Exocrine Pancreas
Matthew A. Wallig1 and John M. Sullivan2, 1University of Illinois at Urbana-Champaign, Urbana, IL, United States, 2Eli Lilly & Company, Indianapolis, IN, United States
Abstract
The exocrine pancreas secretes digestive enzymes into the small intestine and provides a microenvironment for pancreatic islet cells that modulate blood glucose (among other functions). The exocrine pancreas is seldom a prominent target of acute or xenobiotic-induced toxicity in spite of several key observations:
Epidemiologically, pancreatic injury has been associated with both a range of environmental pollutants and consumption of mycotoxins, and has long-standing links to alcoholism and smoking tobacco in humans.
Keywords
Pancreatic acinar cells; pancreatic duct cells; centroacinar cells; pancreatic stellate cells; autophagy; autophagic vacuoles; cholecystokinin (CCK); secretin; amylase; lipase; hyperstimulation; oxidant stress; acute pancreatitis; chronic pancreatitis; necrosis; apoptosis; zymogen; trypsinogen-activated peptide (TAP); carboxypeptidase B activation peptide (CAPAP); trypsin-like immunoreactivity (TLI)
Introduction
The exocrine pancreas is an organ that is seldom prominently involved in toxicity in laboratory and domestic animals. Similarly, it is generally not considered to be a target organ for most xenobiotics and is not routinely considered to be involved in xenobiotic metabolism or bioactivation. Yet, when the pancreas is injured directly in a toxic process affecting islet or exocrine cells, vascular or endothelial injury, or secondarily in concert with damage to the liver, stomach, intestinal tract, or anterior abdomen (via trauma), the consequences can be devastating. Injury can be quite variable, ranging from acute hemorrhagic and necrotizing pancreatitis, with dramatic and rapid clinical onset, to recurrent, subchronic, and low incidence single cell necrosis with slowly evolving subacute to chronic inflammation leading to lobular atrophy and fibrosis that may be clinically silent. The triggers that regulate or modify responses to pancreatic injury have been the object of intensive study over the past decades. Reports link exposure to various drugs or xenobiotics to both acute and chronic pancreatitis. In humans, this is especially true in developing countries, where chronic pancreatitis is endemic. The incidence in these locales parallels environmental pollution (smoke from kerosene stoves, wood fires, and cigarettes); contaminated food supplies (especially by mycotoxins); and consumption of ethanol. These examples highlight the importance of predictive and/or correlative biomarkers for onset of pancreatic injury that ultimately can be predictive of a patient’s prognosis.
Normal Structure and Function of the Exocrine Pancreas
Gross and Microscopic Anatomy
The pancreas can vary widely in location and compactness among species, although microscopic anatomy is remarkably uniform. In most mammals, at least one major component of the pancreas is concentrated along the greater curvature of the stomach near the pylorus and proximal duodenum. This allows the outflow of digestive juices through the major pancreatic duct to empty at or near the same location in the duodenum as the common bile duct. In humans, dogs, cats, ruminants, and horses the pancreas is compact and elongated, and easily distinguished from surrounding tissues. In rodents, major components of the pancreas include a prominent head and body, but also a less distinct and less compact duodenal segment, dispersed within the duodenal mesentery. The main pancreatic duct is generally visible in all species; an accessory duct occurs in humans and multiple ducts may occur in rodents, all entering into the duodenum.
Microscopically, the pancreas is described functionally and morphologically as having two distinct parts, the exocrine pancreas or acini comprising lobules; and the endocrine pancreas or islets of Langerhans that exist within the exocrine parenchyma. The major “functional unit” of the exocrine pancreas is the acinus and the interconnecting ductal network of intra- and interlobular ducts leading to the main pancreatic duct (Figure 16.1). In the acinus, the apical portion of each cell is stained brightly eosinophilic (via hematoxylin and eosin) due to abundant zymogen granules, which contain the precursors (zymogens) of digestive enzymes. Discussion of the pancreas would not be complete without acknowledging the fibrovascular supporting stroma and circulation. The islets are vascularized by afferent arterioles that pass directly through the exocrine pancreas to the islets. The efferent venules exit the islets and bathe the peri-insular exocrine tissue at what has been termed the “endocrine–exocrine interface (EEI).”
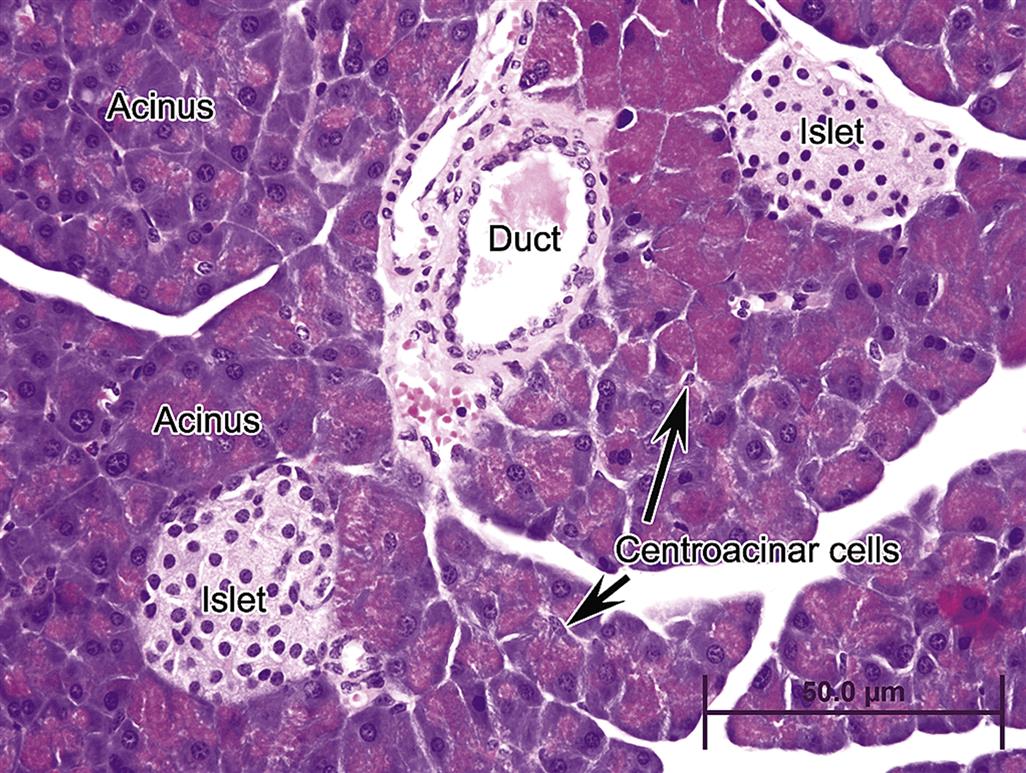
The EEI has been extensively reviewed by Brenneman et al. (2014) and defines the conjunction of islet-acinar portal system (venous outflow from islets) with venous connections of postlobular arterioles that primarily supply the non- or tele-islet acini of the lobule. To date the rat EEI has been characterized the most extensively compared to other species. Capillaries are highly fenestrated in both the exocrine and endocrine pancreas, and the greatest fenestration occurs within pancreatic islets, allowing rapid uptake of endocrine hormones into efferent capillary beds, which then bathe neighboring acini or tele-acinar pancreas as well as distant organs after exiting the pancreas. Digestive zymogens pass from acini along with the ductal secretions directly into the duodenum by arborized pancreatic ducts. Ductular cells (Figure 16.1) form a single layer along a basement membrane beginning at the acinus and are surrounded by minimal to moderate amounts of richly reticulo-vascular stroma depending on the caliber of the duct. Centroacinar cells are aligned with the ductular lumen and acinus.
Physiology
The exocrine pancreas synthesizes and secretes digestive enzymes for lipid, carbohydrates, and protein. Except for amylase, the various proteins within zymogen granules remain inactive until proteolytic cleavage, initiated by enterokinase (enteropeptidase) in the gut lumen. Cholecystokinin (CCK) modulates zymogen release from acini in concert with cholinergic stimulation. Feedback loops involving trypsin and CCK-releasing protein shut down or amplify enzyme release. Secretin similarly modulates submucosal glands in the duodenum and ductal epithelium, triggering the release bicarbonate-rich fluid from ductal cells and gut epithelium. The alkaline pH of ductal secretions (generally 8–9) serves to neutralize the acidity of the chyme entering the duodenum and produce the appropriate near-neutral pH and appropriate ionic balance for maximal activity of chymotrypsin, lipase, and amylase.
Evaluation of Toxicity
Histologic Evaluation
Typically, in most nonclinical toxicity bioassays, a single section of pancreas usually taken as a cross section through the body of the pancreas is examined histopathologically. Evaluation of lipase or amylase measurement is only done if the target has the potential for pancreatic modulation. However, in toxicologic pathology assessment of molecules with known modulation of the acinar or islet pancreas, or in diagnostic cases, correlation of biomarkers with the extent and time course of exocrine injury should include histologic evaluation of the major pancreatic duct and exocrine parenchyma adjacent to the duodenum and surrounding the duodenal papilla, as well as sections through each major component of the pancreas. Particular attention to the peri-insular or peri-islet exocrine tissue should be considered. The peri-islet exocrine pancreas (similar to and extending beyond the EEI) tends to have the highest density of zymogen granules/acinar cell compared to distant exocrine sites. The primate pancreas should be sectioned to include the head, body, and tail. In the dog, sections should include the body of the pancreas (adjacent to the duodenal papilla), the gastric limb (left lobe) and duodenal limb (right lobe). In the rat and mouse, the pancreas is a more diffuse organ that is generally divided into three parts: biliary, duodenal, and gastrosplenic. The gastrosplenic portion along the dorsal aspect of the stomach toward the spleen is the largest section of rat pancreas (analogous to the body of the pancreas in other species). Sectioning oriented perpendicular to the major pancreatic duct is most diagnostic when multiple or step sections are taken. In rodents, islets are heavily distributed along the major pancreatic duct, and recent publications suggest the highest distribution of islets occurs within the tail of the rodent pancreas (Brenneman et al., 2014).
Immunohistochemistry and Histochemistry
In localized mild pancreatic injury, the only evidence of injury may be short-lived increases in either apoptosis as assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling and antiactivated caspase 3 (apoptosis) immunohistochemistry (IHC), or in cell proliferation as evaluated by antibromodeoxyuridine and anti-Ki67 IHC. Routine histochemical stains (periodic acid-Schiff for zymogen granules and Masson’s trichrome for fibroplasia) can be useful in grading subtle injury. In addition, the peri-islet or EEI component of exocrine pancreas should be evaluated for acinar, ductal, and vascular continuity and the presence or absence of inflammation. IHC is useful in identifying proteins specifically expressed by mature acinar and ductal epithelium, and tubular complexes; for example, mature acinar cells uniquely express Reg1, and mature ductal epithelium expresses cytokeratin 19. Similarly, IHC markers may be helpful in identifying specific inflammatory cells such as macrophages and lymphocytes.
In summary, biopsies or postmortem samples, as well as potential immunohistochemical and in situ hybridization techniques are useful in identifying cellular characteristics of affected tissue (ductal and/or acinar), and can guide understanding the pathogenesis of injury and relative significance and mechanism of the biomarker excursions. In most cases the magnitude of exocrine injury can be correlated with the timing of biomarker elevations and biomarker half-life. Postmortem evaluation and excisional biopsies from human surgical cases allow correlations between humans and animals in translating morphological injury with biomarker signals. Definition of chronic injury and regeneration may be aided by IHC identification of unique protein markers in affected cell populations. Importantly, localized or global vascular injury leading to ischemia can be a causative factor that usually drives coagulative necrosis, and/or severe liquifactive injury.
Clinical Chemistry
In acute pancreatitis, previously uninjured or naïve tissue has a greater propensity to elicit rapid elevations in leakage enzymes (biomarkers) based solely on the higher population of acinar epithelial cells proximate to the locus of injury. In cases of prolonged or recurrent pancreatic injury, concentrations of circulating biomarkers may poorly reflect extensive atrophy, fibrosis, or concurrent active inflammation. The most common markers of confined exocrine pancreatic injury are amylase and lipase activity, lipase immunoreactivity, trypsin-like immunoreactivity (TLI), trypsinogen-activated peptide (TAP) concentrations, and carboxypeptidase B activation peptide (CAPAP) concentrations. Micro-RNA shows promise in early and specific prediction of acinar cell injury; however, additional work on precision and translatability is ongoing concurrent shifts in the leukogram, coagulation markers, concentrations of C-reactive protein (CRP), neutrophil elastase activity, phospholipase A2 activity, and concentrations of cytokines interleukin (IL)-6 and IL-8 activity, procalcitonin (a marker of survival in humans), and macrophage chemoattractant proteins (MCP-1 and MCP-2) further define pathogenesis and multiorgan involvement. Similarly, in acute necrotizing pancreatitis, consumption of fibrinogen can suggest fibrin activation, clot formation, and extension beyond the pancreas. A many-fold increase in serum lipase and amylase activities can occur in widespread acute inflammation or acute hemorrhagic pancreatitis, whereas, no or only marginal increases occur in animals or humans with recurring bouts of chronic pancreatitis.
Serum amylase is released from pancreas, liver, small intestine, and parotid salivary gland (man and pig). Typically increases in serum pancreatic amylase are most sensitive for diagnosing exocrine pancreatic injury. Yet total amylase activity has been shown in human diagnostics to be as effective in diagnosing pancreatic injury as the isotype alone. In any case, total amylase is reabsorbed by the renal tubular epithelium and inactivated by the normal nephron, so decreased glomerular filtration or proximal tubular renal injury may increase circulating amylase separately from pancreatic injury by up to 3 times the reference interval for all subspecies of amylase. Therefore, diagnosis of pancreatic injury must be considered in the context of serum lipase elevations, which are most specific. Most serum lipase activity is of pancreatic origin, although hepatocytes and intestinal mucosal cells also contribute to circulating levels. Typically, increases in serum lipase three to fourfold above the reference interval are suggestive of pancreatic injury. Acute injury causes increases in lipase activity within 24 hours, which peaks in 2–5 days. Since lipase concentrations in zymogen granules are approximately 4.5 times those of amylase, recurring injury is more likely to be recognized by leakage of lipase into the circulation. Lipase is less frequently increased by hepatobiliary and intestinal injury or renal failure than is amylase, and is considered more specific for mild localized exocrine injury in dogs. Lipase normally passes through the glomerular filtration barrier and is inactivated by the kidney in the proximal tubules, so hyperlipasemia can occur as a nonspecific finding with severe renal disease. Since pancreatic lipase requires calcium (Ca2+), colipase, and bile salts as cofactors for digestion, elevated lipase with concurrent decreases in serum Ca2+ may suggest accompanying peri-pancreatic injury and abdominal steatitis. Lipase concentrations are usually measured as serum lipase activity; however, measurement of pancreatic lipase immunoreactivity may be more specific for exocrine injury than serum lipase activity alone.
Other markers of pancreatitis consist of TAP as a cleavage product of trypsinogen, and CAPAP as a peptide fragment of procarboxypeptidase B, a large cytosolic protein in acinar cells. Both TAP and CAPAP are released with protease activation. They are uniquely sensitive due to low circulating concentrations in healthy animals. Both pro-peptides are stable but short-lived in circulation; urine presents the preferred matrix for measurement. In a published study in dogs with clinical pancreatitis, urinary TAP, the urinary TAP–creatinine ratio, TLI, and serum lipase activity had the highest specificity for severe versus mild pancreatic necrosis. Procarboxypeptidase B is also present in serum, and with a longer serum half-life is useful in diagnosing necrosis several days after the primary insult is resolved. In combination, urine TAP and CAPAP, serum procarboxypeptidase B, serum amylase, and serum lipase can be diagnostic of the longitudinal nature or chronicity, severity, and progressivity of injury.
TLI measures both circulating trypsinogen and trypsin, which in healthy animals is comprised almost entirely of trypsinogen. TLI is increased in acute exocrine injury; however, rapid excretion by the kidney results in a short circulating half-life. Elevations can occur quickly with necrosis, but serum sampling can easily miss insults of short duration. As such, it is considered an effective marker of exocrine mass, and decreases are highly correlated with exocrine pancreatic insufficiency. Decreased glomerular filtration with impaired renal function can also impair TLI interpretation.
Other Markers of Injury
Whether exocrine injury occurs as activation of digestive enzymes within acinar cells alone, through retrograde efflux of bile acids into the pancreatic ductal system, or due to an acute or subacute ischemic event, biomarkers are defined by increased circulating concentrations of activated pancreatic proteases and their by-products. Concurrent increases in concentrations of acute phase reactants, activated leukocyte proteases, cytokines, chemokines, and traditional clinical chemistry and hematology markers of multiorgan injury express the magnitude and distribution of the lesion. Qualification of a reference interval for each biomarker and the linearity of each assay serve to define both sensitivity (the statistical ability to detect pancreatic injury) and specificity (the statistical ability to discriminate a true negative effect).
Experimental trials in animal species and assessment of diagnostic cases have collectively defined our current knowledge of pancreatic injury. As pancreatic injury develops into peri-pancreatic inflammation and pancreatitis, an influx of activated neutrophils and macrophages releases leukocyte-derived enzymes including neutrophil elastase, phospholipase A2, procalcitonin, peroxidases, and cytokines at the site of injury. Localized lipid peroxidation releases malonyl dialdehyde, and activated macrophages release interleukins (IL-1 and IL-6) and tumor necrosis factor-alpha (TNFα). IL-6 is a potent inducer of acute phase protein synthesis by the liver, and increases in serum concentrations are correlated with disease severity. Increased IL-6 induces acute phase protein synthesis by the liver, CRP being the most prominent. Increases in serum concentrations of CRP, IL-6, procalcitonin, and neutrophil elastase are correlated with progressive abdominal inflammation and are predictive of a poor prognosis.
Animal Models
Studies of normal pancreatic physiology and models of experimental pancreatitis have primarily used rats and dogs although mice are now being used more frequently as transgenic and strains of knock-out mice are developed that exhibit a predisposition to pancreatic injury or pancreatitis. Nevertheless, the rat is still used more often than mouse, because the rat’s larger size makes more pancreatic tissue available for study and permits readier surgical manipulation. Rats also appear to be sensitive to a wider variety of xenobiotics, in particular those that cause hyperstimulation (see later). The guinea pig has also been used in the past in some of the classic in vivo studies of acinar cell biology.
Models of Acute Pancreatitis
Investigators who want to study mechanisms of acute pancreatitis using noninvasive techniques typically use secretagogues that elicit some sort of hyperstimulation, such as supraphysiologic doses of CCK (or CCK analogs such as caerulein). These secretagogues produce many of the early biochemical and morphologic changes observed in acute pancreatitis in humans. Although hyperstimulation models are noninvasive, inexpensive, and relatively easy to implement, the responses can be variable and highly dose and species dependent, unless they are used concurrently with priming or synergizing agents or procedures (e.g., duct ligation or retrograde instillation of bile acids).
Given that overconsumption and abuse of ethanol is the most commonly identified trigger of pancreatitis in humans, numerous ethanol-based animal models of pancreatitis have been proposed and used in rats, cats, and dogs, most notably for studying the impact of oxygen free radical stress on acinar cells and pancreatic microcirculation. Unfortunately, these models tend to be inconsistently reproducible without concomitant hyperstimulation (caerulein), ductal ligation, or retrograde instillation of bile acids into the pancreatic ductal tree.
Models of Chronic Pancreatitis
Noninvasive models of chronic pancreatitis use repeated doses of hyperstimulatory compounds such as caerulein or L-arginine, in essence producing repeated bouts of relapsing acute pancreatitis. This activates pancreatic stellate cells (PSC) to produce the interstitial extracellular matrix (ECM) and elicit fibroplasia. Variations in which additional toxic stimuli such as cyclosporin D, lipopolysaccharide, or ethanol are coadministered enhance or accelerate the generation of chronic pancreatitis. Invasive techniques once used extensively in studying acute and chronic pancreatitis mimicked bile acid reflux and/or increased intraductal pressure, which are prominent features in the pathogenesis of alcoholic and cholelithiasis-induced pancreatic disease. These techniques are less commonly used now that noninvasive techniques can reliably induce pancreatic injury.
A slightly different model utilizes retrograde infusion of the main pancreatic duct at the pancreatobiliary-duodenal opening with bile salts or with bile salts plus activated pancreatic enzymes. While technically less challenging, this model is difficult to reproduce in part because there is poor precision in measuring and maintaining consistent retrograde pressure.
In conclusion, no singular animal model of pancreatitis or pancreatic injury to date meets all the criteria as an optimal model of human disease. A careful assessment of the study goals, for example, elucidation of mechanism, definition of early changes, measurement of impact on regeneration and healing must be made prior to selecting an in vivo model for investigating pancreatic injury by a xenobiotic.
Genetically Engineered Models of Pancreatitis
Enhanced predisposition to enhancement of either acute or chronic pancreatitis has been identified in many knock-out strains, including those with loss of inflammatory mediators. Additional knock-out strains that have an impact on pancreatitis include peroxisome proliferator-activated receptor gamma, metallothionein-1, and phospholipase A2. While the knock-out strains are useful for studying pancreatitis in the context of primary disruption of inflammation, the loss of these genes has a wider impact on other tissues and the body as a whole, making it difficult to ascribe changes specifically to induction of pancreatic injury.
More specific knock-out models, linked to known genetic predispositions to pancreatitis, an array of mutated or knock-out models for the cystic fibrosis transmembrane conductance regulator gene (CFTR), and the pancreatic secretory trypsin inhibitor (Kazal Type I) gene (SPINK1) knock-out models, have allowed for additional targeted research into the pathogenesis of acute recurrent and chronic pancreatitis. The CFTR knock-out mouse has been used to study the role of altered duct secretions in chronic pancreatitis while SPINK1 knock outs have been used to study the role of autophagy and premature intracellular trypsin activation in the evolution of pancreatitis.
Models of Pancreatic Carcinogenesis
Of the various long-term models for chemical induction of pancreatic carcinomas, only two have been extensively characterized and described. The first of these uses azaserine as a pancreatic carcinogen in rats. Azaserine induces microscopically detectable acinar adenocarcinomas in the exocrine pancreas that can become grossly visible nodules 1 mm in diameter or larger.
The second model uses N-nitrosobis (2-oxopropyl) amine (BOP) in the Syrian golden hamster. The dominance of acinar cell tumors in the rat versus the dominance of ductal tumors in the hamster is striking. This disparity in tumor phenotype apparently represents a fundamental difference in the response of pancreatic cells in the two species to initiating agents. Since the vast majority of human pancreatic cancer is ductal in phenotype, the Syrian hamster is the species that has been used more frequently in the past as a model for modeling human pancreatic carcinogenesis. The Syrian hamster, however, has rarely been used in studies not involving pancreatic carcinogenesis, the rat and mouse being the preferred model animals for studying acute and chronic pancreatic injury.
Responses of Exocrine Pancreas to Injury
Response to injury may be localized to individual cells, affect entire lobules, localize uniquely in peri-islet exocrine zones, or involve extensive effacement of the majority of the pancreas, ultimately extending into the anterior abdomen to include peri-pancreatic tissues and organs.
Nonlethal Injury and Autophagy
Beginning at the cellular level, acinar cells may undergo swelling or accumulate lipids in the cytoplasm as a direct or indirect effect of oxidative injury, ischemia, or derangement of cell metabolism. At the ultrastructural level, cell swelling is accompanied by vesiculation of the rough endoplasmic reticulum (RER) and dilatation of RER and SER cisternae, giving the acinar cell a pale, “feathery” appearance histologically. Lipid accumulation and swollen mitochondria may also occur separately or concurrently with free radical injury and oxidative stress. With starvation, acinar cells may fail to synthesize zymogens and lose the apical hyper-eosinophilia of zymogen granules over time; concurrently individual acinar apoptosis can reduce overall pancreatic mass or weight. With overstimulation, acinar cells may “degranulate,” particularly when hypersecretion or supramaximal stimulation by a secretagogue (e.g., caerulein or CCK) is the pathogenesis of the injury. With intense stimulation, inappropriate trafficking of autophagic vacuoles (AV) and zymogen granules to the basolateral membrane occurs and elicits localized autodigestion. Large AV adjacent to the basal lateral portion of the cell are often surrounded by a thin clear halo (Figure 16.2), and in later stages may condense and resemble residual bodies.
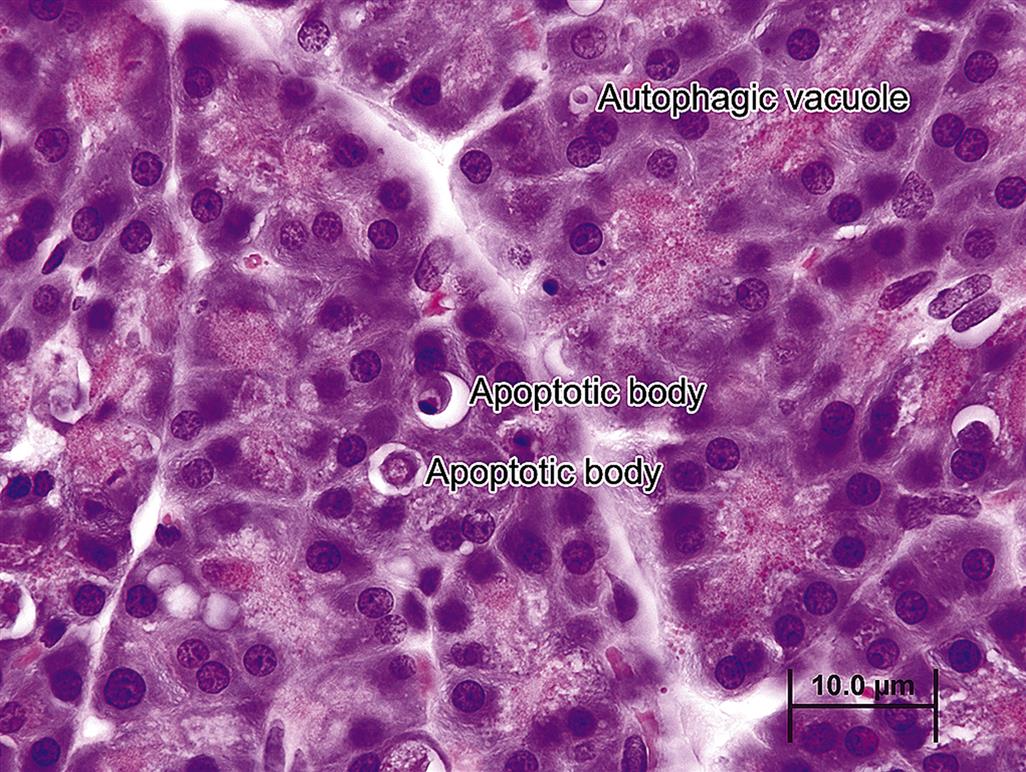
In stable autophagy, cathepsin L released from the lysosome into the newly formed autophagolysosomes degrades any trypsinogen and prevents its activation to trypsin, hence short-circuiting any inappropriate activation of other zymogen proenzymes. However, cathepsin B (another lysosomal hydrolase) can activate trypsin, which could initiate an autodigestive cascade. A balance of cathepsins B and L within the AV during normal autophagy suggests that cathepsin B activity does not predominate; hence, only residual lipids, membranous and granular debris, remain after physiologic or “normal” autophagy. Examples of xenobiotics causing increased AV with dense inclusions in the endoplasmic reticulum (intracisternal granules) have been noted in rats following exposure to puromycin and 1-cyano-2-hydroxy-3-butene (crambene), and other agents that induce acinar cell death. These inclusions represent indigestible cellular remnants from the AV.
Another, somewhat unique process termed “zymophagy” is a modified form of autophagy involving Bcl-1 and VMP1. Zymophagy results in a smaller AV containing only defective zymogen granules and surrounding cytoplasm. It is proposed that this is the process whereby acinar cells regulate their zymogen content. Experimental evidence is accumulating that “normal” autophagy is deregulated in injured acinar cells; dysregulation of zymophagy is at least partially responsible for the inappropriate activation of excessive trypsin, causing acute necrosis. The triggers modulating cellular autophagy versus apoptosis are unclear.
Apoptosis and Acinar Injury
Apoptosis is a more frequent occurrence in the exocrine pancreas than previously supposed, and involves mainly acinar cells, especially in cases of mild injury such as starvation or moderate food restriction, or with persistent low-grade injury. In pigs, for example, the mycotoxins fumonisin B1 and T-2 toxin (vomitoxin) frequently induce vacuolation and “single cell necrosis” consistent with what is now interpreted to be apoptosis (Figure 16.3).
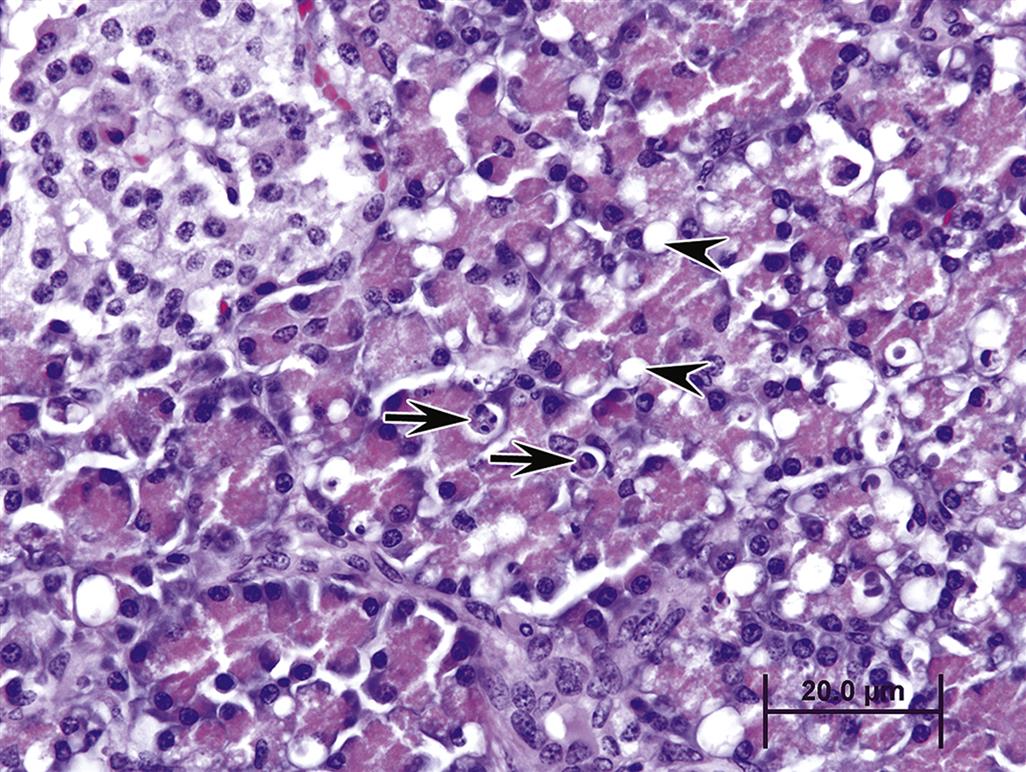
Apoptosis in a sense is the “preferred” response to acinar cell injury since subsequent inflammation is usually minimal or absent, and dead acinar cells are eliminated before they can release their pro-inflammatory contents, which are highly attractive to neutrophils. Acinar cells express high levels of the pro-apoptotic protein Bax (localized in mitochondria) but express very little Bcl-2, which is the prime antiapoptotic protein and regulator of Bax. Acinar cells also express death receptors for FAS-ligand induced apoptosis, including the TNFα receptor and CD95. An inverse relationship between apoptosis and necrosis with acinar cell injury has been described in various experimental models. Stimulation of apoptosis appears to protect against acute necrotizing reactions from cellular toxicants, whereas inhibition of apoptosis leads to a necrosis, severe inflammation, and progression to pancreatitis.
Acinar cell apoptosis is classically manifested in many cases of xenobiotic-induced injury (Table 16.1). Typically only one or two cells per acinus may appear apoptotic at any one time without any neutrophil recruitment. In contrast, engorged macrophages containing apoptotic cells or residual bodies can increase with progressive tissue injury that leads to recruitment of neutrophils and macrophages through cytokine release (Figure 16.4). Even with large-scale apoptosis, inflammation and fibrosis may be minimal and contained locally. Biomarker elevations would not be expected.
Table 16.1
Classification of Pancreatic Alterations with Selected Examples of Causative Agents
| Alteration | Xenobiotic |
| Vacuolation and autophagy (degeneration) | Crambene, ethanol, fumonisin B1, organophosphates, puromycin, T-2 toxin |
| Acinar cell apoptosis | Arginine, crambene, organophosphates, p-chloro-phenylalanine, scorpion venom (Central and South America),T-2 toxin |
| Acinar cell necrosis followed by inflammation | Actinomycin D, arginine, azaserine, crambene, ethioinine, GLP 1 analogs, 4-hydroxyaminoquinoline-1-oxide (4-HAQO), lysine, organophosphates, puromycin, sitagliptin, zinc |
| Acute inflammation with variable acinar cell necrosis | Alcohol, azathioprine, L-asparaginase, chlorthalidone, chlorothiazide, furosemide, 6-mercaptopurine, methyldopa, oral contraceptives, pentamidine, riftampicin, sulindac, tetracycline, thiazide diuretics, valproic acid |
| Chronic interstitial inflammation with fibrosis | Ethanol, dibutyl tin |
| Duct cell degeneration/necrosis | Dibutyl tin, zinc |
| Neoplasia (experimental animals) | |
Well characterized |
Azaserine, 7,12-dimethylbenz[α]anthracene, 4-hydroxyaminoquinoline-1-oxide (rat); N-nitroso (2-hydroxypropyl) (2-oxypropyl) amine, N-nitrosobis (2-hydroxypropyl) amine (rat and hamster), N-nitrosobis (2-oxypropyl) amine (hamster) |
| Weak or sporadic association | Aflatoxin B1 (monkey); colfibrate, nafenopin, nitrofen, N-nitrosodimethylamine (rat); Nδ-(N-methyl-N-nitrosocarbamoyl)-L-ornithine (rat, hamster); N-nitroso-2,6-dimethylmorpholine, N-nitrosomethyl (2-oxopropyl) amine (hamster); N-methyl-N-nitrosourea (guinea pig, mouse), N-ethyl-N′-nitro-N-nitrosoguanidine (dog) |
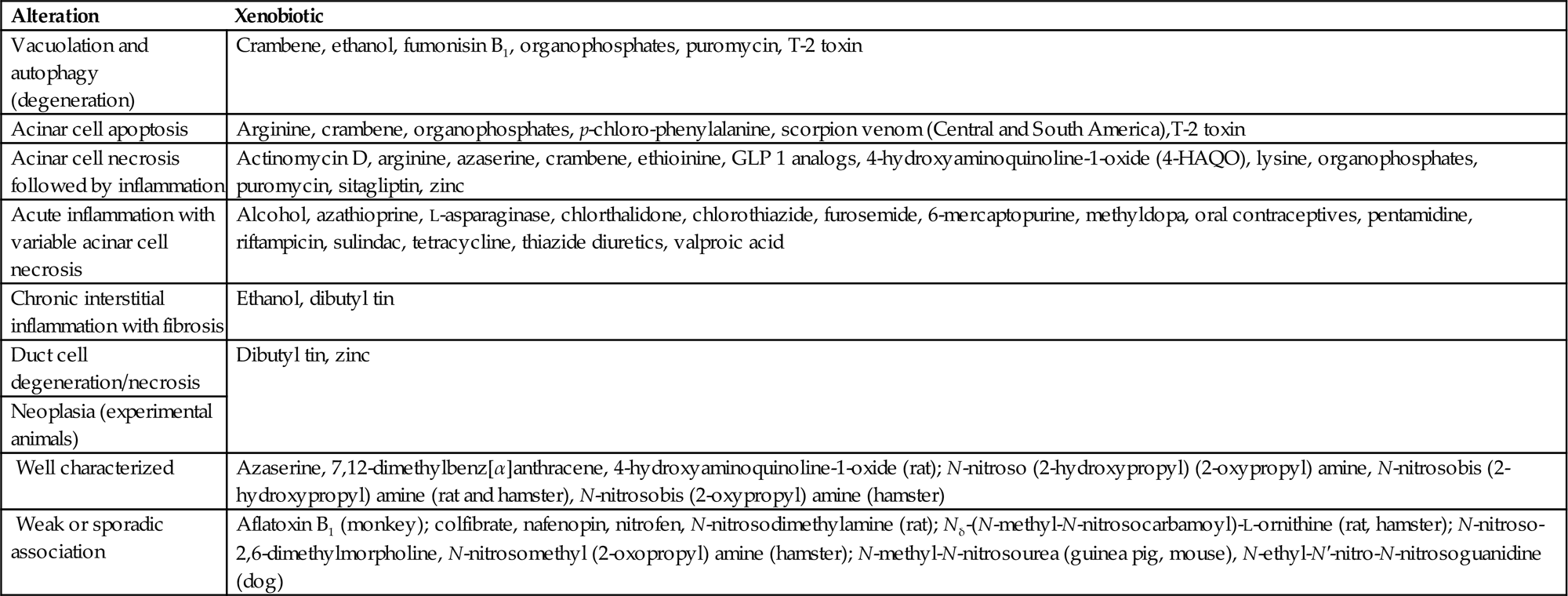
From Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2007. Fundamentals of Toxicologic Pathology, second ed. Academic Press, San Diego, CA, Chapter 10, Table 10.4, p. 244.
Edematous Pancreatitis
The progression of unique mechanisms of acinar or islet injury usually causes apoptosis as defined in the above paragraph. As injury progresses to cause expansion of the pancreatic parenchyma, by edema (initially), followed by influx of inflammatory cells and the cascade of events listed in subsequent sections, it is more appropriate to use pancreatitis as a diagnostic term.
Pancreatitis in its mildest presentation occurs as “edematous pancreatitis (EP),” and has been described as increased numbers of histiocytic macrophages (often engorged with apoptotic bodies), few neutrophils, prominent inter- and intralobular interstitial edema, and expansion of the fibrovascular stroma. Often only apoptosis, edema, and histiocytic infiltrates are present after the initial wave of injury, and release of circulating pancreatic biomarkers does not occur.
Necrosis and Acute Necrotizing Pancreatitis
Necrosis of acinar cells is more widespread throughout single or multiple lobules, often involving large clusters of acini and, in severe cases, the entire lobule. Necrosis often occurs initially at the periphery of the lobule, and progresses centripetally toward the center of the lobule. The changes differ from acinar cell apoptosis such that membrane disruption and release of cellular contents in the adjacent parenchyma initiates localized edema and cytokine recruitment. The context of pathogenesis to higher grade injury has significance in relation to the diffusion of activated zymogen proteins not only into lobular interstitium but also into surrounding peritoneal adipose tissue, triggering fat necrosis and peritonitis. Furthermore, the abundance of venules at the periphery of the lobules allows for rapid accumulation of neutrophils, which enhance the severity of the lesion (see later). With necrosis, acinar cells release zymogen contents within the interstitial stroma, and cell death quickly passes through the morphologic stage of coagulation necrosis and progresses to liquefaction as seen grossly (Figure 16.5A,B). This is because of the high content of hydrolytic enzymes activated and released into interstitial space. An intense neutrophilic inflammation is incited, and the classic microscopic appearance of acute necrotizing pancreatitis (Figure 16.6) can be observed. At this stage of injury, biomarkers of acinar cell injury are released and can be detected peripherally as elevated serum amylase and lipase activities within a time-dependent window. Necrosis is best recognized serologically 12–48 hours after insult, which corresponds temporally with concurrent vascular engorgement (congestion) of the pancreas (especially around the margins of the lobules) and intense chemo-attraction of neutrophils. Intense neutrophil infiltration is likely responsible for the transition between cases of EP to fulminant necrotizing pancreatitis.
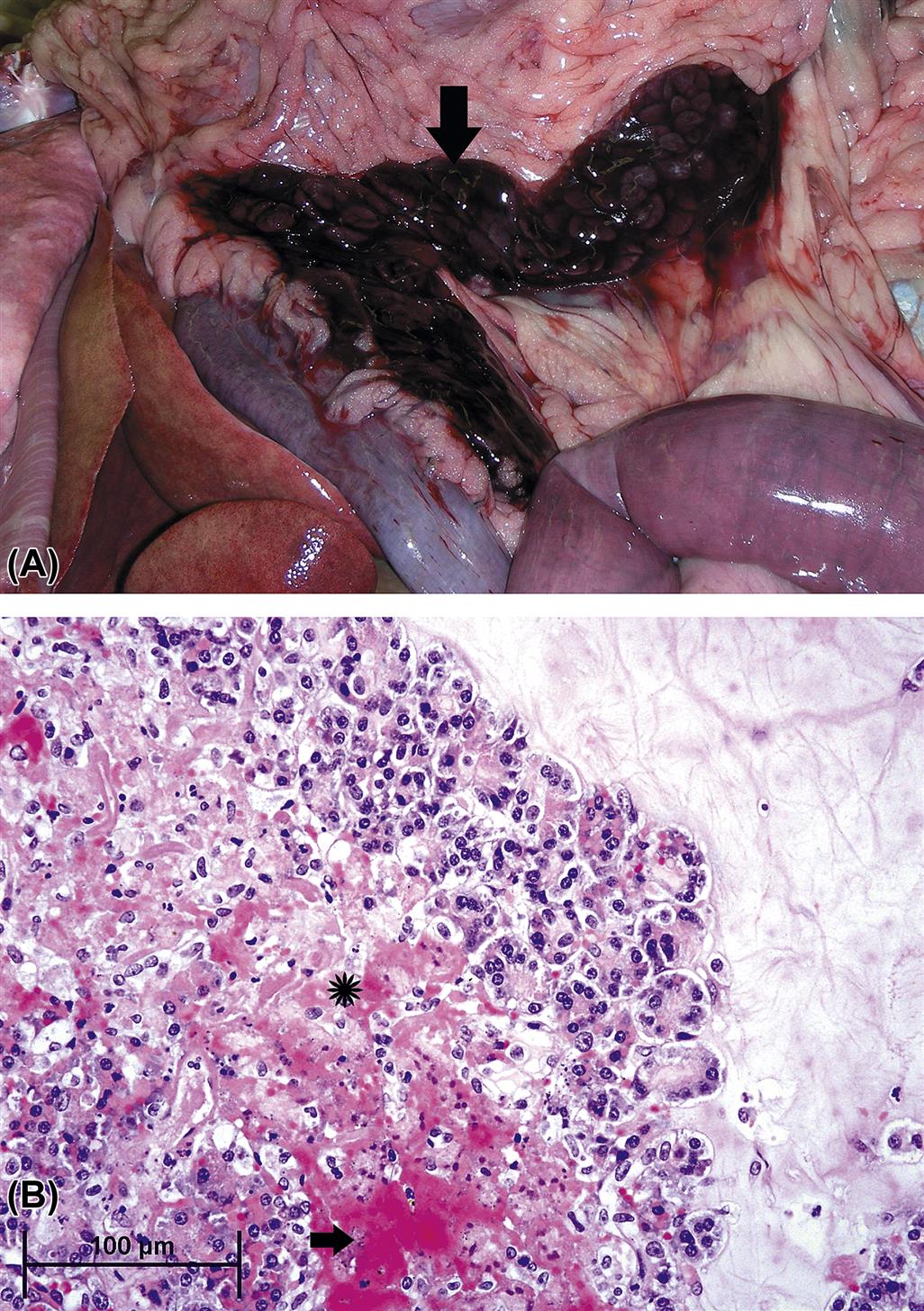
Once necrosis has begun, tissue breakdown products enter the general circulation through the local capillary network, inducing generalized depression of cellular respiration, vasodilation and shock in concert with injury to the myocardium and pulmonary vasculature. Accompanying localized hemorrhage adjacent to the pancreas, with concurrent necrosis of adjacent mesentery and gastrointestinal tissues, leads to localized abscessation and potentially sepsis. If pancreatic necrosis is contained, regeneration follows a defined course of increased mitosis in both acinar and ductal cells; ducts regenerate from acinar cell metaplasia and centroacinar cell proliferation. Acinar cells are regenerated and zymogen granules appear with intact ductal connections. This restorative capacity occurs provided necrosis has not been widespread and damage to preexisting stroma has not been profound.
Interface Pancreatitis
EEI pancreatic injury is a unique type of pancreatitis reported in certain strains of rats (most notably Sprague–Dawley) in response to a variety of test articles. To date, this type of pancreatic injury has not been reported in other species. Initially necrosis is localized around intact islets; sometimes just a few islets are involved, while other times there is widespread involvement of islets. The necrotizing lesions spread centrifugally often to involve the entire lobule, with eventual fibrosis of the entire exocrine portion of the lobule beginning at the peri-islet interface. The pathogenesis is unknown but observations suggest endothelial toxicity or potential extensive congestion at the EEI, with subsequent interactions between damaged venule endothelium and inflammatory cells, which subsequently induces venous dilation, attraction of more inflammatory cells, and localized progressive peri-islet injury (Brenneman et al., 2014).
Chronic Pancreatitis
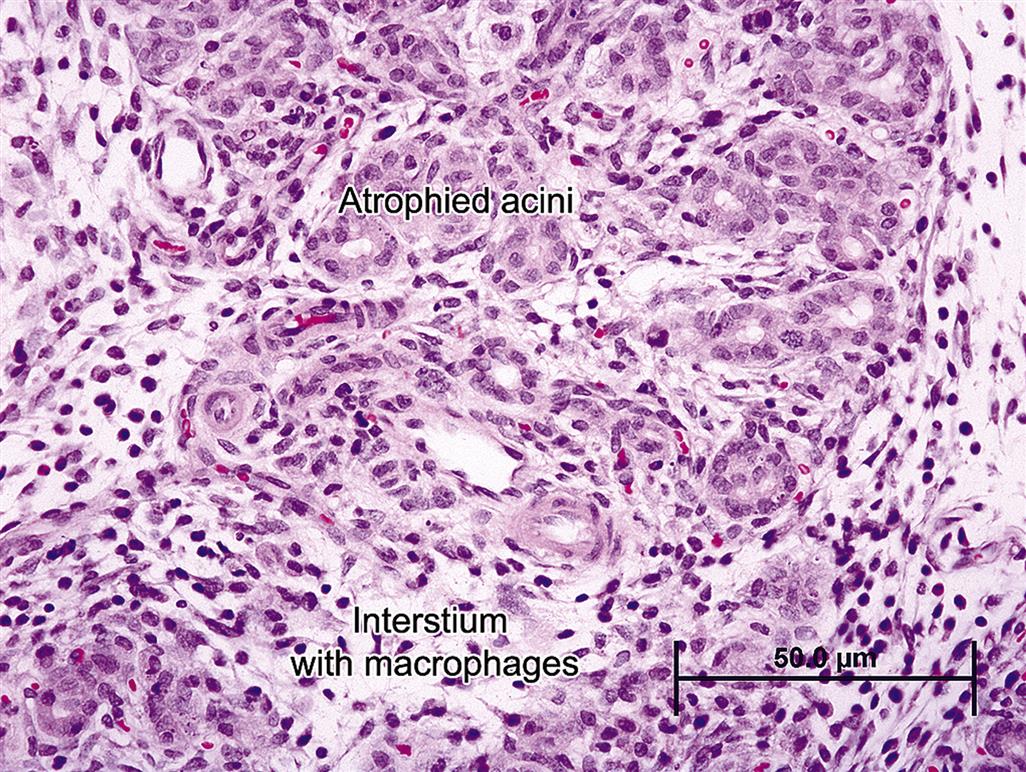
Chronic pancreatitis can be progressive, severe, relapsing, and persistent. The hallmark feature of chronic pancreatitis is marked fibrosis, which is active and progressive. The severity of fibrosis often varies from lobule to lobule and may be relatively mild in one lobule and very profound and exuberant in another. Initially fibrosis is primarily perilobular and periductal, surrounding major interlobular ducts. This fibrosis isolates lobules and traps remnant acinar cells and associated ductules within the shrinking lobule (Figure 16.7). With time, fibrosis becomes more intralobular, centering around small ducts and ductules, with subsequent atrophy of trapped ductal remnants and acinar cells. Much of the fibrosis is mediated by PSC, which share many features with myofibroblasts, in concert with recruited neutrophils and macrophages. In cases of mild pancreatic injury, apoptosis of PSCs occurs with reversal of injury and complete regeneration. However, if injury is severe, active, or relapsing, PSCs continue to actively produce ECM proteins. Long-term relapsing injury increases local pancreatic matrix metallo-protease 2, which is secreted by PSCs. Among its substrates is type IV collagen in lobular basement membranes, and this presumably elicits progressive acinar necrosis and atrophy with accompanying progressive inflammation.

Clinically, chronic pancreatitis manifests in most species as persistent low-grade pain associated with recurrent inflammation. Inflammation is primarily lymphocytic, comprised mainly of T-cells with proportionately fewer plasma cells. Macrophages are also present, as in any chronic lesion with fibrosis. Distribution of the inflammatory cells is concentrated in areas of active necrosis and early fibrosis. Pathogenesis and progression are often complex, compounded by impairment of ductal patency, reflux of biliary or pancreatic secretions, and low-grade adjacent lobular inflammation, necrosis, and fibrosis. As fibrosis progresses, zonal atrophy of ductular epithelium occurs and ducts are distorted (with the epithelium typically appearing folded and irregular).
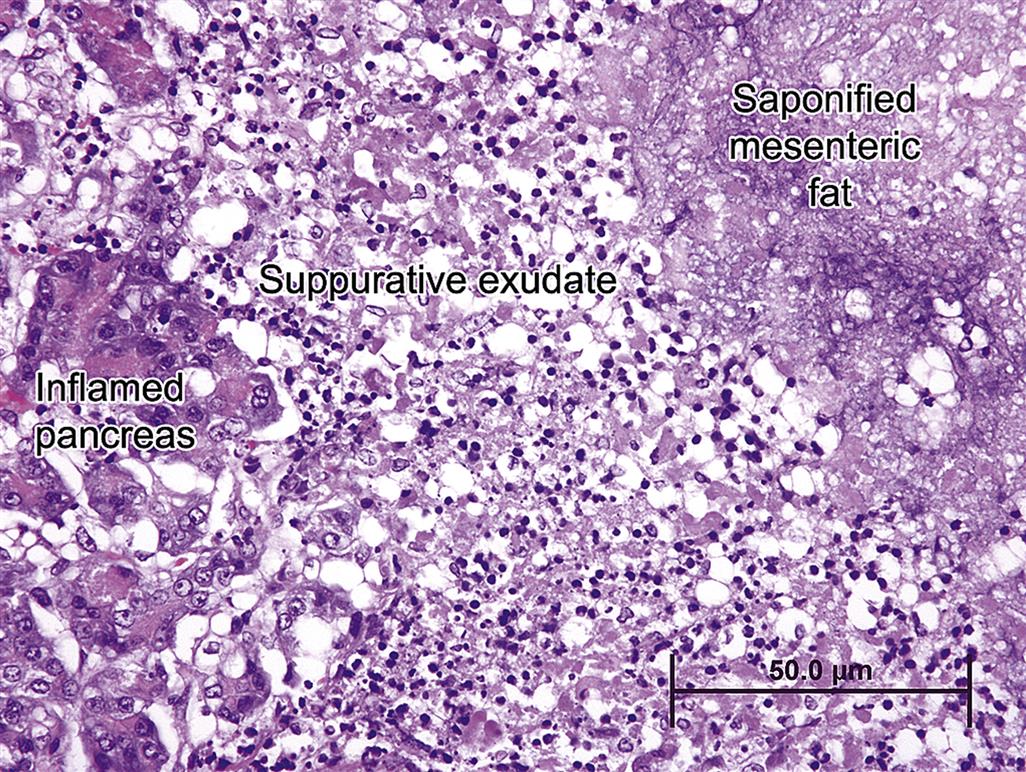
Progression of inflammation and subsequent fibrosis beyond the thin pancreatic extracellular capsule causes perilobular fat necrosis due to the diffusion of activated hydrolytic enzymes released from dead acinar cells and the intense neutrophilic response that follows. Eventually there will be profound fibrosis and formation of pseudocysts (dilated structures adjacent to but separate from the main body of the pancreas). The cysts may be filled with proteinaceous fluid and debris and most likely reflect former foci of fat necrosis. Pseudocysts can become quite large and grossly resemble pancreatic neoplasms.
Immune-mediated pancreatitis in most species is a form of chronic pancreatitis with morphologic features that are somewhat different. Periductal fibrosis is profound and often partially occlusive, pushing the trapped ductal epithelium into star-shaped folds. Inflammation in these cases is centered mainly around large- and medium-sized pancreatic ducts, with degenerative features in ductules and acini reflective of impairment of outflow. The intensity of periductal inflammatory infiltrates is also greater in this condition, with thick cuffs of lymphocytes and plasma cells, sometimes with macrophages and granulocytes present as well. The main biliary ducts are also involved in this condition. Examples of each type of pancreatic injury are included in Table 16.1.
Proliferative Responses to Injury and Carcinogenesis
Benign pancreatic tumors in domestic and laboratory animals are rare, although pancreatic adenomas in dogs and cats do occur. These tumors can be acinar, ductular, or a combination of morphologies; many also elicit a dense fibrotic (scirrhous) stromal response. Aged rats spontaneously develop a low incidence of well-differentiated, apparently benign acinar cell neoplasms (adenomas).
The vast majority of pancreatic neoplasms (excluding rare spontaneous tumors) in laboratory animals occur as malignant neoplasms resulting from treatment with carcinogenic xenobiotics. Rats and mice exposed to pancreatic carcinogens characteristically develop acinar cell neoplasms that exhibit several histologic variants. Hamsters characteristically develop neoplasms with a ductal phenotype. The incidence and number of acinar cell adenomas is often dramatically increased among rats after treatment with carcinogens such as azaserine and 4-hydroxyaminoquinoline-1-oxide. Acinar cell carcinomas in rats have been classified as well-differentiated, poorly differentiated, and undifferentiated. Duct-like, cystic, and microcystic foci can be encountered in tumors in which the dominant phenotype is acinar; mucin secretion is rare in rat pancreatic carcinomas. In contrast, duct-like carcinomas predominate in hamsters and often show evidence of mucin secretion and admixed increases in goblet cells resembling the early preneoplastic changes seen in human patients with developing malignant pancreatic neoplasms. In nonrodents, pancreatic adenocarcinomas are uncommon, and their relation to environmental carcinogen exposure (if any) is unknown. As with adenomas and adenocarcinomas in rodents, pancreatic neoplasms in nonrodents can be acinar or ductular in pattern, often with a marked scirrhous stroma (Figure 16.8).

Background histologic findings accompany pancreatic neoplasia in all species. These incidental changes include necrosis of both normal acinar epithelium and tumor tissue, resulting in separation of cells and loss of distinguishing morphologic features and arrangements between the neoplastic and nonneoplastic phenotypes, sporadic focal lobular atrophy with variable fibrosis, telangiectasia (usually involving islets), mixed mononuclear cell infiltration, sporadic autophagy and apoptosis, and hyperplastic nodules. These hyperplastic lesions seldom evolve into neoplastic foci.
Background or Confounding Changes
Recognition of autolysis is important when assessing microscopic pancreatic appearance. Occurring rapidly after death, this artifact can be reduced through rapid collection and immersion into fixative of the pancreas at necropsy. In cases where pancreatic lesions are expected, collection of this tissue should occur directly after opening of the abdomen.
Both autophagy and apoptosis can be observed sporadically in “normal” pancreas of animals that have been fasted for long periods, have experienced chronic anorexia, or have been starved. Autophagic vacuoles, often containing protein inclusions, in pancreata from fasted or starved animals are substantially smaller than those in animals in the early stages of pancreatic injury and are also much fewer in number. Apoptosis also may be seen in starved animals but in much lower numbers than in animals with xenobiotic injury.
Aged dogs and cats occasionally develop hyperplastic nodules within the exocrine pancreas, composed entirely of acinar cells, which are often somewhat larger and more intensely stained than surrounding normal acinar cells (Figure 16.9). The nodules are irregularly distributed and seldom if ever develop into true neoplasms. The pathophysiologic significance appears negligible, and a connection to toxin exposure has not been established.

Mechanisms of Exocrine Pancreatic Toxicity
Numerous chemical agents are known or suspected to induce acinar cell damage or necrosis, can alter acinar cell or duct cell function, or can induce neoplastic change in the exocrine pancreas. Medical literature contains numerous reports of acute pancreatitis in association with specific therapeutic products (Table 16.2). The risk of pancreatitis, although low, seems well established for some of these agents, for example, asparaginase and azathioprine. For other suspected pancreatic toxicants, however, only rare sporadic cases have been reported, and the connection between the agent and the cause of pancreatitis is often circumstantial.
Table 16.2
Agents and Drugs Reported to Cause Pancreatitis in Humans
| Association with pancreatitis | Agent |
| Strong causative association with pancreatitis | Alcohol (ethanol) |
| Azathioprine | |
| L-Asparagiuase | |
| Corticosteroids | |
| Cytarabine | |
| Didanosine | |
| Estrogens | |
| Furosemide | |
GLP I analogs | |
| Mercaptopurine | |
| Mesalamine | |
| Opiates | |
| Pentamidine | |
| Pentavalent antimonials | |
| Sitagliptin | |
| Sulfasalazine | |
| Sulindac | |
| Trimethoprim-associated sulfonamides | |
| Valproic acid | |
| Probable causative association with pancreatitis | Acetaminophen |
| Carbamazeprine | |
| Cisplatin | |
| Cyclopenthiazide | |
| Enalapril | |
| Erythromycin | |
| Hydrochlorothiazide | |
| Interferon α-2β | |
| Lamuvidine | |
| Methyldopa | |
| Octreotide | |
| Phenformin | |
| Procainamide | |
| Rifampin | |
| Other agents associated with pancreatitis | Clozapine |
| Cyclosporine | |
| 2′,3′-Dideoxyinosine | |
| Lisinopril | |
| Metronidazole | |
| Salicylates | |
| Zalcitabine |

From Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2007. Fundamentals of Toxicologic Pathology, second ed. Academic Press, San Diego, CA, Chapter 10, Table 10.1, p. 242.
Experimentally, rodents are susceptible to pancreatic damage by an overlapping but somewhat different spectrum of xenobiotics (Table 16.3). In contrast, relatively few xenobiotics are clearly linked to pancreatic injury in common domestic animals (Table 16.4) even though there are sporadic anecdotal reports in the veterinary literature.
Table 16.3
Selected Agents Reported to Cause Toxic Effects in Exocrine Pancreas Experimentally in Rodents
| Agent | Species |
| 2-Acetaminofluorene and 4-acetaminofluorene | Rat |
| Aflatoxin | Rat |
| β-3-Thienyl-DL-alanine | Rat |
| Arginine | Rat |
| Azaserine | Rat |
| Carbon tetrachloride | Rat |
| Chloraquine | Rat |
| Crambene | Rat |
| Dibutyl tin | Rat |
| Diethanolamine | Rat |
| Ethanol | Rat |
| GLP 1 analogs | Rat |
| 5-Fluorouracil | Rat |
| Lysine | Rat |
| Manganese | Rat |
| N-Nitrosomethyl(2-oxopropyl)amine | Rat |
| Puromycin | Rat |
| Triparanol | Rat |
| Zinc | Rat |
| Actinomycin D | Mouse |
| Chlorothiazide | Mouse |
| Neutral red | Mouse |
| Vinblastine | Mouse |
| Cobalt chloride | Guinea pig |
| Diazinon | Guinea pig |
| N-Nitrososbis(2-oxopropyl)amine | Hamster |
| Cortisone | Rabbit |
| Ethionine | Rat, mouse, guinea pig, hamster |
| 4-Hydroxyaminoquinoline-1-oxide | Guinea pig, rat |
| Methionine | Hamster, rat |
| N6-(N-Methyl-N-nitrosocarbamoyl)-L-ornithine | Rat, hamster |
| N-Nitroso(2-hydoxypropyl)(2-oxypropyl)amine | Hamster, rat |
From Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2007. Fundamentals of Toxicologic Pathology, second ed. Academic Press, San Diego, CA, Chapter 10, Table 10.2, p. 242.
Table 16.4
Selected Agents Associated with Exocrine Pancreatic Toxicity in Common Domestic Animalsa
| Agent | Species |
| Chocolateb | Dog |
| Corticosteroids | Dog |
| Deoxyvalinol (DON) | Pig |
| Diazinon | Dog |
| Dichloroacetate | Dog |
| Ethanol | Dog |
| DL-Ethionine | Dog |
| Fumonisin B1 | Pig |
| Organophosphates (especially diazinon) | Dog |
| T-2 toxin (vomitoxin) | Pig |
| Trimethoprim-enhanced sulfonamides | Dog |
| Scorpion venom (Central and South America) | Dog |
| Zinc | Dog, cow, sheep, pig, chicken |
aExperimental and “naturally occurring” reports, idiosyncratic occurrences not listed.
bAssociated with the high fat content of chocolate rather than toxicosis.
From Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2007. Fundamentals of Toxicologic Pathology, second ed. Academic Press, San Diego, CA, Chapter 10, Table 10.3, p. 243.
For simplification, xenobiotic-induced injury can be categorized by the morphological characteristics of the injury they induce. These basic mechanisms are:
Altered Zymogen Trafficking
Under normal conditions, secretion of zymogen granules is limited to the apical membrane area of the acinar cell, with tight apical cellular junctions between neighboring acinar cells. Any disruption of these tight junctions results in the leakage of zymogen into intercellular spaces. Apical secretion can be blocked in an acinar cell injury. Disruption of the plasma membrane leads to excessively high intracellular calcium (Ca2+) concentrations, which trigger a signaling cascade that allows release of intact zymogen granules into the intercellular space. Colocalization of cathepsin B from lysosomes at the basolateral membrane or between acinar cells will further acinar cell injury by activating trypsinogen to trypsin and releasing cytokines that recruit inflammatory cells, leading to pancreatic inflammation.
Under physiologic conditions, CCK stimulates both protein synthesis of zymogens and secretion of mature zymogen granules. Secretion generally outpaces protein synthesis, and as a result zymogens tend not to accumulate within the acinar cell. With “supramaximal” doses of CCK (or analogs like caerulein that induce hypersecretion), protein synthesis outpaces secretion. Excessive accumulation of zymogens overwhelms the zymophagy pathway and triggers large-scale autophagy. A potential “autophagic traffic jam” then can lead to formation of dysfunctional autophagolysosomes and uncoordinated activations of lysosomal cathepsins L and B. As previously stated, when cathepsin B predominates, trypsonigen is activated to trypsin, which can activate other pro-zymogen proteins in the zymogen granules. The subsequent autodigestive cascade elicits membrane destruction in the local pancreas. Localized pancreatic autophagy is balanced by continued local membrane destruction by activated digestive enzymes and contained by acinar cell apoptosis at the margin of the lesion with recruitment of inflammatory cells that together contain the injury.
“Prototypical” xenobiotics that cause pancreatic injury via altered zymogen trafficking include caerulein, crambene, carbachol, organophosphates, and scorpion toxin. Ethanol is a “sensitizer,” enhancing the sensitivity of acinar cells to altered zymogen trafficking by enhancing hyperstimulation.
Oxidant Stress
The pancreas, unlike many other nonhepatic tissues, has active glutathione-synthesizing machinery including the inducible, rate-limiting enzyme of glutathione synthesis, glutathione cysteine ligase. Hence, the pancreas can increase its baseline level of glutathione if the oxidant stress evolves slowly. Similarly, both acinar and ductal cells contain phase II enzymes such as the glutathione S-transferases (GSTs). The lower magnitude of GSTs in acinar cells compared to duct cells is postulated to make acinar cells more prone to damage by bioactivated xenobiotics. GSTs are also secreted into pancreatic juice, and this is postulated to play a protective role against xenobiotics present in ingested materials.
Active transsulfuration complements the antioxidation pathway, allowing the acinar cell to derive cysteine from methionine. These can be used to produce more glutathione if needed. Nicotinamide adenine dinucleotide phosphate quinone reductase, another antioxidant enzyme, is also expressed significantly in pancreatic acinar cells and can have increased induction under conditions of oxidant stress, most notably glutathione depletion. The pancreas contains not only phase II drug-metabolizing enzymes but also phase I metabolizing enzymes, although the levels/activities of cytochromes P450 (CYP) and related Phase I enzymes are quite low compared to liver (as little 1% of hepatocyte concentrations). CYP enzyme activity appears to be present in roughly equal proportions in both acinar cell and duct cells, although there is some variation in isoform expression, making both cell types more prone to bioactivation of xenobiotics and injury by reactive intermediates. The low overall activity of CYPs within the pancreas compared to liver makes CYP induction less common in the pancreas. As with CYP enzymes in any tissue, some pancreatic CYP isoforms are inducible. However, variability in isoforms across species requires assessment of respective CYPs important in predicting xenobiotic transformation within pancreatic tissue.
Localized Ischemia
Arteriolar and capillary blood flows are affected by parasympathetic and sympathetic ganglia in the organ and adjacent mesentery. EP in generalhas increased capillary blood flow, and necrotizing pancreatitis is often associated with local pro-coagulant activity and decreased capillary blood flow within the organ. A role for interactions of the renin-angiotensin system in pancreatic necrosis has recently been postulated, and supported by the discovery of angiotensin receptors in pancreatic capillary beds and acini. Binding of angiotensin results in renin-induced vasoconstriction, decreased capillary perfusion, and increases the potential to exacerbate local ischemia.
Impaired Ductal Outflow or Obstruction
Impairment of ductal outflow is a major predisposing factor for pancreatitis both in humans and experimentally in rodent models, in particular in relation to chronic pancreatitis. One of the most important functions of the pancreas is regulation of bicarbonate and chloride flux in acinar and ductal epithelial cells, most importantly the ductal epithelium. Since bicarbonate secretion is important in modulating the pH of ductal secretions and pancreatic juice for appropriate activation of pancreatic digestive enzymes, modest perturbations in the pancreatic microenvironment caused by acute gastritis or alcohol consumption can result in localized activation of digestive enzymes just as blockage of the common bile duct can induce retrograde efflux of bile acids into the pancreatic ductal tree. Collectively, modulation of ductal secretion and/or reflux of bile acids (e.g., with cholelithiasis, or bile duct fluke migration) can induce acute widespread induction of duct injury. Cell membranes are destroyed by bile acids, contents leak into the parenchyma, cathepsin B activates trypsinogen to trypsin, and lipase is activated within the acini of the affected lobule(s) to induce progressive pancreatic necrosis. Predisposing factors include excessive cortisol (endogenous or exogenous sources) and persistent hypercalcemia or hypertriglyceridemia. If ethanol is removed as an etiology of exocrine injury, many of the causes of pancreatitis remain idiopathic.
Immune-Mediated Disease
Immune-mediated pancreatitis is a recently defined entity in humans that originated with characterization of sclerosing cholangitis and sclerosing pancreatitis. These conditions occur with concurrently increased serum γ-globulins (IgG1 or IgG4 autoantibodies), diffuse pancreatic enlargement, obstruction or narrowing of the pancreatic ducts (sclerotic change), and mixed lymphocytic and plasmacytic infiltration of the exocrine pancreas. Typically affected pancreata exhibit stenosis of the major pancreatic duct and common bile duct with capillary thrombosis. An animal model of autoimmune pancreatitis has been developed using the neonatal BALB/c mice that have undergone thymectomy and then are immunized with carbonic anhydrase II (an important enzyme in ductal epithelium) or lactoferrin. These mice express increases in CD4+ Th1 cells (which enhance cell-mediated inflammatory process) as the syndrome develops with subsequent depletion of regulatory T-cells. It is likely that T-helper 2 cytokines (which promote humoral immunity) are involved in the progression of the disease with recruitment and proliferation of B cells and plasma cells.
Exposure to Carcinogens
Nonlethal mutations occur both in acinar and ductal cells exposed to mutagens or carcinogens. The induction of DNA damage in these cells by carcinogens has been documented in hamsters and rats. For example, the β-oxidized di-n-propylnitrosamines are activated to form mutagens by pancreatic microsomal cell fractions. There are significant species differences in the effect of carcinogenic xenobiotics on the pancreas. Short-term in vitro assays for screening pancreatic carcinogens, most of which involve short-term cell culture assays, have been attempted but often are problematic to perform. Similarly, maintaining long-term cell cultures, in particular with acinar cells, has proved challenging, since the precise factors needed for maintenance of viability and differentiation have been difficult to identify. The most intensively studied rodent pancreatic carcinogens, azaserine, 4-hydroxyaminoquinoline 1-oxide, and BOP, besides being directly mutagenic, have poorly characterized cytotoxic effect as well as assessed through measurement of decreased protein synthesis. At present, it is not possible to determine whether or not a focal lesion has the potential to progress to a neoplasm, except by observation in long-term studies, or potentially genetically altered species (transgenic mice, for example). It has been estimated that fewer than 1% of such foci actually complete the progression to neoplasia although recent studies have shown that Kras mutations are present early in pancreatic carcinogenicity, often in preneoplastic lesions, and p16 is mutated early in the progression from benign to malignant, invasive tumors. A wide array of chemical carcinogens with widely divergent structures has been shown to affect the pancreas (Table 16.5).
Table 16.5
Summary of Chemicals that Cause Carincoma in the Exocrine Pancreas of Experimental Animals
| Agent | Species |
| Group A (well-characterized model carcinogens) | |
| Azaserine | Rat |
| 7,12-Dimethylbenz[a]amhracene | Rat |
| 4-Hydroxyaminoquinoline-1-oxide | Rat |
| N-Nitroso(2-hydroxypropyl)(2-oxopropyl)amine | Hamster, rat |
| N-Nitroso(2-oxopropyl)amine | Hamster |
| N-Nitroso(2-hydroxypropyl)amine | Hamster, rat |
| Group B (reported as carcinogens once or in a limited number of studies) | |
| Aflatoxin B1 | Monkey |
| Clofibrate | Rat |
| 4-Hydroxy-3-nitrobenzenearsonic acid (Roxarsone) | Mouse |
| N6-(N-methyl-N-nitrosocarbamoyl)-L-ornithine | Rat, hamster |
| N-Methyl-N-nitrosourea | Guinea pig, mouse |
| Nafenopin | Rat |
| Nitrofuran | Rat |
| N-Ethyl-N′-nitro-N-nitrosoguanidine | Dog |
| N-Nitrosodimethylamine | Rat |
| N-Nitroso-2,6-dimethylmorpholine | Hamster |
| N-Nitrosomethyl(2-oxopropyl)amine | Hamster |
| 2,3,7,8-Tetrachlorodibenzo-p-dioxin | Rat |
From Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2007. Fundamentals of Toxicologic Pathology, second ed. Academic Press, San Diego, CA, Chapter 10, Table 10.5, p. 247.
Summary
The pancreas is a unique and dynamic organ with endocrine and exocrine functions. The location of the pancreas within the anterior abdomen, rich blood supply, and role in synthesis of proenzymes of digestion, along with limited ductal outlets to the intestinal tract, has allowed evolution into a generally stable organ with a variety of adaptions (e.g., autophagy and apoptosis) to limit progression of injury. Disruption or excessive stimulation of digestive proenzyme trafficking to the ductal system can elicit colocalization of zymogen granules and lysosomes within AV eliciting cathepsin B activation of trypsinogen to trypsin leading to localized or global organ injury. Similarly, oxidant stress can induce excessive apoptosis or necrosis in acini and ductal epithelium, and ischemia with reperfusion can elicit thrombosis, localized acidosis, membrane breakdown, and enzymatic digestion. The proximity of the bile duct and bile acids can move retrograde up the pancreatic ducts leading to tissue necrosis and further activation of trypsinogen. Like other tissues, the pancreas is also susceptible to carcinogens and immune–mediated diseases. Its remarkable ability to limit injury through the propensity for acinar cell apoptosis versus necrosis helps manage excessive digestive enzyme activation and minimize injury in many cases. The inherent regenerative capacity of acinar cells and ductal epithelial allows maintenance of organ function in the face of toxicity.
However, the pancreas is very susceptible to recurring injury leading to a loss of ductal patency, fibrosis, and continued attempts to regenerate acinar cells and zymogen granules. Thus, multiphasic and relapsing injury collectively may lead to excessive and permanent glandular or islet destruction. In severe cases, the injury and subsequent inflammation can spread to the peritoneum and liver, causing an acute abdominal crisis. For assessment of pancreatic injury due to toxic insult, a large group of serum biomarkers are available for peripheral evaluation of pancreatic toxicity at in its early stages; however, almost all of these serum biomarkers need to be measured within 24–48 hours after a single-source injury to be effective in detecting pancreatic necrosis, assessing the severity of the insult, or defining clinical or preclinical prognosis.