Phylogeography
Michael E. Hellberg
OUTLINE
1. Direct interpretation of single-locus gene genealogies
2. Comparative phylogeography
3. Lineage sorting and the coalescent
4. Multilocus gene genealogies
5. Testing models of population history
Phylogeography is the study of the history of populations within species. These studies emerged from analysis of mitochondrial DNA sampled from multiple populations, providing a genealogical perspective within species. Early studies helped identify geographical barriers that separated differentiated populations and suggest where recent range expansions had occurred. The development of coalescent theory led to analyses that could not only discern whether populations were isolated, and, if so, for how long, but also identify demographic changes since that point and past gene flow between populations. An emerging multilocus, model-testing framework places greater emphasis on identifying key factors in shaping population history than on estimating parameter values.
GLOSSARY
Coalescent. Genealogy of alleles tracing back in time to a common ancestral sequence.
Haplotype. Stretch of DNA passed across generations without recombination.
Lineage Sorting. Process during which the genealogies of genes within isolated populations move toward becoming reciprocally monophyletic by the differential loss and replication of gene lineages that were shared at the time of isolation.
Mismatch Distribution. Frequency plot of all pairwise sequence differences between sampled haplotypes from a population.
Reciprocal Monophyly. When the members of two defined groups are all more closely related to other members of their own group than to those of the other.
Phylogeography draws its conclusions from the genealogical analysis of genetic data sampled from populations. These conclusions can reveal the history of populations. Phylogeography thus provides insights into the state of populations during the process of their divergence toward forming new species, allowing us to answer such questions as: How large were the populations? When did they diverge? How much migration occurred after the initial population divergence? Phylogeography can also reveal how populations responded to past climatic changes, thereby suggesting possible impacts of future change. Phylogeography can identify long-isolated populations that are not sustained by demographic connections to other regions, and thus may inform conservation and management programs. Phylogeography can also help reveal our human past, via genetic analysis of our own lineage (By what routes did our ancestors populate the world? Did Homo sapiens exchange genes with other hominids?), and through studies of the evolution of pathogens that have helped shape our recent history.
The term phylogeography was coined to mark the incorporation of tree thinking (phylogeny) into the study of genetic variation within species. Phylogeography was born of the study of mitochondrial DNA (mtDNA) variation among animal populations. Analysis of these data resulted in a genealogy of mtDNA haplotypes. The ability to discern relationships among these mitochondrial haplotypes, combined with this molecule’s uniparental inheritance, appeared to provide the means to trace the splitting and coalescing of populations back through time.
This promise stimulated the collection of millions of base pairs of mtDNA sequence data. Spreading PCR technology, automated DNA sequencing machines, and small set of primers that amplified short (350–800 bp) gene regions across a wide phylogenetic range facilitated this explosion. Possessing a dog-eared photocopy of The Simple Fool’s Guide to PCR, put together by Steve Palumbi’s lab in Hawaii in 1989, marked one as a giddy member of a growing club of explorers. Broad geographical sampling commonly revealed distinct mtDNA clades within species, and these were (sometimes fecklessly) associated with present barriers and past events.
The promises of mtDNA phylogeography were eventually exposed, however, not as a lie so much as a tease. For elucidating recent population isolation or for discerning the details of historical events, trees drawn from mtDNA trees were insufficient. For example, the ability to detect population mixing was limited because traditional phylogenetics rarely considered mixing among its units of study (species), even though such mixing (migration) is central to population genetics. The history of phylogeography has been the continuing reconciliation of the perspectives of phylogenetic systematics and populations genetics, combined with the realization that many of the inferences we thought we could make directly from gene trees based on one locus require more complex analytical tools and new sources of data. Since those initial days, the field has been engaged in a search for new analyses and sources of data that allow us to sate our hunger for the possibilities that mtDNA data first suggested but ultimately could not satisfy.
1. DIRECT INTERPRETATION OF SINGLE-LOCUS GENE GENEALOGIES
The typical phylogeographic study entails first acquiring genetic data from an orthologous region of DNA from multiple individuals sampled from different geographic locales, then inferring the genealogy of alleles from those individuals to make inferences about the degree to which populations have been isolated or connected. Initially, the history of mitochondrial haplotypes was equated with that of the populations from which they were drawn. While the analyses used to infer phylogenetic relationships among alleles were often taken straight from those developed by systematists, the connections of these genealogical patterns to geography were far less sophisticated, often amounting to simply placing a tree of haplotypes over a map of their origins.
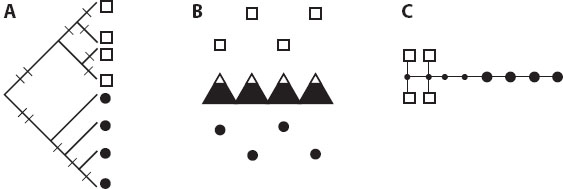
In a simple example (figure 1), mtDNA haplotypes are determined for samples taken from across a species’ geographic range. Phylogenetic analysis reveals that they fall into two reciprocally monophyletic clades. When this genealogy of haplotypes is mapped on a physical map of their origins, the phylogenetic gap between the genes corresponds to a potential geographic barrier to movement between populations, in this case a mountain range.
Figure 1. Phylogeny of alleles (A) taken from individuals sampled from either side of a barrier (B). (C) The same alleles shown in a haplotype network, based on the locations of mutations indicated on tree A.
Many studies followed this same approach, often revealing phylogeographic breaks corresponding with known biogeographic breaks or with geographic features such as rivers (for terrestrial animals) or strong currents (for marine populations). But geography did not always rule. Most remarkable were instances in which animal behavior trumped all else, such as the distinct mtDNA from American and European eels: both species migrate to the Sargasso Sea to spawn before their larval offspring eventually return to their respective ancestral home waters.
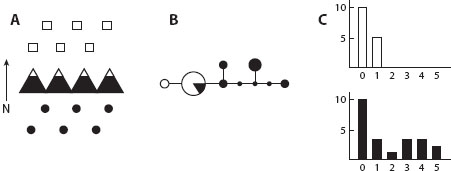
Not every study revealed such patterns, however. In some instances, mtDNA haplotypes were identical across multiple populations, sometimes owing to slow mtDNA substitution rates, as with most plants and some animals, including corals and sponges. For these taxa, chloroplast DNA (like mtDNA, usually with uniparental inheritance) and the internal transcribed spacer (ITS) region between ribosomal RNA genes (whose many copies may be homogenized within a genome) sometimes served as alternative markers for phylogeography. In other cases, some portion of the geographic range was fixed for a single haplotype. In the Northern Hemisphere, the poleward edge of the range of many species showed little genetic diversity (figure 2), a pattern consistent with a recent recolonization following the glacial retreat. The range over which mtDNA diversity was low often coincided with habitat made unsuitable by the equatorial advance of glaciers and cool temperatures during Pleistocene glacial maxima.
Figure 2. (A) Two populations, the northern poleward of a barrier. (B) Haplotype network of the alleles sampled, with area of circles proportional to the frequency of the haplotype. (C) Mismatch distributions for the two populations, with the top pattern consistent with a recent population expansion following a bottleneck.
Data accumulating around this time (the late 1980s) pointed to other instances of population expansion revealed by mtDNA, most notably the “mitochondrial Eve” data for humans. A realization grew: genealogies of haplotypes within species are not strictly the same as those above the species level, and they should not be analyzed the same way. A first step was to realize that in contrast to phylogenetic analysis, in which one deals only with descendant taxa (sister groups descended from a common ancestor; chapter II.1), ancestral and descendant haplotypes commonly coexist, making bifurcating trees a misleading way to illustrate their relationships. Haplotype networks (figures 1 and 2) arose as a means of visualizing the mutational steps among alleles within a species and their frequencies in different populations.
Another useful tool emerged for combining information on the distribution of genetic differences among haplotypes and the frequency of these haplotypes: mismatch distributions (figure 2). These are frequency plots of the number of mutational steps between all possible pairs of sequences in a sample plotted against the frequency of each genetic distance class. Early simulation work showed that these mismatch distributions looked qualitatively different for populations that had maintained a constant size compared with those that had recently undergone a population expansion: while constant-size populations yielded erratic peaks and valleys, growing ones consistently produced a single peak in the mismatch distribution. In the extreme, a population expanding from a population in which all members shared the same haplotype would create a wave in the mismatch distribution over time, beginning with all pairwise differences at zero, then sliding to the right in the graph as mutations accumulated.
The position of the wave’s peak, in mutational units, can (if a molecular clock applies) be translated into the time since the population expansion. Lessios et al. (2001) used this approach to distinguish between two alternative histories for the Caribbean long-spined sea urchin (Diadema antillarum), an important grazer that suffered a massive die-off in 1983. This mass mortality tipped coral reef communities to higher algal cover at the expense of corals. But were these low-coral reefs a newly damaged state, or a reversion to an old form? One view was that the pre-1983 abundance of D. antillarum was a recent condition linked to human activities; an alternative hypothesis was that high urchin densities were the norm even before human impacts. A sample of mtDNA from D. antillarum did show a single wave in the mismatch distribution, but its peak was at about 3 mutational steps for the 642 bp surveyed, equating to a time long before humans appeared, even when calibrated with the fastest molecular clock.
As illustrated by the Diadema example, the direct interpretation of single-locus gene genealogies can provide unprecedented insights into population history. But there are problems. For one, biologists sometimes hope to draw broad conclusions about the history of regional biotas (say, for the purpose of setting conservation priorities), but different taxa sampled from the same populations do not always give the same results. Parameter estimation based on the genealogy of a single gene region often produces high variances and can easily be swayed by locus-specific selection. Furthermore, biologists came to realize that the history of a single gene need not reflect exactly the history of populations or species from which it was sampled. Finally, it became apparent that the human eye is too prone to seeing patterns: a phylogeographic break dropped just about anywhere in a haplotype tree could inspire a post hoc explanation mechanistically linking it to some physical feature or past event. A more explicit hypothesis-testing framework was needed.
2. COMPARATIVE PHYLOGEOGRAPHY
Comparative phylogeography examines the population histories of multiple species sampled from the same communities. These studies might aim to identify major barriers or past events common to many species. Regions themselves thus become the focus of study, with resident species serving as replicate recorders of history. Such work is often aimed at recognizing regions whose distinctive histories warrant special consideration in conservation and management. Patterns of differentiation that share the location of phylogeographic breaks are said to be concordant. Such geographically coincident genetic breaks may reflect a shared history, as when a strong new geographic barrier emerges, simultaneously splitting populations of many taxa. Whole communities can also respond together to shared changes in their environment, as with the many continental European species experiencing northward range expansions following the most recent glacial interlude.
Shared histories among species are not the rule, however. Co-occurring species often show phylogeographic breaks in different places, or some might show evidence of a range expansion while others have maintained constant population size. For instance, among coastal marine animals, those with intertidal distributions appear to have less stable population histories than do sympatric subtidal species, perhaps because exposure to air makes them more vulnerable to climatic change than species thermally buffered by water. The effects of differences in microhabitats can be explored more quantitatively with ecological niche modeling, in which distributions, including past ones, are predicted using a combination of presence-absence data and environmental data such as rainfall and temperature.
Even concordant phylogeographic breaks do not necessarily indicate a common history. Lineages dividing at the same place may have become isolated at different times by that same barrier, or have been in different places before arriving at the common barrier, patterns termed pseudocongruency. Such patterns suggest that community members do not always coevolve over sustained periods, and the patterns can also provide insights into how communities are assembled.
3. LINEAGE SORTING AND THE COALESCENT
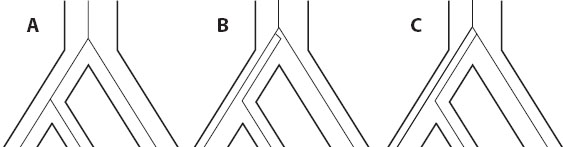
The reciprocal monophyly of alleles, in which all haplotypes are phylogenetically closer to other members of their own population than to any others, is a sure sign that populations are isolated and experience no gene flow between them. But reciprocal monophyly does not emerge at the onset of isolation; far from it. Consider a well-mixed population that is instantaneously split in two (figure 3A). At first, any differences between the two would be due purely to stochastic effects at initial sampling. As time goes on (figure 3B), two independently evolving populations, both of finite size, will lose some haplotype lineages by genetic drift. Over longer periods (figure 3C), new alleles may arise via mutation, further distinguishing the populations. Eventually, one population will become monophyletic (figure 3D), and after a period of paraphyly, the other will follow, at which point lineage sorting is complete.
Figure 3. Sorting of alleles into reciprocally monophyletic groups over time.
As this process of lineage sorting is dictated primarily by genetic drift, the time it takes will be proportional to the effective population size (Ne). For mitochondrial genes, this will take on the order of Ne generations. For diploid nuclear genes with equal numbers of males and females in the population, it will take four times longer.
That such calculations, and others now forming the basis of modern phylogenetic analyses, can be made at all owes to the study of the coalescent: the genealogy of alleles tracing back in time to a common ancestral allele. It is important to recognize that identical alleles still have a nonzero coalescence time, even though we cannot see such coalescences because they are not marked by mutations. Framing things in the retrospective way simplifies calculations, as only the lineages that lead to the haplotypes sampled must be accounted for, not all individuals in the population (as with forward-running simulations).
Coalescent analyses have begun to take full advantage of the richness of information in sequence data from populations. Fundamentally, they provide a null model of what populations that had become recently isolated will look like. From this foundation, additional parameters that impinge on the frequency and phylogenetic relationships of haplotypes can be added, including population size, time of isolation, and levels of migration in each direction (Hey and Nielsen 2004). The statistical validity of adding such parameters can be tested, and their values estimated if addition is warranted.
4. MULTILOCUS GENE GENEALOGIES
While work on the coalescent was motivated by mtDNA data, the large variances that necessarily result from estimating multiple parameters with a single marker led to interest in analyzing multiple loci to infer population history. Multiple markers can improve parameter estimation and increase the power of tests while also allowing for insights that qualitatively exceed anything provided by mtDNA or any single marker.
Multilocus genotypes permit associations among alleles at different loci (linkage disequilibrium) to be the basis for recognizing isolated populations. These clustering analyses have the additional advantage of not requiring that populations or species be defined a priori; identifying these units is part of the analysis. Linkage disequilibrium builds up and breaks down rapidly, allowing populations isolated on the order of dozens of generations to be recognized. Such sensitivity can detect recent phylogeographic breaks not visible to other approaches but, because these analyses are based on genotypes alone and not the relationships among alleles, sensitivity comes at the expense of the ability to infer the timing of the split or to detect hints of any past dispersal between populations.
Different types of loci can also say different things. Because the effective population size of mtDNA is smaller than that of nuclear genes, it is more sensitive to population bottlenecks and recent gene flow. At the same time, bottlenecks recorded by mtDNA may also clear the record of changes in population size that occurred further back in time. Nuclear genes from the roughhead blenny Acanthemblemaria aspera, for example, suggest a population expansion about 400,000 years ago, while mtDNA flags a far more recent expansion beginning just 20,000 years ago (Eytan and Hellberg 2010). Gene regions under selection can increase the breadth of past events that can be inferred. Loci under strong balancing selection, such as the MHC loci in vertebrates or S-alleles in self-incompatible plants, can provide a lower limit for the size of past bottlenecks. The use of sequence data to detect past bouts of locus-specific selection does not usually fall within the purview of phylogeography, but suffice it to say that the combination of tests for selection with reconstructions of population history holds much promise.
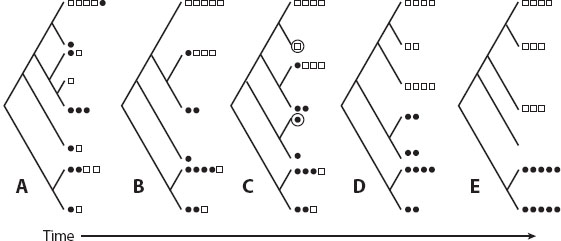
Critically, work with the coalescent has revealed that replicate genes experiencing the same population histories can show high levels of stochastic variation among their gene genealogies. Thus, from first principles, the gene genealogy of any single gene is unlikely to reflect the genealogy of the species or population from which it was sampled (figure 4). In fact, it has been shown theoretically that, under certain conditions of branch length variation, the most commonly occurring gene tree will not be the species tree (i.e., the tree of population splitting). The potential for disagreement between gene trees and species trees is greatest when population sizes are large and when divergence was recent and rapid (Maddison 1997), conditions that match many events in phylogeography. Theory suggest that reciprocal monophyly of nuclear genes is not the most likely outcome for a sample of loci from isolated populations until 1.66 Ne generations after their split (Rosenberg 2003).
Figure 4. Agreement and disagreement between gene trees and species trees. In (A), the gene tree within the species tree perfectly reflects the species phylogeny in terms of both order and timing of splitting events. In (B), the gene topology matches the species tree, but divergence between the sister species’ gene greatly precedes that of the species. In (C), the gene tree disagrees with the species tree.
Analyses that can identify population isolation despite high interlocus variation and long times to monophyly are thus appropriate for the timescales at which most phylogeographic studies are directed. The number of such analyses is growing, with the present pool divided among faster programs requiring resolved gene trees as input, and more accurate ones that take uncertainty in gene trees into account. The use of such approaches generally requires that terminals (either populations or species) be defined a priori, which has turned attention to the difficult problem of genetically delimiting taxa when few, if any, genes are expected to have coalesced.
5. TESTING MODELS OF POPULATION HISTORY
As more and more complex analyses have offered the potential to estimate an increasing number of parameters, the data generated by empirical phylogeography have not kept up with the needs of these data-hungry approaches. Fortunately, however, the crux of a question may hang not on the values that a parameter takes, but rather on alternative hypotheses that can be tested in more tractable ways.
The first steps toward such tests arose from initial inspection of gene trees. Montane grasshoppers live in the northern Rockies, high on mountains in habitat that would have been uninhabitable during recent glacial periods. Knowles (2001) tested two alternative hypotheses for the history of these now-differentiated populations: one in which all sky island populations arose from a single isolation event (having previously been one undifferentiated population), the other in which regional subdivision existed prior to fragmentation. By comparing her mtDNA data against distributions of gene trees generated under conditions matching the two alternatives, she ruled out the null model of a single fragmentation event.
Testing of alternative models is appropriate when prior information suggests a few specific alternatives. A more general approach, and one suitable for a first look at populations with no data on their history, is to test whether particular parameters are needed at all to sufficiently explain the data. For example, one could ask whether isolation alone (and the resultant population-specific coalescent processes taking place) suffices to explain patterns of shared sequence variation in two recently formed populations, or whether migration parameters are also needed. This can again be done by comparing the data to expectations generated by simulation. A conceptually similar approach can be used to ask whether species pairs whose distributions presently are divided by the same barrier were split at the same time. Hickerson et al. (2006) used approximate Bayesian computation (ABC) to estimate the number of different divergence times needed to model splits between eight pairs of urchin species residing on opposite sides of the Isthmus of Panama. Seven of these species pairs, presumed to have been sundered about 3 million years ago, diverged simultaneously, but a final pair appears to have split more recently.
But historical sciences like phylogeography often require many alternative hypotheses. Information theory can enable the simultaneous testing of many alternatives (Carstens et al. 2009). Under this approach, alternative hypotheses are ranked by their power to explain the data, enabling the researcher to identify the particular set of parameters (the model) that best reduces uncertainty about past processes while using the fewest parameters. For example, for two populations that have diverged, likelihoods calculated for a full model that includes ancestral and descendant population sizes and migration in each direction can be compared against submodels from which some of the parameters have been removed. From evidence ratio scores, the information theory metric used to rank hypotheses, the relative strength of each model can be compared. If all the best models have migration parameters set to zero, then the history of the populations is likely one of allopatry. Thus, even when no single model can be identified as best, parameters common to all top models can be found, and a smaller subset of models (now alternative hypotheses) that future work must distinguish can be identified.
FURTHER READING
Carstens, B. C., H. N. Stoute, and N. M. Reid. 2009. An information theoretic approach to phylogeography. Molecular Ecology 18: 4270-4282. Introduces a way to simultaneously evaluate the fit of multiple alternative models to a phylogeographic data set for a salamander found in the northwestern United States.
Eytan, R. I., and M. E. Hellberg. 2010. Nuclear and mitochondrial sequence data reveal and conceal different demographic histories and population genetic processes in Caribbean reef fishes. Evolution 64: 3380–3397. A phylogeographic survey of two hole-dwelling blennies infers different times of past population expansions from mtDNA and nDNA.
Hey, J., and R. Nielsen. 2004. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760. An updated and empirical application of the influential IM model.
Hickerson, M. J., B. C. Carstens, J. Cavender-Bares, K. A. Crandall, C. H. Graham, J. B. Johnson, L. Rissler, P. F. Victoriano, and A. D. Yoder. 2010. Phylogeography’s past, present, and future: 10 years after Avise, 2000. Molecular Phylogenetics and Evolution 54: 291–301. Reviews the rise of model-based phylogeographic analyses and points to an emerging synthesis of ecological niche models, studies of natural selection, and community assembly.
Hickerson, M. J., E. A. Stahl, and H. A. Lessios. 2006. Test for simultaneous divergence using approximate Bayesian computation. Evolution 60: 2435–2453. Employs ABC to test whether multiple pairs of sea urchins divided by the Central American Isthmus were sundered simultaneously.
Knowles, L. L. 2001. Did the Pleistocene glaciations promote divergence? Tests of explicit refugial models in montane grasshoppers. Molecular Ecology 10: 691–701. Early model-based approach to evaluating alternative hypotheses for the divergence of grasshoppers living on Rocky Mountain “sky islands.”
Lessios, H. A., M. J. Garrido, and B. D. Kessing. 2001. Demographic history of Diadema antillarum, a keystone herbivore on Caribbean reefs. Proceedings of the Royal Society B 268: 2347–2353. Employs mismatch distributions of mtDNA to distinguish alternative ecological histories for a Caribbean urchin.
Maddison, W. P. 1997. Gene trees in species trees. Systematic Biology 46: 523–536. Classic discussion of the relationship between these two kinds of phylogenetic trees.
Rosenberg, N. 2003. The shapes of neutral gene genealogies in two species: Probabilities of monophyly, paraphyly, and polyphyly in a coalescent model. Evolution 57: 1465–1477. Eye-opening exploration of the number of generations it takes for samples of nuclear genes from two recently formed species to become reciprocally monophyletic.