Digestive System
Matthew A. Wallig, University of Illinois at Urbana-Champaign, Urbana, IL, United States
Abstract
When considering the potential toxic activity of various agents on the gastrointestinal (GI) tract, a number of mechanisms are possible. For example, acute toxic effects may result from direct irritants (e.g., strong acids and bases), whereas chronic effects maybe manifested as increased muscular layer thickness from bulking agents. Delayed effects maybe expressed years after exposure to the initial ulcerogenic or carcinogenic agents. In addition to an array of tissue responses, the mechanistic relationships between functional abnormalities and morphological alterations can be complex. The focus of this chapter is to examine structural and functional components of the GI tract that are important in understanding mechanisms involved in the toxicologic pathology of this organ system. A major part of the chapter stresses basic mechanisms of toxicologic damage, and how the GI tract responds to toxicologic insult. Also discussed in brief are morphologic approaches that can be used to study selected pathologic mechanisms of toxicological significance.
Keywords
Acetylcholine; arachidonic acid; arsenic; barrier function; β-catenin; bile salts; biotransformation; cadmium; cecum; colon; crypts; constipation; cyclooxygenase (COX); cytochrome P450 (CYP); diarrhea; dioxins; duodenum; emesis; enteric lymphoid system; enteric nervous system; enterocyte; enterohepatic; erosion; esophagus; ethanol; forestomach; fundus; gastric glands; gastrin; glucuronide; glucuronidation; glucuronosyl transferase; glutathione; growth factor; gut-associated lymphoid tissue (GALT); Helicobacter; heavy metals; hypersensitivity; hypoxia; ileum; jejunum; large intestine; malabsorption; mercury; microflora; microbiota; motility; mucin; mucosa; mucosal barrier; mucosal immunity; mucus; muscularis; NSAIDs; proliferation; pylorus; radiomimetic agents; regeneration; stem cells; sulfation; small intestine; steroids; stomach; TCDD; TNF-α; ulcer; ulcerative; ulcerogens; vagus; vomiting
Introduction
This chapter will present the basic mechanisms by which toxic xenobiotics produce their deleterious effects and describe the consequences of toxicity on the integrity of gastrointestinal (GI) structure and function. The intrinsic ability of the GI tract to resist toxic chemicals has led to a paucity of data regarding GI toxicologic pathology, yet this organ system can be readily perturbed, leading to easily identified toxic responses such as emesis or diarrhea. Other perturbations such as insufficiency of enzymes (e.g., lactase, lipase); the presence of localized inflammation, polyps, or neoplasms; changes in function such as excess production of mucus or delayed gastric emptying; or structural damage such as ulcers are more difficult to identify and attribute to toxicologic processes. For these reasons, it is necessary to identify those functions and structures of the GI tract that are subject to direct or indirect chemical toxicity.
When considering the potential toxic activity of various agents on the GI tract, a number of signalments are possible. Acute effects may result from direct irritants (e.g., strong acids and bases), whereas chronic effects may be observed, such as increased muscular layer thickness from bulking agents. Importantly, delayed effects can be expressed years after exposure to ulcerogenic or carcinogenic agents. In addition to the array of tissue responses, interpretation of functional and morphological alterations can be complex. For example, increased mucosal thickness can occur when toxic compounds induce cellular proliferation and hyperplasia (e.g., enterochromaffin cell-like hyperplasia) or when nontoxic foodstuffs such as fiber induce generalized mucosal growth. The focus of this chapter is the examination of developmental, structural, and functional components of the GI tract that are important in understanding mechanisms involved in the toxicologic pathology of this organ system.
The principal functions of the GI tract that are subject to toxic effects of metals, toxicants, toxins, and xenobiotics include storage, propulsion, digestion, absorption, secretion, barrier activity, and elimination. Due to the importance of nervous reflexes and hormones in regulation of the GI tract, this organ system is relatively unusual in that toxic effects of a particular toxic molecule at one site (e.g., stomach) maybe expressed at another site (e.g., colon).
The GI tract is the entry site into the body of orally administered compounds that maybe highly toxic to other internal organs yet have little or no noticeable effect on the GI tract. A distinctive feature of the GI tract is the high proliferative and metabolic rate of the mucosa. In addition, the GI tract mucosa is a complex barrier that must exclude bacteria and their molecular toxins and, at the same time, absorb nutrient molecules that are vital for homeostasis. These two functions alone make the GI tract unique. Furthermore, this organ system cannot sustain widespread toxicity without serious direct and indirect consequences to the rest of the body, if for no other reason than nutrient malabsorption with consequential malnutrition or starvation.
The GI tract is the only internal organ system that contains endogenous biotransforming and toxigenic bacteria, as well as inert drug-binding materials. Consequently, when a compound is present in the GI milieu, the ultimate toxicity to this organ system will be determined by interactions of the chemical with both bacterial and mammalian enzymes, and by the extent of respective detoxification and bioactivation processes. The ability to evaluate genomic, proteomic, biochemical, or morphological changes in the GI tract can be complicated because of the matrix of interactions and the GI tract’s exquisite sensitivity to autolysis and postmortem alterations. Many subtle toxicologic events that occur at cellular and subcellular levels may only be observed by careful and proper handling of the GI tissues immediately after death.
Structure and Function of the Gastrointestinal Tract
Macroscopic and Microscopic Structure and Function
Although many of the basic features of the GI tract are similar for various species (Figures 15.1 and 15.2), major interspecies variations are present in the fore- and hindgut (Table 15.1). This notwithstanding, within each major macroscopic variation, the cell types composing the mucosal lining of the GI tract are remarkably similar (Figure 15.1 and Table 15.2).
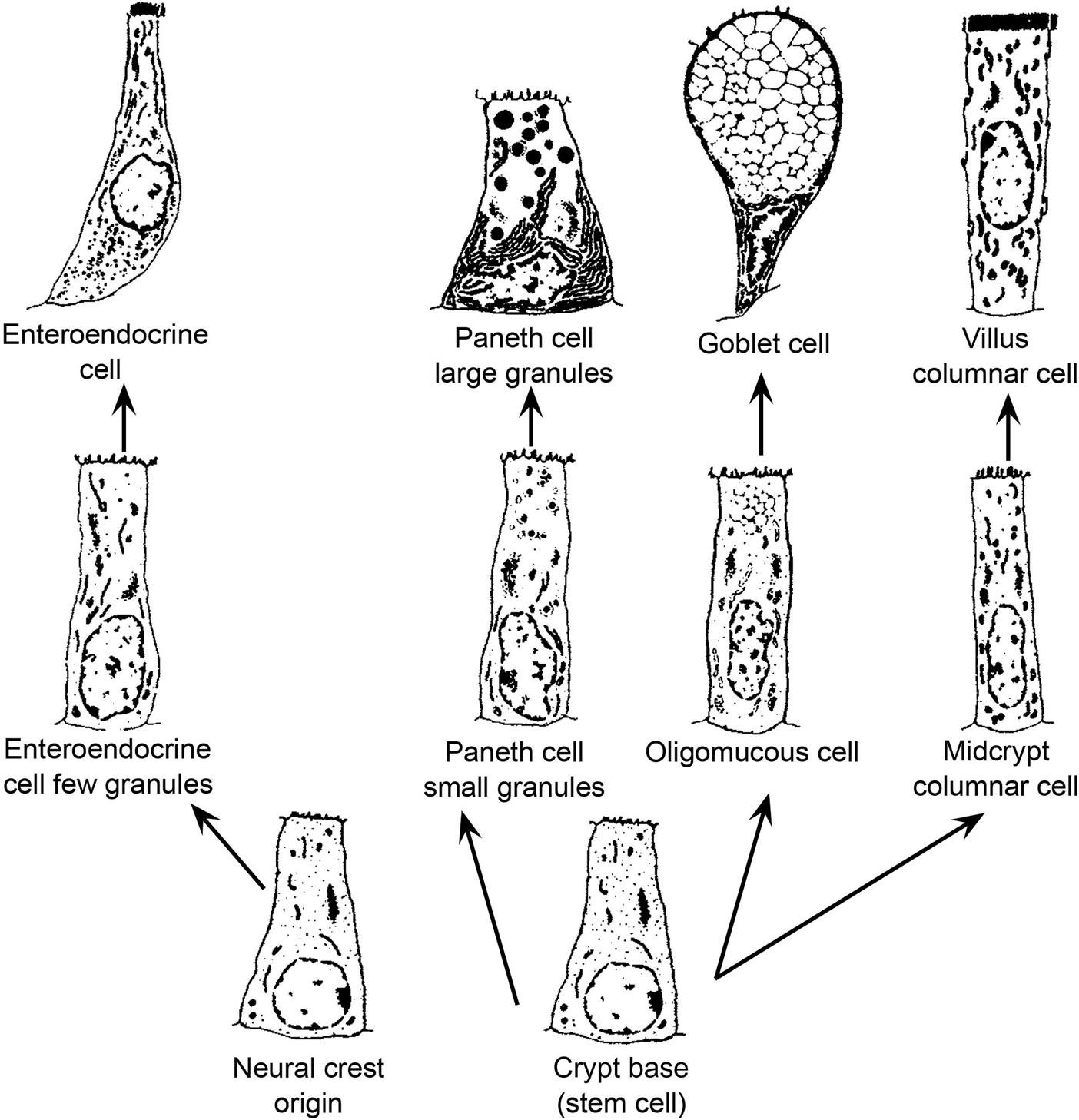
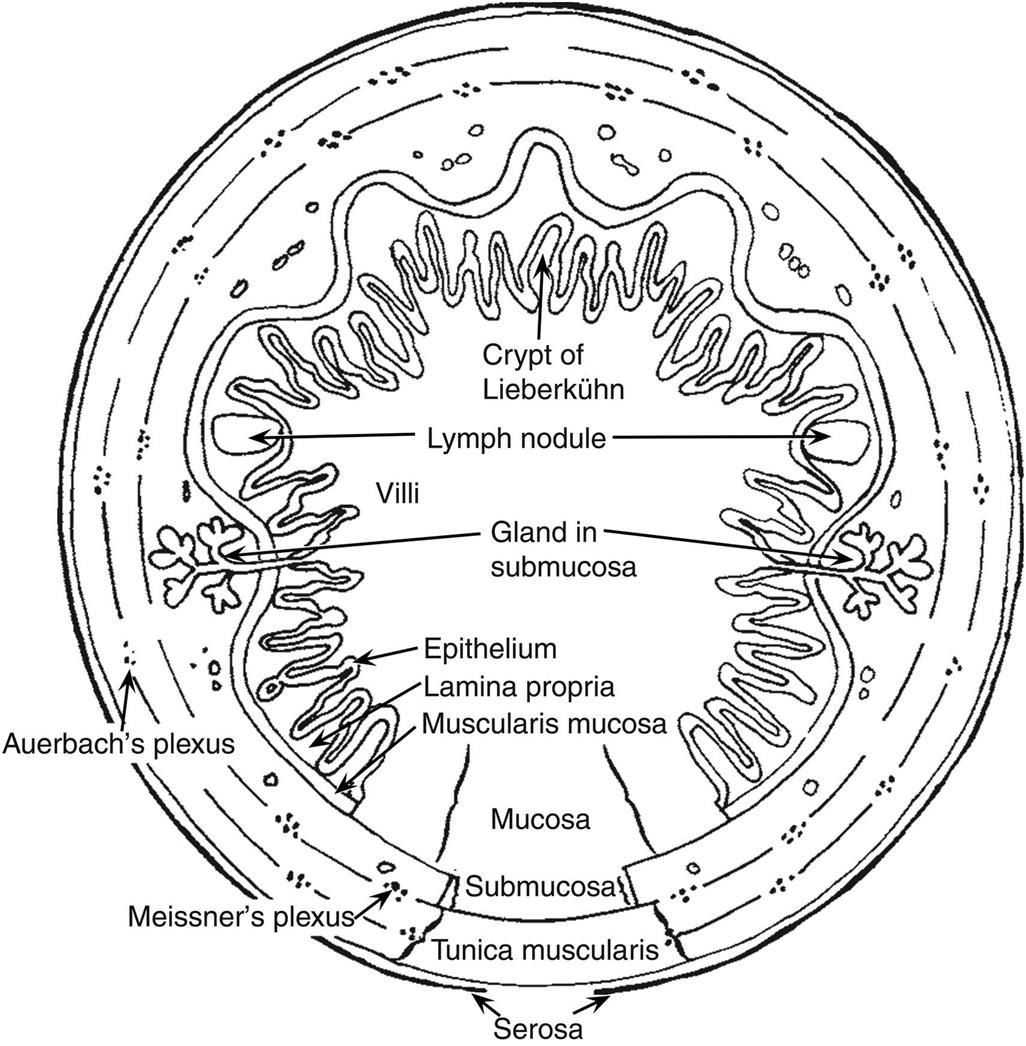
Table 15.1
Variations in Dietary Consumption Related to Gastrointestinal Structure in Various Mammalsa
| Order | Carnivore | Omnivore | Herbivore | Stomach | Large intestine | ||
| Sacculated | Stratified squamous epithelium | Cecum | Sacculated colon | ||||
| Carnivora | + | + | − | − | ± | ± | |
| Rodentia | + | + | + | ± | ± | ± | ± |
| Lagomorpha | + | − | − | + | + | ||
| Primates | + | + | + | ± | ± | + | ± |
| Artiodactyla | + | + | ± | ± | ± | ± | |
| Marsupialia | + | + | + | ± | ± | ± | ± |
| Perissodactyla | + | − | + | + | + | ||
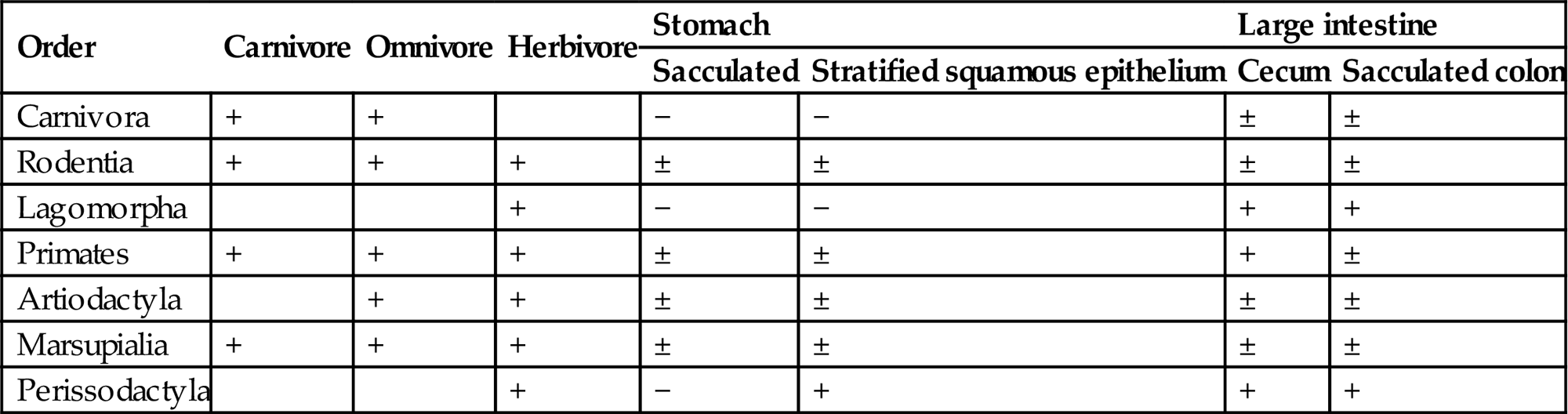
aModified from Stevens (1980).
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table I, p. 124, with permission.
Table 15.2
Cells Composing the Epithelial Lining of the Gastrointestinal Tracta
| Cell type | Shape | Location | Product |
| Absorptive cell (enterocyte or colonocyte) | Columnar | Small intestine to colon | Nutrient absorption |
| Mucous cell | Cuboidal to columnar | Stomach to rectum | Mucus |
| Chief cell | Pyramidal | Stomach (fundic) | Pepsin, rennin, lipase |
| Enterochromaffin cells | Pyramidal | Stomach to rectum | Endocrine (at least 10 types) |
| Goblet cell | Columnar | Small intestine to rectum | Mucin |
| M cell | Membranous | Dome of Peyer’s patches | Processed antigens |
| Paneth cell | Pyramidal | Small intestine | Lysozyme, peptidase |
| Parietal cell | Cuboidal | Stomach (fundic) | HCl, intrinsic factor |
| Undifferentiated crypt cell | Cuboidal | Small intestine to rectum | Progenitor cell |
| Vacuolated cell | Columnar | Colon to rectum | Progenitor cell |
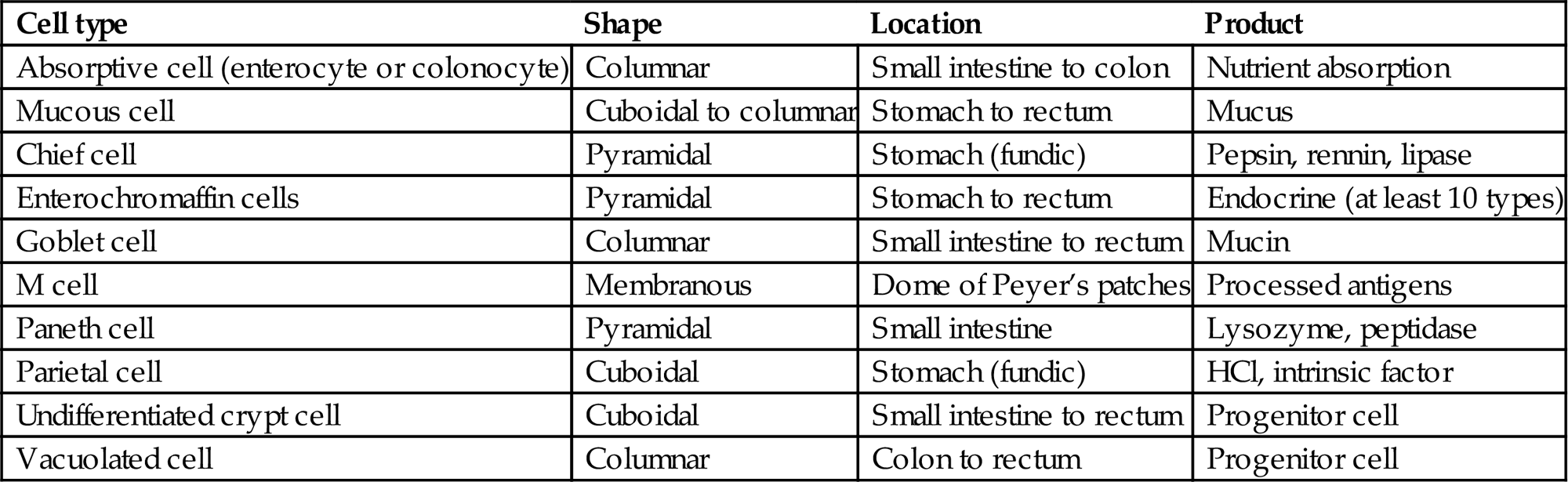
aThe number of these individual type cells in any given anatomical location in the gastrointestinal tract can vary with animal species and diet. Additionally, toxicologic agents can markedly influence the distribution and relative ratios of each cell type.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table II, p. 128, with permission.
Esophagus
The function of the esophagus in all species is to act as a conduit for food materials in the oral cavity to enter the GI tract. Interspecies esophageal variations occur related to the presence and extent of smooth and striated muscle, the gastroesophageal junction, and caudal sacculated adaptations (forestomachs). In all species, the esophagus is lined by stratified squamous epithelium with varying degrees of keratinization.
The extent of keratinization of the esophageal and nonglandular gastric epithelium is dependent on the amount and type of dry foodstuff ingested. Consequently, hyperkeratosis of the mucosa can indicate anorexia or an increase in roughage content of the feed in both ruminants and rodents. This keratinized epithelium imparts a whitish color to the mucosa when the esophagus is viewed macroscopically. Keratinization is also a normal feature of the nonglandular portion of the rodent stomach.
The tunica muscularis of the esophagus has two muscle layers composed of striated, smooth, or a mixture of both types of muscle, depending on the species. Variations in esophageal musculature account for the ability or inability of an animal to vomit or regurgitate. The absence of significant amounts of striated muscle in the esophagus of rats and the presence of a limiting ridge (margo plicatus) in the stomach separating the nonglandular and glandular regions explains why these animals are unable to vomit. The ability of dogs and guinea pigs to vomit and ruminants to regurgitate is dependent upon the presence of striated muscle in the esophagus, which permits esophageal contractions that allow retrograde transit of gastric contents.
Regurgitation, not vomiting, in nonruminants indicates esophageal dysfunction or obstruction. If the esophagus is perforated by caustic compounds, chronic drainage of saliva and ingested foodstuffs into the esophageal submucosa or intrathoracic regions leads to severe inflammatory reactions, fibrosis, and strictures. The esophagus does not heal as rapidly or achieve structural restoration as completely as other portions of the GI tract because of a marginal blood supply and a minimal amount of adventitial and serosal connective tissue.
Stomach
Anatomical and functional variations are important considerations when designing animal studies. Compound absorption and enzyme exposure (e.g., ruminal bacteria or inflammatory cell proteases) often vary from species to species, and sites of storage (e.g., nonglandular stomach) may provide prolonged contact between the host mucosa and a toxic compound.
Function
The stomach functions to store and macerate food—a necessary early phase of food digestion. Some phylogenetic orders have a highly sacculated forestomach (e.g., some artiodactyls and primates). Entrapment of food within saccules aids in food digestion by extending the exposure of ingested materials to digestive acids and enzymes. In ruminants, the ruminal portion (rumen) of the forestomach, which is actually a modification of the esophagus, is highly permeable to volatile fatty acids released from the microbial metabolism of complex carbohydrates and also is capable of active sodium and chloride absorption.
Several mammalian orders, including rodents and perissodactyls (horses, tapirs and rhinoceroses), have a nonglandular stratified squamous portion of the stomach proximal to the fundic mucosa. This squamous portion of the stomach is separated from the glandular stomach by a limiting ridge (margo plicatus) and serves as a storage organ for ingested material. Various inflammatory cells (lymphocytes, plasma cells, and eosinophils) maybe present in the lamina propria of the limiting ridge of rodents as a normal characteristic and should not be interpreted as an inflammatory process.
Structure
The topographical organization of the gastric mucosa varies widely among species. As monogastric simple-stomached mammals, humans and dogs have the cardia as the first glandular portion of the stomach following the esophagus or squamous forestomach; the cardiac mucosa is macroscopically red. This portion of the stomach has foveolae (gastric pits) and tortuous mucous glands. The fundus is the next glandular region and is characterized by mucosal convolutions called rugae. The distal portion of the stomach, the pylorus, also has rugae, but they are smaller than those of the fundus and are arranged obliquely in the direction of the antrum. Unlike other portions of the stomach, the foveolae of the antrum are deeper and make up as much as 50% of the mucosal thickness.
In spite of many macroscopic variations, the microscopic arrangement of the stomach is similar in all species. The mucosa rests on the submucosa, and these two layers are surrounded by a muscular coat (tunica muscularis) that is, covered by the single mesothelial cell layer of the serosa.
The fundic mucosa contains glands that are composed of mucous surface neck cells, parietal (oxyntic) cells, chief (zymogen) cells, and enteroendocrine (enterochromaffin) cells (Table 15.2). Chief cells are cuboidal and have a basally placed nucleus. The apical portion of the cytoplasm is filled with pepsinogen-filled zymogen granules. The primary function of the chief cell is to release enzyme precursors into the gastric lumen; once in contact with the acidic extracellular environment, the enzymes become activated to begin the process of gastric digestion. Parietal cells are larger but generally less numerous than chief cells. Parietal cells have a centrally located nucleus, and the smooth endoplasmic reticulum (SER) and mitochondria-laden cytoplasm stains intensely eosinophilic. Parietal cells release hydrochloric acid (HCl), to maintain gastric pH, and chymosin (or rennin) in young animals, to facilitate digestion of milk. Carbonic anhydrase in the parietal cells acts on carbon dioxide (CO2), thereby producing carbonic acid that dissociates to provide H+ for excretion. Both Cl− and H+ are actively secreted into the lumen, with water following the osmotic gradient. Food material in the stomach, vagus nerve stimulation, gastric distension, and gastrin released from G-cells in the glands of the pylorus and duodenum stimulates the parietal cells to release H+. Stimulation of chief cells to release pepsinogen comes from a combination of vagal nerve stimulation, H+ concentrations, gastrin, and secretin (released from duodenal secretin cells).
In the antrum, the mucosal glands produce mucus. Gastric mucus is a composite of mucin, electrolytes, sloughed cells, enzymes, nucleic acids, lipids, plasma proteins, secreted immunoglobulins, bacteria, and bacterial metabolites. The composition of gastric mucus is 90%–95% water, 5%–10% mucin, 1% electrolytes, and approximately 5% all other components. Mucin, the principal component of gastric mucus, is synthesized by and secreted from mucus-producing cells resident within mammalian gastric mucosa. MUC5AC and MUC6 are the main mucins secreted by surface or glandular mucous cells of the human stomach. Mucins are high-molecular-weight polymers composed of glycoprotein subunits joined by disulfide bridges. Each glycoprotein subunit has a central peptide core flanked by carbohydrate side chains. Hydrogel formation by aqueous mucin polymers leads to formation of a protective layer over the gastric mucosa, which further assists in lubrication of the mucosal surface and digestion. Cells of the gastric glands also release arachidonic acid metabolites (e.g., prostaglandins of the E series) that facilitate protection of the mucosa.
Cellular composition of gastric glands in the fundic mucosa varies among animal species. Intermixed with the gastric gland epithelium are enteroendocrine cells, of neural crest origin. Enteroendocrine cells, which are usually located between the basement membrane and chief cells, synthesize, store, and secrete hormones in response to autonomic and intraluminal stimuli. There are at least 10 different enteroendocrine cell populations in the mucosa. Enteroendocrine cells secrete serotonin, histamine, enteroglucagon (A cells), and gastrin (G cells), among other factors.
Replication of the mucosal cells in the stomach is somewhat different from that of the rest of the GI tract. Unlike the replication of crypt cells of the intestines and basal cells of the esophagus, gastric mucosal cell replication occurs in the neck of the gastric glands.
The next layer of the gastric mucosa, immediately below the epithelium, is the lamina propria, which is separated from gastric epithelial cells by a basement membrane. The lamina propria of the cardia and pylorus contains high numbers of lymphocytes and plasma cells. These immune cells are abundant throughout the gastric mucosa and submucosa, and the pyloric lamina propria may contain numerous lymphoid follicles even in a healthy animal. This lymphoid tissue can markedly enlarge in disease states where there is antigenic stimulation (Figure 15.4).
The lamina muscularis mucosae separate the mucosa from the submucosa. The submucosa is composed of a loose connective tissue matrix supporting many nerves and blood and lymphatic vessels. Three smooth muscle layers constitute the tunica muscularis, which encircles the stomach in overlapping layers and functions to mix food and move contents from storage in the stomach into intestine for continued digestion and nutrient absorption. The muscle fibers are oriented in circumferential (middle layer), longitudinal (outer layer), and oblique (inner layer) bundles to massage the ingesta from as many angles as possible, which facilitates physical disruption of intragastric solids.
Small Intestine
Function
This segment is primarily responsible for secretion and absorption of nutrients. In addition, the small intestine functions to biotransform compounds, resulting in bioactivation or detoxification (Table 15.3), as a barrier to luminal contents (bacteria and nonabsorbed compounds), and as a conduit for indigestible ingesta to pass out of the body. Numerous anatomical modifications increase the functional capacity of the small intestine, including its long length, linear plicae, circular plicae, villi, and microvilli. These characteristics influence mucosal surface area and can modify the transit time of a compound through the GI tract. Relative to the stomach and large intestine, passage time through the small intestine is relatively rapid (i.e., a few hours).
Table 15.3
Mucosal Metabolic Conjugation of Selected Chemicals
| Chemical | Glucuronidation | Sulfation |
| o-Aminophenol | + | + |
| Anthranilic acid | + | + |
| Buprenorphine | + | |
| Dihydromorphine | + | |
| Ethinylestradiol | + | |
| Etorphine | + | |
| Isoprenaline | + | |
| Midaglizole | + | |
| Morphine | + | |
| p-Nitrophenol | + | + |
| Phenol | + | + |
| Salicylamide | + | + |
| Salicyclic acid | + | + |
| Thyroxine analogs | + | + |
| Xamoterol | + | + |
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table III, p. 129, with permission.
Between the proximal and distal small intestine, a functional gradient of ion and water transport occurs, which controls the movement of fluids and electrolytes across the mucosa. In the proximal small intestine, passive movement of sodium and water is from the blood to the GI tract lumen. In contrast, fluid and sodium movement is from the lumen to the blood in the distal small intestine. Net secretion occurs in the ileum and jejunum of guinea pigs, the ileum of rabbits, and the proximal portion of the jejunum in neonatal swine. The jejunum absorbs sodium, chloride, and bicarbonate against an electrochemical gradient; however, this decreases as the animal ages. Bile salts are primarily reabsorbed in the ileum.
Structure
The small intestine constitutes the majority of the GI tract’s length. Major structural features of the small intestine vary little among species. The general microscopic organization of the small intestine is similar to that of the stomach, with three distinct layers (mucosa, submucosa, and tunica muscularis) surrounded by the serosa.
Small-intestinal morphology reflects its absorptive function and can be artificially divided into two zones: villi for absorptive and enzyme release, and crypts for secretion and mucosal replication/replacement (Figure 15.2). Crypt-depth-to-villus-height ratios vary from species to species but tend to remain constant within a species; hence, they can be used to assess the degree of intestinal damage resulting from exposure to a toxic compound. The distance from the base of the crypt to the tip of the villus is divided by a “shoulder” at the crypt–villus junction (Figure 15.3D). This juncture is the transition between the crypt and villus functionally as well as anatomically. The crypt-depth-to-villus-height ratio in the proximal small intestine ranges from a small 1:7 ratio in the pig to a larger 1:2 ratio in the dog. Within a species, this ratio also will vary with the amount of food material in the lumen, luminal distension, and diet.
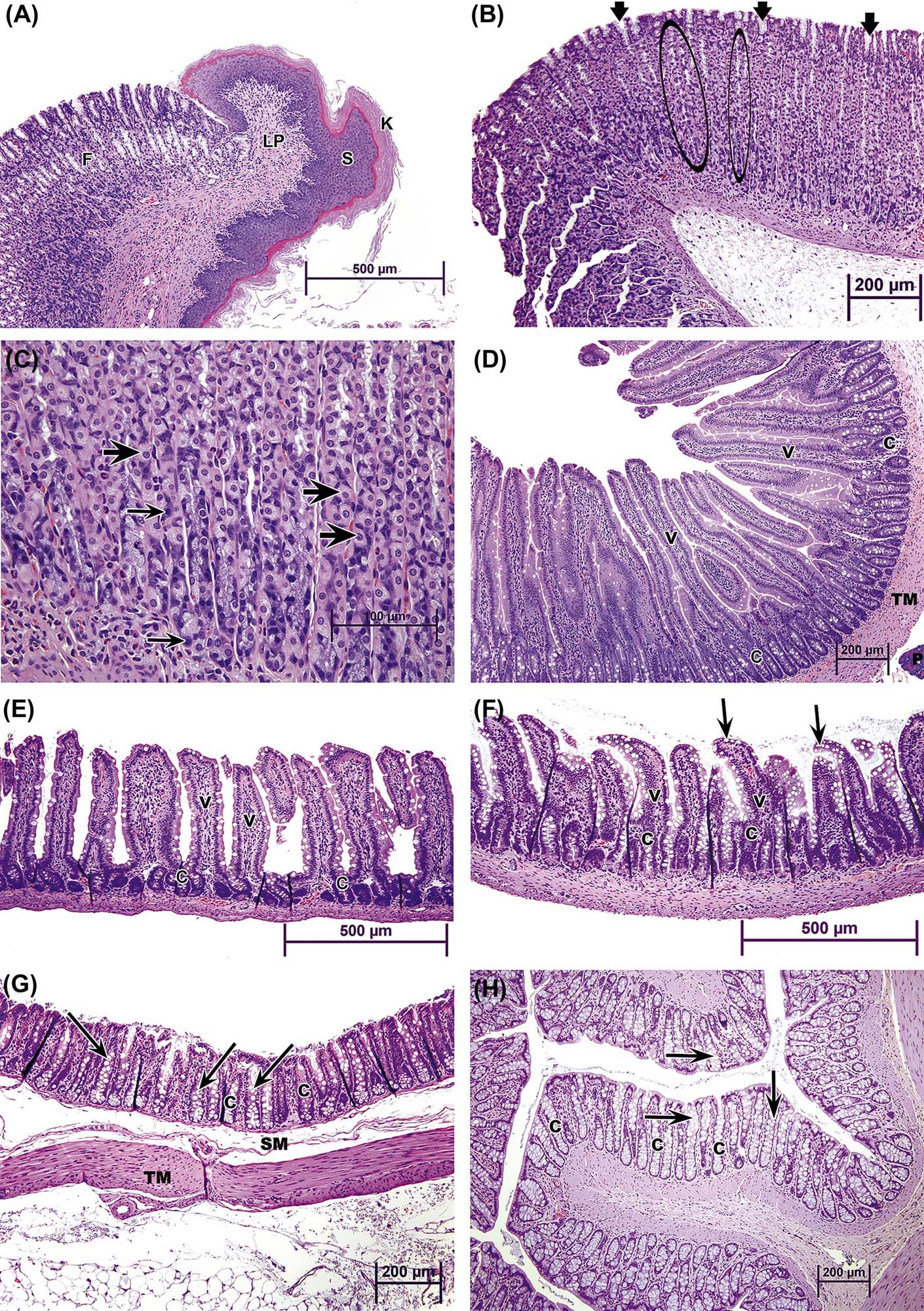
Villus height progressively decreases from proximal to distal small intestine. Villi are covered by mature but senescent cells that have migrated along the basement membrane up from the crypts and are supported by a connective tissue core, the lamina propria. The center of the villus has blind-ended lymphatic vessels, or lacteals (Figure 15.3D), that are surrounded by an elaborate capillary bed; both these vascular beds are located subjacent to the epithelial basement membrane. The lacteals serve to carry fat-soluble compounds to the systemic circulation, thus bypassing hepatic metabolism.
Cells lining the mucosa of the small intestine are primarily composed of simple tall columnar epithelium (enterocytes) on the villi and cuboidal epithelium in the crypts (Table 15.2). Villous epithelial cells have a thick microvillar apical membrane (approximately 11 nm in height) and have multiple biotransforming and metabolizing enzymes at the luminal surface. The thickness and composition of this enzyme-rich apical membrane is maintained by cytoskeletal elements (microtubules) and the presence of tight junctions at the lateral membrane–junctional complexes near the apices of the cells. Enterocytes absorb simple carbohydrates, amino acids, and some xenobiotics and then actively transport them, with little processing, into subjacent capillaries for transportation to the liver.
Each villus consists of 2000–8000 enterocytes and is surrounded by 6–14 crypts. The crypt is the proliferative unit of the intestinal mucosa, as cell division is confined to the crypts. Each crypt generates four types of terminally differentiated cells: enterocytes, goblet cells (to secrete mucus), enteroendocrine cells, and Paneth cells (to produce lysozymes); all these cells serve as a barrier to bacteria. Each crypt produces 300–400 cells per day, and each epithelial cell has an average life of 3 days. Most extensive replication occurs in the cells immediately above the bottom four to six cells. Each crypt has committed stem cells that divide rapidly to produce daughter cells. Daughter cells may themselves divide several times in the lower and middle portions of the crypts, but their mitotic capabilities are limited. Negative feedback mechanisms coordinate the rate of cell proliferation in the crypts. The cells differentiate and mature during an orderly and rapid migration from the crypt to the apex of a surrounding villus. As the cells migrate up the villus, they differentiate both structurally and functionally. Enterocytes (absorptive cells), by far the majority cell type, have a microvillous “brush border” on their apical surface. Goblet cells secrete mucus, and their apical cytoplasm is typically distended with mucus-filled secretory granules. Closely related to goblet cells are the epithelial cells lining submucosal (Brunner’s) glands present in the cranial duodenum of most mammalian species. These highly branched glands produce mainly mucin but also serve an endocrine function (see below). Enteroendocrine cells (themselves composed of many individual subtypes) are smaller and secrete various digestive hormones, such as catecholamines. Once these three cell types reach the apex of a villus, they are exfoliated. This process of proliferation, upward migration, and subsequent exfoliation is completed in 2–5 days.
Relative to other cell types in small intestinal crypts, Paneth cells exhibit several unusual traits. They remain anchored in the lower portion of the crypt rather than migrating up villi. They are long-lived, surviving for approximately 21 days. Finally, Paneth cells are a critical component of the mucosal defense system because they secrete antibacterial proteins (lysozyme, defensins) that provide local control of luminal microbiota. Paneth cells are found in monkeys, mice, rats, hamsters, guinea pigs, ruminants, and horses, but not in dogs, cats, swine, or raccoons. Paneth cell numbers increase in number from duodenum to ileum. The Paneth cell secretes mercury and other heavy metals into the intestinal lumen. These cells become necrotic in chronic methylmercury intoxication of primates.
Intestinal crypt cells secrete fluids and electrolytes, which are reabsorbed by intestinal villus cells. The transport of electrolytes across epithelial cell apical membranes occurs by multiple mechanisms. Uniport mechanisms move a single ion (e.g., sodium), symport systems move two ions simultaneously in the same direction (e.g., sodium and chloride), and antiport elements are ion exchangers which move two ions with the same charge in opposite directions (e.g., sodium and hydrogen). These systems are “active” processes that frequently require energy (ATP) and are stimulated by cAMP, cGMP, or increased levels of intracellular calcium. Fluid transport is also modulated by neurotransmitters such as serotonin (increases secretion) and neuropeptide Y (increases absorption).
Toxicant-induced damage to membrane-bound proteins can influence the viability of the mucosal epithelial cells and as a consequence the nutritional status of the animal. Within the apical membrane are proteins that consist of intimately membrane-associated calcium–magnesium-dependent ATPases and alkaline phosphatases, and less tightly held disaccharidases (i.e., lactases, sucrases, and maltases), and leucine aminopeptidases. The less tightly held enzymes are responsible for digestive processes, while the more tightly held ATPases control cellular homeostasis and viability. In the thinner lateral membranes (approximately 7 nm thick) of mucosal epithelial cells, ouabain-sensitive sodium–potassium ATPase is found in concentrations that are higher than in the apical surface. This enzyme is tightly linked to glucose absorption.
Less numerous goblet cells are scattered among enterocytes of the villi. The numbers of goblet cells increase in the villous mucosa from proximal to distal small intestine. Lysozyme- and peptidase-rich Paneth cells are found near the base of the crypts, associated with the proliferating cells, but there is no known dietary or environmental factor that controls this distribution (Table 15.2).
M cells are located in the surface epithelium overlying the lymphoid tissues (Peyer’s patches, see below) of the intestinal tract. These cells are recognized histomorphologically by the microfolds on their luminal surface (folds are not present in rats), and they are highly phagocytic. M cells are responsible for sampling antigens from the lumen contents and for transfer of the antigen to T lymphocytes and dendritic macrophages. The M cells can also function as an access route for pathogenic microbes and particulate toxic agents (e.g., asbestos).
Enterocytes may also function as antigen-presenting cells, especially for soluble proteins. Enterocytes express Class II major histocompatibility complex antigens and are capable of stimulating T lymphocytes to activate and proliferate. While enterocytes may process soluble antigens, M cells seem to be primarily responsible for processing particulate antigens.
The lamina propria is the neighboring loose connective tissue layer that nourishes the mucosal epithelium and its associated mucosal glands. Lymphocytes, plasma cells, and to a lesser extent, mucosal mast cells and eosinophils are present throughout the lamina propria, both within the villi and around the crypts. The numbers of these cells increase with age in all species. Although most of the immune cells in the lamina propria function in a similar manner to those in other regions of the body, the mucosal mast cell is functionally distinct from mast cells in other tissues (Section 2.3, Enteric Nervous System).
The muscularis mucosae separates the mucosa from the underlying submucosa. The muscularis mucosae is a thin smooth muscle layer that functions to move luminal contents in a single (distal) direction by modulating the sizes and shape of small intestinal mucosal folds (rugae). Motor activity in the muscularis mucosae exhibits considerable variations among regions and across species. Intracellular signaling pathways to control motor activity in this layer differ from those of smooth muscle in the tunica muscularis (muscularis externa). Since the submucosal area is a major source for eicosanoid production, abnormality of muscularis mucosae motor activity may link with abnormality of mucosal absorption and secretion functions.
The submucosa is found between the muscularis mucosae and the tunica muscularis. This layer consists of loose connective tissue and contains comparatively large vascular and lymphatic elements.
The tunica muscularis is usually a thin, double layer of smooth muscle. The smooth muscle cells are oriented circularly or helically within the inner layer, and longitudinally in the outer layer. This is typically the most substantial layer of the intestinal wall.
The adventitial layer on the outer surface of the small intestine is covered by mesothelium derived from the peritoneum in which the organ is suspended and hence is called the tunic serosa. This layer is composed of loose collagenous and elastic tissue in which adipose tissue can be found in well-nourished animals. The serosa is also the entry point for major arteries originating from the mesenteric arterial system. These arteries arborize longitudinally within the serosa but also penetrate the muscularis to form a second longitudinal arborization in the submucosa. Smaller arteries radiate outward into muscularis and mucosa, respectively, from these two major vascular supplies. Veins and lymphatics that drain the intestine follow the arterial supply back to the serosa to mesenteric venous and lymphatic drainage systems. There are extensive anastomoses between the various branches of the mesenteric arterial system and similar anastomoses between the various branches on the venous side as well.
Large Intestine
Function
Major functions of the large intestine include storage of digesta, water and electrolyte absorption, and secretion. One of the main electrolyte-absorbing processes is through the Na+/K+-dependent ATPase pathway of the mucosal epithelium. Herbivores secrete large volumes of salivary, pancreatic, and biliary fluids, and in perissodactyls (horses), the large intestine secretes additional fluids equivalent to 40% of the extracellular fluid volume. However, 98% of the fluid and ions secreted in the upper GI tract is reabsorbed in the cecum and colon. It is critical that this reabsorptive process is taken into account when attempting to investigate toxicant-induced diarrheas.
The colon has protein-absorbing activity. However, relative to the small intestine, the large intestine absorbs a small proportion of total body protein needs. The large intestine instead serves as the major site of digesta retention; however, the duration and primary site of retention varies between species. The rate of passage is inversely related to the degree of colonic compartmentalization. Additionally, retrograde propulsions of the colon, associated with absorption of water and electrolytes, may help delay the passage of a toxic compound and prolong exposure of a toxicant to various biotransforming enzymes.
The high concentration of bacteria in the colon facilitates roughage digestion and compound biotransformation. Although bacterial metabolism is critical for nutrition and influences toxicologic processes, the role of bacteria in colonic physiology has received limited study (Table 15.4).
Table 15.4
Metabolic Reactions by Intestinal Microfloraa
| Reaction | Representative substrate |
| HYDROLYSIS | |
| Glucuronides | Bilirubin glucuronide |
| Glycosides | Cycasin |
| Sulfamates | Cyclamate |
| Amides | Methotrexate |
| Esters | Acetyldigoxin |
| DEHYDROXYLATION | |
| C-hydroxy groups | Bile acids |
| REDUCTION | |
| Nitro groups | P-Nitrobenzoic acids |
| Double bonds | Unsaturated fatty acids |
| Azo groups | Food dyes |
| Aldehydes | Benzaldehydes |
| Alcohols | Benzyl alcohol |
| N-Oxides | 4-Nitroquinoline-1-oxide |
| Decarboxylation | Amino acids |
| Deamination | Amino acids |

aModified from Simon and Gorbach (1984) Intestinal flora in health and disease, Gastroenterology 85, 144–193, Table p. 175, with permission.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table IV, p. 132, with permission.
Structure
Macroscopic morphology of the large intestine varies widely among species. Anatomical modifications of basic structure include marked variations in relative length, diameter, volume, and compartmental complexity of this organ. The secretory and absorptive capacity of the large intestine is related to both its anatomical complexity and the need for the animal to conserve water.
Cecum
Although considered separately here, the cecum is an extension of the colon arising at the ileo–eolic junction. Cecal structure varies considerably among different animal species, as a rule quite small in carnivores and very large in herbivorous animals that rely heavily on ceco-colonic breakdown of cellulose for energy needs (e.g., rabbits and to a lesser extent rodents). The cecal lumen contains many bacteria that are metabolically active in detoxifying or bioactivating ingested compounds and producing essential vitamins. Some aspects of antimicrobial toxicity are directly related to the modification of normal cecal microflora.
The cecal mucosa is anatomically similar to that of the colon (see below), and the submucosa contains abundant lymphoid nodules that function like Peyer’s patches of the small intestine. The primary function of the cecum is for microbial fermentation and storage of ingesta. Intraluminal ingesta can be passed back and forth from the cecum to the proximal colon several times before continuing its passage down the remaining distal segments of colon.
Animals with large and functionally active ceca (e.g., rats, ruminants, swine) may have significantly different passage rates for ingesta and toxic compounds than do species with a rudimentary cecum (e.g., dogs, primates). Such information must be incorporated into the design of animal model studies and considered when interpreting toxicokinetic and drug metabolism data.
Colon
The mucosa of the colon and cecum is significantly different from the mucosa of the small intestine. Goblet cells are abundant in the colonic mucosa and are responsible for adding mucus to the dehydrated ingesta. Inflammation of the colon can lead to epithelial Paneth cell metaplasia, which reduces mucus production and renders the mucosa prone to bleeding.
The submucosa, tunica muscularis, and serosa of the large intestine are similar to those of the small intestine. The terminal end of the large intestine (rectum) is located retroperitoneally in the pelvic canal and is not covered by a serosa.
Enteric Lymphoid System
The GI immune response is multifactorial and involves both cellular and humoral immune mechanisms. Immunologic response of the GI tract is predominantly mediated by immunoglobulin isotype A (IgA) with and without secretory component (sIgA). The GI tract mucosa contains many IgA-producing plasma cells. Additionally, cell-mediated immune mechanisms are involved in the mucosal response to toxic compounds. Cell-mediated immunity of the mucosa is distinctly different from that of nonmucosal sites. This difference is exemplified by the enterocytes of the small intestine, which can function as antigen-presenting cells, IgA-antigen carriers, and activators of T lymphocytes. Consequently, immune mechanisms in the GI tract involve multiple pathways for response to toxic compounds, which may include hypersensitivity.
Located throughout the small intestine are deep mucosal/submucosal lymphoid aggregates called Peyer’s patches (Figure 15.4). Peyer’s patches represent the organized portion of the GI immune system and are part of the gastrointestinal (gut)-associated lymphoid tissue (GALT). GALT composes over 25% of the body’s total lymphoid organ mass and is most extensive in the small intestine. Peyer’s patches can be composed of only a few lymphocytes or maybe well-developed lymphoid nodules with many active secondary follicles. Well-developed nodules have both follicular and parafollicular regions. Follicles are composed of B cell-rich germinal centers located in the lamina propria or submucosa. Germinal centers are surrounded by parafollicular T cells and are capped by a dome of small lymphocytes that extends into the specialized M cell-rich mucosal epithelial covering. The stronger the antigenic stimulus, the more extensive will be the response and development of the nodules.
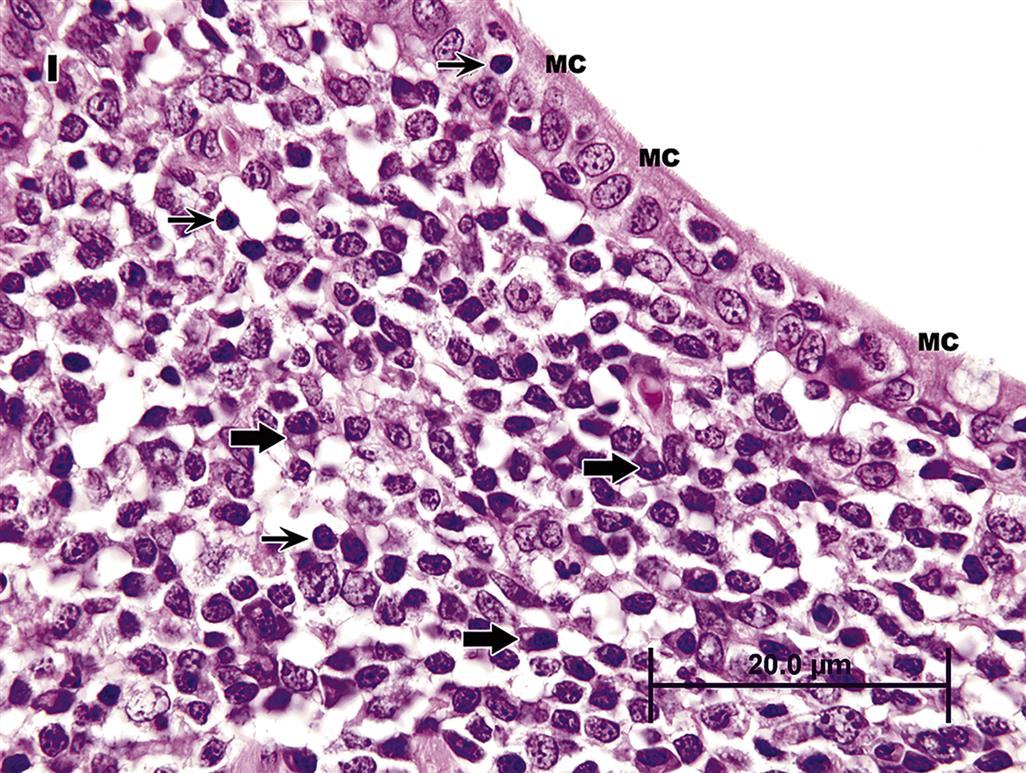
Lymphoglandular complexes of the colon are an important part of the GALT complex. Lymphoglandular complexes have germinal centers in the submucosa with deep mucosal epithelial invaginations projecting into them. As in Peyer’s patches, M cells partially line the surface of these complexes, and lymphocytes are closely apposed to these phagocytic cells. Epithelial cells lining true Peyer’s patches in the rat colon function like M cells but do not have the characteristic morphological appearance of microfolds on their luminal surfaces.
When GALT is activated, lymphocyte traffic through Peyer’s patches increases. Primed and activated T and B lymphocytes migrate to mesenteric lymph nodes via the thoracic duct to postcapillary venules lined by high endothelial cells (high endothelial venules, HEV), and then into intestinal lymphoid tissue. Tissue specificity of the T and B cells is determined by interaction with the endothelial cells of the HEV. The ability of the immune system in the GI tract to respond to microbial, chemical, and dietary antigens helps prevent these agents from entering the body. GI tract mucosal hypersensitivity can be induced by circumventing the normal process in which a toxic compound is handled by GALT. This can be done, for example, by coadministering a mucosa-damaging agent and the antigenic compound concomitantly.
Lymph flows from the central lacteal to Peyer’s patches and then to many different lymph nodes, including mesenteric, pancreatic, gastric, hepatic, splenic, and colonic nodes. Intestinal lymph contains absorbed lipids, fat-soluble xenobiotics, and recirculating lymphocytes. Some of the circulating lymphocytes have been primed by antigen exposure and are migrating to other mucosal sites, including respiratory and genital tracts. This allows immune cells exposed to antigens in the GI tract to localize at other sites of the common mucosal immune system that may also be exposed to environmental toxicants.
The T-lymphocyte population in the GI lamina propria consists primarily of CD4 helper/inducer cells. These cells play a major role in the development of the initial immunologic response to a new antigen, which is consistent with the concept that the gut represents a site of primary exposure of the host to many new antigens. Cytotoxic/suppressor (CD8) cells are less frequently seen in this location. For more specific information regarding GALT, and the immune system as a whole, and their responses to toxic substances, refer to Chapter 12, Immune System.
Enteric Nervous System
The nervous tissue of the GI tract is highly organized but diffuse in nature. These elements are components of the autonomic nervous system (ANS). Various motor and sensory neurons ramify throughout the wall of the GI tract and form multiple plexuses. Nerve fibers emanate from these plexuses and vary in thickness, carrying information from one ganglion to another and from intrinsic to extrinsic neurons. The nervous tissue of the GI tract differs from other portions of the ANS because many of its neurons do not receive direct input from the central nervous system. However, neural information does come from autonomic motor neurons that are both sympathetic and parasympathetic, and from GI sensory neurons. This results in reflex activities that act independently of the brain or spinal cord. Yet the central nervous system can regulate the rate of turnover of GI mucosal cells.
Neurons of the parasympathetic ganglia are in both the submucosal (Meissner’s) plexus and myenteric (Auerbach’s) plexus. The myenteric plexus is responsible for the electrical rhythms of the GI tract but is not needed for propagation of the myoelectric complex that controls intestinal function. The myenteric and submucosal plexuses are interconnected to form a single functional unit, so integration of electrical activity occurs at multiple locations in the GI tract.
Parasympathetic stimulation of the GI tract leads to increased blood flow, enhanced secretion, and increased muscular activity. Stimulation by the sympathetic nervous system has the opposite effects. The enteric nervous tissue is also composed of integrative circuits that consist of interneurons within ganglia that process information from intramural and mucosal sensory receptors. Sensory neurons detect fluidity, volume, chemical composition, and temperature of the luminal contents. The appropriate motility of the GI tract is affected via motor neurons. Specific motor neurons release neurotransmitters in the proximity of mucosal effectors, blood vessels, and muscle layers. In addition, receptors for neurotransmitters are present on and near epithelial cells. The spatial density of myenteric neurons decreases with age. Additionally, toxic substances such as anthraquinone injure the nerve fibers and may alter the number of neurons. Topical application to the GI tract of cationic surfactants, such as benzalkonium chloride (a mixture of compounds) or benzyldimethyltetradecylammonium chloride, destroys intrinsic neurons in the myenteric plexus of the small intestine as well as damages smooth muscle. After nearly complete regeneration of the smooth muscle, the damage to nerves persists.
The small intestine contains small-diameter sensory nerve fibers in the mucosa and tunica muscularis, which can be stimulated by capsaicin to release substance P and calcitonin gene-related peptide (CGRP). These sensory nerve fibers have the potential when hyperstimulated to initiate a cascade of proinflammatory events and transmit nociceptive information to the central nervous system. The contractile effects of intragastric capsaicin via release of substance P and CRGP in the colon can be inhibited by muscarinic antagonists, implying that cholinergic neural pathways are involved in this sensory-motor pathway of reflex motility.
There are three principal motility patterns of the GI tract: storage, mixing, and propulsion. Movement of a swallowed bolus from the mouth to the stomach and into the intestinal tract is a propulsive event, caudally progressing in front of a contraction wave of the circular muscle layers in various digestive tract segments. Effective gastric emptying requires coordinated propulsive contractions in the antrum that progress to the pyloric canal, as well as properly timed relaxation of the upper duodenum. Some drugs and toxicants reduce the rate of gastric emptying by producing contractions of the duodenum that abolish the antral-duodenal pressure gradient required for effective emptying.
Contractions of the circular muscle occur more or less randomly but are somewhat fixed in timing and location by the electrical slow waves or electrical control activity generated initially in interstitial cells (of Cajal), which serve as bioelectrical pacemakers for smooth muscle. Clusters of propulsive contractions associated with contractile rings migrate 5–30 cm caudally, thereby propelling content toward the cecum. Toxic agents that induce excessive migrating clustered contractions would abnormally speed propulsion through the small intestine, which can impact electrolyte, nutrient, and water absorption.
Migrating motor complexes are bands of contractile activity that move caudally over the stomach and small intestine during fasting to sweep digested food remains out of the stomach. These motor complexes are active during periods of fasting and continue until another meal is consumed. The central nervous system exerts some degree of control over the activity of these complexes, but the actual complexes are initiated in the enteric nervous system. Premature migrating motor complexes can be induced by opiates and erythromycin.
Biotransformation
The mucosa of the GI tract is a site of high enzymatic activity and compound conjugation. The mucosa is exposed to the highest concentration of orally administered compounds and can modify these compounds prior to their entry into the blood. The consequences of mucosal biotransformation can be compound activation or deactivation (detoxification) (Figure 15.5).
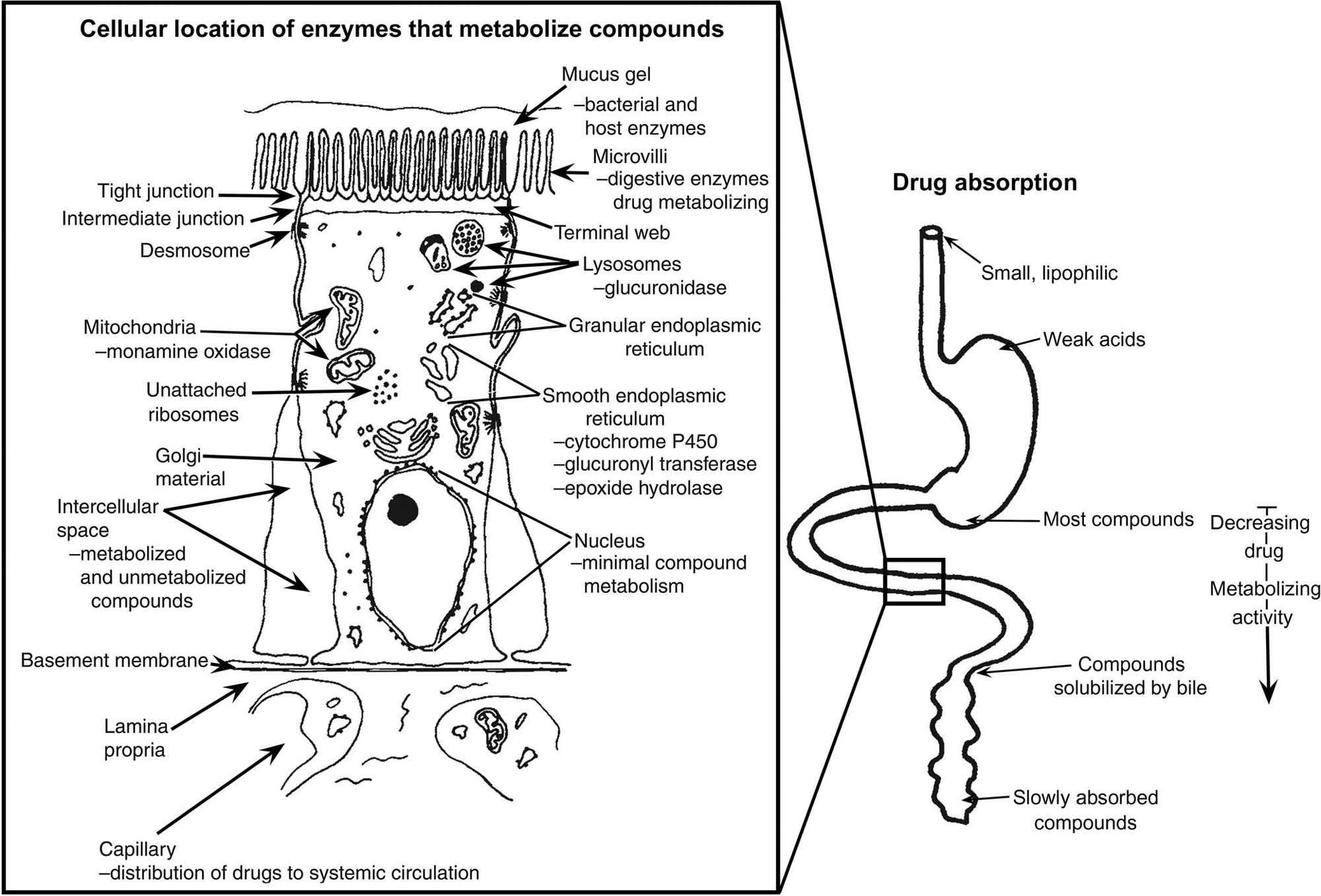
Intestinal mucosal enzymes that metabolize xenobiotics can prevent systemic absorption of many potentially toxic substances such as peptides (via peptidases), esters (via esterases), and alcohols [via alcohol dehydrogenase (ADH)] present in the gastric mucosa. Xenobiotic metabolism also can be carried out by luminal microorganisms; furthermore, luminal organisms can affect mucosal enzyme activity. Factors affecting the metabolic activity of the intestinal microflora must be taken into account in studies of the biotransformation of orally ingested xenobiotics. Marked differences exist in microbial composition and metabolism of the gut flora of different species of animals, and environmental factors such as drugs (especially antibiotics), diet, and xenobiotics can modify microbial metabolism, and thus the toxicity, of foreign compounds. Presystemic clearance can occur for some toxicants either within the enterocyte or within the gut lumen itself. This gut-associated first-pass effect represents the irreversible extraction and/or biotransformation of toxicants passing through enterocytes on their way into the lacteals or portal venous blood. Metabolites produced by enterocyte biotransformation can enter the intestinal lumen, the portal venous system, lacteals, or simply remain stored in the cell. Conjugated water-soluble compounds formed during transport into enterocytes tend to be excreted relatively quickly into the intestinal lumen, and therefore are cleared from the body (Table 15.3). After oral administration, when the concentration of a xenobiotic within the enterocyte is very high, intestinal biotransformation reactions will generally be capacity-limited.
The colon is three- to fivefold more active than the small intestine in certain enzymatic processes (e.g., demethylation). Compared to the liver, jejunum has a higher monoamine oxidase activity. Several compounds, such as polychlorinated biphenyls (PCBs) and phenobarbital, increase cytochromes P450 (CYP) levels in intestinal mucosa 2–4 days after exposure; the effect is greatest after oral administration of the compound. This augmentation of enzyme activity is similar to that which occurs in the liver. As occurs in the liver, chronic intake of ethanol will also increase the level of activity for several intestinal enzyme pathways.
Several biotransforming and toxicant-metabolizing gradients exist in the GI tract (Figure 15.5). Monooxygenase (CYPs) and uridine diphosphate (UDP)-glucuronosyl transferase activities are higher in the upper duodenum than in the lower small or large intestines. Sulfation proceeds more rapidly in the proximal than in the distal small intestine and colon.
CYP activity in the small intestine provides the principal, initial biotransformation of ingested xenobiotics. Enzymes of enterocytes are fully competent to carry out oxidative, reductive, hydrolysis, and conjugation reactions. The oxidative reactions are largely catalyzed by CYP isozymes. The intestinal mucosa also contains nonspecific esterases and amidases, (UDP)-glucuronosyltransferases, and reductases. Some enzyme activities, such as nitroreductase and dechlorinase, maybe attributable to both mucosal enzymes and luminal microflora. Most CYP isozyme activity increases in enterocytes during their migration from crypt to villus. Nearly all CYP activity is attributable to villous cells, and NADPH CYP 450 reductase is expressed constitutively only in villus cells. Both glucuronidation and sulfation reactions increase solubility of xenobiotics and thus play a major role in intestinal first-pass clearance for various xenobiotics. Intestinal presystemic elimination of a dopamine prodrug, N-(N-acetyl-L-methionyl)-O,O-bis(ethoxycarbonyl)dopamine, indicates that catechol ester hydrolysis, amido hydrolysis, and catechol O-methylation can also occur in enterocytes. A biotransformation gradient from the apical to the basal surface of enterocytes is present; it is controlled by enzyme-rich drug-metabolizing organelles (e.g., SER) and active transport systems in the apical cell membrane. However, the gradient varies with the cellular location in the GI tract and route of exposure. Compounds entering from the blood (basal) side can be found in the GI tract lumen independent of enterohepatic circulation. Many toxicants enter the intestinal contents by direct transfer from blood or when released out of the enterocyte. In general, intestinal excretion is a relatively slow process that is important for chemicals having low rates of biotransformation and/or low renal or biliary clearance.
Although passive diffusion is an important mechanism for intestinal excretion, active secretion of organic acids and bases has been demonstrated in the gut. The transepithelial elimination of ciprofloxacin in rabbits and rats is probably due to active transport. It has been shown that P-glycoprotein (Pgp) mediates efflux of etoposide out of intestinal cells, and this efflux is inhibitable with quinidine. Pgp is the 170-kDa product of the ABCB1 gene in humans and is an ATP-powered efflux pump which can transport hundreds of structurally unrelated hydrophobic amphipathic compounds, including therapeutic drugs, peptides, and lipid-like compounds. An organic cation transporter, originally identified in kidney and liver that is responsible for translocation of hydrophobic and hydrophilic organic cations of different structures has also been identified in the intestine. As an adaptive response to renal failure, the intestine can excrete chemicals such as oxalate. In addition, epithelial cells of the GI tract can absorb and export compounds from the circulating blood and the intestinal lumen, indicating that many intestinal transport systems are likely “two-way streets.”
Disposition of highly lipophilic chemicals in an organism often requires consideration of lipid transport. The two important mechanisms that contribute to the nonbiliary intestinal excretion of lipids are (1) exfoliation of intestinal cells and (2) exudation of lipids across the mucosa.
Besides altering the biological activities of toxicants, biotransformation reactions in enterocytes may influence the postabsorptive fate of xenobiotics. Metabolites maybe excreted by enterocytes into the intestinal lumen and eliminated as fecal matter, thereby permitting escape from enterohepatic circulation. Metabolites maybe either excreted across the mucosal membrane, back into the lumen, or secreted across the serosal membrane into portal venous blood.
Blood supply to the mucosa is a critical component of mucosal biotransformation. Provision of oxygen to the epithelium is important in oxidation and reduction reactions. The microvascular anatomy of the mucosal villi provides a countercurrent exchange system, which can reduce entry of a toxicant into the portal circulation. As the toxicant is picked up in the villus and moved to the crypt, exchange with blood going to the villus occurs, resulting in slower compound absorption and increased time for biotransformation (Figure 15.6).

Fecal excretion is a major route of elimination for many lipophilic chemicals, with most toxicants probably being transferred by passive diffusion and a number excreted into the feces by nonbiliary pathways. Direct mucosal-to-serosal transport into the feces occurs for some nonpolar, lipophilic xenobiotics that undergo little or no biotransformation. However, rapid exfoliation of intestinal cells may also contribute to fecal excretion of some toxicants. The intestinal excretion rate of some lipophilic chemicals can be substantially enhanced by increasing the lipophilicity of the GI contents by, for example, adding mineral oil to the diet.
Enterohepatic Circulation
Enterohepatic circulation allows for recycling of metabolized and nonmetabolized compounds, and is of critical importance in toxicologic processes involving the GI tract. This circulatory route is active when ingested compounds that are absorbed in the GI tract enter the portal circulation, go to the liver, and then return to the GI tract via biliary excretion. The enterohepatic circulatory pathway can also be utilized by dermally absorbed or inhaled materials that are excreted in the bile.
A compound leaves the enterohepatic circulation if it passes in the feces before being reabsorbed or into the urine before being cleared by the liver. The ultimate destiny of a compound is dependent on its chemical composition and the species. The importance of species differences is best illustrated by the nonsteroidal antiinflammatory drug (NSAID) indomethacin, which undergoes enterohepatic circulation; it is excreted in the feces of dogs but in the urine of rats. The duration of enterohepatic circulation is most extensive for this drug in dogs and rats, and least extensive in rabbits and humans. This observation impacts resulting species-specific variability in the toxic response of the GI tract to NSAIDs, with dogs being less tolerant of NSAID administration when compared to rats, rabbits, or humans.
The amount of a compound that is excreted in the feces is controlled by the lipophilicity of the chemical and the extent of metabolism that alters this lipophilic character. Processes that increase the aqueous nature of a compound include dealkylation, glucuronidation, and sulfation. One process that increases lipophilicity is glucuronide hydrolysis, often by microbial glucuronidases. Increasing lipophilicity is associated with higher excretion of the compound in feces.
The rate at which a chemical is excreted in the feces is limited by the time it takes for a compound to be excreted in the bile and reabsorbed by the intestine. Increasing metabolism of the chemical will raise the rate of excretion. Factors that modify this excretion rate include motility of the intestine, distance of the site of (re)absorption from the major duodenal papilla (site of common bile duct entry), rate of conjugate hydrolysis by GI bacteria, transport rate across the intestinal wall, and motility of the gall bladder (in species which have this structure). With the exception of gall bladder motility, all factors influence intestinal transit time.
Combined biotransformational processes in the liver and intestine can substantially affect the toxicity of a compound. For example, the activation of diphenolic laxatives by microbial metabolism in the GI tract, and conjugate hydrolysis by these microbes has been clearly established. Bacteria can also modify dinitrotoluene by nitro-reduction and give rise to elevated hepatic levels of the carcinogenic metabolite dinitrobenzyl alcohol. Arylamines formed from the biliary metabolite of chloramphenicol maybe responsible for the goitrogenic effect of this antibiotic in rats. Hydrolysis of polycyclic aromatic hydrocarbon glucuronide metabolites demonstrates how enterohepatic circulation can reactivate a detoxified compound. These are just a few of many instances of how enterohepatic circulation can affect toxicity.
During enterohepatic circulation, compounds may interact with intestinal contents. This is demonstrated by the binding of bile salts to dietary fibers. Such binding will decrease the reabsorption of bile salts, and maybe partially responsible for the healthful effects of soluble fibers. Alteration of bile acid circulation can influence the hepatobiliary level of several compounds that are bile-soluble (cholephils). In addition, the bile salt taurocholate promotes motor activity in the colon, thereby reducing intestinal transit time. Bile salts also increase the transport of compounds across the intestinal mucosa, and may consequently enhance the toxic properties of a compound.
Enterohepatic circulation will increase the toxicity of a compound to organs in the enterohepatic circuit if the compound remains active during circulation. The concentrating capacity of enterohepatic recycling may play an important role in the ulcerogenic effects of NSAIDs (such as indomethacin) in dogs. This same process maybe important in the carcinogenic effects of 3,3-dimethoxybenzidine and tris(2,3-dibromopropyl)phosphate in the colon. Biliary excretion and enterohepatic circulation have a role in colon carcinogenesis of rats induced by 2,3-dimethyl-4-aminobiphenyl (DMAB). Rats treated orally with this compound excrete mutagenic agents in the bile. However, rats injected subcutaneously with DMAB do not develop colonic neoplasms.
The Microbiome
Ingested materials are metabolized not only by digestive and intestinal enzymes but also by resident bacteria (Table 15.5). These bacteria have metabolic activities that include reductases, hydrolases, demethylases, β-glucuronidases, and β-glucosidases. Since there are approximately 109–1012 bacteria per gram of feces in humans and animals, the potential enzymatic activities of this compartment of the GI tract cannot be ignored. The microfloral composition in mammals depends on the nutritional and health status of the host, and also the host’s dietary composition.
Table 15.5
Microbial Density in Different GI Compartments of Humans and Mice
| Organ/tissue | Human | Mice | ||
| pH gradient | Microbial mass (cells/mL) | pH gradient | Microbial mass (cells/g) | |
| Stomach | 1.5–5.0 | 102–103 | 3.0–4.5 | 107–109 |
| Duodenum | 5–7 | 103–104 | 4.5–5.0 | 107–109 |
| Jejunum | 7–9 | 104–105 | 4.5–5.5 | 107–109 |
| Ileum | 7–8 | 108 | 4.5–5.5 | 107–108 |
| Cecum | – | – | 4.3–5.0 | 107–108 |
| Colon | 5–7 | 1011–1012 | 4.5–5.0 | 109–1010 |
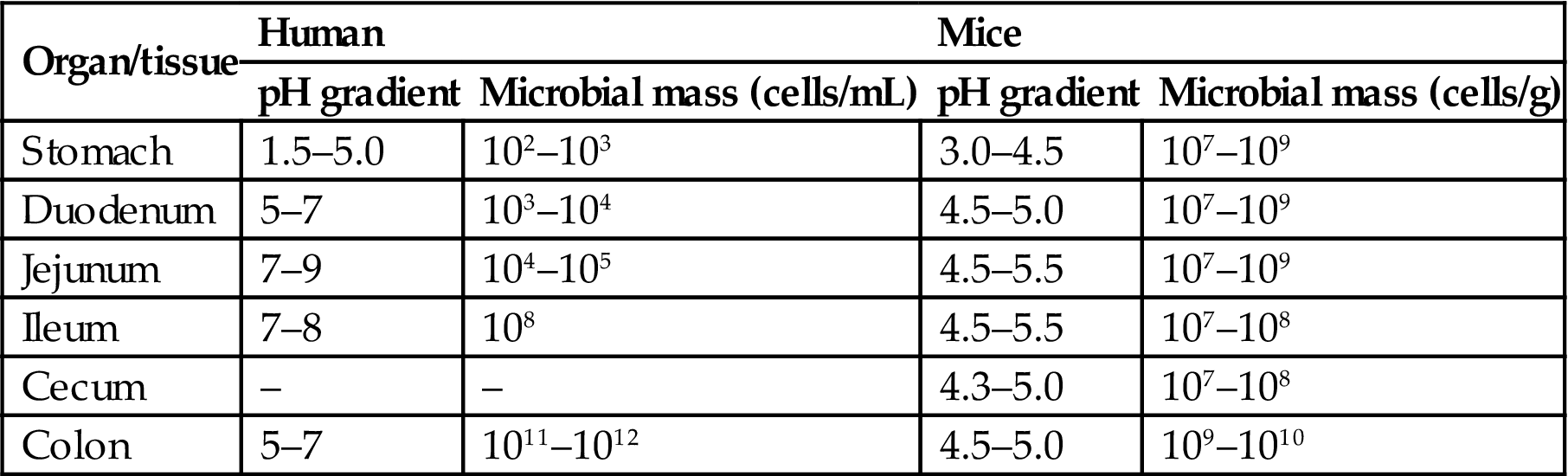
Table adapted from Handbook of Toxicologic Pathology, third ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2011) Academic Press, Table 56.5, p. 2298, with permission.
Microorganisms have an active metabolic function promoting a wide variety of biochemical reactions important in normal vertebrate as well as bacterial physiology. With the exception of ruminants, most bacteria are present in the lower small intestine, cecum, and colon. There is a gradual transition from sparse gram-positive microflora in the stomach to a mixture of gram-positive and gram-negative bacteria in the ileum and finally a preponderance of gram-negative bacteria in the large intestine. Bacterial concentrations in excess of 1012/mL of ingesta are common. Anaerobic bacteria outnumber aerobic species by a factor of 102–104. Frequently identified anaerobic microorganisms include Bacteroides, Bifidobacteria, and Eubacteria, anaerobic gram-positive cocci, and Clostridium sp. Aerobic isolates include species of Enterobacteriaceae, enterococci and other streptococci, staphylococci, and also the fungus Candida.
The influence of intestinal microbes on the host’s nutritional status has been clearly demonstrated. Weight of the GI tract and mucosal thickness are markedly reduced in animals without bacteria in their gut contents. Conversely, bacterial overgrowth can modify lipid and carbohydrate absorption. Overgrowth of intestinal bacteria can lead to steatorrhea due to hydrolyzing bile-acid conjugates and altering micelle-forming abilities of the microflora. Bacterial proteases also remove maltase from brush border membranes, which results in carbohydrate malabsorption. Consequently, compounds altering microbial populations can lead to altered nutritional status. The composition of the GI microbiome may influence the onset and progression of degenerative processes in more distant systems (e.g., neurodegenerative diseases).
Bacteria produce and release compounds that have local effects or, if absorbed, systemic impacts. Mammalian metabolic pathways generally require oxygen, so injurious compounds are generally detoxified by oxidation and conjugation pathways. However, gut bacteria are active in oxygen-free environments and thus utilize reduction and hydrolysis reactions, resulting in different metabolites with potentially harmful side effects. The role of bacteria in modifying host responses is most marked in the lower segments of the GI tract. For example, the pharmacologic activity of digoxin is dependent upon bacterially mediated hydrolytic removal of a trisaccharide, which releases digoxigenin. In some individuals, however, there is a further reduction in the double bond of the lactone ring by Eubacterium lentum present in the colon, which results in the formation of a pharmacologically inactive substance, dihydrodigoxigenin. Diet appears to play a role in the presence of this bacterial species and the frequency of digoxin inactivation in humans.
Bacterial deamination is another important metabolic activity that is mediated by the bacterial flora. The breakdown of urea into carbon dioxide and ammonia is catalyzed by bacterial urease. Approximately, 40% of the urea synthesized by the liver is degraded by a variety of aerobic and anaerobic bacteria.
The role of bacteria in GI toxicity is most clearly defined for carcinogen activation. Many indirectly acting chemical carcinogens require enzymatic activation before they can cause cellular transformation. Bacterial β-glucuronidases can deconjugate glucuronides and lead to the release of carcinogenic aglycones. Additionally, fecal flora nitroreductases can activate procarcinogens. Bacteria also have a direct role in the detoxification process. Bacteria can deactivate carcinogens by N-dehydroxylation.
Antibiotics can not only modify bacterial populations in the GI tract but also depress neuroeffector and neuromuscular transmission in the walls of digestive organs. In vitro studies have demonstrated that ampicillin, lincomycin, erythromycin, and clindamycin depress contractions of the muscularis mucosa. Clindamycin and erythromycin depress the responses of the muscularis mucosa to acetylcholine. As an example, impaired GI motility after administration of oral antibiotics can facilitate the proliferation of Clostridium difficile in the lower GI tract and lead to pseudomembranous colitis.
Microbial metabolism in the gut also serves to detoxify many toxic xenobiotics and protect or suppress the harmful effects of the xenobiotic metabolites on the host. Examples of such metabolized drugs/additives that are degraded by microflora include digoxin, diethylstilbesterol, estrogens, cyclamate, azulfidine, 3,4-dihydroxyphenylalanine, amygdalin, metronidazole, caffeine, propachlor, morphine, buprenophine, oxazepum, phenolphthalein, warfarin, and dichlorodiphenyltrichloroethane (DDT).
Intestinal microbiota, however, also have many bacterial enzymes that can catalyze the production of mutagens, carcinogens, and tumor promoters. Examples of these enzymes include β-glucuronidase, β-glucosidase, β-galactosidase, nitroreductase, azoreductases, tryptophanase, and 1-α-steroid dehydrogenase, to name a few. These enzymes can act on a variety of substrates, including nonnutritive plant material, as well as metabolize administered drugs/toxicants and supplements/additives. Some examples of substrates that are acted upon by gut microfloral enzymes to become mutagenic include cycasin (a plant-derived β-glucoside; carcinogenic), 2-nitofluorene, trypan blue, tryptophan, and dimethylamine.
Evaluation of Gastrointestinal Toxicity
The ability of the GI tract to adapt to various diets and nontoxic compounds is well established. Both adaptational and toxicologic processes can be manifested by altered structure or function. Evaluation of these processes requires a basic understanding of the mechanism or suspected pathogenesis of the toxic injury or response. Routine approaches involve in vitro and in vivo methods. Because alterations in numerous other organ systems can occur as a result of GI toxicity, whole animal studies are generally required in order to properly interpret GI toxicity. As a result of this complex interrelationship among organ systems and the inherent complexity of the GI tract, animal models have been developed to study various GI diseases and toxicities. This section will focus on conventional morphologic assessments of injury as applied to animal models (including knockout/transgenic models) of GI toxicity.
Morphological Methods
Evaluation of the GI tract for toxicity should be conducted using macroscopic, microscopic, and ultrastructural methods. Macroscopic evaluation includes identification of ulcers, enlarged lymphoid tissues (e.g., Peyer’s patches), neoplasms, and foreign bodies. Microscopic studies should be conducted on all lesions observed macroscopically, and also at preselected sites in macroscopically normal organs. Proper tissue fixation is essential for structural studies. In an attempt to standardize communications, specialty organizations are adopting harmonized nomenclature for the description of microscopic lesions in certain portions of the GI tract (Table 15.6). The most current terminology, including representative images of common lesions, developed by the International Harmonization of Nomenclature for Diagnostic Criteria in Rats and Mice, is now available on the Society of Toxicologic Pathology website (http://www.toxpath.org/inhand.asp).
Table 15.6
Nomenclature for Describing Lesions of the Gastrointestinal Tract
| Location | Term | Definition | Term | Definition |
| Oral cavity | Proliferative lesions | Inflammatory and ulcerative lesions | ||
| Hyperplasia | Broad areas of thickened and differentiated epithelium especially the keratin layer; which may have prominent papillary endophytic projections | Stomatitis | Initial lesions frequently begin as ulcerative processes followed by neutrophilic exudative phase; established lesions have lymphocyte and plasma cell accumulation; advanced lesions can be associated with alveolar bone loss; dietary content influences extent; severity and frequency of lesion | |
| Esophagus | Proliferative lesions | Degenerative lesions | ||
| Hyperkeratosis/parakeratosis | Thickened mucosal layer with retraction of nuclei; absence of cellular atypia | Megaesophagus | Esophageal enlargement; degeneration of muscle and nerve cells in the wall | |
| Stomach | Degenerative lesions | |||
| Mucosal atrophy | Results from ulceration, inflammation, mineralization, or infarction; glandular ectasia and inflammation or fibrosis of lamina propria | |||
| Chief cell atrophy | Age-related decrease in size and number of chief cells | |||
| Intestinal metaplasia | Focal crypt and villous formation with goblet cells and Paneth cells; complete forms have a small intestinal morphology while incomplete forms have a large intestinal morphology | |||
| Nonneoplastic lesions of the glandular mucosa | Neoplastic lesions of the glandular mucosa | |||
| Erosion | Area of superficial necrosis of the mucosa that does not extend beyond the muscularis mucosa | Adenoma | Adenomatous polyps are usually located in the antrum and are composed of basophilic columnar epithelium organized into glandular structures; mass maybe pedunculated or endophytic and arise in areas of reactive hyperplasia | |
| Ulceration | Area of mucosal necrosis that extends to or through the muscularis mucosa | Adenocarcinoma | Lesions invade into the submucosa and may metastasize or locally infiltrate; cytologically, cells range from dysplastic to anaplastic with pleomorphic nuclei and increased mitotic index | |
| Glandular dilation | Distention of the basal portion of the gastric gland; distention maybe sufficiently large to form a microcyst | Neuroendocrine cell tumor | Also called carcinoids; rarely spontaneous; characteristic lesion of potent gastric antisecretory agents causing hypergastrinemia; agyrophilic cells forming the tumor are reactive for nerve specific enolase and chromagranin-A; maybe intramucosal or invasive | |
| Nonneoplastic lesions of the nonglandular mucosa | ||||
| Eosinophilic chief cells | Infrequent spontaneous lesion that maybe associated with lymphoma of gastric mucosa or other disease; induced by antisecretory drug treatment; reversible, nonprogressive alteration only reported in rats | Hyperplasia | maybe a focal or diffuse thickening characterized by increased numbers of one or more cell types (basal, spinous, or granular); focal hyperplasia is differentiated from papilloma by the absence of a connective tissue core containing blood vessels and the presence of an intact muscularis mucosa | |
| Hyperplasia (focal) | Most common type is associated with erosions or ulcers and found primarily in the antrum; hyperplastic glands may extend through the muscularis mucosae forming persistent adenomatous diverticula | Hyperkeratosis | Increased thickness of nonnucleated keratin layer | |
| Neoplastic lesions of the nonglandular mucosa | ||||
| Hyperplasia (fundic) | Also called hypertrophic gastritis or adenomatous hyperplasia; associated with administration of antisecretory compounds; result of endocrine stimulation of the oxyntic region; generalized increase involving all cellular compartments | Papilloma | Pedunculated with a prominent connective tissue core; evidence of localized invasion through the muscularis mucosa near base of papilloma should be considered evidence of carcinoma formation | |
| Hyperplasia (neuroendocrine) | Specific neuroendocrine cell hyperplasia in response to endocrine alterations; in rats, enterochromaffin-like (ECL) cells are the main cell types responsive to hypergastrinemia; in mice; basal portions of gastric glands may normally be lined by pure populations of neuroendocrine cells | Adenoma | Circumscribed areas of dysplastic epithelium that distorts adjacent normal mucosa and is confirmed by a basement membrane; same tumors maybe sessile or pedunculated | |
| Carcinoma | Locally invasive lesion that is a neoplastic proliferation of squamous epithelium | |||
| Small and large intestine | Proliferative lesions | Neoplastic lesions | ||
| Reactive hyperplasia | Epithelial cells are basophilic and depleted of mucus with enlarged nuclei containing prominent nucleoli; crypt herniation may occur especially if the muscularis mucosae has been disrupted; in the small intestine, reactive hyperplasia can be accompanied by villous atrophy | Adenoma | Circumscribed areas of dysplastic epithelium that distort adjacent normal mucosa and are confirmed by a basement membrane; same tumors maybe sessile or pedunculated | |
| Focal atypical hyperplasia | Crypts are elongated and have dilated lumens and may have a tortuous contour; architecture of the adjacent mucosa is not distorted by compression; epithelial cells lining the crypts are normal to markedly dysplastic and may forma single layer or be pseudostratified with many mitotic figures; goblet cell numbers are reduced; severe inflammatory reactions maybe associated with these foci | Adenocarcinoma | Dysplastic epithelium with clear evidence of invasion past the basement membrane into the lamina propria or submucosa; the invasive characteristics of adenocarcinomas must be differentiated from tangential cuts of glands in the lamina propria found at the base of adenomas; there maybe a scirrhous (desmoplastic) and/or inflammatory response which can help differentiate invasive epithelial nests from cryptal herniation; adenocarcinomas of the small intestine are more invasive and metastasize more frequently than those of the large intestine | |
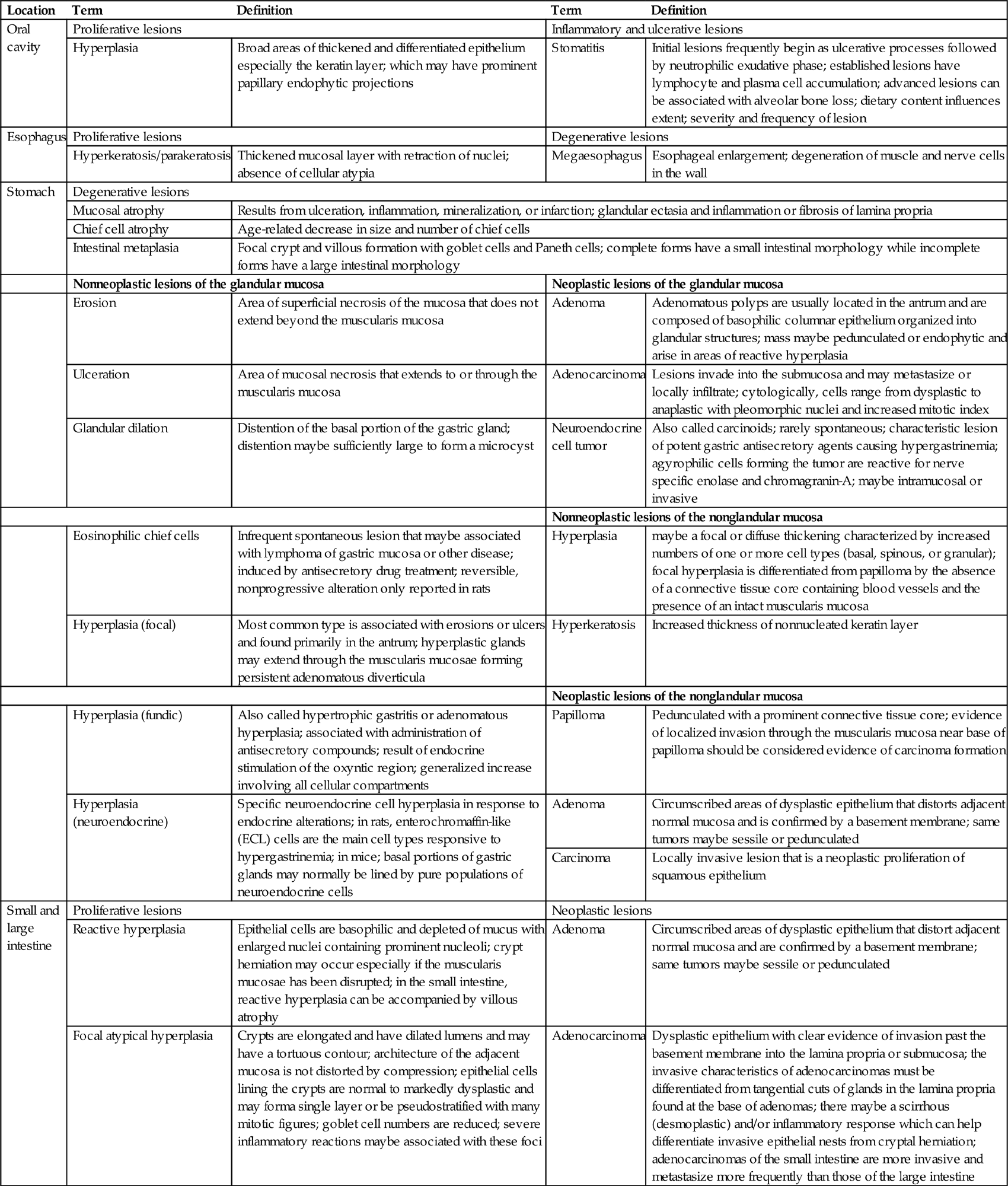
Information adopted and modified from Nolte, T., Brander-Weber, P., Dangler, C., Deschl, U., Elwell, M.R., Greaves, P., Hailey, R., Leach, M.W., Pandiri, A. R., Rogers, A., Shackelford, C.C., Spencer, A., Tanaka, T., and Ward, J.M. (2016). Nonproliferative and proliferative lesions of the gastrointestinal tract, pancreas and salivary glands of the rat and mouse (review). J. Toxicol. Pathol. 29 (1 Suppl), 1S–124S.
Assessment of morphological alterations of the GI tract should consist of close evaluation of the mucosa and its specializations. The mucosa consists of surface epithelium, crypts/glands, lamina propria, and a thin layer of muscle (the muscularis mucosae) separating the mucosa and submucosa. Specializations of the mucosa include glands of the esophagus, foveolae of the stomach, villi of the small intestine, and glands of the large intestine. The submucosa and the tunica muscularis (outer muscle layers) also should be examined for changes in thickness and cellularity.
Villi should be evaluated critically when assessing small intestinal toxicity. Since villi bend in various directions, and have shapes that vary with species (e.g., tongue-like in rats and finger-like in humans) and location (longer in the duodenum than in the ileum), close comparisons with control animals is required to prevent misinterpretation.
Changes in the lamina propria will be detected by assessing alterations in the normal cell population. Neutrophil, eosinophil, lymphocyte, and plasma cell populations may change. An increase in any of these populations is a potential indication of an underlying toxic or disease process. Inflammatory cell infiltrates frequently occur secondary to epithelial cell toxicity. Lymphoid follicles may develop and be associated with an extensive increase in lymphocytes and plasma cells. These follicles may occur in the lamina propria or submucosa. Additionally, lymphomas maybe indicated by a substantial number of abnormal lymphocytes expanding the lamina propria. Additional sites of potential injury in the lamina propria include blood vessels and lacteals (lymphatic capillaries). Common lesions associated with GI toxicity include vascular blockage (thrombosis), dilation (e.g., lymphangiectasia), and rupture.
Changes in the submucosa may involve blood vessels, nerves, and lymphatics. Alterations in these structures in this region are frequently characterized by lymphangiectasia and inflammatory or neoplastic cell infiltrates.
Different fixatives are used for histologic evaluation of GI organs, depending on the purpose of the study and the technique being used. A routinely used multipurpose fixative is neutral buffered 10% formalin. Because of rapid postmortem autolysis, GI tract tissues must be placed into the fixative within 1–2 minutes of death for optimal preservation. In some laboratories, it is routine to immerse the entire segment of GI tract of interest overnight in neutral buffered 10% formalin. Upon completion of fixation, the GI tract is opened longitudinally and flushed extensively with sterile water to remove any fecal matter. Segments of the fixed GI tract may then be dissected out and embedded in polymer resin prior to sectioning and histological analysis. Alternatively, fecal matter maybe removed from the mucosa prior to fixation by flushing the surface with physiological saline (rather than water, which may promote osmotic rupture of epithelial cells).
Ultrastructural studies can be conducted using scanning or transmission electron microscopy (TEM). Scanning electron microscopy provides information on surface alterations. This technique is particularly useful for examining altered villus structure in the small intestine. TEM, although not utilized as extensively as in the past, is valuable for identifying subtle changes in organellar structure that precede later, more “generic” histologic changes and thus give an indication of possible mechanism(s) of injury. Morphometric and stereological analyses at the light microscopic and electron microscopic level are powerful morphological methods that can combine biochemical and morphological data.
Normal cellular proliferation, differentiation, and senescence are processes that have distinctive phenotypic and genotypic characteristics. These molecular alterations can be examined phenotypically by using a variety of histo- and immunohistochemical marker. For example, for enterocytes various disaccharidases, Intestinal Fatty Acid Binding Protein, Enterocytin, MDM4, Epithelial Cell Adhesion Molecule (EpCAM) and various cytokeratins (CK 8, CK 18, & CK 20) can be used, whereas in colonocytes Carbonic Anhydrases 1 & 2 (CA-1 & CA-2) as well as the plant lectin DBA have been utilized to identify these “mature” nonproliferating cell populations. For proliferative crypt cell populations, the nonspecific Proliferating Cell Nuclear Antigen as well as more specific Lysozymes 1 & 2 (Lyz1 & Lyz2), α-defensins (Defa 5 & Defa 22), β-catenin and Epbh2 can be used of identification. Paneth cells express a combination of markers seen in both proliferative crypt cells and nonproliferative enterocytes, including MDM4, α-defensins and Epbh2. An understanding of the genetic control of proliferation and differentiation via Wnt and Notch signaling pathways, for example, can be obtained using techniques such as in situ hybridization and rtPCR.
Animal Models
General Considerations
Variations in mammalian GI tract morphology show closer correlation with diet, body weight, and the need for water consumption than with taxonomical classification. The capacity of the GI tract to hold digesta decreases with decreasing body weight in herbivorous animals; however, the rate of metabolism increases with decreasing body weight. Smaller animals may have various strategies to compensate for this phenomenon. These adaptive mechanisms include an increase in cecal volume or the practice of coprophagy by lagomorphs and rodents. Because of these modifications, the GI tract in these species may render them unsuitable as animal models for humans.
Nutritional issues must be considered when extrapolating results from GI studies performed in healthy animals to humans. Generally, compounds of therapeutic importance are administered to human patients who are ill and consequently malnourished. In contrast, toxicologic effects of therapeutic or other beneficial compounds are tested in healthy animals. These animals are fed nutritionally balanced commercial diets for their lifespan, and are allowed to grow and develop under ideal conditions of lighting, temperature, and humidity. Since macronutrients in the diet markedly affect the drug-metabolizing enzyme systems associated with the GI tract, such modifications may alter the maximally tolerated dose in test animals relative to a tolerable dose for sick humans.
Evaluation of the GI tract for toxicity must involve consideration of dietary effects on the mucosa. All diets support normal growth and health conditions of rats, yet the absorption site (mucosa) in the animals fed a semipurified diet varies significantly among the various formulated diets. This indicates that structural and functional results that are observed maybe independent of a compound’s toxic or biological effects.
Since many mammalian systems (including humans) have similar mechanisms of response to toxic compounds, animal models serve as ideal test systems for the evaluation of toxic potential, pathophysiological responses, and systemic complications of an ingested or injected compound. In contrast to toxicity testing of compounds for human or animal use, animal models of human diseases are generally defined only on the basis of specific pathologic criteria. The specific lesions may be subtly or markedly different, but the general character of the disease process is usually similar to that observed in the human disease; otherwise, the animal model would be discarded.
The few examples below demonstrate the significance and problems of using models for investigating the toxicity and efficacy of any compound that could produce lesions in the GI tract. Animal models allow the testing of the structure–function activity of chemical compounds, as well as the identification of chemical therapeutics for human and animal disease. Additionally, basic information on the pathogenesis of the underlying cellular and biochemical processes important to lesion development can be identified. Finally, animal testing provides a substantial and significant bridge between in vitro testing and the ultimate application of any compound for human use.
Specific Models
Ulcerative Lesions of the Stomach and Small Intestine
Propionitrile, cysteamine, 3,4-toluenediamine, and 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine produce acute and chronic gastric and duodenal ulcers in rodents. The morphological appearance of duodenal ulcers produced by these compounds in rats is similar to that seen in humans (Table 15.7).
Table 15.7
Selected Characteristics Comparing Cysteamine-Induced Duodenal Ulcers of Rats to the Natural Disease of Humansa
| Rat duodenal ulcer | Human duodenal ulcer | |
| Location of ulcer | Anterior and posterior wall | Anterior and posterior wall |
| Tendency to perforate | + | + |
| Occurrence of massive bleeding | + | + |
| Chronic and active ulcers | + | + |
| Pyloric ulcers | + | + |
| Increased gastric acid release | + | + |
| Elevated gastrin levels | + | ± |
| Responds to therapy: | ||
| Antisecretory agents | + | + |
| H2 receptor antagonists | + | + |
aModified from Cheville, N.F. (1980) Discussion: Period 3, from “Criteria for development of animal models of diseases of the gastrointestinal system.” Am. J. Pathol. 101, S77–S89.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table XIV, p. 179, with permission.
Colitis and Typhlitis
Experimental models of colitis have been created in a number of laboratory animals (e.g., rats and guinea pigs) with ricinoleic acid, bile acids, poligeenan, melphalan, formaldehyde, alcohol, dextran sodium sulfate, and acetic acid. Some of the lesions observed in these models maybe mediated by mechanisms involving hypersensitivity or alterations in prostaglandin synthesis, and are frequently associated with colon cancer if the compound induces an inflammatory process that is longstanding (e.g., poligeenan). Intracolonic administration of the hapten trinitrobenzene sulfonic acid (TNBS) will also lead to an immunologically based colitis. This model is best demonstrated if damage to the mucosal barrier occurs prior to compound exposure (Figure 15.7).
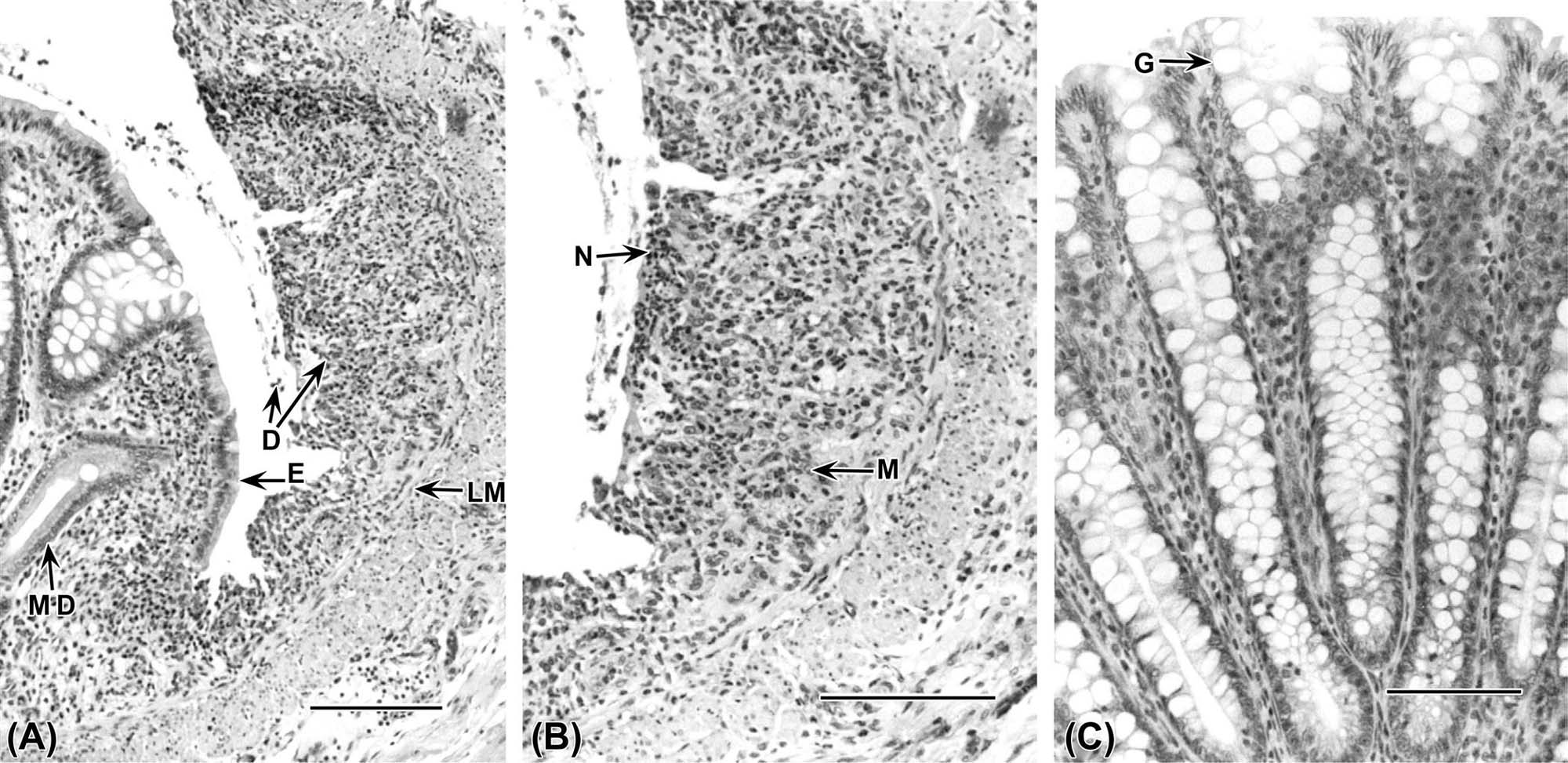
Colon Cancer
Colon neoplasia represents a leading cause of cancer-related death in Western civilization, but is relatively rare as a spontaneous lesion in laboratory animals. Consequently, a number of experimental models have been generated to evaluate this process. In addition, various animal species and dosing regimens have been used to produce colon cancers in animals (Figure 15.8). Both genotoxic and nongenotoxic models of colon cancer are used, but the chemically induced genotoxic models are most consistent (Table 15.8).
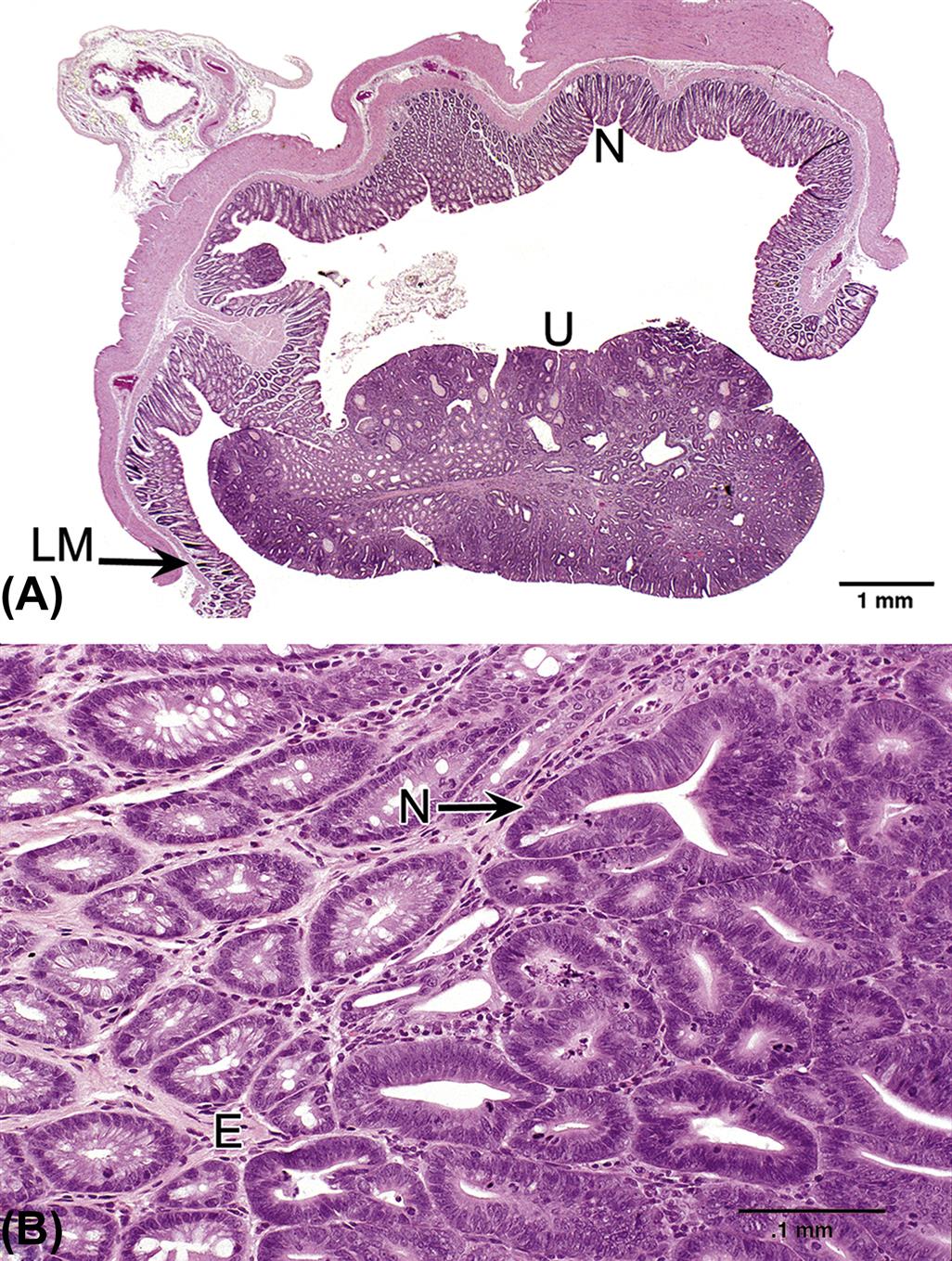
Table 15.8
Animal Models of Colon Carcinogenesis
| Chemical | Dose (mg/kg) | Animal |
| Aflatoxin B1 | 0.1a | Rat |
| 3,2-Dimethyl-4-aminobiphenyl (DMAB) | 20 | Rat |
| 100 | Hamster | |
| 1,4-bis(4-fluorophenyl)-2-propynyl-N-cyclooctylcarbamate (FPOC) | 125–500a | Rat |
| N-Methyl-N-nitrosourea | 2–2.5b | Rat, mouse, guinea pig |
| N-Methyl-N-nitro-N-nitrosoguanidine (MNNG) | 2 | Rat |
| Methylazoxymethanol acetate (MAMA) | 35 | Rat |
| 1,2-Dimethylhydrazine (DMH) | 40–200 | Rat, mouse, hamster |
| Azoxymethane | 8 | Rat |
aDose in ppm.
bDose is in mg/week.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table XV, p. 183, with permission.
Of the various chemical models available, the hydrazine derivatives have several properties that make them useful for evaluating the two-step mechanisms of colon carcinogenesis. Unlike the skin, colon carcinomas can be obtained in 1,2-dimethylhydrazine (DMH)-treated rats without a preliminary benign tumor stage. The incidence rate is constant and directly proportional to total dose, with an average of one carcinoma per colon per year. This hydrazine derivative produces the promutagenic DNA lesion of O-6-methylguanine. In general, hydrazine models demonstrate that dysplastic or adenomatous changes are not obligatory stages before cancer; however, these lesions do indicate an increased risk of cancer development. The models may also be used to determine whether the major factors in determining malignancy are ones that cause benign lesions to become larger, or ones that initiate specific procancerous changes in the DNA.
The rat is widely used as a rodent model of colon carcinogenesis. The F344 strain has a colon with many of the same structural and histochemical features of the human colon. The rat model maybe better than the mouse since colon structure and epithelial-cell histochemical composition in the mouse differs from that of the human. In F344 rats, azoxymethane produces colon cancer within 3 months, and the neoplasms readily metastasize to regional lymph nodes and the liver. This biological behavior is similar to that of certain human colorectal carcinomas.
Enterohepatic Circulation
Species differences in enterohepatic circulation are primarily determined by variations in biliary excretion. Since the rat is a very good biliary excretor, extrapolation of toxicity data on drugs undergoing enterohepatic circulation from rats to other species may result in over-estimation of a compound’s safety or toxicity.
The metabolism and toxicity of drugs maybe influenced by enterohepatic circulation, especially in species, such as rats, deer, and horses, that lack a gall bladder. Because of the concentrating capacity of the gall bladder, compounds maybe concentrated to levels 10 times greater than in hepatic bile. Such concentrating activity, coupled with prolonged biliary retention, may favor passive reabsorption of compounds through the gall bladder mucosa. Additionally, reabsorption would be markedly enhanced by concomitant mucosal injury to the gall bladder epithelium. These factors must be taken into consideration when considering selection of a model for compounds that may enter the enterohepatic circulation.
Knockout/Transgenic Models
Transgenic and targeted gene recombination have been used to study the basic biology and toxicology of the gut in a wide variety of systems. These animal models have been used to study cell growth and regulation, metabolism, mutagenesis, and carcinogenesis.
Cell Growth and Regulation
Transforming Growth Factor-Alpha
In the small intestine and colon, there is regional variation in the production of epidermal growth factor (EGF) and transforming growth factor-alpha (TGF-α). Transgenic mice expressing TGF-α regulated by a metallothionein-inducible promoter/inducer have been created which have a phenotype similar to human Menetrier’s disease, including hypertrophic gastric folds with foveolar hyperplasia and cystic dilation, increased neutral mucin staining, and reduced basal and histamine-stimulated rates of gastric acid secretion. Overexpression of TGF-α in the mouse duodenal epithelium results in a pronounced increase in crypt epithelial cell proliferation and increase in crypt–villus dimensions, and suggests that TGF-α maybe a physiological regulator of small intestinal epithelium proliferation.
β-Catenin
A critical component of the Wnt/Wingless signal transduction pathway and an important effector of cell–cell adhesion through cadherins, β-catenin has been implicated in human colorectal carcinogenesis via its association with the APC (adenomatous polyposis coli) gene product. However, attempts to genetically engineer mice homozygous for the null allele resulted in early embryogenic death. Recently, chimeric mice were generated with amino-terminal truncated β-catenin to study the effects on proliferation, cell fate specification, adhesion, and migration within the intestinal epithelium. The resulting mice showed marked increases (fourfold) in cell proliferation and apoptosis in the small intestinal crypts, augmentation of cell–cell junctions, and interference with normal cellular migration along the crypt–villus axis, occasionally resulting in abnormal architecture. The significance of these abnormalities to the pathogenesis of colon cancer is still being explored as elevated neoplastic transformation was not observed.
Mutagenesis and Carcinogenesis
Investigations of the mutagenicity and carcinogenicity of benzo(a)pyrene [B(a)P; 75 and 125 mg/kg orally for 5 days] in transgenic lacZ mice (MutaMouse) have demonstrated that the mutation frequency (MF) is increased 37-fold in colon, followed by increases in MF in the ileum > forestomach > bone marrow, spleen > glandular stomach > liver, lung > kidney and heart cells 14 days after the last dose of B(a)P. The main target organs for carcinogenicity in this transgenic mouse strain were the forestomach and lymphatic organs. These studies demonstrate that mutation data reflect carcinogenicity outcome, but not all organs with high frequencies of induced mutation in the lacZ transgene develop tumors, nor does the magnitude of the induced MF in the different target organs correlate with the their carcinogenic responsiveness. Although there is no clear rationale for these discrepancies between MF and cancer susceptibility, possible explanations include (1) the nature of the target gene and type of mutations detected by the transgenic assay (the lacZ gene is neutral and is mainly sensitive to point mutations, which may not reflect the mutations in the cancer genes associated with these tumors); (2) inadequate selection of target cells within the target organs; and (3) the importance of factors such as cell proliferation, turnover, and apoptosis in tumorigenesis at specific organ sites.
Transgenic and knockout models for a wide range of cancer genes, such as p53 (tumor suppressor gene), APC (a tumor suppressor gene), and ras (oncogene), have also been created and used to investigate both spontaneous and environmentally induced tumorigenesis in the intestine. Although these models allow for understanding of specific gene changes within the context of a genomic environment of the model species, it is clear from the complexity of gene regulation in different species that extrapolation of carcinogenic vulnerability from one species to another requires information which is currently beyond state-of-the-art interpretation of genomic and proteomic information.
Metabolism
Heavy Metals
Metallothioneins are low-molecular-weight-inducible proteins, rich in cysteine (~33%), that bind heavy metals. They are associated with the homeostasis of essential heavy metals such as zinc, and also provide protection from exposure to toxic heavy metals such as cadmium. Metallothionein knockout and transgenic mice have been developed to study the expression, distribution, regulation, and function of these proteins, and to investigate heavy metal metabolism and toxicity. Mice transgenic for metallothionein have elevated levels in many tissues, including liver and intestine, and are resistant to dietary zinc deficiency. Knockout mice, in contrast, have delayed renal development and increased sensitivity to zinc and cadmium toxicity.
Cytochrome P450
Transgenic and knockout mice of various CYP genes (e.g., CYP1A2, CYP2E1) and the aryl hydrocarbon (Ah) receptor have been constructed to investigate the role of these proteins in phase 1 metabolism of various tissues, including intestine, and their contribution to embryonic development. Proposals have been made to use genetic modification to “humanize” rodent models with human P450s for toxicological studies in vivo.
Response of the Gastrointestinal Tract to Injury
Most responses that occur in GI intoxication are ulcerative, proliferative, or inflammatory (Table 15.9). Because of variations in structure and function, each segment of the GI tract is affected by toxic compounds in a slightly different manner. In addition, each segment has a different range of pathophysiological responses to a toxic compound. Clinically, most alterations of the GI tract are manifested clinically as abnormal function, such as vomiting (if the animal is capable of it), diarrhea, constipation, or nutrient malabsorption. Additionally, occult bleeding or large amounts of hemorrhage into the GI lumen may occur and blood, maybe present in the stool. Of these possible manifestations, diarrhea is the most commonly observed sign.
Table 15.9
Gastrointestinal Reaction to Various Chemical and Elemental Toxins
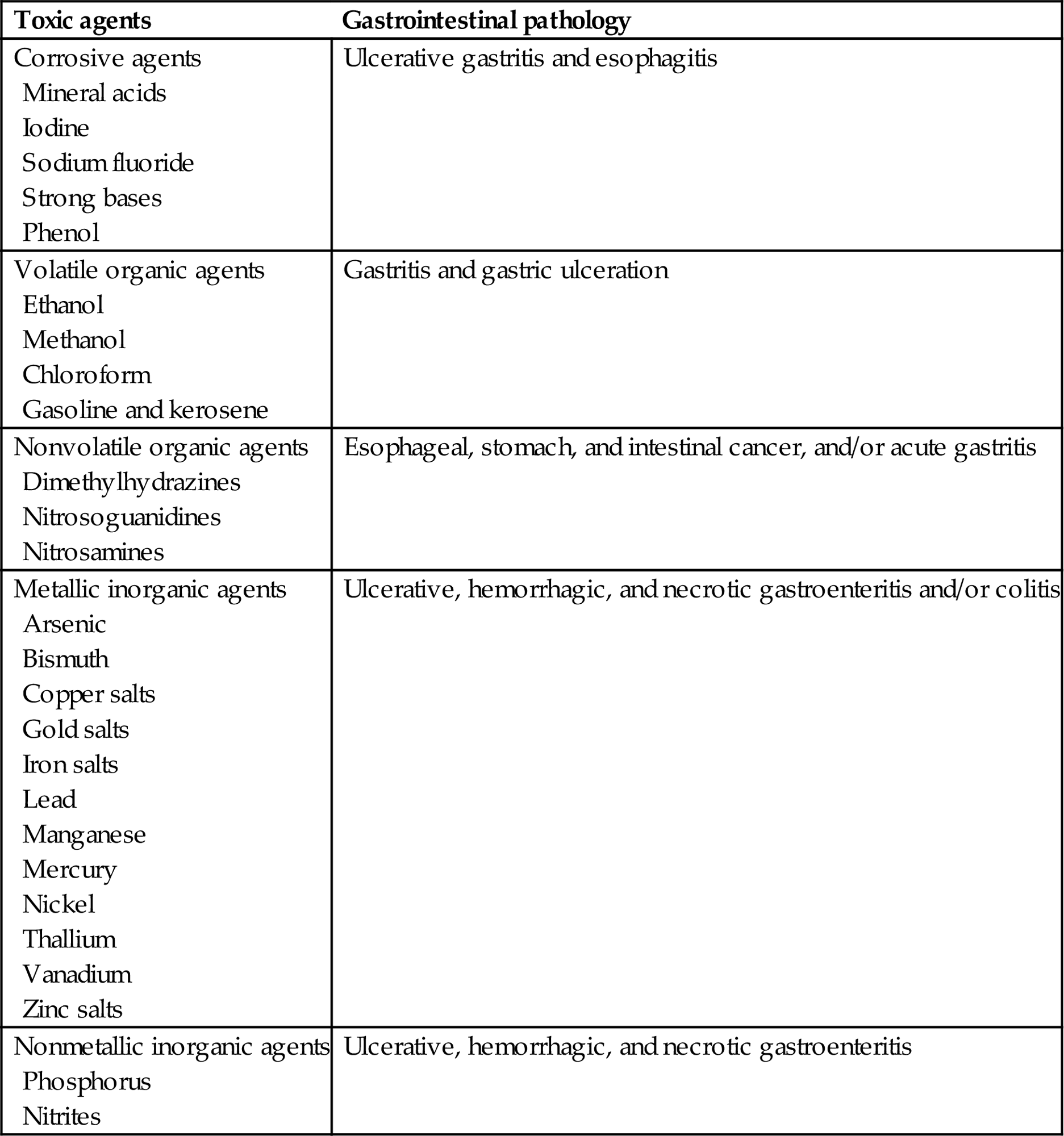
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table XI, p. 155, with permission.
Responses of the GI tract to toxic compounds must be considered on an organ, tissue, and cellular basis. Two specific processes, the inflammatory response and the immune response, are discussed in detail here because many of the disease processes resulting from chemical injury involve these two host reactions. Finally, the response of the enteric nervous tissue is discussed. Although the GI tract has a large amount of autonomic nervous tissue, most neural responses are manifested only on a functional basis.
Pathophysiological Responses
Diarrhea
Identification of mechanisms responsible for diarrhea should focus on the small and large intestine separately as well as collectively. Small intestinal diarrheas are generally associated with increased mucosal permeability, hypersecretion, and/or malabsorption. Major mechanisms of large intestinal diarrhea include hypersecretion, large-bowel malabsorption, and/or direct colonic mucosal injury. Small intestinal malabsorption allows fermentable nutrients to enter the colon, where bacteria generate osmotically active products that hold water in the lumen. Mucosal damage and inflammation lead to release of prostaglandin E2 (PGE2), which stimulates electrolyte secretion, and activation of mast cells, with subsequent release of histamine to enhance vascular permeability. Inflammatory cells can also release mediators that stimulate nerve activity, which may lead to localized GI tract hypermotility. Consequently, the pathogenesis of diarrhea can be generally divided into four major categories: (1) increased mucosal permeability and exudation; (2) hypersecretion; (3) malabsorption; and (4) abnormal GI motility.
The exact pathogenesis of diarrhea can vary, but some of the most injurious mechanisms involve toxins or toxicants that affect the GI tract’s ability to transport fluid. Since a major function of the GI tract is both to secrete and then absorb large amounts of fluid, such intoxications are life-threatening even though little morphological damage maybe present. This process, and the extent of the resulting diarrhea, is best demonstrated with cholera toxin. Cholera toxin activates adenylate cyclase, resulting in the secretion of large amounts of fluid into the small intestinal lumen, overloading the large intestine’s ability to absorb intraluminal water. This process leads to severe diarrhea and death, with little morphological evidence of mucosal damage.
Malabsorptive diarrheas associated with loss of enterocytes, crypt cells or both, often accompanied by mucosal hemorrhage and inflammation, are commonly associated with GI toxicosis. This type of diarrhea is more often accompanied by morphologic changes, although they are relatively nonspecific. These lesions maybe site-specific, however, providing a clue to the type of toxicant involved. For example, corrosive agents are far more likely to cause necrosis and inflammation in the pharynx and esophagus than lower down in the GI tract.
Coordinated peristalsis can be altered by bulking agents such as fiber. By causing distension, these compounds elicit contractions that occur in either direction along the bowel. In a few cases, for example, organophosphate-insecticide toxicosis, diarrhea is via prolonged stimulation of muscarinic receptors due to inhibition of acetylcholinesterase at the synapse. The greatly enhanced peristaltic activity, combined with stimulation of secretion, leads to decreased transit time through the GI tract and resultant diarrhea.
The consequences of diarrhea are systemic in nature, and include dehydration, acidosis, and electrolyte alterations. Direct loss of bicarbonate in the feces is a major cause of acidosis. Intracellular hydrogen ion concentrations increase and potassium concentrations decrease. This electrolyte imbalance leads to improper maintenance of intracellular pH ranges, and reduces the activity of multiple enzyme systems. Reduced intracellular potassium is the result of a failure in cellular electrolyte transport. Consequently, there is inadequate maintenance of electrochemical gradients, which leads to increased extracellular potassium and mass excretion of this electrolyte in the urine and feces.
Vomiting
Vomiting is a clinical response that occurs in some animal species. Vomiting requires the presence of skeletal muscle in the wall of the esophagus. Vomiting maybe stimulated by direct mucosal irritation or by stimulation of the vomiting center in the central nervous system. Vomiting may also occur if the esophageal or pyloric lumen is obstructed by a scar or neoplasm. Toxic substances can act on the GI mucosa to induce emesis via activation of sensory nerves that travel over vagal and sympathetic afferent pathways to brain medullary centers that control vomiting. One mucosal sensory emetic pathway involves activation of 5-hydroxytryptamine 3 (5-HT3) receptors at the peripheral ends of these sensory nerves in the GI mucosa. 5-HT3 receptors are ligand-gated ion channels that mediate depolarization of nerves, and the vagus nerve is densely populated with 5-HT3 receptors. Cyclophosphamide, carmustine, dactinomycin, and cisplatin interact with mucosal enterochromaffin cells to promote release of large quantities of 5-HT, which leads to emesis. Bilateral abdominal vagotomy and bilateral splanchnic nerve transection completely inhibit emesis induced by these anticancer drugs. Emesis induced by cancer chemotherapeutic agents can be reduced or prevented by administration of 5- HT3 antagonists, such as granisetron, or those that block both dopamine D2 receptors and 5-HT3 receptors, such as metoclopramide. However, 5-HT3 antagonists are not effective against some other emetogenic substances, such as copper sulfate, protoveratrine, or apomorphine.
Cisplatin appears to act by effects on the central nervous system that promote release of 5-HT and subsequent activation of 5-HT3 receptors. Evidence for this mode of action comes from observations that combined vagal and splanchnic nerve transections do not completely prevent vomiting in response to peripherally administered cisplatin, whereas emesis is reduced by administration of 5-HT3 antagonists. The highest concentration of 5-HT3 receptors in the mammalian brainstem is located in the area postrema of the brain. Activation of vagal sensory neurons by 5-HT3 receptor-mediated events causes release of proemetic neurotransmitters from the central terminals of sensory fibers in the solitary nucleus and in the area postrema. 5-HT3 receptors in the brainstem appear to be associated with presynaptic sites and serve primarily to modulate release of neurotransmitters. Presumably, activation of the central 5-HT3 receptors enhances release of proemetic substances, thereby activating the chemoreceptor trigger zone of the area postrema and the nearby emetic center.
Agonists of dopamine D2 receptors such as apomorphine, L-dopa, and bromocryptine act in the chemoreceptor trigger zone of the brainstem area postrema. Phenothiazine drugs with significant dopamine D2 antagonists properties, such as chlorpromazine and promethazine, block the emetic actions of dopamine D2 agonists. Emetine, the principle ingredient of ipecac, and opiates, such as morphine, act nonspecifically at the chemoreceptor trigger zone to initiate an emetic response. The area postrema is unprotected by a complete blood–brain barrier, thus allowing chemicals in blood to penetrate with relative freedom into this brain area.
Constipation
Constipation is both a structural and a functional disease. Compounds that bulk the stools (e.g., fiber) may lead to constipation if there is concomitant reduction in water intake. Polyps and neoplastic masses induced by carcinogenic agents as well as partly or completely circumferential scars may result in physical obstruction. The constipating effects of certain analgesic compounds (e.g., morphine) have a neurological component to their pathogenesis.
Gastrointestinal Nervous Tissue and Motility
Nervous tissue responses are generally identified by functional abnormalities that are initially detected clinically. Normal intestinal motility consists of peristaltic activity, which moves intraluminal contents down the GI tract. This activity represents contraction of both longitudinal and circular muscle layers. Neuronal networks involved in peristalsis are complex and incompletely understood; however, cholinergic excitation mechanisms play a major role. Such coordinated peristaltic activity can be altered by bulking agents. These compounds cause intestinal distension and elicit contractions that occur in either direction along the bowel.
Opiate (e.g., morphine) toxicity is manifested clinically as a nonpropulsive and constipating pattern of segmentation motility. Segmentation movements accomplish mixing of intraluminal contents. This activity involves reciprocal neural inhibition and disinhibition of adjacent muscle segments; it maybe preprogrammed into the internuncial circuitry of the GI nervous system. Endogenous opioid peptides maybe involved in segmentation motility, since morphine locks the intestine into a continuous segmentation pattern of motility. In morphine-dependent rats, diarrhea, which is the opposite of the acute effects of morphine, is a primary withdrawal event. PGE2 and 5-HT may also contribute to a secretory type of diarrhea.
Extrinsic nervous input affecting motility includes both stimulatory and inhibitory nerve fibers. Both vagal and sacral innervation to the large intestine is especially active during defecation. Sympathetic nerves are active in reducing blood flow and motility of the GI tract. Inhibitory nerve input into the GI tract leads to reduction or cessation of muscle motor activity (ileus). Tonically active inhibitory neurons can account for a low responsiveness of circular muscles to myogenic pacemakers. A model toxicity of inhibitory nerve input that leads to suppression of both cholinergic and serotoninergic synaptic transmission is norepinephrine overdose. Peritoneal irritation can also cause this effect and lead to ileus.
Spasm is a functional disorder, the opposite of ileus, and consists of accelerated activity of circular muscles with no activity on inhibitory neurons. Intoxication by various cholinergic agents can cause spasms of the GI tract. The model for this disorder is aganglionic megacolon of piebald mice. In these animals, the terminal segment of the large intestine lacks inhibitory neurons.
Inflammatory Response
Because of the high number of bacteria and the physicochemical nature of the luminal contents, inflammation is frequently involved in many lesions of the GI tract regardless of the underlying mechanism of injury. However, the inflammatory response is generally less severe in primary toxicologic lesions than in primary infectious (bacterial and viral) diseases.
Inflammation of the stomach (gastritis) is essentially a process that is restricted to the mucosa. Gastritis is usually catarrhal (with large amounts of mucus), and may involve ulceration, hemorrhage, and lymphoid hyperplasia. “Gastritis glandularis,” a disease of primates, is characterized by mucosal hyperplasia and mucus-filled cysts in the mucosa and submucosa. This lesion maybe induced by ingestion of PCBs.
Inflammation of any part of the intestinal tract can be termed enteritis, but in practice this term is frequently used to designate only small intestinal inflammation. In contrast, the term colitis is used to designate large intestinal inflammation. Direct irritants usually cause more severe inflammation of the proximal intestine (duodenum) and less inflammation of the distal tract (ileum and large intestine). However, mercury can cause lesions of the large intestinal mucosa as a result of transport from the blood into the colonic lumen.
Chronic inflammatory reactions can be a primary or secondary effect in toxicologic lesions. Immune-mediated responses are characterized by accumulation of chronic inflammatory cells (lymphocytes, plasma cells, and macrophages), although they may have an active cellular component consisting of neutrophils and eosinophils.
Diseases that result in chronic inflammation or injury to the lamina propria or lymphatic vessels cause malabsorption of fatty acids and weight loss. Fatty acids and monoglycerides are packaged into chylomicrons by the enterocytes before being exported to the central lacteal and into the lymphatic circulation. Consequently, longstanding damage to the lymphatic circulation can result in significant malabsorption-related disorders. Systemic complications such as septicemia and bacteremia may develop as a result of chronic inflammation and ulceration of the GI tract. Secondary lesions maybe present in the liver (e.g., abscesses), skin (e.g., perianal ulcers that develop secondary to chronic diarrhea), or urinary tract (e.g., females can develop an ascending infection of the urethra from malabsorption-induced diarrhea).
Mucosal Response
General
The intestinal mucosa separates the body from the GI tract’s luminal contents (including bacteria and nonabsorbed toxic compounds). This lining is also responsible for selective absorption of ingesta to obtain the proper nutrients that will maintain homeostasis. The mucosal lining is the first site of exposure to an ingested toxicant, and the cells lining the GI tract have the capacity to respond to these toxigenic compounds.
The exposure of the rapidly proliferating GI mucosal epithelial cells to toxins would suggest that the GI tract should be a frequent site of toxicologic injury. The actual frequency of toxicity in commonly used laboratory animals is lower than one might expect, primarily due to the high rate of cellular proliferation that can occur in the reparative response to any loss of mucosal epithelium. Factors that account for the GI tract’s ability to escape damage include its capacity for compound biotransformation and the large surface area that permits extensive contact between the toxic compound and multiple toxin-metabolizing enzymes. Additionally, mixing an injurious compound with luminal contents dilutes the toxin and its effects. Finally, mucosal barrier components (e.g., mucus) are protective, and the short half-life of GI epithelial cells rapidly removes those cells that have suffered molecular damage (e.g., DNA mutations).
Since the epithelium of the GI tract is the first layer of host cells to contact ingested compounds, these cells can respond to toxic compounds before they enter the circulation. Epithelial cells can also undergo biochemical changes that allow them to functionally reconstitute mucosal integrity after a toxic insult. Modification of several enzyme pathways—including the CYP 450, ADH, monoamine oxidase, epoxide hydrolases, esterases, amidases, glucuronidases, sulfatases, and various conjugation pathways—allows epithelial cells to maintain a barrier function. Many of these same enzymes are involved in compound metabolism and biotransformation. Interestingly, transgenic mice that carry a mutant dihydrofolate reductase (DHFR) gene display resistance to methotrexate toxicity of the GI tract. Methotrexate interferes with DNA replication via inhibition of DHFR bioactivity and consequent reduction of de novo thymidine and purine biosynthesis.
Mucosal epithelial cells interact with reactive compounds via both membrane-bound and cytoplasmic enzymes. Although the CYP 450 pathway can generate cell-damaging and reactive intermediate epoxides, mucosal cells can form nontoxic dihydrodiols and glucuronide conjugates from these intermediates. This process occurs via mucosal cell enzymes, including epoxide hydrolases and glucuronosyl transferases.
Unique forms of adaptive mucosal protection occur after exposure of the mucosa to mild irritants. Under these conditions, increased levels of PGE2 are elaborated and protect the mucosa from strong irritant damage by increasing blood flow and stimulating local bicarbonate secretion in the small intestine.
Sublethally injured GI epithelial cells are able to reseal damaged membranes and can participate in covering discontinuities in the epithelial barrier. Repaired cells at the margins of ulcers and erosions become active participants in GI barrier restitution by “flattening,” then spreading out and migrating over denuded basal lamina. Membrane resealing is a key process in maintaining an intact epithelial layer if the injury does not cause widespread and severe loss of epithelial cells. GI epithelial cells are protected from injury by an apical membrane enriched with viscosity-enhancing glycosphingolipids, a microvillar surface that can be sloughed if damaged, and a circumferential apical cytoskeleton that can contract and seal off the apical surface of enterocyte, preventing further injury. Resealed or healed cells may remain viable for up to 24 hours in the stomach and 48 hours in the intestine.
Rapid epithelial restitution is one of the mucosa’s primary defense mechanisms throughout the GI tract (esophagus to anus). Each segment of the GI tract has a basal rate of mucosal cell proliferation, which varies with species, age, diet, and disease state. Under normal dietary conditions and health, the range of proliferation rates for the most actively dividing mucosal cells (stomach to colon) is 3–6 days. When the intestine encounters a noxious agent, enterocyte half-life is reduced to quickly replace injured cells. If the damage is transient, mucosal replacement, and normal microarchitecture will recover within 3 days.
Arachidonic acid metabolites from the cyclooxygenase (COX) and lipoxygenase pathways are elevated during GI inflammation and after acute colonic injury in which an inflammatory process has not yet developed. These inflammatory mediators have many effects on the structure and function of the GI tract, particularly with regards to blood flow.
Bile salts (e.g., deoxycholate) induce the release of arachidonic acid and COX and lipoxygenase metabolites of this fatty acid, and lead to the generation of active oxygen radicals Bile salts also induce increased colonic secretion and permeability, but this occurs by mechanisms independent of endogenous arachidonic acid metabolism. However, prostaglandins of the E series suppress enzyme release and superoxide anion production by neutrophils. Thus, the interaction of these chemical mediators may be important in controlling the inflammatory response of the GI mucosa and the final outcome of injury.
Nitric oxide (NO) can be beneficial or toxic, depending on circumstances. It is synthesized from the amino acid L-arginine by at least two different NO synthases. The two major isoforms of NO synthase are a constitutive enzyme, which is calcium- and calmodulin-dependent, and an inducible form, which is calcium-independent. The constitutive form has been identified in endothelium, nerves, and brain. It seems to be active continuously, generating small amounts of NO. The inducible form has been identified in macrophages and is probably present in other tissues, such as GI and vascular smooth muscle and vascular endothelium.
NO in low concentrations is thought to protect the GI mucosa from injury and to enhance restitution of injured mucosa. It produces vasodilation of gastric microvessels and exerts an antiaggregation effect on platelets. These actions tend to maintain adequate mucosal blood flow. It also stimulates secretion of mucus by surface mucous cells and helps maintain protection against luminal acid. Laboratory studies indicate that functional repair of the epithelial barrier after acute injury is enhanced by NO. While NO confers many benefits to the GI tract by protection of the mucosa, maintenance of mucosal blood flow, and regulating contractions of smooth muscle and propulsion, it can also be responsible for GI toxicity. This dual nature of NO appears to be related to the relative expression of the two major isoforms of NO synthase. In general, the NO produced by the constitutive form of the enzyme produces beneficial effects, while the NO produced by the inducible form of the enzyme has often been implicated in vascular or epithelial cell injury. Excess production of NO in the GI mucosa by the inducible form of NO synthase is linked to initiation of secretory diarrhea. Diarrhea induced in rats by castor oil or bile salts (e.g., sodium choleate) is blocked by inhibitors of NO synthase. Castor oil and bile salts also induce direct mucosal damage, but the damage is ameliorated by coadministration of NO synthase inhibitors. These results suggest that NO mediates, at least in part, the diarrheal effect of these compounds, presumably by increasing secretion of fluid into the intestinal lumen, even though, simultaneously, NO exerts a protective effect on the intestinal mucosa. This suggests that bile acids and castor oil directly damage intestinal mucosa and activate the inducible isoform of NO synthase to produce large amounts of NO linked to production of diarrhea. Although laxatives, such as phenolphthalein and bisacodyl, are also associated with electrolyte secretion, changes in mucosal histology, and abnormal motility, their effects have yet to be linked to NO production remains to be established.
Stomach
Mucosal defense and protection, initially termed cytoprotection, was originally described as the ability of prostaglandins to prevent macroscopic evidence of gastric mucosal injury. This protective phenomenon is partially dependent on the antisecretory activity of prostaglandins, and is dose- and route-dependent.
It is currently understood that several mechanisms besides prostaglandin-mediated pathways are responsible for preventing mucosal damage by both normal digestive processes and injurious compounds. These include increased amounts or modifications of mucous gel covering the mucosal epithelial surface, increased secretion of bicarbonate, increased resistance to acid back-diffusion, and increased blood flow. Several of these processes are mediated by prostaglandin synthesis in mucosal and submucosal tissues (Figure 15.9). Additionally, mucosal protection is mediated in part by lipids (neutral lipids and phospholipids) within the mucous gel layer. These lipids increase the hydrophobicity of the mucous gel, leading to repulsion of water-soluble compounds (including many toxicants).
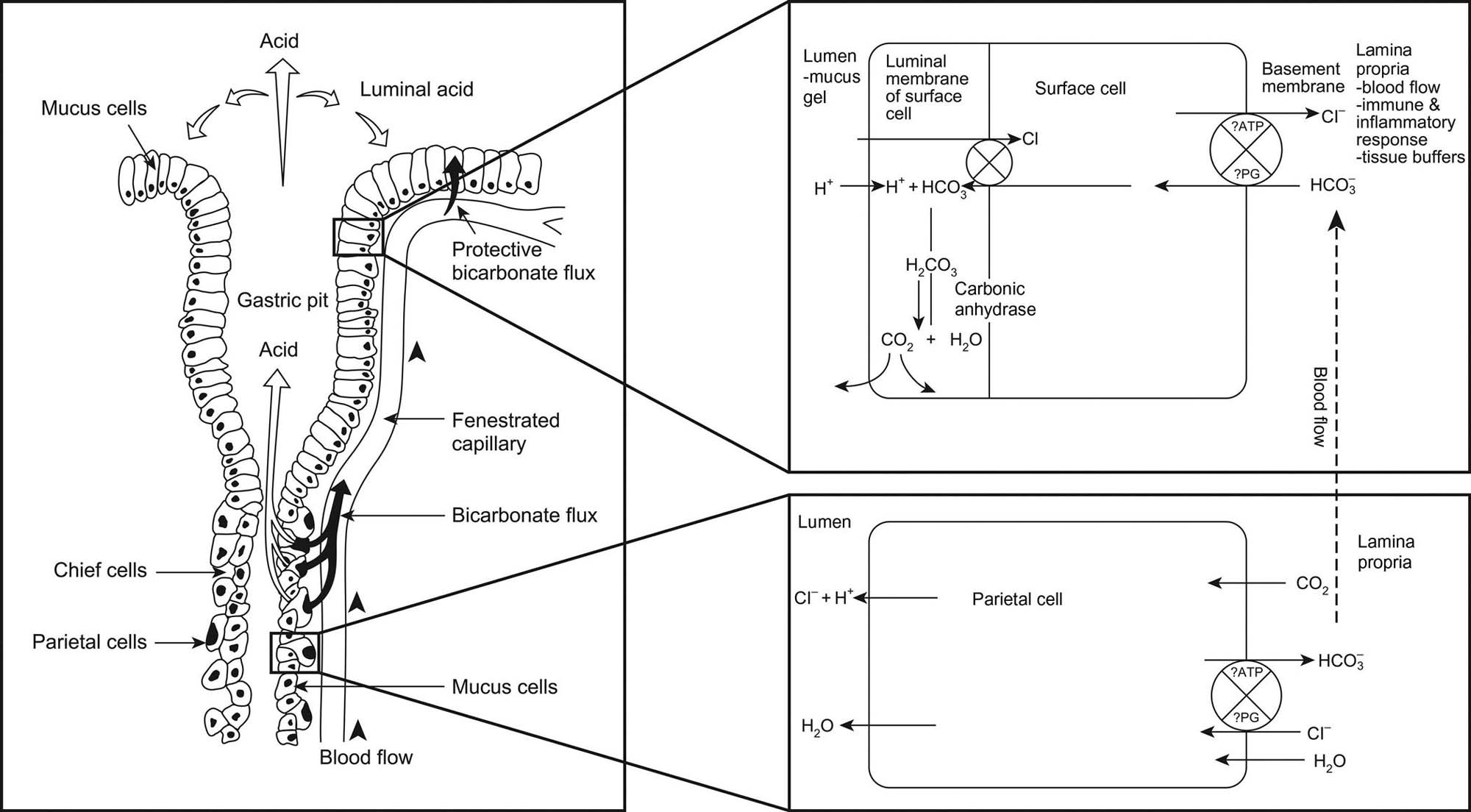
Mucosal damage can occur with or without mucous-layer damage. The adherent mucous gel is not extensively disrupted by mucosal-damaging agents such as dilute HCl or indomethacin (an NSAID). These agents permeate the mucous barrier and directly damage the underlying epithelium. Since the mucous layer remains intact, it facilitates epithelial repair in these situations. However, mucus is lost when the stomach is exposed to mucosal-damaging agents such as pepsin, bile acids, and ethanol, and also mechanical trauma (Figure 15.10). With persistent mucosal damage, mucous-gel secretion is impaired and mucous-gel loss will exceed production. Additionally, mucus composition can be modified by epithelial cell metaplasia, leading to chemical changes in the mucous gel and loss of functional integrity.
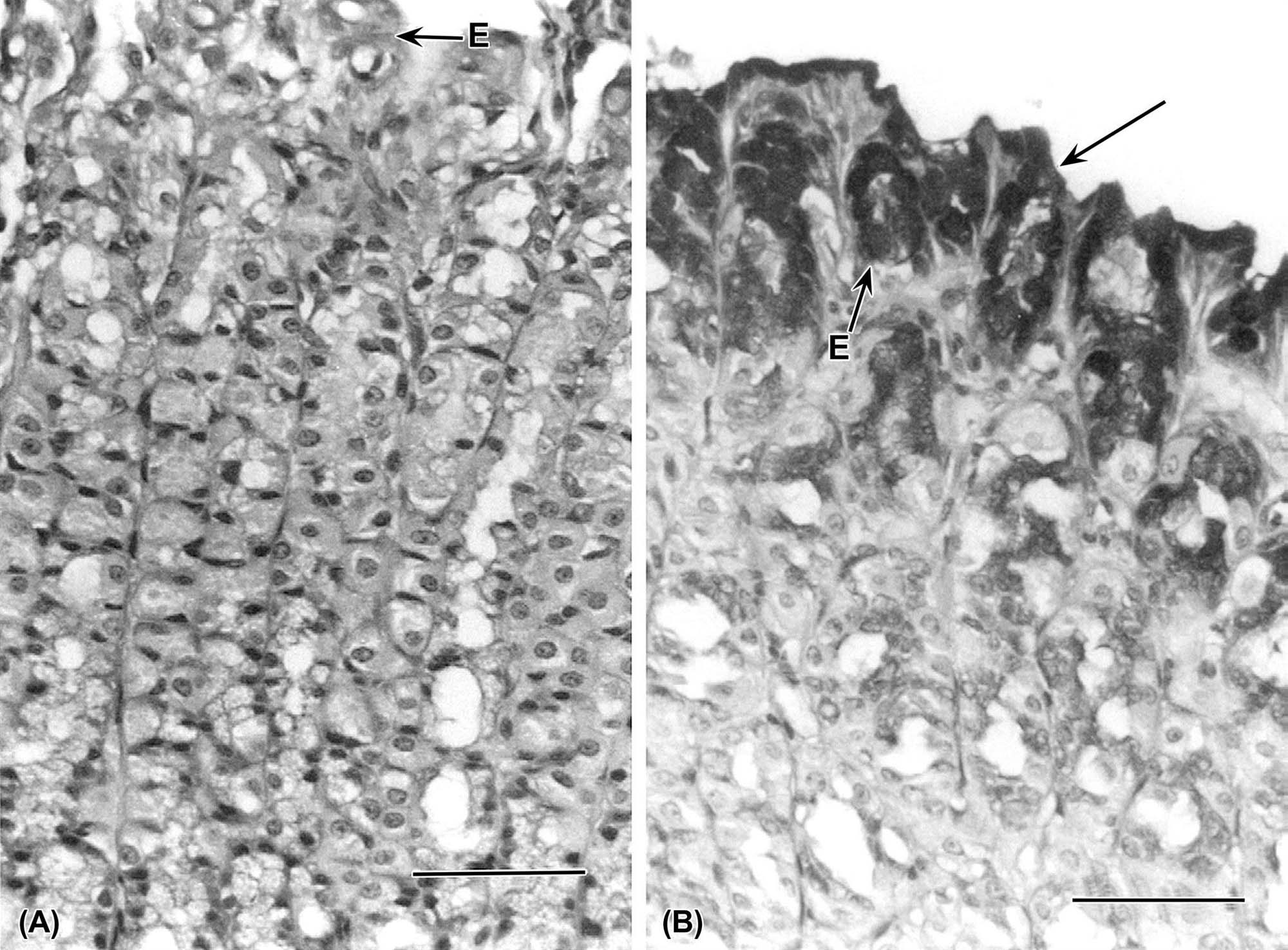
These types of direct injuries will ultimately lead to collapse of the mucous barrier because the mucous gel alone, without constant renewal, cannot protect the mucosa and support rapid recovery after epithelial cell damage. The damaging effects of mucus loss are manifested by further loss of surface epithelial cells, reduced mucus release, vascular occlusion in underlying lamina propria, and, ultimately, ulceration and scar formation (Figure 15.11).
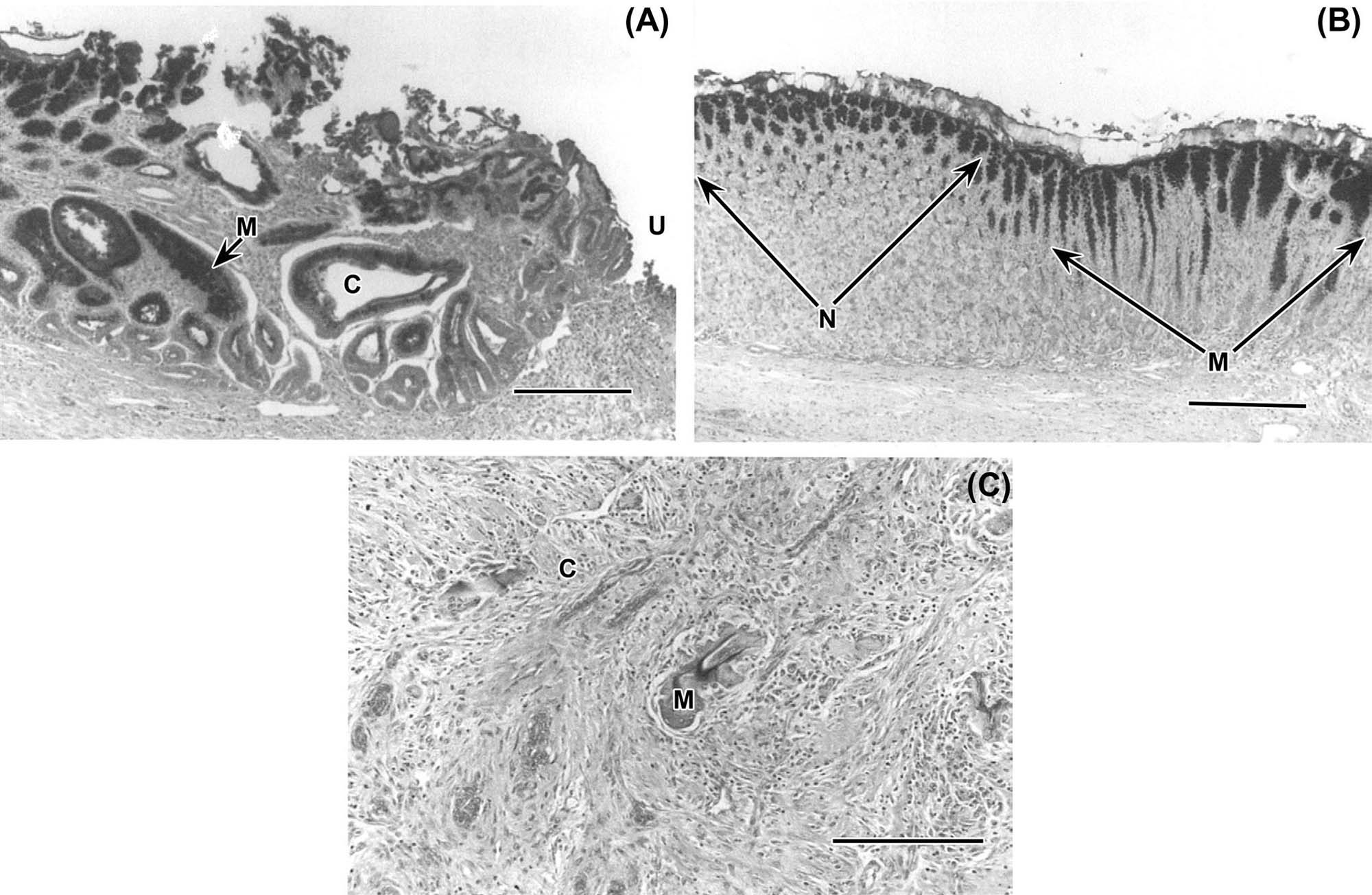
Repair of chemically or mechanically induced gastric epithelial cell discontinuities can be complete within 30–90 minutes of injury. The rate of repair will vary with location in the GI tract (stomach, small intestine or colon) and extent of initial damage. Repair can be very rapid. For example, gastric mucosa experimentally damaged by exposure to high concentrations of sodium chloride, which produces direct structural damage, can be repaired over a period of 6 hours by a gradual process of restitution of epithelial integrity via migration of cells from the gastric glands.
Cellular proliferation is a key epithelial cell mechanism in maintaining mucosal barrier function. The extent of general mucosal damage resulting from an insult can be anticipated based on the amount of damage the compound inflicts on proliferating cells of the mucosa. Minor damage to the proliferative compartment leads to mild gastritis or enteritis, and any increased loss of surface cells is readily compensated by the undamaged proliferative cells that remain in the glands. However, severe mucosal damage will occur when the proliferating unit of the mucosa is destroyed. Such injury develops after irradiation, exposure to some mycotoxins, or administration of cytotoxic drugs. One of the mechanisms by which PGE2 enhances healing of the mucosa is by protecting the cells in the isthmus of the gastric pits and allowing these replicative cells to reconstitute the surface epithelium.
Modifications of epithelial cell proliferation maybe the only morphological indication of mucosal injury. Low doses of indomethacin and aspirin (an NSAID) increase epithelial proliferation in rat gastric glandular mucosa but have no effect in the antrum and duodenum; they do not cause inflammation. In contrast, corticosteroids depress epithelial cell proliferation in fundic, antral, and duodenal mucosa of rats, and hydrocortisone predisposes to gastric ulcers. Proliferative rates are also modified by starvation, and pharmacologic and toxicologic doses of mineralocorticoids, glucocorticoids, and adrenocorticotropic hormone.
Intestines
Damage to small intestinal mucosa by toxicants results in a variety of changes depending on the degree and extent of injury. Damage to enterocytes without damage to underlying lamina propria or to the proliferative epithelial cell population lining the crypts will result in a temporary shortening of villi and decrease in height of remaining enterocytes as they flatten out to cover the denuded mucosal surface. Restitution of the lost enterocytes can happen within a matter of a few days. As proliferative rate of crypt cells increases and the cell cycle time decreases, crypts will elongate as the number of newly formed epithelial cells increase. Complete cell differentiation and restoration of function generally takes longer than cell replacement since newly formed epithelial cells must leave the proliferative compartment and develop the specialized biochemical and morphologic features associated with absorptive enterocytes.
Damage to the crypt cells by toxicants is a more serious matter, leading to “mucosal collapse.” The mechanisms of restitution after proliferative unit ablation are not well understood. Death of crypt cells without underlying damage to lamina propria, in particular the vasculature, will eventually result in complete restitution of cryptal and villous epithelium, provided that complicating factors associated with loss of the mucosal barrier are minimized. Secondary events such as fluid loss from the denuded mucosal surface, leakage of ingesta into the lamina propria, colonization of the lamina propria by resident gut bacteria (Figure 15.11), and the inevitable inflammatory response are minimized. Any one of these complicating factors, if unchecked, can lead to severe systemic consequences and defective restitution of the mucosal barrier. Damage to underlying vasculature, especially if thrombosis occurs, can exacerbate injury even further and lead to ulceration and hemorrhage as well. It is thought that maintenance of regional blood flow in areas of damage is essential for the minimization of ongoing damage and enhancement of repair and restitution in all sections of gut.
By contrast, the colonic mucosa is covered by relatively flat mucus-secreting cells and crypts. Several substances serve as growth factors that can positively stimulate epithelial growth. These include gastrin, TGF-α, and transforming growth factor-beta (TGF-β). The influence of these growth factors is exerted on the stem cell. The ingestion and digestion of food appear to be important in maintaining growth of the intestinal mucosa. Mucosal toxicity can be exhibited by decreased cell production or increased cell loss, which can lead to atrophy or ulceration. Increased cell production in excess of the rate of sloughing can lead to hyperplasia. This latter response is characteristic of mucosal repair, where it is transient, or certain infectious (bacterial) and preneoplastic conditions.
Rapid epithelial restitution is now considered one of the primary defense mechanisms of not only the stomach and small intestine but also colon. However, this occurs only under conditions in which damage is confined to the superficial mucosa. In the colon, for example, epithelial restitution is associated with migration of cells at a speed of approximately 2 μm/minute. On the other hand, regions of the mucosa with gross hemorrhagic lesions heal by a lengthy process of tissue replacement involving cell mitosis. It is thought that maintenance of regional blood flow in the area of damage is important for repair of lesions (as well as prevention thereof). Indirect damage to epithelium may occur as a result of hypoxia when blood flow to the area of damage is compromised.
Three mechanisms influence restitution of the mucosal barrier after toxic damage: (1) the rate of increased epithelial cell production; (2) the reduced cell cycle time (the period between two successive divisions of proliferating cells); and (3) an increased proliferative compartment via an increase in the proportion of cells in the proliferative cycle or an increase in the absolute number of cells that are replicating at any given time. In contrast, under adaptive conditions (e.g., intestinal resection or dietary change), only one mechanism is operative: increased cell production rates via increased numbers of cells that are proliferating. The intestinal mucosa can also be induced to proliferate as a healing response near areas of cellular toxicity and ulceration. PE2 and fermentable fibers (e.g., guar) are examples of inducers of mucosal proliferation in the intestinal tract. Mechanisms of restitution after proliferative unit ablation are not well understood.
Organ-Specific Response
Esophagus
Various species have unique anatomical adaptations of the esophagus (e.g., forestomachs of ruminants), but the lining mucosa and its response to injury are similar among species. Both neoplastic and ulcerative processes can lead to a reduction in the esophageal lumen by the physical obstruction of a space-occupying mass (neoplasm) or a stricture from scar contraction. If highly caustic agents are fed to animals, severe mucosal damage is followed by ulceration, inflammation, fibroplasia, and scar formation. Ulceration can be restricted to the esophagus, or may involve the stomach, small intestine, and/or large intestine.
Spontaneous esophageal cancer is rare in animals and humans living in Western civilizations. However, esophageal cancers are commonly found in humans living in regions of China, reportedly due to high levels of aflatoxins in contaminated grains. When rats are exposed to chemical carcinogens (N-methyl-N-nitrosoaniline), their esophageal mucosae undergo a progression of changes, a sequence of hyperplasia and hyperkeratosis leading to dysplasia, papillomas, and finally to carcinoma. Esophageal cancer can also be induced in rats with dihydrosafrole, and in mice with gamma irradiation. Additionally, zinc-deficient rats treated with carcinogens may develop multiple neoplasms of the esophageal mucosa.
Stomach
Ulceration and Inflammation
Gastric ulceration and associated inflammation and mucus loss are responses to stress (unrelated to compound administration) and various mucolytic agents (Figure 15.12). Active ulcerogens include NSAIDs, alcohol, taurocholate (bile acids), nitriles, thiols, and amines (Table 15.10). In addition to these direct GI irritants, which also affect the stomach, antimitotic and antineoplastic agents (e.g., colchicine and 5-fluorouracil) cause ulceration in various parts of the GI tract. Mechanisms of ulceration are discussed extensively in Section 5.
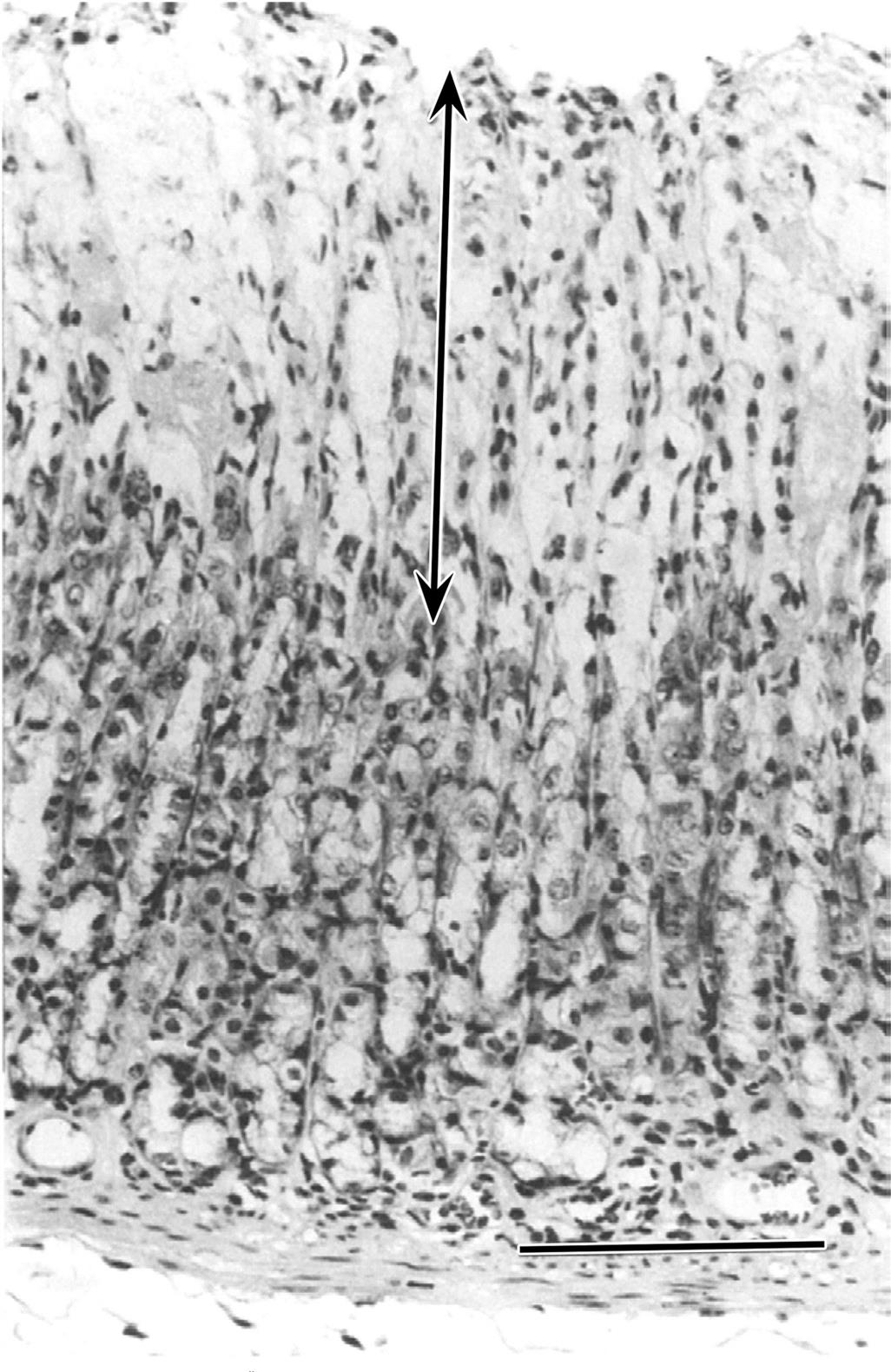
Table 15.10
Selected Mechanisms of Toxicity to the Gastrointestinal Tract
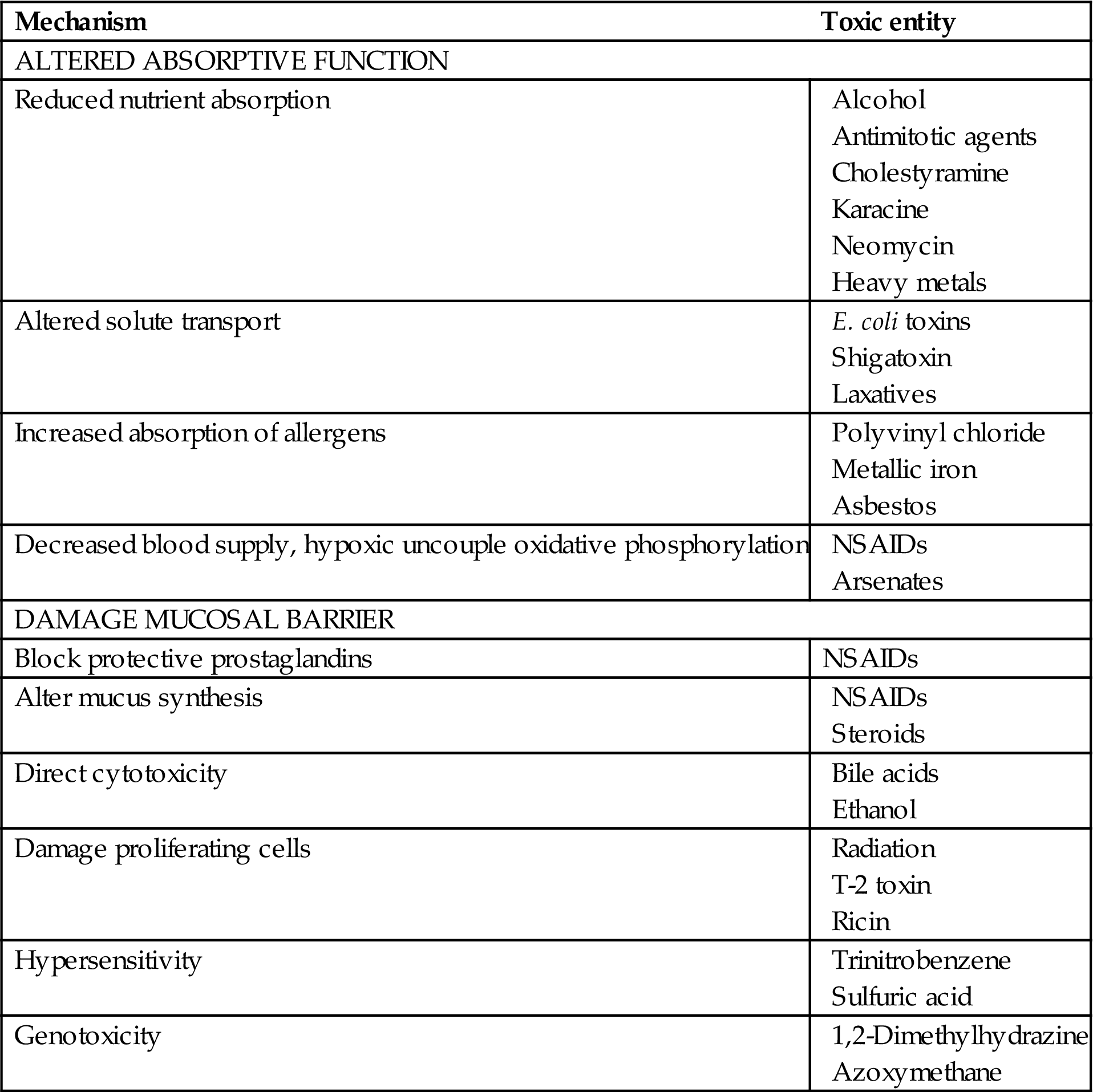
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table V, p. 143, with permission.
Proliferative Response
Epithelial cells maintain normal anatomical boundaries (i.e., do not infiltrate into submucosal tissues) in hyperplastic conditions. However, if hyperplasia is of sufficient duration, it may in certain situations increase the risk of a neoplastic process developing. Adenomas are the result of a benign proliferative response that is neoplastic, and they can potentially progress to malignancy. Determination of the malignant potential of adenomas should include evaluation for evidence of epithelial dysplasia, proliferative activity, cysts, blood capillary organization, stromal infiltration of epithelial components, and inflammation. Intestinal metaplasia can occur in adenomas, as can tissue invasion and distortion. If the neoplasm being examined has morphologically demonstrable tissue invasion or areas of severe epithelial atypia, metaplasia, or dysplasia—with or without extensive proliferative activity—the lesion should be considered malignant (i.e., an adenocarcinoma).
A hormone-mediated proliferative response of the stomach is demonstrated by hyperplasia of enterochromaffin-like cells and an increase in neuroendocrine-cell tumors (“carcinoids”), which develop after prolonged gastrin release (Figure 15.13). Hyperplasia of these enteroendocrine cells has been demonstrated after exposure to ranitidine and substituted benzimidazoles like omeprazole, both of which produce hypergastrinemia. Animals with gastric hyperplasia have abnormal mucosal maturation that is characterized by a decrease in the number of cells with cytoplasmic zymogen granules, a decrease in mature surface cells, glandular atrophy, loss of regular parietal cell distribution, and an increase in cellular proliferation. The initiation of a carcinoma is demonstrated by loss of cellular differentiation, abnormal gland structure, invasion of mucosal elements into surrounding tissue, abnormal glycoprotein expression, and displacement of normal tissue.
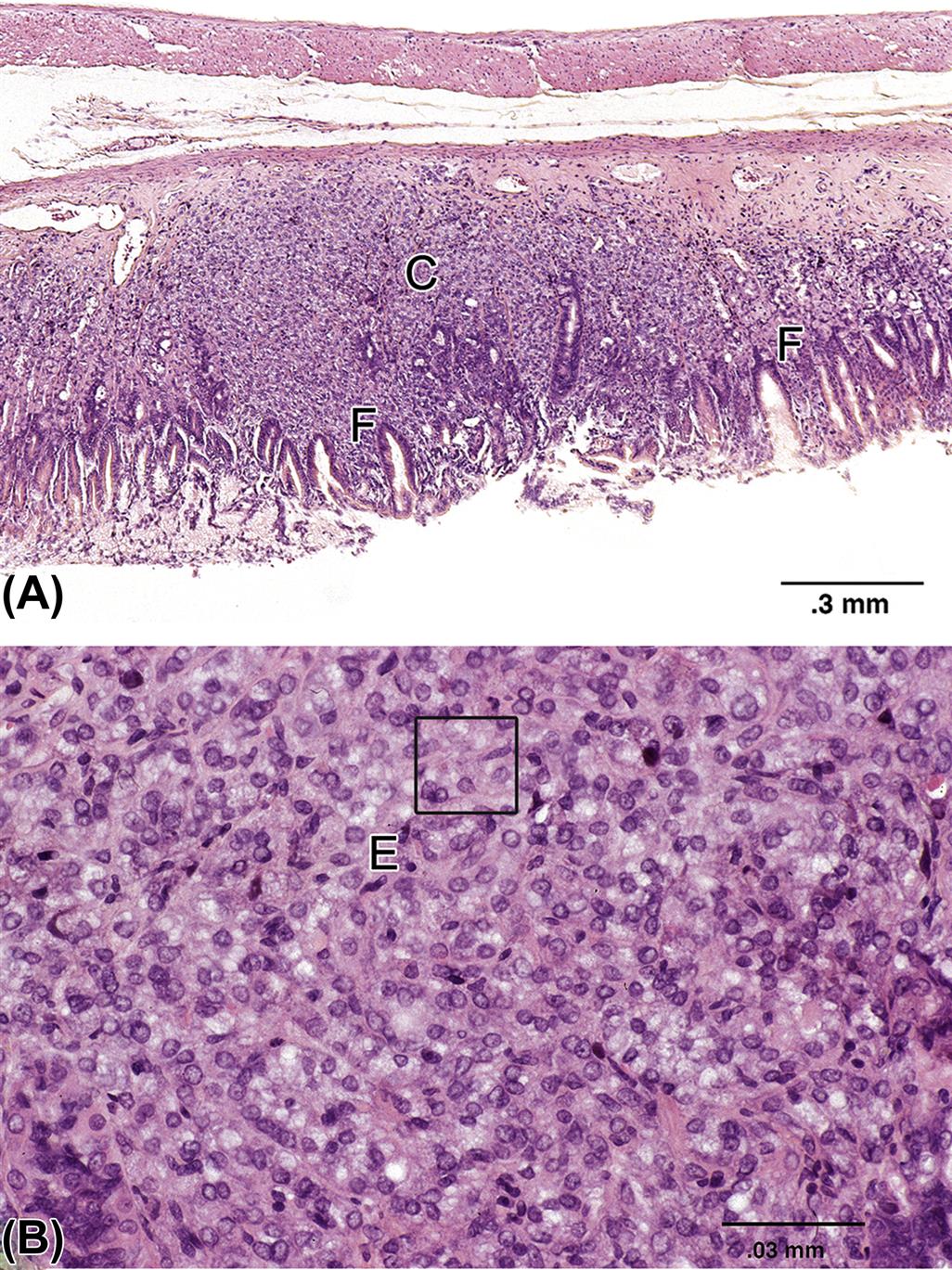
Small Intestine
Damage to the small intestinal mucosa frequently results in villus atrophy and crypt-cell hyperplasia. If ulceration occurs, an associated inflammatory reaction ensues. Duodenal sites are more frequently found to have ulcers than other small intestinal segments. The same ulcerogens that affect the stomach frequently damage the duodenum. The distal small intestine is a frequent site of functional abnormalities, such as diarrhea, rather than a location for morphological damage. The small intestine is an infrequent site of neoplastic formation. However, lymphosarcomas may originate in the lymphoid nodules of the lamina propria and submucosa. Adenocarcinomas induced by a carcinogenic agent or natural causes may originate from the mucosal epithelium and invade the submucosa and tunica muscularis mucosa (Figure 15.14).
Large Intestine
Ulceration and Inflammation
The response of the large intestine to toxic injury can be studied using models that induce acute and chronic lesions. Acute erosive injury to the colonic mucosa can be induced in rat and pig. using the bile salt deoxycholate. Damage to the surface cells is mediated by reactive oxygen species, and complete ablation of the surface epithelium occurs within 8 minutes. Mucosal permeability is regained after 40 minutes, and recovery of absorptive activities occurs when the epithelium is restored to a columnar phenotype (2 hours). The reparative process of the mucosa occurs by active cell migration from the proliferative zone to the surface. This process will be delayed or is unable to take place if damage to the mucosa is severe enough to damage stem cells, in which case ulceration and inflammation will be observed.
Proliferative Response
One common response seen in the colon is mucosal hyperplasia and polyp formation. Hyperplastic polyps of the colon maybe either inflammatory or regenerative in nature. Benign lymphoid polyps, not necessarily associated with toxicity, also occur in the colon or rectum as a result of lymphoid hyperplasia. Proliferative polyps of epithelial origin can be classified as adenomatous polyps or adenomas. Adenomatous polyps are composed of tubules of neoplastic epithelium with little stroma (Figure 15.15). In contrast, villous adenomas have multiple projections of epithelial-lined lamina propria. Regardless of classification, the mucus content of neoplastic epithelial cells is reduced, and mitotic figures are common in these polyps. The presence of multiple polyps should be regarded as an early cancerous event in rodents, since there is an established adenoma–carcinoma sequence in the colon.
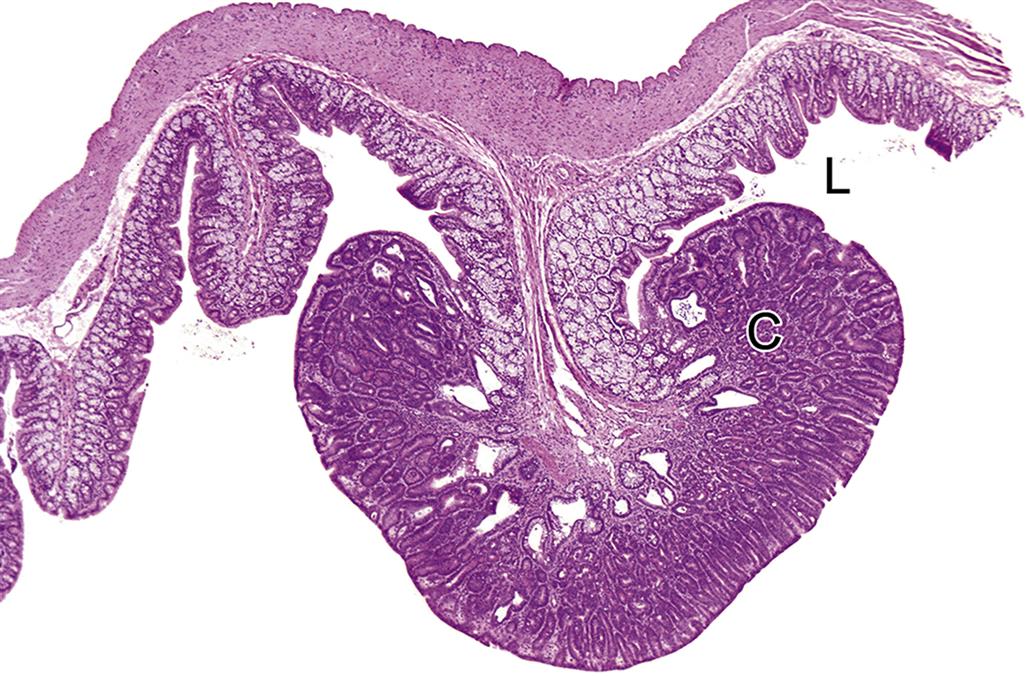
Cecal Enlargement
Cecal enlargement is a response in several rodent species to various compounds and food additives in the digesta. These materials include antibiotics, modified starches, polyols (sorbitol and mannitol), some fibers, and lactose. Cecal enlargement, for example, is associated with increased death losses in rats fed raw potato starch. Enlargement of rodent ceca has been interpreted as both a toxic and an adaptive phenomenon.
Compounds that are poorly absorbed and are osmotically active are frequently associated with cecal enlargement. The mechanism for the distension has been proposed to be the attraction of fluid into the lumen. However, when the luminal contents are removed, tissue weights remain elevated, so other mechanisms are also operative. Other processes involved in cecal enlargement and dilatation include mucosal hypertrophy and hyperplasia. This morphologic response is associated with functional changes that lead to soft stools, diarrhea, and increased large-bowel mucosal permeability. These functional alterations are likely to be mediated by the increased osmotic activity of the cecal contents. Morphological changes probably represent an adaptational process, since the changes are reversible when the diets are returned to normal.
Large intestinal enlargement is a common change observed with incompletely digested and poorly absorbed substances that are subjected to microbial metabolism in the cecum and colon. The increased microbial metabolism leads to an increase in osmotically active material, and results in soft stools and cecal distension. One functional change in rats fed sugar alcohols and lactose is increased absorption of calcium. Sequelae to this process are increased calcium excretion in the urine and nephrocalcinosis.
Regenerative Response
Repair vs Regeneration
Tissue response to injury maybe divided into two classes: repair and regeneration. Repair refers to the physiologic alteration of an organ after injury for the purpose of restoring stability without exact replacement of lost or damaged tissue. Repair often results in fibrosis. In contrast, regeneration signifies the replacement of lost or damaged tissue with a precise duplicate, such that both morphology and functionality are completely restored. Conditions required for regeneration are sometimes in direct contrast to those favoring repair. For example, a prolonged inflammatory response needed for repair will not allow for regenerated tissue to form due to the granulation bed that forms at the injured site. Likewise, contraction at the site during repair will further inhibit replacement with duplicate tissue. Many tissues are not overtly capable of regeneration, so the only response to injury is repair.
Injuries involving the inner mucosal layer of the small intestine can result from chemical and radiation exposure during treatment for cancer, changes in gut microbiology in response to antibiotics, and inflammation or necrosis due to parasitic or autoimmune disease. In contrast, blunt force injuries to the small intestine, due to automobile accidents, stabbings, or gunshot wounds, often compromise the entire structure of the organ, involving both the inner and outer tissue layers. Since the inner mucosal layer is crucial for the nutrient absorption function of the small intestine, the following subsections will provide a broad overview of some of the key components involved in repair and regeneration of the epithelial cells lining this organ.
Stem Cells
It is reasonable to assume that any injury to the small intestine will require repopulation of the injured area by one or more cell types. Adult stem cells are multipotent and can differentiate into a limited number of cell types. These are capable of maintaining, generating, and replacing terminally differentiated cells within their own specific tissue in response to physiological cell turnover or tissue injury. The multipotent properties of adult stem cells makes them easier to coax into replacing lost or damaged adult tissue with exact copies of defective cells, thereby reconstituting original function. Pluripotent stem cells from embryonic sources require more controls during the differentiation process, to reduce the occurrence of neoplastic transformation.
Stem cells may differentiate into fibroblasts and myofibroblasts in response to connective tissue growth factor (CCN2) and platelet-derived growth factor, respectively, thus contributing to tissue repair of GI epithelial layers. Multipotent adult stem cells may ameliorate colitis by exerting an antiinflammatory effect in the affected tissue by reducing expression of cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), and COX-2.
Activation and proliferation of stem cell reservoirs within the crypts of the intestine are modulated by Wnt signaling. Proliferation appears to be dependent upon activation of nuclear β-catenin/T-cell factor transcriptional activity. Expression of Ephrin B receptors and ligands, critical for establishing the migratory path from the crypt to the villus, is modulated by the β-catenin/T-cell factor transcriptional complex. Without Wnt signaling, β-catenin, needed by resident epithelial cells for regulating growth and adhesion between cells, becomes targeted for destruction, thereby contributing to fibrosis. Transcription factor 3 (TCF3, also known as E2A immunoglobulin enhancer-binding factors E12/E47) is another TCF regulated by Wnt signaling. TCF3 can repress expression of the embryonic stem cell gene, nanog, potentially downregulating stem cell pluripotency and self-renewal.
Signaling Pathways
In addition to Wnt, which has been discussed above, bone morphogenic protein (BMP) signaling also plays an important role in regulating intestinal development and epithelial homeostasis in normal, nondiseased tissue. BMPs generally function as a negative regulator of cell proliferation in the small intestinal crypts. In so doing, they effectively act as a brake to intestinal epithelial cell regeneration in normal gut. A BMP antagonist, called noggin (NOG), is expressed in the submucosal region adjacent to the crypts, providing a feedback mechanism for proliferation control. NOG binding to BMP receptors removes the negative effect of BMP, resulting in an increase in intestinal stem cell proliferation, thereby removing the brake to tissue regeneration. Under normal circumstances, BMP inhibits intestinal fibrosis by downregulating TNF-α and by inhibiting TGF-β-mediated epithelial-to-mesenchymal transition. Without such BMP inhibition, activated fibroblasts would be generated, which are a key component in tissue repair and scarring.
Hormones
Endocrine cells can be found throughout the mucosa of the GI system, in particular gastric glands and crypts of the cranial small intestine (duodenum and jejunum). Peptides released by these cells not only regulate secretion, absorption, digestion, and motility but also affect the pathogenesis of several GI diseases, such as cancer. The hormones discussed below have been implicated in regulating mucosal cell growth and regenerative responses either positively or negatively.
Gastrin-Releasing Peptide and Gastrin
Gastrin-releasing peptide (GRP) functions primarily as a stimulator of gastrin release and subsequent gastric acid secretion. Gastrin also stimulates gastrointestinal mucosal cell growth in the small intestine. Recent evidence suggests that gastrin can modulate the function of cells involved in immune and inflammatory responses. This proinflammatory function must be downregulated during repair of the intestinal epithelium resulting from colitis, perhaps by affecting the activity of another proinflammatory factor, TNF-α.
Ghrelin
Ghrelin is produced largely in the stomach, and to a lesser degree in the small intestine and colon. This peptide plays an important role in regulating food intake, gastric emptying, and gastric acid secretion. Reports in the recent literature support a role for ghrelin in suppressing inflammatory and apoptotic pathways activated by GI injury, enhancing intestinal motility, and promoting mucosal epithelial cell proliferation. Mechanistically, ghrelin has been shown to inhibit cellular apoptosis by regulating the ratio of the apoptotic regulator protein Bcl2 with the proapoptotic protein BAX.
Somatostatin
Somatostatin (SST) is arguably the master controller of all GI hormones, capable of inhibiting gastric acid secretion, motility, and mucosal cell growth. SST accomplishes this feat either directly via interaction with specific receptors, or indirectly by antagonizing the function of other trophic hormones, such as gastrin. It has been demonstrated that radiation injury of intestinal mucosal cells can be alleviated by administration of an SST analog. This may imply that SST functions in a “proliferative capacity” by inhibiting apoptosis to enable maintenance of the surface epithelium.
Growth Factors
Epidermal Growth Factor
EGF consists of a family of peptides, of which EGF and TGF-α are two of the most studied members. EGF is produced by epithelial cells located in the submucosal (Brunner’s) glands of the duodenum, while TGF-α is produced by epithelial cells lining the small intestine. Both EGF and TGF-α are mitogens stimulating cell division in multiple cell types within the gastrointestinal tract. Hence these growth factors enhance mucosal healing after injury by increasing cell proliferation and stimulating angiogenesis.
Fibroblast Growth Factors
The family of fibroblast growth factors (FGFs) also regulates growth and differentiation of intestinal epithelial cells, in addition to the proliferation of stem cells during the process of tissue regeneration. Acting as a mitogen, FGFs promote mucosal and epithelial cell proliferation in the small intestine in addition to mediating angiogenesis. FGFs stimulate fibroblast and endothelial cells, two key building blocks for angiogenesis and granulation tissue development. Together, angiogenesis and granulation tissue increase blood supply to the area and fill the injured site with well-oxygenated cell mass during the tissue repair process.
Insulin-Like Growth Factors
There are two members of the insulin-like growth factor (IGF) family, designated IGF-I and IGF-II. Following interaction with their receptors, these growth factors upregulate epithelial cell and fibroblast proliferation while downregulating apoptosis. During the tissue repair process, IGF also stimulates intestinal epithelial cell migration so that cells can quickly be redistributed over a damaged area. In so doing, the barrier between the intestinal lumen and submucosa is rapidly restored while epithelial cell proliferation is being initiated to more completely repair the damaged area with new cells.
Transforming Growth Factor-Beta
The TGF-β family functions to inhibit the growth of GI mucosal cells. Following epithelial cell injury in the small and large intestine, increased levels of this growth factor can be found in the mucosa, resulting in an inhibition of epithelial cell proliferation. There appears to be a synergistic relationship between TGF-β and GRP, in that together they negatively regulate intestinal epithelial cell proliferation and differentiation better than each does individually. Similar to IGF, TGF-β also stimulates intestinal epithelial cell migration to restore a barrier quickly during tissue repair.
Mechanisms of Gastrointestinal Toxicity
Basic functions of the GI tract include acting as a barrier, digesting and metabolizing ingested material, secreting enzymes, and absorbing needed nutrients (including water). Any impairment of these basic functions will result in functional or structural alterations and disease (Table 15.10). Because the GI tract is involved in transport of nutrients, it is especially prone to injury by processes that alter absorptive functions. Additional mechanisms that can cause severe GI pathology include reduced blood supply or hypoxia, acid build-up with damage to the mucosal barrier, hypersensitivity reactions, and genotoxicity, which potentially leads to neoplasia. At a cellular level, injury to the plasma membrane and mitochondria of mucosal epithelial cells represents an irreversible loss of cellular viability from which there is little likelihood of return. At the tissue level, the difference between development of a superficial or a deep mucosal lesion depends on the extent of involvement of subepithelial capillaries. In this section, general toxicologic mechanisms are discussed, and several model toxicants are used to illustrate these processes.
Intestinal Barrier Function
Most ingested toxins enter the systemic circulation through the small intestine, either by passing through the enterocytes or by passive paracellular diffusion. Contents of the gut are mainly in an aqueous phase, with a 35-μm unstirred water layer next to the mucous layer of the surface epithelial cell. The ability of a xenobiotic to traverse the mucosal barrier depends upon its solubility in water for diffusion through the unstirred water layer, its size and charge for paracellular flow, and its lipid solubility for transcellular diffusion. Large and polar molecules pass poorly through epithelial tight junctions unless the epithelial barrier is disrupted (as with high doses of ethanol). Small electroneutral molecules pass easily around the epithelial cells and into the portal circulation, but polar molecules cannot pass through the lipid barrier of the cell. Weak acids or bases are in equilibrium, with both ionized and nonionized states present, making them simultaneously soluble in water and lipids. The nonionized molecules can diffuse through the membrane into the enterocyte. Once in the enterocyte, a xenobiotic maybe pumped out of the cell by a multipurpose transporter (e.g., Pgp) on the luminal surface of villus cells, metabolized by various enzyme systems within the cell to either a toxic or nontoxic metabolite, or transferred into the portal blood or lymph. Intestinal metabolism may play an important role for some medications, including lidocaine and cyclosporine, or maybe the site of drug interactions. Some compounds also have direct toxic effects on the enterocyte without being systemically absorbed, such as toxins produced by the blue green algae Microcystis.
Substrates that do not have specific transporters are absorbed passively around epithelial cells. Tight junctions are very permeable in the proximal intestine, becoming less permeable in the ileum. Electrolytes are absorbed either by paracellular bulk flow or by electrogenic transport and exchange processes, depending upon the permeability of the tight junctions of the specific segment of the intestine. Net secretion of fluid is quite large; for example, in the human there is a net secretion of approximately 7 L of fluid into the jejunum, originating from biliary, pancreatic, and intestinal secretions. Fluid secretion via intestinal tissue is due to the paracellular flow of water drawn into the lumen by the high osmotic load of ingesta. This fluid is then absorbed in the ileum and colon as nutrients and electrolytes are absorbed against a concentration gradient. A variety of ion pumps, exchangers, and channels are involved in the electrogenic transport of electrolytes in the distal small and large intestine. As electrolytes are transported out of the gut, water is also reabsorbed passively to maintain electrochemical gradients. When nutrients or electrolytes are not absorbed, there is an increase in luminal liquid volume that results in diarrhea.
Intestinal Malabsorption
A number of transport pathways exist in the GI tract to carry materials across the mucosal epithelium. These mechanisms include active transport, facilitated diffusion or solvent drag, passive diffusion, pinocytosis, and phagocytosis. Most nutrients are absorbed by active transport mechanisms, in contrast to most toxicants, which are transported by a passive diffusion process. Consequently, greater lipid solubility of a toxicant will enhance absorption, smaller molecules will diffuse more rapidly, and the nonionized forms of acids and bases will be absorbed more rapidly than the ionized forms. A significant exception to this generalization includes the active transport of inorganic ions, as typified by calcium carrier mechanisms present in the GI tract.
Malabsorption results from alterations in epithelial transport mechanisms, reduction in surface area (e.g., villus blunting from antimitotic agents), or the binding of nutrients or compounds to unabsorbed intestinal contents (e.g., modified bile salt absorption by cholestyramine) (Table 15.11). Reduced nutrient absorption can be mediated by various toxicants, including heavy metals and plant extracts. Cadmium interferes with or inhibits the absorption of calcium and alters digestion of protein and fat. Tobacco-leaf extracts reduce the activity of the loosely held intestinal brush border enzymes lactase, sucrase, maltase, and alkaline phosphatase. When enzymes involved in the metabolism of complex carbohydrates are damaged, the GI epithelial cells are unable to absorb carbohydrate-derived nutrients. Malabsorption results in malnutrition, vitamin deficiencies, and diarrhea.
Table 15.11
Intestinal Malabsorption Induced by Drugs and Chemical Agentsa
Surface active agents that block fat and vitamin absorption: alcohol, cholestyramine
Antibacterial agents that block fat, protein, electrolyte, and vitamin absorption: kanamycin, neomycin, polymycin
Miscellaneous agents that block fat, vitamin, protein, and carbohydrate absorption: calcium carbonate, clofibrate, colchicine, indomethacin, methotrexate, phenformin, phenytoin, phenolphthalein, quinacrine, sulfasalazine, and triparanol
aModified from Banwell (1979) Environ. Health Perspect. 33, p. 111.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table VI, p. 144, with permission.
Toxic compounds can alter solute transport across or between mucosal epithelial cell membranes. By damaging junctional complexes between enterocytes, interfering with hydrostatic pressure gradients, or causing high luminal osmotic pressure, a toxic compound can contribute to net water loss from the body into the feces and lead to diarrhea. Some bacterial toxins (E. coli toxins and shigatoxin) and laxative compounds act as secretagogues and promote active sodium and water loss into the lumen. Toxic doses of these secretagogues eventually lead to diarrhea.
GI toxicity can also be mediated by an increased absorption of nutrients or toxic compounds. Glycogen content increases in the midgut epithelium of cockroaches fed pyrethrum. Degeneration of fish intestine is observed as a consequence of water and electrolyte transport alterations which occur from exposure to DDT. Increased toxicity of organophosphates in young mice compared with older mice is the result of an increased rate of absorption of the toxic compound.
Particulate materials maybe taken up by pinocytosis (nanometer-sized particles) or phagocytosis. In mice, phagocytosis is limited to particles smaller than 6 μm in diameter. Particulate uptake plays an important role in pathological responses to polyvinyl chloride, metallic iron, and asbestos. The passage of these particles through the protective mucosal epithelium of the GI tract can lead to allergic hypersensitivity reactions or the entry of unmetabolized compounds directly into the lymphatic and blood circulations.
Hypoxia
Hypoxia is a key factor in the pathogenesis of GI mucosal injury. This is typified by the development of mucosal lesions in various types of shock. The degree of mucosal damage in shock is correlated with the extent of reduced GI blood flow. Decreased blood flow and oxygen exchange increases the susceptibility of the mucosa to injury. A local reduction in blood flow can occur with vascular thrombosis; this mechanism is a major process in gastric injury induced by absolute ethanol.
Stomach
Hemorrhagic shock in rats leads to uniform blanching of the glandular mucosa of the stomach and a generalized reduction in blood flow. Small, white, ischemic foci develop on the gastric mucosa, which will ulcerate and bleed as necrotic tissues slough after the restoration of blood pressure or flow. Ischemia predisposes the stomach to HCl-mediated mucosal lesions because blood flow is sufficiently reduced to cause a build-up of hydrogen ions in the tissue. A decrease in local blood flow or an increase in acid back-diffusion can lead to mucosal injury and erosion. This combination of events may cause severe mucosal damage (Figure 15.9).
Arachidonic acid metabolites are inflammatory mediators that can induce gastric damage. Thromboxane A2 (TXA2), formed by platelets, is a potent vasoconstrictor and causes extensive mucosal damage in the presence of topical taurocholate. Platelet aggregation is also promoted by TXA2 and can lead to vascular thrombosis and mucosal infarction. Both mechanisms are active when there is tissue hypoxia, and both are involved in the ulcerogenic effects of TXA2. In contrast, some prostaglandins protect the mucosa from injury. Protective processes mediated through prostaglandins are thought to be increased mucosal blood flow, and therefore an improved supply of oxygen. Modification of blood flow, prostaglandins, and the mucosal barrier, coupled with tissue-damaging bile acids and activated neutrophils, are the basis for gastric ulceration observed with NSAIDs (Table 15.12).
Table 15.12
Chemical Agents and Drugs That Can Induce Gastrointestinal Ulcers
Antiinflammatory agents (steroids and NSAIDs): corticosteroids, phenylbutazone, indomethacin, flunixin, oxyphenbutazone, sulindac, flurbiprofen, tolmetin, ketoprofen, fenoprofen, naproxen, and ibuprofen
Inflammatory mediators: histamine, serotonin
Antihypertensive agents: reserpine
Hormone analogs: gastrin-like compounds
Catecholamines: epinephrine
Antimicrobial agents: polymyxin B
Antimetabolic agents
Sympatholytic agents: priscoline
Antihistamines: dimaprit (H2 blocker)
Amines: ethylamine, cysteamine, and cystamine
Nitriles: propionitrile and butyronitrile
Short-chain alkanes and alkenes
Miscellaneous agents: caffeine, KCl, gold thioglucose, and haloperidol
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table VII, p. 146, with permission.
Intestines
Ulcerative mucosal lesions can develop as a result of impaired villus microcirculation during hypotension. Hypoxia develops as a result of increased mean transit time for blood in the villus vascular loop, which increases the efficiency of the countercurrent exchange mechanism in the villi of the GI mucosa (Figure 15.6). When blood flow is sluggish, the time available for oxygen diffusion back into the blood is increased, resulting in reduced availability of oxygen at the villus tip. Rheological factors such as intravascular aggregation of erythrocytes and platelets can contribute to compromising oxygen transfer, especially when blood flow is already significantly reduced. Hypoxic injury is compounded by epithelial and intraluminal enzymes, such as trypsin, that contribute to mucosal lesion development under these conditions.
Mucosal Barrier Damage and Cytotoxicity
Nonsteroidal Antiinflammatory Drugs
NSAID-induced mucosal damage follows a temporal course of events, starting early with neutrophil-independent toxicity and progressing later to neutrophil-dependent toxicity. The time course of the cascade is approximately 6 hours. Early changes include alterations in mitochondrial oxidative functions and inhibition of COXs. Once absorbed into the mucosa, NSAIDs interact with epithelial cell mitochondria to cause uncoupling of oxidative phosphorylation, leading to energy depletion and decreased mitochondrial enzyme activity. Energy depletion leads to disruption of ATP-dependent epithelial cell junctions, thus increasing intestinal epithelial permeability.
The increase in permeability due to altered epithelial barrier functions reduces protection from hostile gastric and/or duodenal luminal factors such as bile acids, hydrogen ions, and bacteria. The presence of bacteria will attract and activate neutrophils, which increase the ulcerogenicity of NSAIDs. Activated neutrophils attracted to the mucosal microvessels and lamina propria release reactive oxygen species, lysosomal proteases, and leukotriene B4 (LTB4). Myeloperoxidase is a hemoprotein peroxidase released by activated neutrophils into the extracellular medium, where it interacts with hydrogen peroxide (H2O2) to form an enzyme–substrate complex with great oxidizing potential. Activated neutrophils produce large quantities of hypochlorous acid (HOCl), a powerful oxidizing agent, leading to OCl− generation by means of myeloperoxidase-catalyzed oxidation of Cl−. Active oxygen species and lysosomal enzymes cause direct damage to epithelial cells, especially membranes. The LTB4, released by neutrophils, is a powerful chemoattractant for additional neutrophils. In addition, LTB4 causes vasoconstriction of arterioles. As a result of this cascade, GI epithelial cells become targets of attack by bile acids, hydrogen ions, active oxygen, and lysosomal enzymes.
NSAIDs are also directly cytotoxic to the mucosal epithelium through reduction of cytosolic ATP as well as by blocking COX activity and the synthesis of mucosal protective prostaglandins. NSAIDs stimulate membrane-bound sodium ion pumps and alter acid production by the gastric mucosa. Lesions occur after oral or parenteral administration of NSAIDs. The lesions caused by these compounds are erythema, hemorrhage, erosions, and ulceration of the GI mucosa, especially in the stomach and throughout the small intestine. When NSAIDs are administered at toxic levels, the same types of mucosal lesions are present regardless of route of administration or anatomical location.
The two major COX isoforms, COX-1 and COX-2, differ in their sensitivity to inhibition by individual NSAIDs. COX-1 is the constitutive form of the enzyme found in healthy tissues, while COX-2 is an inducible form that can be stimulated by several cytokines and mediators of inflammation. Most NSAIDs inhibit activity of both isoforms of COX. Inhibition of COX-2 maybe associated with most of the beneficial effects of NSAIDs, while inhibition of COX-1 maybe associated with many of their adverse effects. Inhibition of COX by NSAIDs results in two significant toxicological effects: reduction in formation of prostaglandins, and increased formation of leukotrienes. The loss of endogenous prostaglandin protection may then render the stomach prone to damage by other agents that are normally only mild ulcerogens, and leaves lipoxygenase metabolites like hydroperoxyeicosatetraenoic acid and leukotrienes without the counterbalancing effects of endogenous prostaglandins.
PGE2 and prostacyclin are vasodilators, so blockade of PGE2 and prostacyclin synthesis may favor some degree of vasoconstriction and oppose prostaglandin-mediated tonic vasodilation. Increased metabolism of arachidonic acid by the 5-lipoxygenase pathway with concomitant inhibition of COX may contribute to NSAID-induced GI toxicity, since leukotrienes C4 and D4 are vasoconstrictors and LTB4 is a powerful chemoattractant of neutrophils. LTB4-stimulated attraction and activation of neutrophils leads to release of lysosomal enzymes and microvascular occlusion. Cellular damage to epithelial cells is further exacerbated by a reduction in mucosal blood flow, brought about by a combination of vasoconstriction caused by leukotrienes, and occlusion of microvessels by activated neutrophils.
As potent vasoconstrictors, lipoxygenase metabolites indirectly deprive the mucosa of oxygen. This relative hypoxia may then predispose the mucosa to other damaging agents. Support for this pathogenesis is demonstrated by studies in which lipoxygenase and COX coinhibition and antioxidant agents decrease the incidence of gastric ulcers compared with COX-inhibitor-only exposed controls.
Another important process that contributes to damage when these agents are given by an oral route is direct mucosal irritation by the chemical itself. NSAIDs also directly decrease mucus and bicarbonate release, independently of prostaglandin inhibition. The mucous layer and presence of bicarbonate contribute to mucosal protection from endogenously produced acids. Damage to the mucosal barrier also causes intramucosal histamine release by mucosal mast cells, with resultant vascular congestion, edema, and plasma exudation.
Aspirin inhibits COX activity and is rapidly deacetylated to salicylate. Both aspirin and salicylate are toxic toward mucosal epithelial cells but with differing potencies; furthermore, salicylate is toxic to mucosal epithelial cells at different gastric sites. Salicylate also affects mucosal barrier function, reduces cellular ATP, stimulates sodium ion pumps, and increases proton (H+) loss. Thus, NSAIDs like aspirin and salicylate reduce production of protecting substances and increase production of damaging substances.
Species differences exist with regard to NSAID-induced GI toxicity. Dogs are more sensitive than rats, which are more sensitive than monkeys. However, although monkeys do not develop lesions after oral exposure to ibuprofen (300 mg/kg/day), gastric ulcers occur when the same dose is given intravenously. Species differences maybe related to the plasma half-life of the active compound, since the propionic acid NSAID flurbiprofen has a half-life of approximately 40 hours in dogs, 6 hours in rats, and 3 hours in monkeys, which correlates with the relationship to ulcerogenic sensitivity.
Alcohol (Ethanol)
Ethanol, like NSAIDs, causes hemorrhagic erosions in the gastric mucosa. The rate-limiting step in this lesion development is the extent of microvascular damage. Vascular injury is the result of cell membrane injury, mast cell degranulation, leukotriene release, and increased mucosal permeability. As occurs with other mucosal-damaging agents, injured epithelial cells are rapidly replaced if blood flow is maintained and the basement membrane remains intact.
Gastrotoxic effects of alcohols like ethanol are related to their ability to increase cell membrane fluidity. Osmolality and lipid solubility are also involved, but to a lesser extent. Depletion of intracellular glutathione (GSH) has been implicated in alcohol injury to mucosal cells. The levels of GSH decline in proportion to the degree of alcohol injury, while treatment with PGE2 can essentially abolish alcohol injury. N-ethylmaleimide, which causes profound GSH depletion, abrogates prostaglandin-induced protection against alcohol injury.
Chronic administration of alcohol is also associated with enhanced expression of a number of growth factors, including EGF and TGF-α. These growth factors are thought to protect the gastric mucosa against acute injury, and may explain the observation that adaptation of the gastric mucosa to chronic alcohol administration is associated with increased cell proliferation and increased expression of mucosal EGF and TGF-α. The ability of chronic alcohol exposure to lead to hyper-regeneration of the gastric mucosa could be responsible for the suspected carcinogenic effect of alcohol in the stomach, and likely is mediated by peptide growth factors. Generation of acetaldehyde (which can alkylate nucleotides in DNA) by endogenous CYP isozymes, has also been implicated, thus suggesting a potential role of gastric mucosal ADH in deleterious effects of alcohol on GI mucosa.
Alcohol increases the permeability of the mucosa and causes back-diffusion of H+ and a rise in luminal Na+ concentrations. At low alcohol concentrations (10%), mucus synthesis and bicarbonate secretion are inhibited. At higher concentrations (12%–15%), alcohol releases surface mucus, depletes intracellular mucus, and promotes leakage of bicarbonate and electrolytes into the gastric lumen. At concentrations above 20%, the severity of gastric erosions increases with rising concentrations of alcohol. At concentrations above 40%, there is dose-dependent damage to the mucosal blood vasculature.
Steroidal Compounds
Steroids, like NSAIDs, also induce gastric and large intestinal mucosal alterations and damage by altering cytoprotective mechanisms and the mucosal barrier. Long-term or high-dose steroid administration induces gastric ulceration. Dogs given toxic levels of dexamethasone, a phospholipase inhibitor, develop gastric bleeding, erosions, and melena (black, tarry stools due to gastric hemorrhage). These findings indicate that the mechanism for steroid-induced gastric lesions is partially mediated through inhibition of prostaglandin synthesis. Since the prostaglandin synthetase (COX) substrate arachidonic acid is reduced by inhibiting phospholipase activity, the mucosal protection provided by prostaglandins (PGE2) is lost and gastric acid activity proceeds without inhibition. This mechanism of gastric damage is in distinct contrast to that demonstrated by cysteamine, which inhibits somastatin activity but enhances gastrin production, leading to hyperacidity and delayed gastric emptying (due to altered duodenal motility), followed by mucosal damage.
Bile Acids
Bile acids are synthesized from cholesterol, and can damage the GI mucosa. Bile acids are usually ionized and occur in two forms: monomeric and micellar. Of the excreted bile acids, over 97% are reabsorbed in the ileum and returned to the liver via the enterohepatic circulation. The remaining 3% undergo bacterial degradation in the colon and are excreted in the feces or reabsorbed in the colon. GI bacteria deconjugate and desulfate bile salts, leading to the production of toxic and even carcinogenic metabolites. Bile salt malabsorption during certain ileal diseases is implicated in colonic mucosal damage and diarrhea. Bile salts in the stomach break down gastric mucosal permeability and solubilize the outer lipid bilayer of surface epithelium; deoxycholate inhibits active sodium ion transport from mucosa to submucosa. The basic mechanism of mucosal barrier damage is similar for ethanol and deoxycholic acid. Bile salts stimulate colonic epithelial cell proliferation and are capable of acting as tumor promoters in the colon.
Radiomimetic Agents
Radiomimetic compounds result in substantial cytotoxicity in mucosal epithelial cells, and the mitotic mucosal cells of the crypt are at high risk. Ingestion of a trichothecene mycotoxin, T-2 toxin, results in widespread crypt epithelial necrosis and mucosal injury that resembles the effects of radiation exposure (Figure 15.16). Contributing to the mucosal injury is necrosis of proliferating crypt cells. This eventually leads to loss of remaining mucosal epithelium lining the villi as a result of continued cell senescence in the absence of replacement. Consequently, there is collapse of the mucosa, ulceration, hemorrhage, and secondary inflammation along with bacterial invasion. Chemotherapeutic agents such as the fluropyrimidines floxuridine and 5-fluorouracil as well as plant toxins (e.g., ricin) can cause similar lesions.
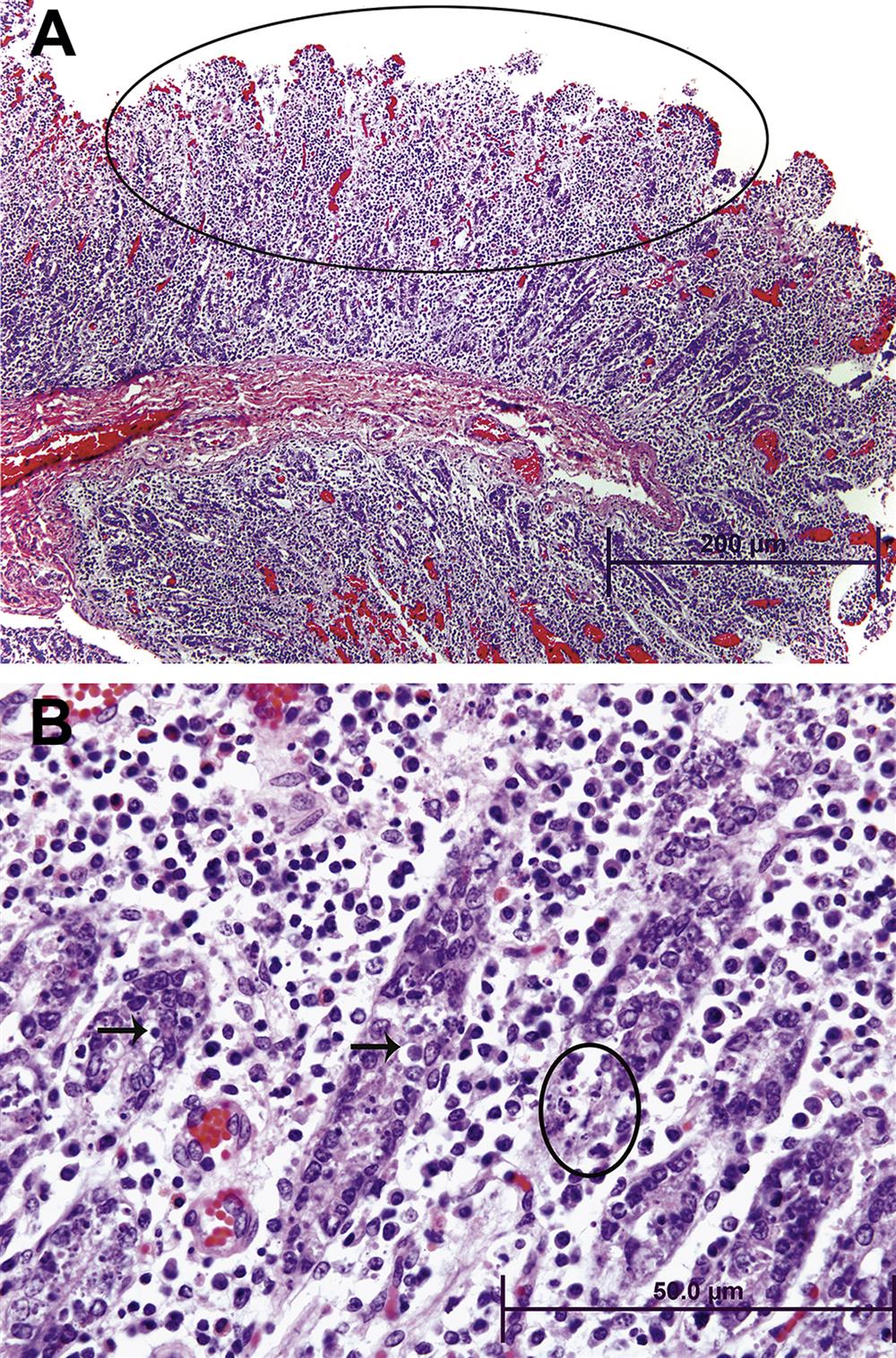
Dioxins
Gastrin, EGF, TGF-α and other endogenous growth factors can stimulate GI crypt cells to proliferate. Mucosal hyperplasia can be associated with ingested chemicals, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and probably other related polychlorinated dioxins. TCDD binding converts the Ah receptor to its activated functional form, which binds to DNA of the CYP1A1 gene and increases the rate of CYP1A1 transcription; this CYP isozyme is an important player in phase I xenobiotic and drug metabolism. The intestinal mucosa contains significant levels of Ah receptors and CYP1A1. TCDD can induce mucosal hyperplasia in some animal species, possibly by exerting a disinhibitory effect on gastrin release, which can then stimulate crypt cell proliferation and result in mucosal hyperplasia. TCDD intoxication also induces a “wasting syndrome” that is characterized by hypophagia and severe loss of weight. Most species given lethal doses of TCDD die within 2 weeks.
Cytotoxic Agents and Heavy Metals
Compounds that react by 1,4 addition at the β-olefinic carbon of some cellular nucleophiles (e.g., ethyl acrylate) are cytotoxic. These compounds are typically biotransformed/detoxified by conjugation to GSH, which is the dominant nonprotein sulfhydryl-containing constituent of epithelial cells that protects them from oxidative damage. Cytotoxicity occurs because GSH is consumed in the biotransformation process for inactivating the compounds. The cell is therefore more susceptible to the oxidation products that are produced during normal metabolism. Ethyl acrylate, for example, causes forestomach damage through depletion of GSH and other sulfhydryl-bearing (cysteine) groups found in thiol-containing peptides.
Cellular damage can also occur by inhibiting oxidative metabolism. Arsenates uncouple oxidative phosphorylation in the mitochondria, possibly by substituting for inorganic phosphate and forming unstable esters. Arsenic is stored in several body sites, one of which is the wall of the GI tract. Inorganic arsenicals cause hyperemia of the GI blood vessels, which, coupled with endothelial cell damage, leads to submucosal hemorrhage. Suppression of epithelial cell proliferation accentuates the damage, and hemorrhagic enteritis develops.
Cadmium causes irritation of GI epithelial cells leading to vomiting, salivation, and diarrhea. Ingestion of ionic inorganic mercury leads to precipitation of mucosal proteins and a corrosive effect on the GI mucosa. Acute inorganic mercury intoxication causes vomiting.
Hypersensitivity
Immune-mediated hypersensitivity mechanisms of toxicity require as a predisposing factor some form of damage to the mucosal barrier. Mucosal damage leads to inadequate clearance of an antigenic or haptenic compound by the mucosal immune system. Although multiple examples of immune-mediated GI toxicity exist, specific antigen models provide the clearest evidence of the interaction between the mucosal barrier and GI tract-associated immune responses. Antigens can enter enterocytes by pinocytosis or by interactions with nutrient transport systems, or can cross the mucosal barrier by paracellular pathways to interact with immune cells in the lamina propria. Antigen-presenting cells release IL-1, which activates T cells to express IL-2 and release a number of cytokines, including TNF-α, and several interferons, including interferon-gamma (IFN-γ). Activated macrophages also release IL-6, which activates lymphocytes; IL-8, which attracts neutrophils; colony-stimulating factors that activate immune cells; and prostaglandins that maintain blood flow. B cells are stimulated by antigens and interleukins to proliferate and differentiate into plasma cells that synthesize and secrete immunoglobulins. Immunoglobulin-E is one of a number of regulators (cytokines, complement 3a) that can activate GI mast cells. Activated mast cells release neurotransmitters (substance P, CGRP), histamine, interleukins, and platelet-activating factor.
These proinflammatory and immune system regulatory factors can induce changes in mucosal transport and GI motility. Increased fluid secretion stimulated by immune mediators maybe involved in regulating stimulatory effects on enteric nerves, with subsequent neural-mediated activation of mucosal secretory mechanisms. Secretory products of mast cells may act both directly on muscle fibers and indirectly by means of enteric neurons to increase contractile activity of GI smooth muscle. Cytokines released from immune and epithelial cells during the GI tract immune response may affect mucosal blood flow, induce a chronic inflammatory response, and/or promote generation of reactive oxygen species.
Experimental hypersensitivity in the GI tract is best exemplified by using ethanol to break down the mucosal barrier and increase permeability toward luminal antigens. By administering trinitrobenzenesulfonic acid (TNBS, which acts as a hapten) after ethanol preadministration, a severe transmural granulomatous inflammation (a model of ulcerative colitis) develops in the distal colon of mice (and rats). The inflammatory response is characterized by mucosal and submucosal infiltrations of neutrophils, macrophages, Langhans-type multinucleated giant cells, lymphocytes (T>B), and mast cells, and represents an example of delayed (Type IV) hypersensitivity. Such immune-mediated inflammatory responses lead to severe colonic ulceration. Once an animal is sensitized to an antigenic compound, an immune reaction can be generated on subsequent hapten exposures without first causing damage to the mucosal barrier. Such “intact barrier” reactions are likely the result of hapten transport through the barrier by mucosal epithelial cells or leukocytes that engage in transepithelial migration through the mucosa. How such low-molecular-weight luminal antigens gain access to the intestinal lumen across the epithelial barrier remains unclear. One mechanism of action may involve paracellular diffusion across pores in tight junctions connecting epithelial cells.
Acetylcholinesterase Inhibitors
Acetylcholinesterase is an enzyme normally responsible for inactivation of the excitatory neurotransmitter, acetylcholine, at synaptic and neuroeffector endings of cholinergic motor and secretomotor neurons in the enteric (autonomic) nervous system. Inhibition of enzyme activity allows accumulation of acetylcholine, leading to increased motor activity in the GI tract via stimulation of smooth muscle M3 muscarinic receptors. The accumulated acetylcholine also acts at M1 and M3 muscarinic receptors in other digestive tract domains to increase salivary, gastric, pancreatic, and intestinal secretions. Extensive inhibition of acetylcholinesterase leads to the secretion of large volumes of fluid and electrolytes into the lumen of the intestine, which results in profuse, watery diarrhea. Anticholinergic drugs such as neostigmine, edrophonium, and pyridostigmine; organophosphate insecticides that include parathion, malathion, and paraoxon; and toxic nerve gases including tabun, sarin, and soman all are capable of causing severe diarrhea and death as a result of reversible or irreversible inhibition of acetylcholinesterase.
Microfloral Effects
Administration of antibiotics has a profound effect on the colonic and fecal flora, depending upon the specific antimicrobial activity of the agent involved, the route of administration, and the local luminal concentration of the drug. A marked reduction in the concentration of intestinal bacteria can be achieved with oral antibiotics, although this effect is usually short-lived. The effect of antibiotics in reducing the bacterial concentrations leaves a void in the bacterial ecosystem that can be filled by pathogenic bacteria. For example, a toxin-producing anaerobic bacterium, Clostridium difficile, colonizes the large intestine and produces pseudomembranous colitis, usually after prolonged treatment with antibiotics. Clindamycin and ampicillin are most frequently implicated, but virtually any antibiotic can cause this syndrome. The organism elaborates protein toxins that cause ulceration and necrosis of the intestinal mucosa.
The plethora of gut microbiota is mostly beneficial to the host by virtue of the various symbiotic physiological associations between the microflora and the host. However, this association can also be detrimental to the host under conditions in which gut microbial homeostasis is disturbed, such as in immunodeficient states, after exposure to antibiotics, toxins/carcinogens, or copathogens, or after mechanical damage to the GI tract (especially the mucosa). Subtle but important differences in microfloral composition among individuals may determine the outcome and severity of many pathological conditions and the host’s subsequent response to therapy. Increasingly, the GI microflora is thought to be an important determinant in the pathogenesis of many human and similar animal conditions such as inflammatory bowel disease (IBD), celiac disease, type 1 (insulin-dependent) diabetes, obesity, cardiovascular disease, atherosclerosis, autoimmune disease (rheumatic disorders), allergy, cancer, and some neurological and psychiatric diseases and viral diseases.
In IBD (e.g., Crohn’s disease and ulcerative colitis) of humans and similar experimentally induced conditions in laboratory animals, the disruption of regulatory T-cell functions and associated abnormal mucosal immune (T-cell) responses to normal intestinal commensal bacterial flora are considered as key elements in sustaining chronic immune-mediated intestinal inflammation and injury. Genetically manipulated rodents such as IL-10 knockout or IL2 knockout mice are common models of chronic intestinal inflammation. Interestingly, under germ-free conditions these mice do not develop chronic colitis, highlighting the vital role of GI microflora in aggravating immune-mediated GI injury.
The intestinal microbiota are vital for GI physiology, including the development and functionality of the gut–brain axis, a bidirectional communication nexus composed of neural, immunological and endocrine mechanisms that aid the brain in monitoring and modulation of GI function. As a result, the gut microbiota is now increasingly being explored for its roles in some autoimmune neurological and demyelinating diseases like multiple sclerosis, Parkinson’s disease, and autism.
Carcinogenicity
Stomach
Naturally occurring tumors of the forestomach are rare in rats and mice (1%), although hamsters can have an incidence as high as 12%. Many agents are capable of inducing or modulating forestomach neoplasia in laboratory animals. For induction of carcinogenic activity, nongenotoxic carcinogens must be in contact with the epithelium of the forestomach for extended periods of time. The absence of a forestomach in humans complicates translational decisions when conducting risk assessments for rodent forestomach neoplasms.
Morphologically, both genotoxic and nongenotoxic agents lead to dysplastic areas of the forestomach (Table 15.13). However, early lesions induced by the prototypical forestomach nongenotoxic carcinogen, butylated hydroxyanisole, are reversible, whereas those induced by genotoxic agents are irreversible. Epithelial dysplasia and metaplasia with glandular distortion is a consistent feature of chemically induced precancerous lesions in rodents. The metaplastic process is also associated with changes in epithelial cell enzymes (alkaline phosphatase, β-glucuronidase) and glycoprotein (neutral and acid mucopolysaccharides) content. Glandular atrophy occurs near neoplastic sites as a result of compression and expansion of the adjacent neoplastic process. In humans, “intestinalization” should be considered a precancerous condition if it is part of a longstanding chronic process; it is not established if the same criteria exist in laboratory animals. The intestinalization process is characterized by gastric-gland neck-region elongation. These regions are replaced by a metaplastic mucosa composed of goblet cells and tall columnar absorptive-type cells of the intestine.
Table 15.13
Compounds that Induce Forestomach Neoplasia in Rodentsa
| Chemical | Class | Species affected |
| N-Ethyl-N-nitrosourea | Nitroso | Rat |
| Butylated hydroxyanisole (BHA) | Aliphatic/aromatic hydrocarbon | Rat, hamster |
| 8-Nitroquinoline | Nitro | Rat |
| Benzo[a′]pyrene | Polycyclic/aromatic hydrocarbon | Hamster, mouse |
| Allyl chloride | Halogenated hydrocarbon | Mouse |
| Sodium saccharin | Miscellaneous | Rat |
aNo genotoxic properties have been demonstrated for BHA, allyl chloride, or sodium saccharin.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table VIII, p. 152, with permission.
Intestines
Spontaneous intestinal tumors in laboratory rodents are rare. However, the high incidence of colon cancer in humans nonetheless has led to the development of animal models that utilize chemical carcinogens to initiate colon tumors (Table 15.14). Chemically induced tumors of the colon are polypoid or sessile. The more dangerous tumors are the sessile variants, which are usually mucinous and can progress to malignancies that are characterized by local invasion, metastasis to mesenteric lymph nodes, lung, or liver, and intussusception.
Table 15.14
Experimental Compounds for Induction of Colon Cancera
| Chemical class | Compound |
| Cholanthrenes | |
| Aromatic amines | |
| Hydrazine derivatives | |
| Alkylnitrosamides | |
| Miscellaneous agents |

aModified from Maskens (1983).
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table IX, p. 152, with permission.
Several aromatic amines induce intestinal cancers in laboratory animals through genotoxic processes (Table 15.15). However, extensive metabolism is generally required before many of these chemicals become carcinogenic. Target specificity is associated with chemical structure and animal species. Many nitrosamines induce tumors of small and large intestine in rats, hamsters, or guinea pigs. Additionally, the rat esophagus is sensitive to the carcinogenic effects of some nitrosamines. The organ specificity for many nitrosamines may relate to the affinity of these compounds for enterocyte or nonenterocyte receptors or alternatively site-specific cellular biotransformation pathways (Figure 15.17). Duration of mucosal exposure to the chemical is also critical. Azoxymethane is an alkylating agent that only methylates DNA in colonic epithelium of rats and hamsters, which likely partially accounts for the site-specific carcinogenic activity of this compound. In the large intestine of the rat, tumors induced by certain genotoxic carcinogens (e.g., dimethylhydrazine) are associated with lymphoid aggregates, with at least two distinct neoplastic processes occurring in the colonic mucosa. If the target epithelial cell is not associated with a lymphoid aggregate, polypoid adenocarcinomas and adenomas develop; if the target cell is near lymphoid tissue, sessile tumors develop. The reason for this dichotomous lesion pattern is not understood.
Table 15.15
Compounds That Induce Intestinal Neoplasia in Rats
| Agent | Duodenum | Jejunum/ileum | Colon |
| Azoxymethane | − | − | + |
| Ethylazoxymethane | − | + | + |
| Nitroso-hydroxypropyloxopropylamine | − | − | + |
| bis-Oxopropylamine | − | − | + |
| bis-2-Hydroxopropylamine | − | − | + |
| Ethylureaa | + | + | + |
| Hydroxyethylureaa | + | + | + |
| Methoxyethylureaa | + | − | + |
| Phenylethylureaa | + | + | − |
| Allylureaa | − | + | + |
| Butylureaa | − | − | + |
| Amylureaa | − | + | + |
| Hexylureaa | − | − | + |
| 3-Hydroxypropylureaa | + | + | + |
| Diethylureaa | + | + | + |
| Ethylmethylureaa | − | + | + |
| Ethyl-hydroxyethylureaa | − | − | + |
| Hydroxyethyl-ethylureaa | − | − | + |

aThe proportion of animals that will develop tumors of the large and small intestine after exposure to the nitrosourea compounds ranges from 10% to 50%; females have the lowest and males the highest frequency of lesions. The median time to death ranges from 25 to 70 weeks.
Table adapted from Handbook of Toxicologic Pathology, second ed. W. M. Haschek, C. G. Rousseaux and M. A. Wallig, eds. (2002) Academic Press, Table X, p. 153, with permission.
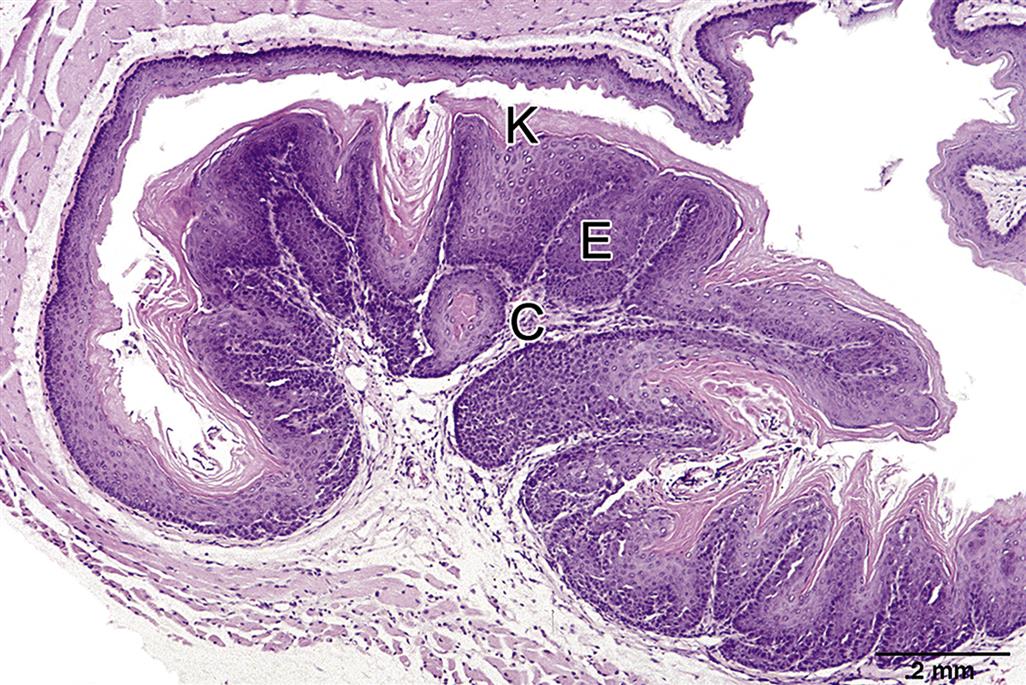
Organ-specificity of cancer development in the GI tract also relates to the sites of specific genetic mutations. An example is the dominant mutation that occurs in the germline of mutant, APCmin/+ mice, which predisposes the animals to multiple intestinal tumors. The propensity for tumor development is dependent on a single allele, and tumors develop in the duodenum, ileum, and colon. Since all cells of these mice carry the mutation and tumors occur only in the intestinal tract, somatic events as well as genetic predisposition are needed for neoplasia to develop. Mutation of this gene may involve loss of function at a genetic site important for normal intestinal development, gain of function in a gene that has an unknown activity, or a complex interaction of both gene suppression and activation.
Genotoxic carcinogen-induced changes in rat colonic epithelium are similar to those observed in spontaneously developing colorectal cancer in humans. In rats, sulfomucins are the primary glycoprotein of the normal colonic epithelium. Shortly after treatment with azoxymethane and N-methyl-N-nitro-N-nitrosoguanidine, cryptal epithelium mucus changes to express primarily sialomucins. Normal intestinal biopsies are characterized by the predominance of sulfomucins. These features support the de novo histogenesis of colon carcinoma.
Aberrant crypt foci (ACF), which represent individual glands or clusters of glands lined by hypertrophic and hyperplastic epithelial cells, are present in the carcinogen-treated rodent colon. The two most common colon-specific carcinogens, azoxymethane and DMH, have been used in rats and mice to induce ACF. Aberrant crypts are observed topographically on whole mounts of colonic mucosal surfaces stained with methylene blue, where these foci are easily distinguishable from the surrounding normal crypts because they take up increased amounts of blue stain.
ACF are purported to be preneoplastic lesions. This is supported by a number of studies into the biology of ACF. A carcinogenic dose of azoxymethane or 1,2-dimethythydrazine induces a large number of ACF. A systematic and sequential analysis of the number and growth features of ACF has demonstrated that ACF appear in the colon of rats and mice within 2 weeks after carcinogen injection, and that their number increases with time. At early time points, ACF contain one or two crypts (i.e., crypt multiplicity of 1 or 2); however, as time progresses many of the foci expand clonally and contain several crypts in the foci. ACF display proliferative atypia and dysplasia, a preneoplastic phenotype, and also show biological heterogeneity both among individual ACF and within a focus. Some ACF expand clonally without exhibiting dysplasia, whereas others start exhibiting dysplasia with or without clonal expansion; not all crypts in foci exhibit dysplasia, however. Crypt “budding” (the fission and multiplication of intestinal crypts) is evident in both types of foci. These findings support the hypothesis that dysplasia arises in ACF as a result of clonal selection. A number of genotypic atypias also occur in ACF. These genetic differences are indicated by variable resistance to apoptotic cell death induced by azoxymethane and sometimes by elevated levels of GSH-S-transferase isoforms.
Compounds that are carcinogenic to the GI tract of laboratory animals may act by direct or indirect actions. Direct-acting (genotoxic) carcinogens lead to initiated cells without prior metabolic activation, with subsequent persistence of neoplastic cells that can grow to become morphologically verified tumors. Indirect-acting (nongenotoxic) compounds, requiring biotransformation or additional promotional interactions to incite a carcinogenic response, may result in a prolonged stimulus of proliferation leading to a substantial increase in the number of dividing (stem) cells. Intestinal stem cells maybe identified by expression of the stem cell markers Lgr5 and EphB2. Single Lgr5+ stem cells isolated from intestine are capable of forming intestinal organoids recapitulating the three-dimensional crypt/villus organization in vitro. Expression of Lgr5 and EphB2 has been shown to define a cancer stem cell niche within colorectal tumors, and is predictive of disease relapse in colorectal cancer patients; consequently, a tissue containing such stem cells is more vulnerable to background initiating stimuli. The exact relationships between the genetic alterations and the phenotypic expression of cancer for nongenotoxic carcinogens are incompletely understood. Regardless of mechanism, carcinogenic compounds can act on all tissues of the GI tract.
Gut Microflora and Cancer
The role of gut microflora in the promotion of inflammation-associated cancers, like Helicobacter pylori (Hp)-associated gastric cancer and IBD-associated colorectal cancers, is now an active area of research. In Hp-induced gastric ulcers, gastritis, and its subsequent promotion to gastric cancer, the role of other gastric microflora, at least in experimental models, is now believed to play an important role in the severity of initial gastric lesions and also lesion progression over time. Of note, in the hypergastrinemic transgenic insulin-gastrin mouse model [INS-GAS (where the rat insulin 1 promoter drives overexpression of human gastrin in the pancreas)] of Hp infection, the lack of commensal flora in germ-free mice resulted in a decreased severity of Hp-induced gastritis and delayed the progression to GI intraepithelial neoplasia (GIN) as compared to their infected conventional specific pathogen-free (SPF) counterparts. Interestingly, Hp-infected SPF mice showed a significant increase in the amount of Frimicutes and a decrease in the levels of Bacteroidetes in their stomach as compared to non-Hp-infected SPF mice. This microfloral signature implies a potential role of gut microflora in the copromotion of Hp-associated gastritis and gastric cancer. Intestinal bacteria are capable of degrading dietary components into toxic byproducts with genotoxic, carcinogenic and tumor-promoting activity, and hence are also considered to play a role in the development of colon cancer. Germ-free rats and mice have lowered abilities to activate dietary procarcinogens or chemical tumor initiators like DMH and induce DNA adduct formation as compared to their conventional counterparts, which is indicative of the role that a complex intestinal microbiota may play in carcinogenesis. The microbiota in general is important in bile metabolism and formation of secondary bile acids (which are considered to be possible tumor promoters) and other toxic dietary metabolites like N-nitroso compounds which are associated with an increased risk for colon cancer. Intestinal bacteria, depending upon the species, can cause intestinal tumor promotion or suppression based on their ability to express enzymes such as β-glucuronidase, β-glucosidase, and nitrate- and nitro-reductases. Intestinal Bacteriodes, Eubacteria, and Clostridia are associated with enhanced carcinogen formation and metabolism, whereas some Lactobacillus spp. and Bifidobacterium spp. have beneficial tumor protective effects.
Summary
For many toxic substances, the GI tract is the first portal of entry. Many of these pass through the GI tract without causing harm, but a significant number directly or indirectly damage the GI tract itself. When considering the potential toxic activity of various agents, one must take into account a number of potential mechanisms and the various cell populations (and their unique physiologies) present in each GI segment. For example, acute toxic effects may result from direct irritation (e.g., strong acids and bases), whereas chronic effects maybe manifest as increased muscular layer thickness (e.g., bulking agents). Delayed effects also maybe expressed years after exposure to the initial ulcerogenic or carcinogenic toxic agents. In addition to an array of tissue responses, the interplay of toxicant-induced functional abnormalities and subsequent morphological alterations can be complex and must be taken into account when assessing the proximate cause of the injury. In this chapter, structural and functional components of the GI tract important for understanding mechanisms involved in the toxicologic pathology were discussed, including the role of gut microflora in the genesis, evolution and resolution of lesions caused by toxic substances. Hence, a thorough knowledge of core structures and functions, basic mechanisms of toxicologic damage and specific responses to toxicologic insult is essential before tackling the complex issues of determining whether the GI lesion observed in a particular situation is a primary, secondary or even tertiary effect of the substance to which the animal (or cohort) was exposed.
Acknowledgments
Timothy A. Bertram
RegenMed (Cayman) Ltd., Grand Cayman, Cayman Islands
John W. Ludlow
JWLudlow Consulting, LLC, Carrboro, NC, United States
Joydeep Basu
RegenMed (Cayman) Ltd., Grand Cayman, Cayman Islands
Sureshkumar Muthupalani
Massachusetts Institute of Technology, Cambridge, MA, United States