Interpretation of Phylogenetic Trees
Kevin E. Omland
OUTLINE
1. Introduction to phylogenetic trees
2. Misreading trees with species-poor lineages
3. Reading trees correctly: Ancestral state reconstruction
4. Understanding the process of evolution: We are all cousins
All organisms on earth share common ancestry; we are related to every species that has ever existed. Evolutionary biologists since Darwin have sought to infer a “tree of life,” a phylogenetic tree showing how all species are related to one another. The concept of phylogeny as the evolutionary history of organisms—and phylogenetic trees as a depiction of that history—is central to evolutionary biology. Phylogenies form the basis for our understanding of relationships among organisms, and they are key tools of modern evolutionary research; however, phylogenetic trees are frequently misinterpreted because of fundamental misconceptions about what trees can and cannot tell us. In particular, people frequently misinterpret trees by reading trees “laterally” from one extant species to the next. This tendency results partly from mistakenly thinking of evolution as a “ladder of progress.” Ultimately, a well-informed interpretation of phylogenetic trees goes hand in hand with a clear understanding of the process of evolution.
GLOSSARY
Ancestral State Reconstruction. A procedure that uses a phylogeny as well as character data from extant species to infer likely ancestral character states.
Character State. Alternative states for a given biological character (e.g., brown, blue, hazel are character states of the character eye color; A, C, G, or T are character states of the character corresponding to base position 12 in the actin gene).
Chronogram. A phylogenetic tree with branch lengths scaled to represent time (related to phylogram, a tree in which branch lengths are scaled to the amount of character evolution).
Cladogram. An unscaled phylogenetic tree that shows relationships among organisms, but in which branch lengths are meaningless. The key information retained in a cladogram is topology, which refers to the composition of clades/monophyletic groups and how they are related to one another.
Derived. Generally the opposite of ancestral; a derived character state is one that has evolved recently relative to an ancestral character state (e.g., scaly skin is an ancestral character state for reptiles, whereas feathers in avian reptiles represent a derived character state).
Monophyletic Group (Clade). A group of species including a common ancestor and all its descendants; a “natural group” with all members more closely related to each other than to any other species (compare with paraphyletic).
Paraphyletic Group. A nonmonophyletic group that includes a common ancestor but leaves out some descendants (e.g., “reptiles” leaving out birds). Sometimes contrasted with polyphyletic groups, which include two or more different ancestors and their descendants, for example, bats plus birds without their common ancestor.
Phylogenetic Tree. A branching diagram showing relationships among organisms (e.g., frequently among different species, or among different individuals).
Phylogeny. The evolutionary history of a group of species (or any set of taxa, genes, or tips).
Root Node. The ancestral node at the base of a tree, representing the most recent common ancestor of all species included in that tree.
Shared Derived Character State (Synapomorphy). A character state that defines a monophyletic group (e.g., the presence of mammary glands for mammals).
Species Tree. Phylogenetic tree showing relationships among species, generally based on multiple independent genes (distinguished from gene tree, a tree showing relationships based on one gene from multiple individuals, or showing relationships among multiple paralogous genes).
1. INTRODUCTION TO PHYLOGENETIC TREES
Phylogeny is a fundamental concept, and phylogenetic trees are important tools in evolutionary biology; thus, it is crucial to read phylogenetic trees in a way that aligns well with an accurate understanding of the overall process of evolution. Although the next two chapters focus on how to construct trees, this chapter is about what those trees represent and how to interpret them. The challenge of interpreting trees is present even if we know the correct tree (e.g., for animal breeds), or when we have inferred a well-supported uncontroversial tree. Most biologists need not know how various phylogenetic algorithms work, but all biologists should know what trees represent and how trees inform our understanding of the process of evolution.
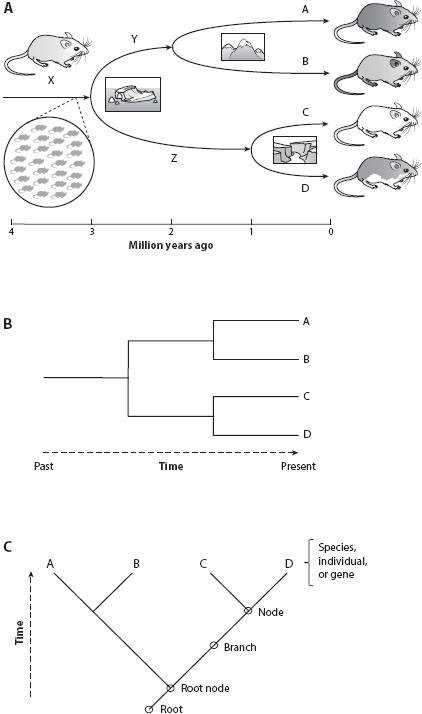
What does a phylogenetic tree represent? What can a phylogeny tell us? Figure 1A depicts a series of hypothetical speciation events. The arrow at the left labeled X represents a mouse species that lived 4 million years ago. The mouse populations evolved as a result of mutation, genetic drift, and selection for 1 million years as time moved from left to right. At 3 million years ago, an evolutionary fork in the road occurred, some geological change such as a rise in sea level divided the ancestral species into two descendant species, species Y and Z. These species evolved for another 1 million years before a mountain chain formed in the range of species Y, leading to the formation of species A and B. Later, a river formed in the range of species Z, leading to species C and D. Thus, four species are present today. The drawing in figure 1A includes extra information about ancestral species and speciation events, but basically it is a phylogenetic tree. In this tree, the branch lengths are drawn to scale based on time, so this is a scaled tree, specifically a chronogram.
Frequently, phylogenetic trees focus on living species. In figure 1B, only species A, B, C, and D are shown, and the branch lengths are not drawn to scale. Such unscaled trees are known as cladograms, which focus on the most important information, the evolutionary relationships or tree topology. There are essentially only two such pieces of information in this tree. First, A and B are more closely related to each other than either is to C or D. Second, C and D are more closely related to each other than either is to A or B. Crucially, in the language of phylogenetic systematics, “more closely related” means “shares a more recent common ancestor.” Cladograms depict the tree topology, which shows the monophyletic groups or clades. Clades are composed of organisms that are more closely related to each other than to any organisms outside the clade, Two clades are shown in figure 1B: (A, B) and (C, D). (The tree in figure 1B also shows that A, B, C, and D all share a common ancestor, but this third piece of information is trivial: we already know that all species on earth share a common ancestor.) Thus, there are only two useful data points on this tree: the two monophyletic groups. Anything else that one thinks can be inferred from this cladogram is overinterpretation.
Many botanical terms used for real trees are used metaphorically for phylogenetic trees. Figure 1C shows the same information as figure 1B, although now as a slanted vertical tree. The “root” is at the base of the tree, representing the furthest point in the past. The path of evolution can then be traced along “branches” (“internodes”), always moving away from the root toward the present. Branches split at “nodes” (“internal nodes”) that represent speciation events. Finally, terminal branches lead to “tips” (“leaves” or “terminal nodes”), which frequently represent extant species that exist in the present (species A–D in this case). The tips of the tree generally represent a single species or a species as a representative of a larger group; however, tips sometimes represent an individual organism or an allele in a gene tree. The trees in figure 1 show each ancestral node with two descendant branches—these trees are bifurcating trees. (Note that trees can have nodes with three or more descendant branches; such nodes are called unresolved.)
Evolutionary Trees Have No Trunks
One key botanical term is missing from phylogenetic trees: there is never a trunk, never a main stem, never a main branch. To look at a phylogeny and see a “main path” of evolution is to be misled. Understanding that from every node there are two descendant branches, both of which lead to continued evolutionary change, is key to understanding phylogenetic trees. More important, understanding that there is no trunk is crucial for a clear understanding of the very process of evolution.
Other aspects of the tree metaphor can also lead to misinterpretation, especially the idea that some extant species might be “lower down on the tree,” either within a branch, or at the tip of a branch that terminates low down, closer to the root. It is important to trace history starting from the root of the tree and moving toward the branch tips; thus, the lineages are evolving as time moves from the past toward the present. For example, on a vertical tree such as figure 1C, evolution moves from the bottom to the top along the y-axis. Critically, the axis perpendicular to time has no meaning whatsoever (the apparent “x-axis” left to right on a vertical tree). That this axis perpendicular to time has no meaning and that there is no trunk are best illustrated by the concept of branch rotation.
Figure 1. (A) Hypothetical speciation scenario showing one ancestral mouse population repeatedly diverging, leading to four extant species. Arrows represent the paths of evolution from the common ancestor to the descendant species. These arrows form a phylogenetic tree showing relationships among extant species A–D. (B) Relationships among species A–D shown as a horizontal cladogram, an unscaled tree with no information about time of divergence. (C) Vertical slanted cladogram of the same species showing key botanical terms used to label phylogenetic trees.
Branches Rotate around Nodes
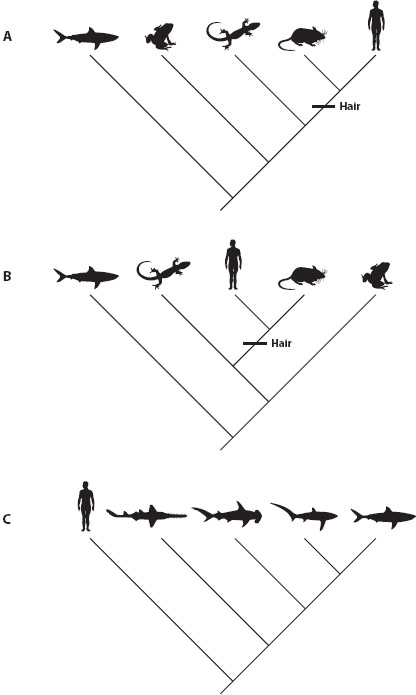
One of the most important points to understand about phylogenetic trees is that branches can be rotated around any node. David Baum and colleagues (2005) published a perspective article in Science titled “The Tree-Thinking Challenge” that made this very point about branch rotation. The simple figures of Baum et al. illustrate how even the most basic information in trees can be misinterpreted (redrawn in figures 2A and 2B). In figure 2A, frogs are next to sharks, so one might think that frogs are more closely related to sharks than to humans; however, figure 2B shows the exact same evolutionary relationships, yet frogs are now furthest from the shark, illustrating that proximity on the page has no meaning when reading evolutionary trees. Clearly, frogs share a more recent common ancestor with other four-limbed animals (tetrapods) than they do with sharks.
Figure 2. (A) Cladogram showing relationships among five vertebrate lineages: human (Homo sapiens), mouse (Mus musculus), lizard (Anolis carolinensis), frog (Rana pipiens), and great white shark (Carcharodon carcharias). (B) Rotated cladogram showing the same relationships among the same species, but with branches rotated around each node. (C) Cladogram showing relationships among five vertebrates, but this time the focus is on sharks, with humans included simply as an outgroup. Sharks depicted left to right are sawshark (Pristiophorus cirratus), hammerhead (Sphyrna zygaena), thresher shark (Alopias vulpinus), and great white shark (Carcharodon carcharias). (Note that the great white shark lineage and the human lineage are equally old: since they last shared a common ancestor approximately 450 million years ago, both lineages have continued to evolve.)
In figure 2A, one might think the straight line going from the root to humans would represent a trunk or a main branch; however, because the branches have been rotated in figure 2B, there is no straight line, making it more apparent that there is no trunk. There is no way to determine where evolution is heading. Many people incorrectly think of humans as the end point of evolution, but clear tree thinking can help dispel this notion. In this tree, humans could go in any one of the five positions left to right. Think of trees as being like a mobile, with each of the species able to spin around and appear in any position, yet their connections to each other remain the same. Branch rotation emphasizes the fact that there is no way to find a “main path” of evolution (whether leading to humans or to any other favorite organism) because there is no main path.
2. MISREADING TREES WITH SPECIES-POOR LINEAGES
Phylogenetic trees are most confusing when the trees are “unbalanced,” when one side of the tree has few species, but the other side has many species. For example, in figure 2A, the shark branch to the left of the root node is depicted as leading to one descendant species, whereas the branch to the right of the root node is shown leading to four descendant species. Many people mistakenly associate the “species-poor lineage” (e.g., the shark at the left in figure 2A) with the root node (also known as the basal node). This misconception occurs largely because no nodes are depicted that separate this basal node from the extant species. A group of biologists centered in Canberra, Australia, published a series of papers explaining the problem of this “basal fallacy.” Frank Krell and Peter Cranston (2004) asked, “Which Side of the Tree Is More Basal?” The answer is neither: all extant species are equally distant from the base of the tree, so no extant species should ever be considered basal. For example, with reference to figure 2, the great white shark and humans shared a common ancestor approximately 450 million years ago. We should remember that both lineages have been evolving for the same amount of time since that common ancestor.
Mike Crisp and Lyn Cook (2005) went on to highlight the most serious problem with this way of misreading a phylogenetic tree (e.g., left to right on vertical trees). Because many people associate the species-poor lineage with the base of the tree, they incorrectly assume that these species retain more ancestral characteristics (e.g., the species on the left in figure 2). Crisp and Cook focused on these species-poor lineages that may seem to “branch off early.” They asked, “Do early branching lineages signify ancestral traits?” They answered no—both lineages that descend from the ancestral node continue to evolve by mutation, drift, and selection. As both lineages continue to encounter new environments, food sources, predators, pathogens, etc., they will continue to evolve adaptations—no lineage of organisms stops evolving. Every species is a mix of retained ancestral, shared derived, and uniquely derived characteristics. As an example, Crisp and Cook highlight the platypus, which retains the ancestral tetrapod trait of egg laying, but has evolved many derived character states, including the flattened bill with electrosensory capabilities and a complicated system of 10 pairs of sex chromosomes. In contrast, humans and other placental mammals retain many ancestral tetrapod traits, including several unspecialized jaw characteristics and a single pair of sex chromosomes.
Misreading Trees as Ladders of Progress
Omland, Cook, and Crisp (2008) pointed out that deep-seated biases in the way people think of evolution feed into the misreading of phylogenetic trees. Many people think of evolution as “progressing” linearly from “simple organisms” to more “complex species,” then eventually to “the most advanced species,” which, unsurprisingly, humans consider to be humans. This flawed view of evolution as a “ladder of progress” can be traced back to Aristotle and Linnaeus, but it is especially apparent in work of the early evolutionary biologist Ernst Haeckel. Although he coined the word phylogeny, and he helped bring trees to the study of evolution, his “Pedigree of Man” (1866) furthers the mistaken viewpoint that humans are the end point of evolution. Haeckel’s tree has a trunk containing extant taxa depicted as ancestral to other extant taxa: “Monera” ancestral to “Amoebae” at the bottom, later with “Amphibia” ancestral to “Pouched Animals” then “Semi-Apes” (lemurs), eventually leading to “MAN” at the very top.
This ladder-of-progress view of evolution is still prominent in biology; species that are mistakenly considered “primitive” are generally shown at the left of vertical trees, whereas species thought of as “advanced” are shown at the right. A major reason species are considered primitive is that they are members of species-poor lineages: monotremes such as the platypus in mammals, ratites such as the ostrich in birds, tuataras relative to lizards and snakes, and mosses among land plants.
Modern genome comparisons and studies of molecular evolution have revealed that our perceptions of which species are “primitive” versus “advanced” (frequently based on a small number of morphological characters) are not borne out at the level of the genome. For example, despite the tendency to assume that humans are “advanced” and chimps are “primitive,” a comparison of 14,000 genes revealed that the chimp genome has substantially more genes with evidence of positive natural selection. Likewise, classic work by Alan Wilson, John Avise, and others found no evidence that species considered to have slow morphological evolution (e.g., horseshoe crabs) have slower rates of molecular evolution. Furthermore, no overall genome measures (e.g., total genome size) correspond well to typical human perceptions of which species are considered “more advanced,” “more evolved,” or “more complex.” Many of these words or concepts are also hard to define or are value laden. The word primitive carries a strong connotation of inferior, and advanced carries the opposite connotation. The words ancestral versus derived are related terms that can be used for individual character states. But as Crisp and Cook (2005) pointed out, even ancestral and derived should not be applied to an extant species for two related reasons.
Phylogenies Do Not Indicate Which Extant Species Are “Ancestral” or “Older”
Generally it is best to assume that no extant species is ancestral to another extant species. For example, a common misconception is that chimpanzees are ancestral to humans. The human and chimp lineage shared a common ancestor roughly 6 million years ago (figure 3), but that common ancestor was neither a chimp nor a human (as recent fossil finds of the extinct hominid Ardipithecus strikingly demonstrate). Thinking that chimps are ancestral to humans assumes that the chimp lineage stopped evolving either before or right after the human/chimp split. In fact, at least as measured by number of genes subject to selection, the chimp lineage has evolved more than the human lineage since their split.
For similar reasons, phylogenies do not indicate which species are “older” versus “younger.” Perhaps one might think the age of a species can be inferred by determining when it last shared a common ancestor with another species. Species at the left of typical vertical trees could seem “older,” and species at the right with other close relatives on the tree could seem “younger.” A dated chronogram of African apes shows the chimpanzee/bonobo split at the far right at approximately 2 million years ago, and the human/chimpanzee lineage split at approximately 6 million years ago (figure 3; see chapter II.4 for the way such dates are inferred). Does that deeper split mean that humans must be “older” than the other two apes? Does that deep split mean that the species Homo sapiens is 6 million years old? Furthermore, if bonobos went extinct, would that suddenly make common chimpanzees several million years older? The answer to all these questions is no—one can tell very little about the “age of species” from a phylogenetic tree. Evolution is a continuous process—allele frequencies are constantly changing, and new character states are evolving—so it would be difficult to determine when one species ends and another begins. Even if one were somehow able to directly observe the process, the origins of livings species would be hard to associate with a specific moment in time.
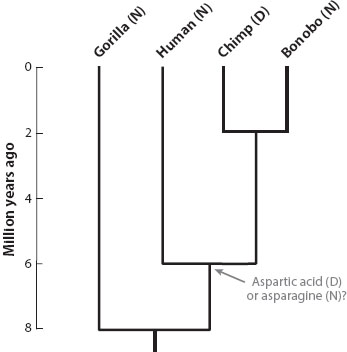
Figure 3. Chronogram showing rough divergence times of the four main lineages of African apes. Scientific names left to right: Gorilla gorilla, Homo sapiens, Pan troglodytes, and Pan paniscus. Also shown, amino acids at position 71 in the green sensitive opsin: N = asparagine, D = aspartic acid. (To practice tree thinking, replace the apes in figure 3 with members of the present generation of your family—siblings and cousins. In particular, in place of the gorilla, put one of your second cousins—your parent’s cousin’s offspring. Can you tell from the tree whether your second cousin is any more “primitive,” older, or more ancestral than you are? This analogy emphasizes how flawed it is to even ask such questions about extant species based on a tree.)
Taxon Sampling, Tree Focus, and Extinction Affect Which Species Seem “Primitive” or “Older”
Which species are sampled (“taxon sampling”) can dramatically affect a tree’s appearance. For example, a systematist might include only a few species from a species-rich but difficult to sample genus from central Africa. Because of incomplete taxon sampling, this lineage would appear to be species poor and might thus be shown on the left side of a vertical tree; hence, it might seem to be “earlier branching” or “older.” In fact, every tree ever drawn suffers from incomplete taxon sampling, so there are always missing tips and missing nodes. For a complete tree, at the very least you would want to include every one of the millions of extant species on the planet.
During phylogenetic inference (see chapter II.2), the position of the root of a tree is frequently inferred by comparing the focal group (the in-group) to a few closely related taxa (the out-group). As a result, out-groups frequently appear at the left side of vertical trees, and might therefore seem “primitive,” “simple,” or “ancestral.” But as Omland et al. (2008) detail, out-groups are not chosen because they are “ancestral” in some way. Out-groups are chosen because they are closely related to, but not within, the focal in-group.
Returning to figure 2, we can compare a tree focused on humans to a tree focused on sharks. For a systematist interested in relationships among tetrapods, the in-group includes two mammals, a lizard, and an amphibian, with a great white shark as an out-group (figure 2A); thus, the shark would appear at the left and might in some way seem “ancestral to tetrapods.” However, a marine biologist interested in shark relationships would include multiple sharks in the in-group, and humans could serve as a valid out-group (figure 2C). Thus, in a tree focused on sharks, Homo sapiens would appear at the left, which by the same incorrect logic might make humans seem “primitive” or “ancestral to sharks.” As in this example, humans will be on the species-poor side of many trees; knowing which lineage is species poor does not indicate how “old” or “primitive” the species are in that lineage.
Over time, the process of evolution can lead to changes in which one of two sister lineages has more species. Rates of speciation can speed up and slow down, and extinction can dramatically reduce the number of species in a lineage. Extinction can cause a lineage that once was species rich to become the species-poor lineage millions of years later. Thus, a species-poor sister group can at a later time be the more species-rich lineage. To summarize, left to right order on a vertical tree has no meaning because branches can be rotated around nodes, and because taxon sampling and extinction strongly influence which lineage appears species poor. More generally, the number of species in a lineage does not indicate whether a given characteristic of a species will be ancestral versus derived.
3. READING TREES CORRECTLY: ANCESTRAL STATE RECONSTRUCTION
Given that phylogenetic trees cannot indicate which species are “primitive” versus “advanced,” ancestral versus derived, or older versus younger, we can now focus on what it is that trees can tell us. Evolutionary trees are tremendously powerful tools for helping us understand evolutionary history, but the focus must be on learning about characters. Although trees cannot tell us which species are ancestral versus derived, trees can be used to infer which character states are ancestral versus derived.
Consider the amino acid sequence of a visual protein, green-sensitive opsin. In amino acid position 71, humans have asparagine (N), whereas chimps have aspartic acid (D) (figure 3). Clearly there has been an evolutionary change somewhere along either the chimp or the human lineage, but did the common ancestor have N or D? Using outdated ladder-of-progress thinking, we might think that chimps would retain the ancestral state, so that the chimp/human ancestor would have had D. Using a phylogenetic approach, we know gorillas are the sister to the human/chimp clade, and gorillas have N. This observation makes it more likely that N is the ancestral state—one change in the chimp lineage is probably more likely than two independent changes in the human and gorilla lineages. This inference is further supported because bonobos and other apes also have N at that site.
Figure 2A illustrates another example of ancestral state reconstruction involving the presence or absence of hair. To be precise, the “character” is body covering, and the “character states” are hair versus no hair. Three of the species have no hair, whereas both mice and humans have hair. On this tree, mice and humans are each other’s closest relatives (they form a monophyletic group), so it seems reasonable to infer that hair evolved in the common ancestor of the two mammal species (as indicated by the horizontal line). This reconstruction is more parsimonious than the alternative hypothesis—it requires fewer evolutionary changes (only one change). In many cases, evolutionary biologists use the principle of parsimony to infer ancestral characteristics. Parsimony, which is based on the principle known as Occam’s razor, assumes that the simplest explanation is the one most likely to be true. It is possible that the two mammal lineages evolved hair independently, but such a reconstruction would be less parsimonious because it would require two evolutionary changes. Data from thousands of other species and from fossils support parsimony in this simple case of mammalian hair. It is worth noting that the ancestral state is directly determined not by which state is the most common but by the distribution of states across the tips (see chapter II.8 for other ways of studying ancestral states).
A useful exercise is to look for patterns of nesting of taxa with certain character states. In figure 2A, the species with hair form a monophyletic group nested within a paraphyletic assemblage of vertebrates without hair. Frequently, the ancestral state can be inferred when character state A is shared by a series of species-poor lineages, with a nested focal clade having character state B. That mammals are nested within nonhairy tetrapods may be even more apparent when the nodes are rotated in a zigzag fashion (figure 2B).
Shared Derived versus Shared Ancestral Traits
Hair is a good example of a trait that is a shared derived character state (synapomorphy) of mammals. Hair marks a clade, which means that all organisms with hair are closely related to each other; however, a trait can lead one astray if it is a shared ancestral trait (symplesiomorphy). Scaly skin is an ancestral characteristic shared by lizards, turtles, and crocodiles. This trait contributed to misleading taxonomy for hundreds of years. Data from morphology, fossils, and DNA sequences now strongly indicate that “scaly reptiles” do not form a clade; rather, crocodiles are more closely related to birds than they are to lizards and snakes, for example. The data show that birds are clearly part of the reptile clade. This example illustrates that shared derived states provide evidence of evolutionary kinship, whereas shared ancestral states do not. (Figure 2A shows the series of lineages on the left with the shared ancestral trait of “no hair.”)
It is important to emphasize that one lineage at the “left” of a vertical tree does not necessarily retain the ancestral state for any given character. For example, in figure 2A, sharks have a cartilaginous skeleton, whereas all the other lineages have bony skeletons. But that fact alone does not mean that the common ancestor at the root node had a cartilaginous skeleton. In fact, considering the bony skeleton of other living and extinct chordate species indicates that the common ancestor had a bony skeleton that became cartilaginous in the shark and ray lineage. Again, it is crucial to avoid the fallacy of thinking of the species at the left as “basal” or “primitive.”
Figure 2C shows another example with a species that lacks a tail—human—as the out-group; the four shark species all clearly have a tail. But that tree does not indicate that the common ancestor of these five vertebrates lacked a tail. Presence or absence of a tail at the root node are two equally parsimonious reconstructions (each requires one change of character state). In fact, in this case we know from fossils and additional taxon sampling that the common ancestor had a tail, which was recently lost in the lineage leading to humans and other apes.
4. UNDERSTANDING THE PROCESS OF EVOLUTION: WE ARE ALL COUSINS
In conclusion, phylogenetic trees have a wide range of uses in evolutionary biology and systematics, and subsequent chapters focus on many of these topics, including molecular dating, biogeography, and phylogenetic comparative methods (see chapters II.4–II.8). A central goal of phylogenetics is to define monophyletic groups and sister group relationships, which can be used by taxonomists to develop stable and predictive classifications (chapter II.9 and subsequent chapters); however, knowing how to interpret the basic structure of trees and how to avoid the many widespread misconceptions about trees is a necessary first step for all of these applications.
More generally, knowing how to interpret trees leads to a better understanding of the process of evolution. One might incorrectly think that evolution is goal directed—that natural selection is “advancing” evolution in some way toward “more evolved” characteristics. Understanding phylogenetic trees provides an excellent opportunity to understand that evolution is not proceeding from one extant species to the next toward species with humanlike characteristics. Phylogenetic trees are frequently depicted with species unlike humans off on the left side, with humans on the far right. By understanding tree thinking, we know that node rotation and taxon sampling can make species appear almost anywhere left to right on a vertical tree; however, some lineages really are species poor; such species-poor sister groups will always be off on the side. But, it is crucial to emphasize that such side lineages do not represent “primitive” early evolutionary stages; side lineages need not retain the ancestral state for any given character.
Focusing on the extant species at the tips of the branches, it is important to remember that relationships among extant species are cousin relationships, not ancestor-descendant relationships. Thus, phylogenetic trees of extant species emphasize relationships among cousins. Our closest cousins are chimpanzees and bonobos, followed by gorillas, then other primates, other mammals, etc. Chimpanzees are our cousins, not our ancestors. Natural selection has molded adaptations that have enabled each of our cousin species to persist. Each extant species depicted at the tips of phylogenetic trees has a unique combination of ancestral and derived characteristics. Knowing how to read the tree of life—which Darwin first conceived of more than 150 years ago—enables us to better understand evolutionary history and better appreciate our common ancestry with all life on earth.
FURTHER READING
Avise, J. C. 2004. Molecular Markers, Natural History and Evolution. 2nd ed. Sunderland, MA: Sinauer.
Baum, D. A., S. D. Smith, and S. S. Donovan. 2005. The tree-thinking challenge. Science 310: 979–980.
Baum, D. A., and S. D. Smith. 2013. Tree Thinking: An Introduction to Phylogenetic Biology. Denver, CO: Roberts & Company.
Brooks, D. R., and D. A. McLennan. 1991. Phylogeny, Ecology, Behavior: A Research Program in Comparative Biology. Chicago: University of Chicago Press.
Crisp, M. D., and L. G. Cook. 2005. Do early branching lineages signify ancestral traits? Trends in Ecology and Evolution 128.
Maddison, D. R., and W. P. Maddison. 1992. MacClade: Analysis of Phylogeny and Character Evolution. Version 3.0 (user’s manual). Sunderland, MA: Sinauer.
Omland, K. E., L. G. Cook, and M. D. Crisp. 2008. Tree thinking for all biology: The problem with reading phylogenies as ladders of progress. BioEssays 30: 854–867.