final step in primary hemostasis.
Platelet Swelling
This occurs as soon as platelets attach to the injured surface. Activated platelets undergo a conformational change of shape from spherical to stellate (star-shaped). These changes help make them stickier by increasing the surface area for fibrinogen to cross-link adjacent platelets via the GP IIb/IIIa receptor. The end result is a large accumulation of platelets and formation of a platelet plug (primary hemostasis) over the site of injury. This plug is strong enough to stop bleeding until the final product of coagulation cascade, fibrin, helps stabilize the platelet plug via the formation of fibrin mesh (secondary hemostasis).
The coagulation cascade (Fig. 11-4) is initiated when there is trauma to blood vessels or tissues or if circulating blood comes into contact with subendothelial collagen. There are two basic pathways, both involving plasma clotting proteins—the extrinsic pathway and the intrinsic pathway. Both these pathways culminate in the activation of prothrombin (factor II) to thrombin (factor IIa) and fibrinogen to fibrin.
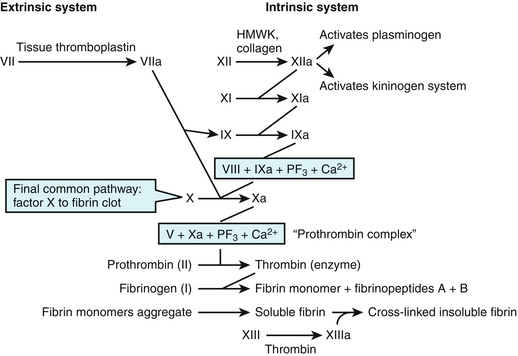
Figure 11-4 The intrinsic and extrinsic coagulation cascade. The “a” next to the factor name refers to the activated form of that clotting factor. (From Goljan EF. Rapid Review Pathology Revised Reprint. 3rd ed. Philadelphia: Elsevier; 2011.)
The extrinsic or tissue factor (TF) pathway begins with trauma to the endothelium of the blood vessel and is the most prominent pathway involved in the clotting cascade. Following damage to blood vessels, two initiators of clotting are released, tissue factor (TF), factor III, and tissue phospholipids. Factor VII (FVII), a plasma-clotting factor in the circulation, comes into contact with the exposed TF and forms an activated complex (TF-FVIIa). FVII is one of the vitamin K–dependent clotting factors and requires calcium to form a complex with TF. The TF-FVII complex, along with tissue phospholipids and calcium, then acts on factor X to form activated factor X (FXa). The activation of FX to FXa is the point at which the extrinsic and intrinsic pathways converge into the common pathway. FXa, along with tissue phospholipids, calcium, and factor V (which acts as a cofactor to FXa), form a complex that activates prothrombin (factor II) to thrombin (factor IIa).
The intrinsic, or contact activation pathway, is initiated on the activation of factor XII (Hageman factor) to factor XIIa (FXIIa). It is known as the contact system because it was discovered that FXII can autoactivate when it comes into contact with a negatively charged surface, such as a glass tube or exposed collagen from a damaged blood vessel. The activation of FXII is also facilitated by circulating high-molecular-weight kininogen (HMWK) in this setting. FXIIa simultaneously activates coagulation and anticoagulation cascades. The coagulation cascade works by amplification and the creation of large amounts of thrombin; activation of the anticoagulation cascade helps localize the thrombin production. The clotting cascade is continued by FXIIa’s activation of factor XI to factor XIa (FXIa), which in turn activates factor IX (Christmas factor) to factor IXa (FIXa). FIXa forms a four-component complex with factor VIII (FVIII), platelet phospholipids, and calcium. This four-component complex merges into the first step of the final common pathway by activating FX to FXa. FXa, along with tissue phospholipids, calcium, and factor V, forms a complex that activates prothrombin to thrombin.
The anticoagulation cascade is initiated by FXIIa via the activation of plasminogen and of the kininogen system:
 Plasminogen is activated by tPA and produces plasmin, an enzyme that works to cleave the fibrin meshwork. This cleavage produces fibrin(ogen) degradation products (FDPs) and insoluble fibrin monomers called D-dimers.
Plasminogen is activated by tPA and produces plasmin, an enzyme that works to cleave the fibrin meshwork. This cleavage produces fibrin(ogen) degradation products (FDPs) and insoluble fibrin monomers called D-dimers.
The kininogen system produces kallikrein and bradykinin. Kallikrein activates the fibrinolytic system and promotes the activation of plasminogen into plasmin, whereas bradykinin acts as part of the body’s inflammatory response to increase vasodilation, vessel permeability, and pain.
Coagulation Cascade: Key Points
Prothrombin (factor II) is a vitamin K–dependent plasma protein that is produced in the liver. In the liver, epoxide reductase converts vitamin K to activated vitamin K, which acts as a cofactor for the vitamin K–dependent coagulation proteins.
The vitamin K–dependent coagulation proteins include factors II, VII, IX, and X, protein C, and protein S. Warfarin inhibits the vitamin K epoxide reductase enzyme and blocks production of these cofactors.
All vitamin K–dependent proteins require calcium as a cofactor.
Vitamin K deficiency leads to decreased production of factors II, VII, IX, and X, protein C, and protein S. Neonates require supplemental vitamin K at birth because their bowel lacks the bacteria that produce vitamin K.
The main actions of thrombin (Fig. 11-5) include:
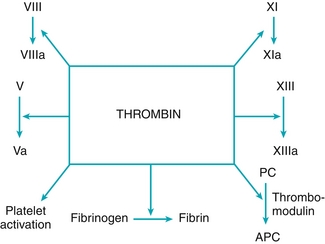
Figure 11-5 Main actions of thrombin (factor IIa). (From Hudnall SD. Hematology. Philadelphia: Elsevier; 2011.)
Factors that are consumed in a clot are factors I, II, V, and VIII. A person will be deficient in these factors if they are constantly activating the coagulation cascade (e.g., sepsis).
Factors that enhance formation of a thrombus in vessels include the following:
TF (factor III) is a noncirculating protein that is released from injured tissue and initiates the extrinsic pathway by activating factor VII.
vWF acts as the ‘glue’ that allows platelets to adhere to exposed collagen on injured endothelium (platelets bind vWF via GP Ib receptors on their surface). It is made by endothelial cells and is also carried within platelets in their alpha granules.
TXA2 is made within platelets; when it is released, it acts as a vasoconstrictor and enhancer of platelet aggregation by improving fibrinogen attachment to its receptor on the platelet.
Platelet and Coagulation Tests
The ristocetin cofactor assay helps evaluate the function of vWF by promoting platelet agglutination. Platelet agglutination caused by ristocetin (an obsolete antibiotic) can only occur in the presence of functionally normal vWF multimers.
Bleeding time (BT) is used to evaluate the function of platelets. Normal BT is from 2 to 7 minutes. Below are causes of prolonged BT:
Aspirin irreversibly inhibits the platelet cyclooxygenase (COX) enzyme, thereby halting production of TXA2. Platelet count will remain normal in these patients.
Thrombocytopenia will result in abnormal BT because of the decreased number of platelets in the circulation. An increase in BT will be seen when the platelet count falls below 90,000/mm3.
von Willebrand disease is an autosomal dominant disorder in which individuals lack vWF or have defective vWF protein. Patients will also have a decrease in FVIII (remember that vWF complexes with FVIII to prevent its degradation), resulting in a combined platelet and coagulation factor disorder. BT and the partial thromboplastin time (PTT) will be increased. Patients will also have a correctable, abnormal, ristocetin cofactor assay result. The ristocetin test result will normalize after the addition of normal plasma (which contains normal vWF).
Bernard-Soulier syndrome results when platelets lack the GP Ib receptor on their cell surface. The GP Ib receptor helps bind the platelet to exposed collagen, using vWF as an intermediate; the lack of the receptor results in inappropriate platelet adhesion. This disease is characterized by prolonged BT, thrombocytopenia (because of decreased platelet survival), and increased megakaryocytes. These patients will have an abnormal ristocetin cofactor assay result that is not correctable by the addition of normal plasma; the issue is not with vWF but with abnormal circulating platelets.
Glanzmann’s disease is an autosomal recessive disorder in which platelets lack the GP IIb/IIIa receptor on their cell surface. This receptor binds fibrinogen and promotes interplatelet aggregation. BT is significantly prolonged (think of this as a so-called congenital abciximab syndrome).
Uremia or renal failure results in the accumulation of toxic products in the blood that leads to inhibition of platelet phospholipids, thereby producing a platelet aggregation defect. BT prolongation can be reversed with dialysis and desmopressin acetate.
The clinically important coagulation tests include PTT and prothrombin time (PT). The PTT is used to assess the function of the intrinsic pathway and to follow heparin therapy. Remember that PTT has an extra T inside it and measures the intrinsic pathway. A normal PTT ranges from 25 to 40 seconds.
The PT is used to assess the function of the extrinsic pathway and to follow warfarin therapy. A normal PT ranges from 11 to 15 seconds.
Anticoagulant System
The coagulation cascade is regulated by two anticoagulant systems that help regulate (by inhibiting) clot formation. These systems are the protein C and protein S systems and the antithrombin (serine protease inhibitor) system.
Protein C is a vitamin K–dependent protein that, when activated, will inactivate factors Va and VIIIa, thereby decreasing production of thrombin. Protein C is activated by thrombomodulin, which is just thrombin bound to an endothelial cell membrane.
Protein S is a vitamin K-dependent protein that works as a cofactor for activated protein C (APC) and helps direct APC to the required site of action.
Antithrombin III (ATIII) is a serine protease inhibitor; therefore, it inhibits all activated serine proteases (basically inhibits all coagulation proteins):
Fibrinolytic System and Tests
Factors that prevent formation of a thrombus (fibrin clot) in vessels:
Prostaglandin I2 (PGI2), also known as prostacyclin, functions as a vasodilator and inhibitor of platelet aggregation. This prostaglandin is constitutionally produced by intact endothelial cells to prevent thrombus formation in vessels.
Heparin-like molecules that act to enhance ATIII activity, thereby inhibiting the function of factors II, IX, X, XI, and XII.
Tissue plasminogen activator (tPA) is an enzyme that helps activate plasminogen to plasmin. Plasmin works by degrading coagulation factors and fibrin clots.
Proteins C and S are vitamin K–dependent factors that work to deactivate coagulation factors V and VIII.
The fibrinolytic system (which degrades clots) is initiated by various activators, and all work by converting plasminogen to its active form, plasmin.
Plasmin cleaves the fibrin meshwork created by the coagulation cascade and creates FDPs and D-dimers. D-dimers are a type of FDP, so-called because it contains two cross-linked D fragments of fibrinogen. Plasmin also degrades coagulation factors I, V, and VIII.
Activators of plasminogen include tPA, Hageman factor (factor XIIa), urokinase, and streptokinase.
An important inhibitor of plasminogen activation is aminocaproic acids, used clinically to stop excessive postoperative bleeding or overdoses of tPA and streptokinase.