Nervous System
Brad Bolon1, Robert H. Garman2, Mark T. Butt3 and David C. Dorman4, 1GEMpath, Inc., Longmont, CO, United States, 2Consultants in Veterinary Pathology, Inc., Murrysville, PA, United States, 3Tox Path Specialists, LLC, Frederick, MD, United States, 4North Carolina State University, Raleigh, NC, United States
Abstract
Clinical diseases characterized by significant neurological deficits are among the most devastating outcomes produced by exposure to toxicants. The exquisite sensitivity of the nervous system results from its limited capacity, especially centrally, to reverse or compensate for destruction as well as its neural cell–specific mechanisms and manifestations of toxicity. Present regulatory approaches for detecting and managing neurotoxic risk generally regard any substantial toxicant-induced change in neural chemistry, function, or structure as harmful (i.e., adverse). Neurotoxicant-induced injury may appear as an acute illness or death, typically pointing to severe disruption in CNS function, or structure-associated with a high-dose exposure. Alternatively, early declines in neurological function (i.e., premature senescence) and/or the quality of life often follow chronic and low-level neurotoxicant exposure. The practice of toxicologic neuropathology requires an integrated understanding of comparative neurobiology (the major likenesses and dissimilarities among species, strains, ages, and genders) and correlative neurobiology (the relationship among neural structure, function, and chemistry) as well as a solid grasp of neurotoxicant classes, targets, and mechanisms.
Keywords
Nervous system; neurotoxicity; morphology; biomarker; neurotransmission; neuropathology; neurohistology
Introduction
The linked central and peripheral components of the nervous system (a.k.a., the CNS and PNS) are the instruments by which an animal regulates all other body systems and interacts with the external environment. Neurotoxicants injure (structurally and/or functionally) one or more neural cell types, often acting regionally to produce subtle to calamitous dysfunction. Characterization and prevention of neurological deficits related to xenobiotic exposure is essential to the protection of human and animal health. This chapter largely focuses on methods that are used in regulatory studies designed to assess neurotoxicity in animals. A discussion of other techniques used in neuroscience research such as the use of light to selectively control neural activity (optogenetics), electrophysiological approaches, brain circuit analyses, neural genomics and proteomics, use of in vitro techniques and alternative animal models (e.g., Caenorhabditis elegans), and other experimental approaches are beyond the scope of this work.
Many agents have been demonstrated to elicit neurotoxic effects in humans and/or animals (Table 21.1). Humans may encounter neurotoxic chemicals at both home and work, particularly such agents as agrochemicals, metals, and solvents. Some therapeutic agents produce neurotoxic side effects, including cytotoxic (e.g., antineoplastic agents) and neuroactive small molecules (e.g., antidepressants, antiepileptics) and some biomolecules (e.g., natalizumab, an antibody directed against the cell adhesion molecule α4-integrin). “Life style” chemicals like alcohol (ethanol), nicotine, and “recreational” drugs (e.g., cocaine) tend to have very powerful neurotoxic actions, and many microbes, plants, and animals generate potent neurotoxins.
Table 21.1
Common Neural Targets and Neurotoxicants
| Site of action | Neurotoxic effect/mechanism | Selected examples |
| NEURON | ||
| Stem cell, neuronal | Altered migration and/or differentiation | Ethanol |
| Methanol | ||
| Methylazoxymethanol (MAM) | ||
| Methylmercury | ||
| Mature cell | ||
| Body | Cytotoxicity | |
| Alkylating agent | Doxorubicin (alkylating agent) | |
| Energy depletion (disrupted electron transport) | 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) | |
| Excitotoxicity | Domoic acid (marine algal toxin) | |
| Kainic acid (seaweed toxin) | ||
| Quinolinic acid | ||
| Trimethyltin | ||
| Mitotic inhibitor | Vincristine | |
| Protein conjugation/inactivation | Methylmercury | |
| Trimethyltin | ||
| Axon | Axonopathy | |
| Central (unknown mechanism) | Clioquinol | |
| Proximal (altered intracellular transport) | β,β′-Iminodipropionitrile (IDPN) | |
| Distal (macromolecular cross-linking) | Acrylamide | |
| Carbon disulfide | ||
| n-Hexane | ||
| Synapse | Altered neurotransmission | |
| Decreased neurotransmitter release | Botulinum toxin | |
| Tetanospasmin | ||
| Persistent presence of neurotransmitters | Selective serotonin reuptake inhibitors (SSRI) | |
| St. John’s wort (Hypericum perforatum) | ||
| Reduced neurotransmitter metabolism | Carbamate insecticides | |
| Monoamine oxidase inhibitors (MAOI) | ||
| Organophosphorus insecticides | ||
| Termination of transmembrane ion gradients | Pyrethrin insecticides | |
| Pyrethroid insecticides | ||
| Tetrodotoxin | ||
| GLIA | ||
| Stem cell, glial | Neoplasia (mutation by DNA alkylation) | Acrylonitrile |
| Ethylnitrosourea (ENU) | ||
| Ethylene oxide | ||
| Methylnitrosourea (MNU) | ||
| Astrocyte | Swelling (Alzheimer Type II cells) | Ammonia |
| Myelinating cells | Abnormal protein production—Schwann cell | Isoniazid |
| Abnormal protein production—Oligodendrocyte | Lead | |
| Triethyltin | ||
| Anoxia (insufficient oxygen delivery) | Carbon monoxide | |
| Edema—Oligodendrocyte | Cuprizone | |
| Hexachlorophene | ||
| Membrane disruption | Lead | |
| Triethyltin | ||
| Metabolic disturbance | Coyotillo (Karwinskia humboldtiana) | |
| Uncoupling oxidative phosphorylation | Bromethalin | |
| Hexachlorophene | ||
| ENDOTHELIUM (CAPILLARY) | ||
| Physical penetration of blood–brain barrier elements | Arsenic | |
| Protein kinase dysfunction | Lead | |

Neurotoxicity may present as anatomic, chemical, or functional alterations affecting the CNS, PNS, or various effector organs (e.g., muscle). Anatomic indices of neurotoxicity may be observed as structural abnormalities at the macroscopic, microscopic, or ultrastructural levels. Common chemical lesions induced by neurotoxic agents include changes in neurotransmitter levels and/or neural cell enzyme activities. Functional shifts caused by neurotoxicants include neurobehavioral and neurophysiological (i.e., electrophysiological) fluctuations. Abnormalities in neural chemistry or function secondarily alter neural structure, while structural alterations in the CNS or PNS frequently produce functional deficits. Neurologic alterations produced by chemical exposure are not necessarily adverse. Some chemical actions are pharmacologic, including expected but excessive reactions to neuroactive drugs (i.e., supra-pharmacology) and temporary perturbations in global or regional neurochemistry. Receptor blockade or alteration may result in long-lasting functional effects that may even have structural impacts on neurons, but the therapeutic result (example: analgesia) may justify those alterations. Whether intended or not, local, regional, or widespread effects on nervous system tissues induced by xenobiotic exposure or implanted cells or devices may elicit changes that do impair an individual’s ability to survive and prosper in their environment, therefore producing an adverse neurotoxic event.
The nervous system as a whole is predisposed to toxic damage for many reasons. The key factors are the intricate interconnectivity, extensive regional specialization, and restricted ability for cell and tissue repair, especially within the CNS. Locally discrete CNS lesions may produce significant effects in more distant parts of the nervous system due to the close linkage of structure and function in a particular region; neural lesions may affect other body systems since the CNS and PNS act to directly or indirectly regulate the physiological activities of nearly all peripheral organs. The extensive nutritional needs of neural cells embody another key area of neurotoxic vulnerability. The brain is almost entirely reliant on aerobic, glucose-dependent metabolism, which requires approximately 15% of the total cardiac output and 20% of the body’s oxygen-carrying capacity, even though the brain represents only 1%–2% of the total body weight. The limited antioxidant capacity and high quantities of polyunsaturated fatty acids in CNS tissue predisposes neural cell membranes, particularly myelin, to oxidation. Furthermore neural lipids concentrate lipophilic agents—many of which have neuroactive properties—in the CNS, thereby potentiating their possible neurotoxic impact.
The nervous system also features many structures with amplified sensitivity to neurotoxicant exposure. Key factors explaining such site-specific vulnerability include regional variations in biochemistry, metabolism, and vascularization. Another major influence is the capacity for repair following a neurotoxic insult. Injury to the PNS may be repaired, yielding a full or nearly full restitution of function. In contrast, damage to the CNS is met with a minimal ability for structural renovation and functional renewal; instead, brain “repair” typically relies on the recruitment of compensatory processes mediated by other, less affected brain regions. These attributes are impacted by age with older individuals exhibiting less robust structural rebuilding (due to lower neuronogenesis and synaptic plasticity) as well as diminished functional restoration (arising from the inability of adaptive behaviors to fully compensate for deficits produced by damage to other regions). Therefore neurotoxicant-induced CNS damage is likely to engender permanent and often progressive deficits.
Neuroanatomic Considerations in Toxicologic Neuropathology
Mammals (chiefly rats, dogs, and nonhuman primates) are the favored animal models for neurotoxicity testing because the functional and structural characteristics of their nervous systems are very similar to those of humans. Pathologists require a thorough and integrated knowledge of the connection between regional anatomy, chemistry, and function (i.e., correlative neurobiology) as well as the divergence that exists among species, strains, ages, or genders (comparative neurobiology) in order to effectively identify neurotoxic hazards, assess neurotoxic risks, and diagnose neurotoxic conditions. A detailed consideration of these topics is beyond the scope of this chapter, but further information is readily available in the Further Reading list.
Neural Cell Populations: Key Targets for Neurotoxicity
At the cellular level, neurons are the key functional elements of the CNS and PNS. The adult human brain (weight, 1400 g) holds approximately 130 billion neurons forming 150 trillion synapses. Different populations of CNS neurons may be recognized by many traits, including cell conformation (e.g., pyramidal neurons are large and polygonal, granule cells are small and round; Figure 21.1); axonal length (longer for pyramidal projection neurons, shorter for granule cell interneurons); function (afferent, efferent, or interneuron); neurotransmitter composition; and/or location. In general, neurons, once damaged, do not regenerate, although their processes in the PNS may be rebuilt over time if the cell body remains viable. Neurons are highly vulnerable because they use about 10-fold more oxygen than other neural cells. Typical neuron responses to toxic agents are cell body degeneration leading to necrosis and/or disintegration of its peripheral processes (i.e., axonal degeneration).
Glia are essential support cells in the CNS and PNS, with each CNS neuron being sustained by 10–50 glial cells. Multiple glial cell lineages have been identified. Astrocytes maintain neuronal processes, remove neurotransmitters from synapses, control chemical and ionic gradients within the brain parenchyma, and form the major protective layer of the blood–brain barrier (BBB). Astrocytes react to neurotoxicants by swelling or vacuolation (if the astrocyte is damaged) or hypertrophy and hyperplasia (reactive astrogliosis) when reacting to structural damage in the CNS. Astrocytes also play a key role in the formation of the “glymphatic system,” a recently discovered series of perivascular channels that eliminate soluble proteins and metabolites from the CNS.
Myelinating glial cells are the oligodendrocytes of the CNS, which envelop multiple axons, and Schwann cells of the PNS, which encircle only one axon. The presence of myelin sheaths is required for fast transmission of neural impulses down axons, Myelinating cells respond to neurotoxicants by degeneration and necrosis or by vacuolation due to fluid accumulation between myelin lamellae. Disruption or loss of myelin retards the speed of impulse propagation, especially in thickly myelinated axons in the CNS and somatic (or sensorimotor) PNS required for rapid responses to environmental stimuli.
Unlike other glial cells, which originate from neural stem cells, microglia (the resident CNS immune surveillance and phagocytic cells) arise in peripheral hematopoietic organs from monocytic precursors. Some microglia are also derived from the yolk sac and may be identified in the brain primordium as early as embryonic Day 8. Microglia react to neurotoxicants with hypertrophy and hyperplasia, and they serve as local phagocytic cells clearing debris from the injury of many cell types.
Other classes of glia have been identified. For example, satellite cells in PNS ganglia are thought to serve many of the same metabolic support and barrier functions that their astrocyte counterparts provide in the CNS. In general, the responses of these other glial cell types to neurotoxic agents have yet to be characterized in detail.
Other major cell types in the CNS include choroid plexus epithelium, ependymal cells, and meningeal fibroblasts. The choroid plexus epithelium covers the ventricular surface of the small fibrovascular tufts of choroid plexus that reside, primarily, in the lateral and fourth ventricles. The choroid plexus epithelium actively secretes cerebrospinal fluid (CSF) and also serves as a depot for sequestering neurotoxic heavy metals to prevent their accretion in the brain parenchyma or distribution via the CSF to other portions of the CNS. This epithelium commonly responds to xenobiotic exposures by either vacuolation or degeneration, which may culminate in necrosis and sloughing. Ependymal cells are a special glial derivative that line the surfaces of the brain ventricular system and the spinal cord central canal; the ependyma appears to be resistant to many toxicants (likely because chemicals do not enter the CSF at high levels), although responses similar to those of the choroid plexus epithelium are possible. Meningeal fibroblasts also are resistant to toxicants.
Comparative Neuroanatomy: Basic Principles
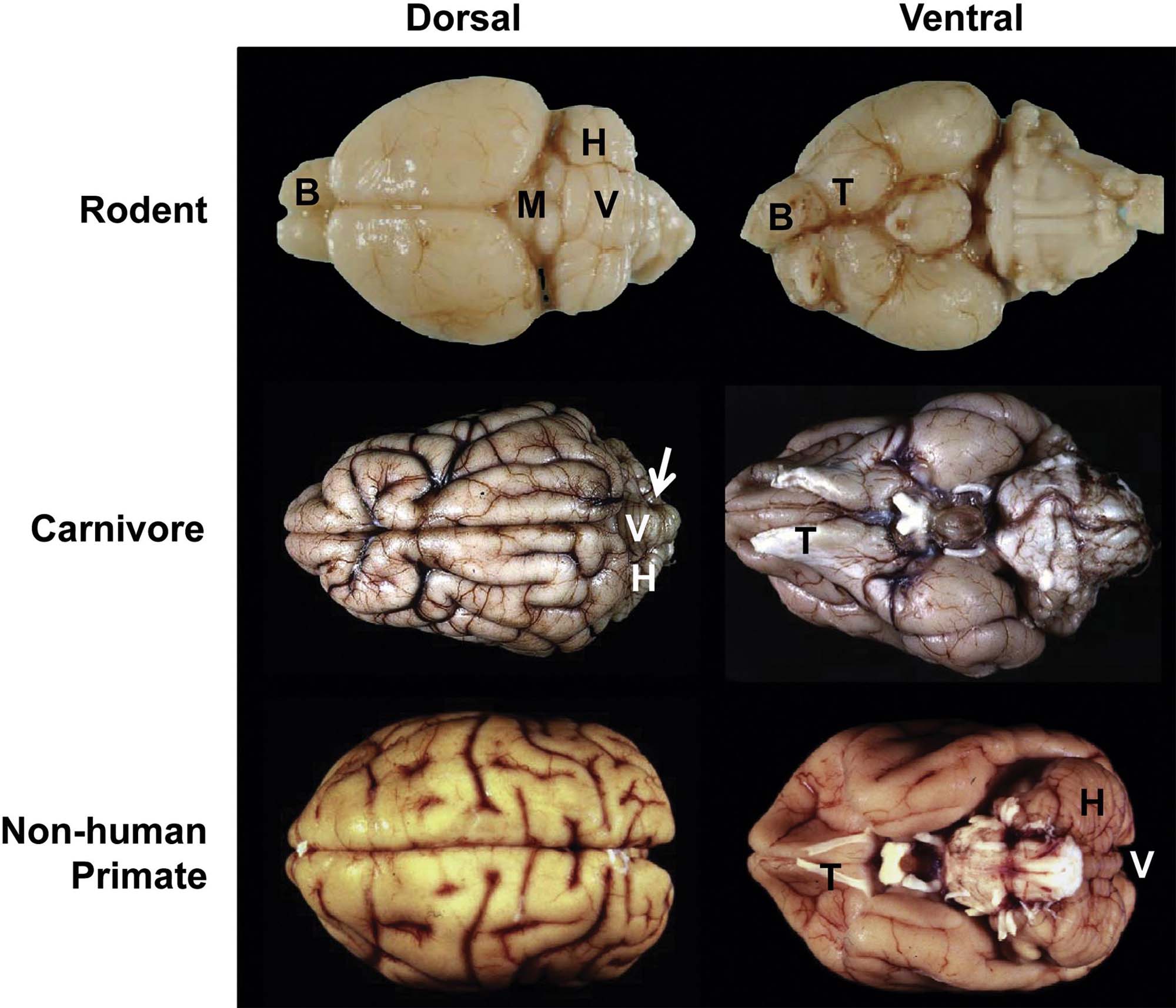
Mammalian species used for neurotoxicological hazard identification and risk assessment may be divided into three categories based on reproducible neuroanatomic features. These classes are Rodent (including true rodents like mice and rats but also lagomorphic rabbits), Carnivore (cat and dog), and Primate (monkeys and humans).
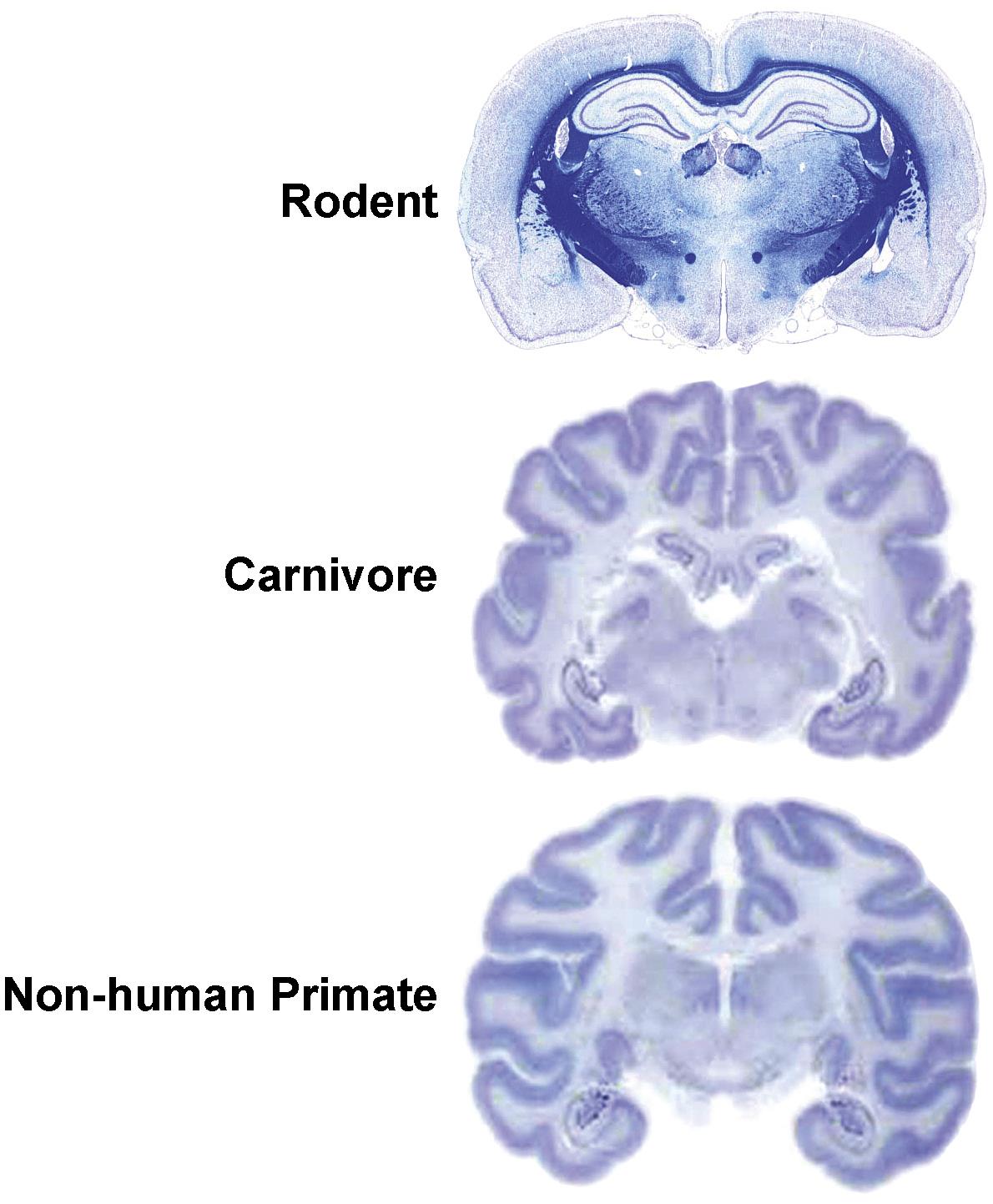
While regions and their connections are quite similar across mammalian species, some interspecies variations in neuroanatomic structure are prominent. For example, the weight of this organ increases substantially as the phylogenetic tree is ascended: 1300–1400 g (representing about 1.5%–2% of total body weight) for an adult human versus 1.5–2 g (about 0.8% of total body weight) for an adult rat. The single most conspicuous gross difference among species is the surface contouring of the cerebral hemispheres, which are lissencephalic (smooth) for Rodents but gyrencephalic (convoluted) in Carnivores and Primates (Figure 21.2). The pattern of cerebral gyri and sulci is specific to each species and may be asymmetric between the two hemispheres. Old World primates (e.g., baboon, cynomolgus monkey, rhesus monkey) may be better models for human neurotoxic risk since all these species have bigger brains as well as more and larger gyri relative to New World monkeys (e.g., marmoset, squirrel monkey). Relative to Rodents, brains of gyrencephalic Carnivores and Primates have comparatively more white matter (Figure 21.3) and many more interneurons. The structure–function correlations for major brain domains are similar across mammalian species, but their importance diverges among species. For example, the cerebral surface area mapped to olfactory, motor, and sensory tasks is approximately 80% in Carnivores but 20% in Primates, while the cortical area dedicated to signal integration is nearly 80% in Primates and 20% in Carnivores. Similarly the cerebellar hemispheres (Figure 21.2) and the inferior olivary nucleus are much larger in Primates relative to Carnivores and especially Rodents. Taken together, this species-specific divergence reflects the more elaborate circuitry needed for fine sensorimotor control and efficient cognition (higher thought) in Primates.
The basic neuroanatomic pattern of the brain within a species is relatively constant, but several factors may produce structural variations that must be recognized as normal background findings. For example, many Rodent strains exhibit grossly different brain anatomy relative to the expected wild-type configuration, particularly a smaller brain size and a reduced or absent corpus callosum; these architectural changes may or may not be associated with behavioral abnormalities, so it is important to understand the background neuroanatomy and function of specific species and strains/breeds when interpreting data sets for neurotoxic risk assessment. Similarly the sex of the individual may impact regional neuroanatomy. For instance, males tend to have larger numbers of neurons and oligodendrocytes overall, which translates into larger amygdaloid and hypothalamic nuclei (i.e., sexually dimorphic nuclei) and wider white matter tracts. Neural anatomy also evolves throughout development, including final maturation of protective elements like the BBB and glial cell detoxification pathways well after birth as well as the gradual loss of BBB integrity with age. The complexity of these differences indicates that scientists who design neurotoxicity tests in animals must review the literature and, for novel methods or uncommon endpoints, perform suitable pilot studies with appropriate control animals to confirm that the chosen test species, strain, sex, and age are appropriate.
The spinal cord of mammals is generally comparable, although two species-specific features do need to be considered when extrapolating neurotoxicity data from animals to humans. First, relative to Rodents, the ratio of white matter to gray matter is higher in Carnivores and Primates due to their greater surface areas and larger numbers of interneurons, which require a correspondingly larger supply of axons to innervate the tissue. The volume of white matter is greater in the dorsal funiculus (i.e., proprioceptive and tactile tracts) of Carnivores and Primates compared to Rodents because the extra fibers are predominantly dedicated to sensation in the digits. Second, the organization of the spinal motor tracts that support voluntary motor activities varies among species. In Rodents the main corticospinal tract is located within the ventral portion of the dorsal funiculus, while the equivalent tract in Carnivores and Primates is localized to the lateral funiculus. The rubrospinal tract (which carries subcortical signals from the red nucleus to spinal cord motor neurons) is much smaller in Primates than in Rodents and Carnivores. The spinal cord white matter devoted to such spinal motor activities rises from approximately 10% in Carnivores to between 20% and 30% for nonhuman Primates and humans, respectively.
Evaluation of Neurotoxicity
Neuropathology assessment is an accepted “gold standard” for identifying neurotoxicants because visible neuroanatomic defects in a critical, minimally reparable system generally are considered to be adverse. Additional methods for identifying neurotoxic risks have emerged (Table 21.2), which can be used together with or occasionally in place of routine neuropathology endpoints. The choice of method(s) used to define neurotoxic risk (Table 21.3) depends on the kinds of effects that investigators suspect and/or follows a menu of regulatory guidelines.
Table 21.2
Flow Chart for Toxicologic Neuropathology Endpoints in Conventional Nonclinical Toxicity Testing

Table 21.3
Potential Endpoints of Neurotoxicity

Some modalities for assessing neurotoxicants and their effects are performed during life, like neurological and behavioral evaluations as well as electrophysiological testing. Others are usually confined to the postmortem setting, such as macroscopic and microscopic assessment (routine neuropathology). A few can be used in life as well as after death, like noninvasive imaging and assessments of nerve conduction velocities. This section provides an overview of conventional anatomic pathology techniques as well as a brief introduction to other key methods in which toxicologic neuropathology expertise may be important.
Morphologic Evaluation
Basic Patterns for Neurotoxicant-Induced Lesions
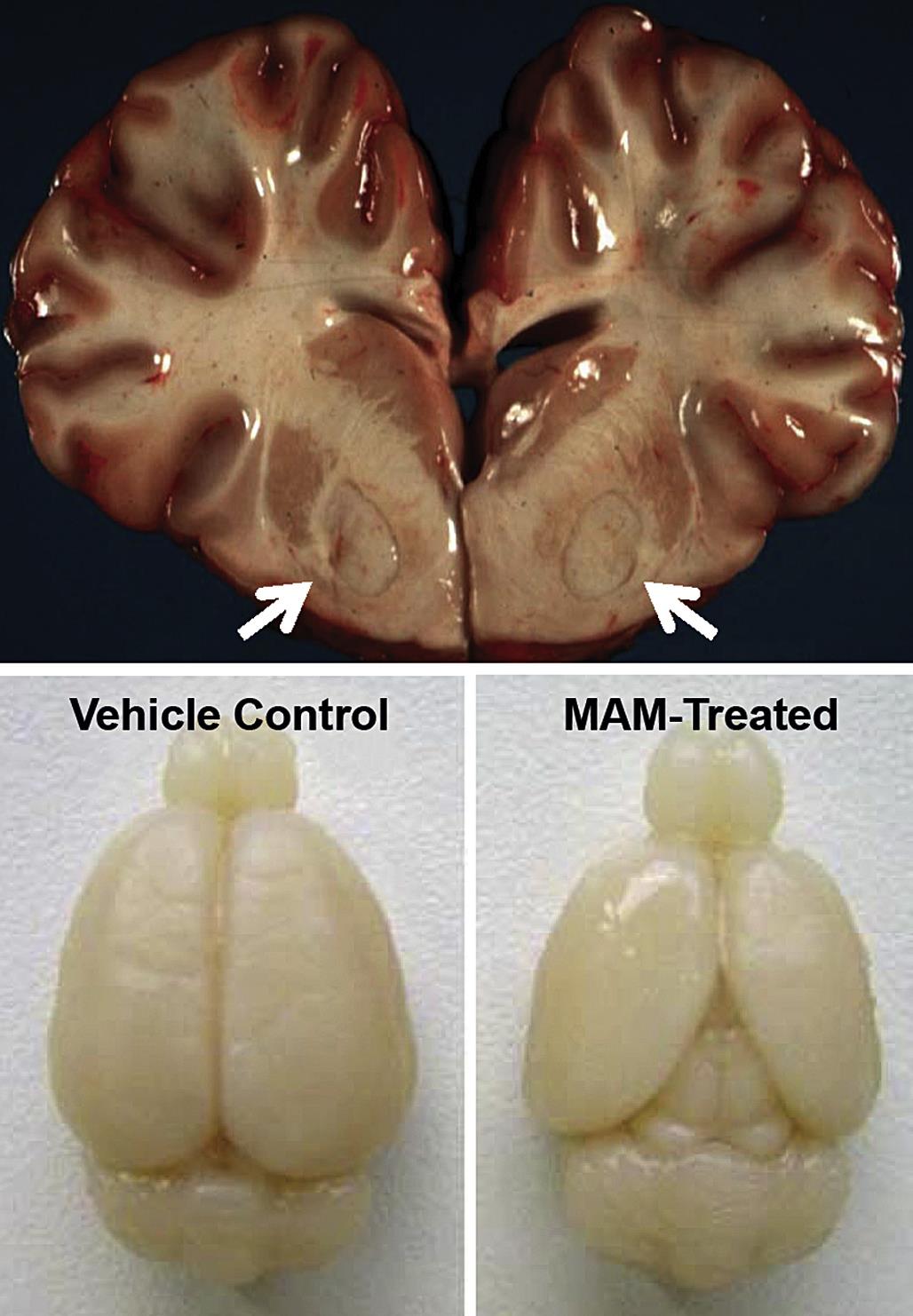
Gross observations may reveal toxicant-induced alterations in neural structure at the time of necropsy. An obvious qualitative change that may reveal a neurotoxic effect is discoloration, which demonstrates the presence of necrosis (Figure 21.4) or a neoplasm. More quantitative gross evidence of a neurotoxic outcome is indicated by reduced brain weight (measured at necropsy immediately after organ removal or in fixed organs prior to tissue trimming) or alterations in the dimensions (length, area, or volume) of the brain or a major subregion (Figure 21.4). Generally abnormal absolute brain weights or brain dimensions are judged to be biologically significant markers of neurotoxic damage.
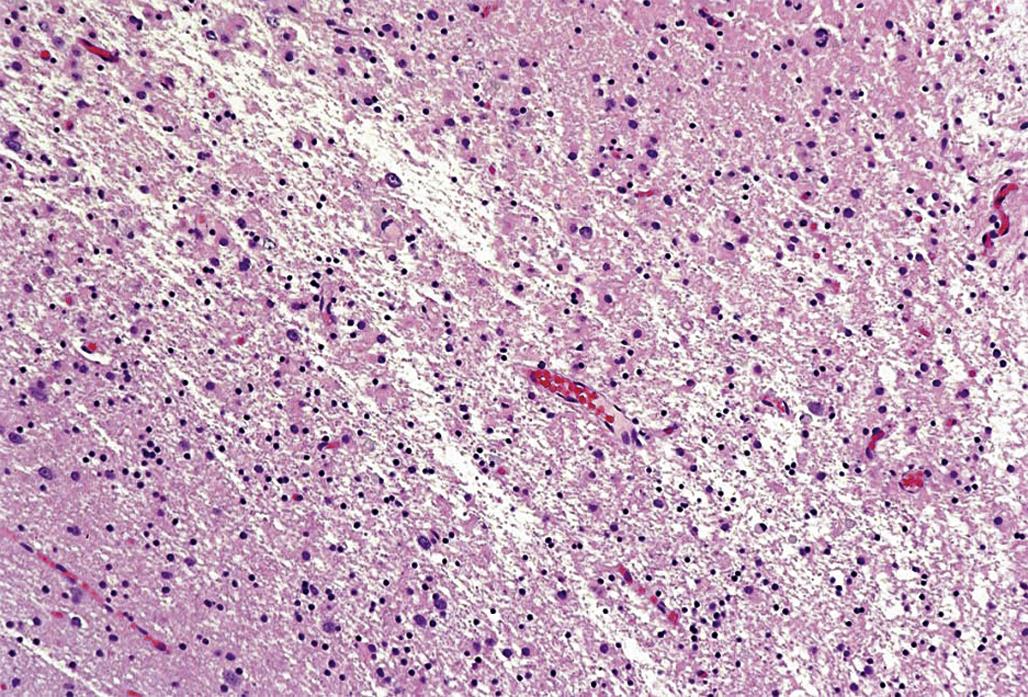
Histopathologic lesions are a common manifestation of neurotoxicant exposure, and indeed microscopic assessment is often the most effective means for identifying many neurotoxicants. The type of histopathological finding will depend on many factors. Perhaps the most important is the age of the lesion. Acute cell death (neuronal necrosis) usually presents as shrunken neurons with hypereosinophilic cytoplasm and dark, condensed, or fragmented nuclei (i.e., classic “red dead” neurons; Figure 21.5). In contrast, chronic lesions typically result in neuronal loss (which may go unrecognized if subtle and in the absence of a glial reaction); coagulative necrosis of the parenchyma (Figure 21.6); neuropil cavitation (i.e., the end-stage finding caused by liquefactive necrosis of the parenchyma); or a prominent glial response (gliosis). The difficulty in detecting small numbers of dead neurons or disintegrating axons microscopically in the “vast pink wasteland” of a large brain section has driven the development of special techniques designed to preferentially label toxicant-injured cells. The two most common methods utilized for this purpose are Fluoro-Jade dyes and silver degeneration procedures (Figure 21.7). These two procedures work best when the cell bodies remain intact, and thus are of most use for detecting acute damage.
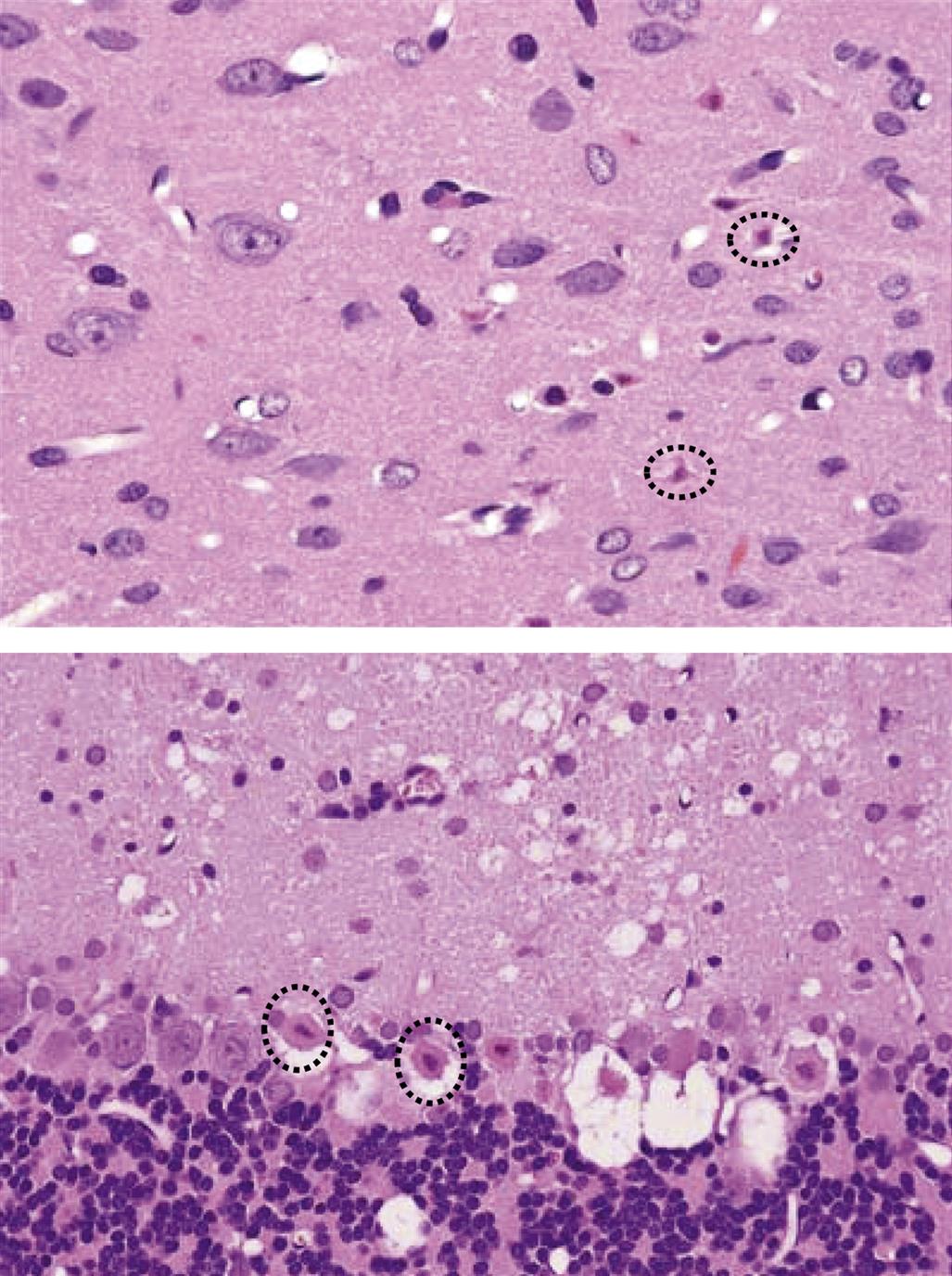

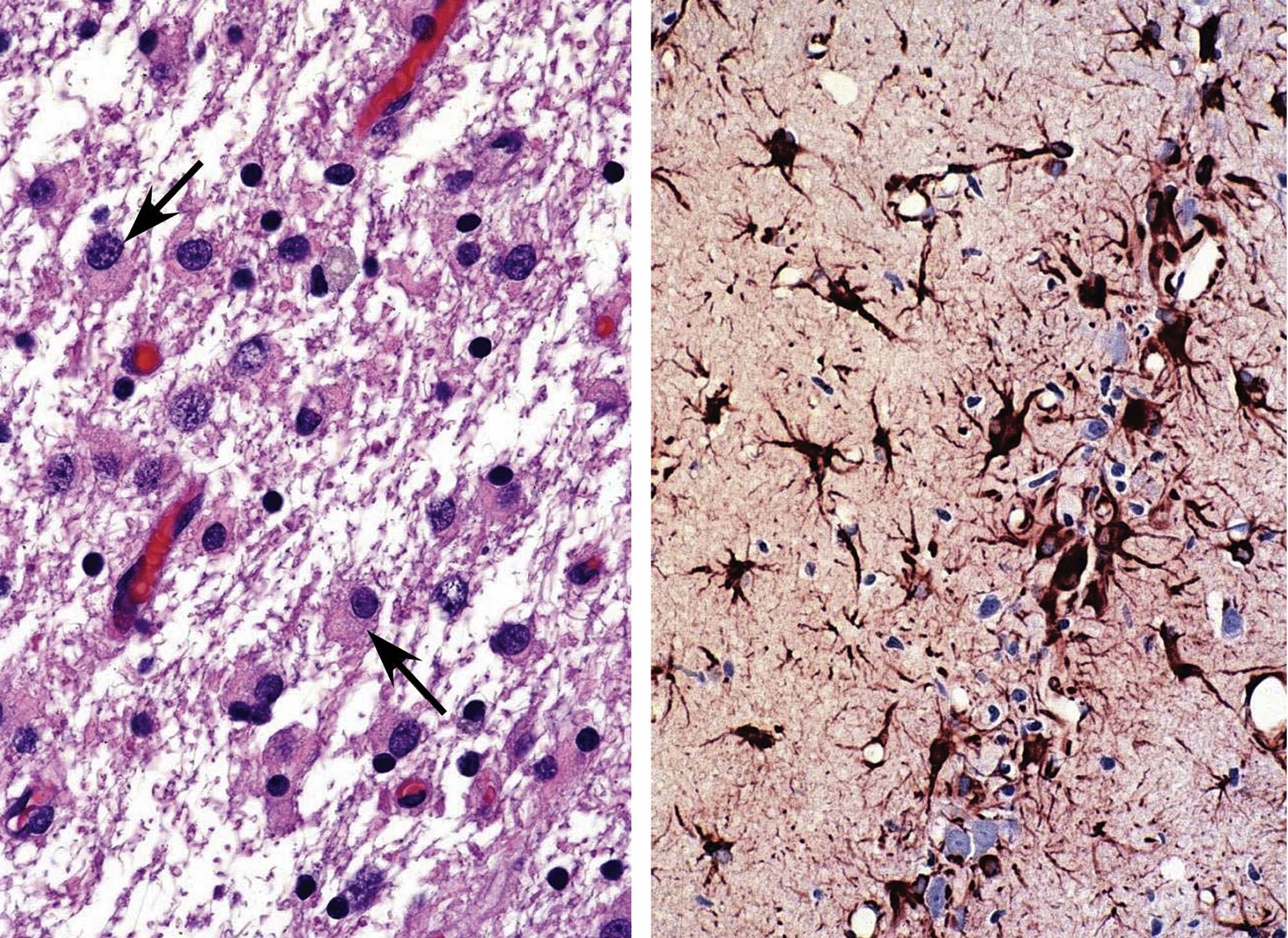
Damage to a neuronal population usually is accompanied by an increase in the number of reactive glia in the immediate vicinity of the stricken cells. The typical glial response features astrocytes (Figure 21.8) and/or microglia (Figure 21.9), either or both of which may become larger (hypertrophy) and/or proliferate (hyperplasia). These reactions take place to render the glial cells more adept at filling defects, supporting other local cells, or efficiently removing necrotic neuronal, axonal, and myelin debris. As with injured neurons, reactive glia may be detected more easily if visualized using a special method to reveal the responding cells. The most common means for detecting cell type–specific markers on reactive glia are conventional immunohistochemical (IHC) techniques to detect the astrocyte-specific glial fibrillary acidic protein (GFAP; Figure 21.8) and the microglia-specific ionized calcium-binding adaptor molecule 1 (Iba1; Figure 21.9)
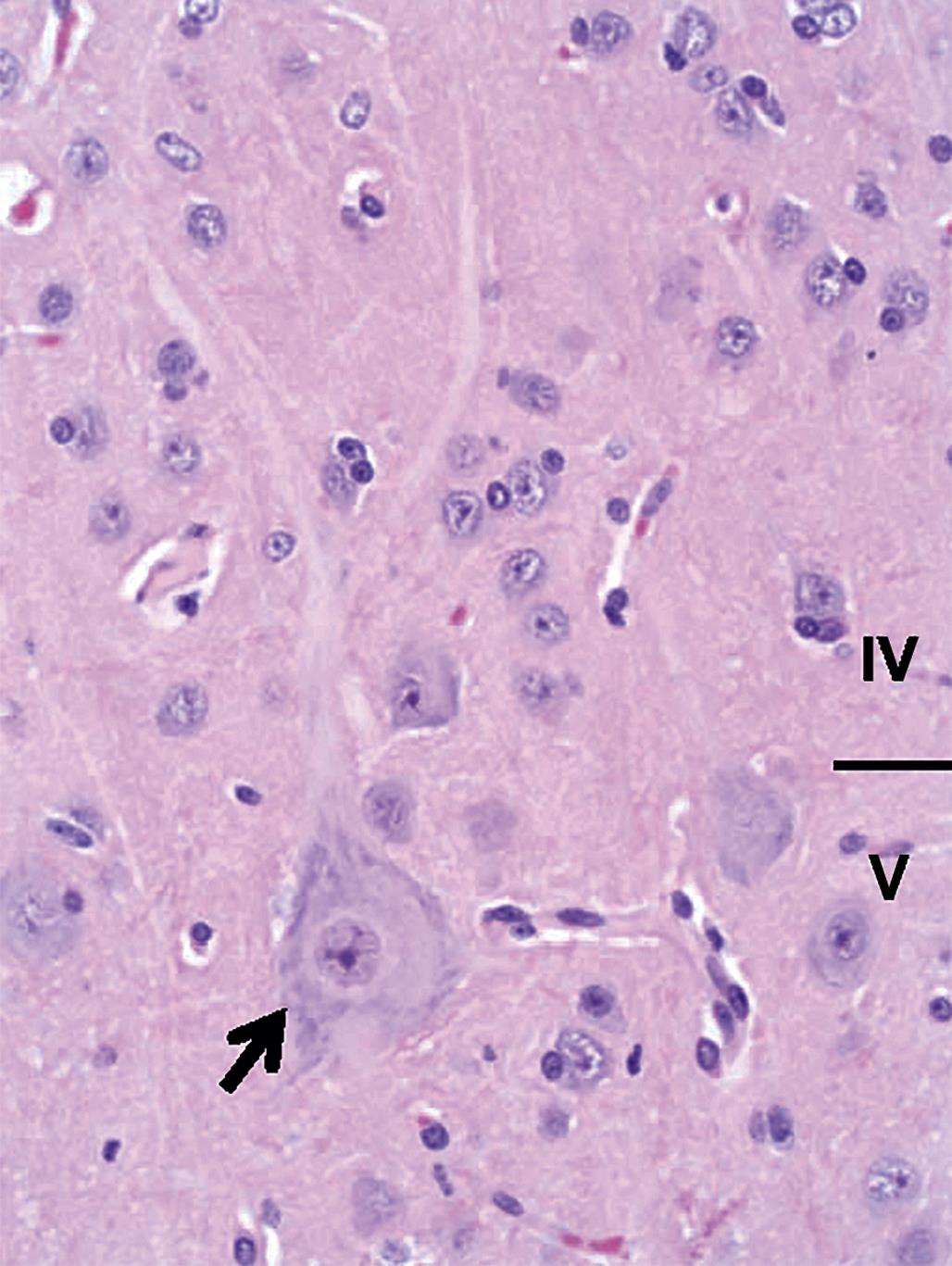
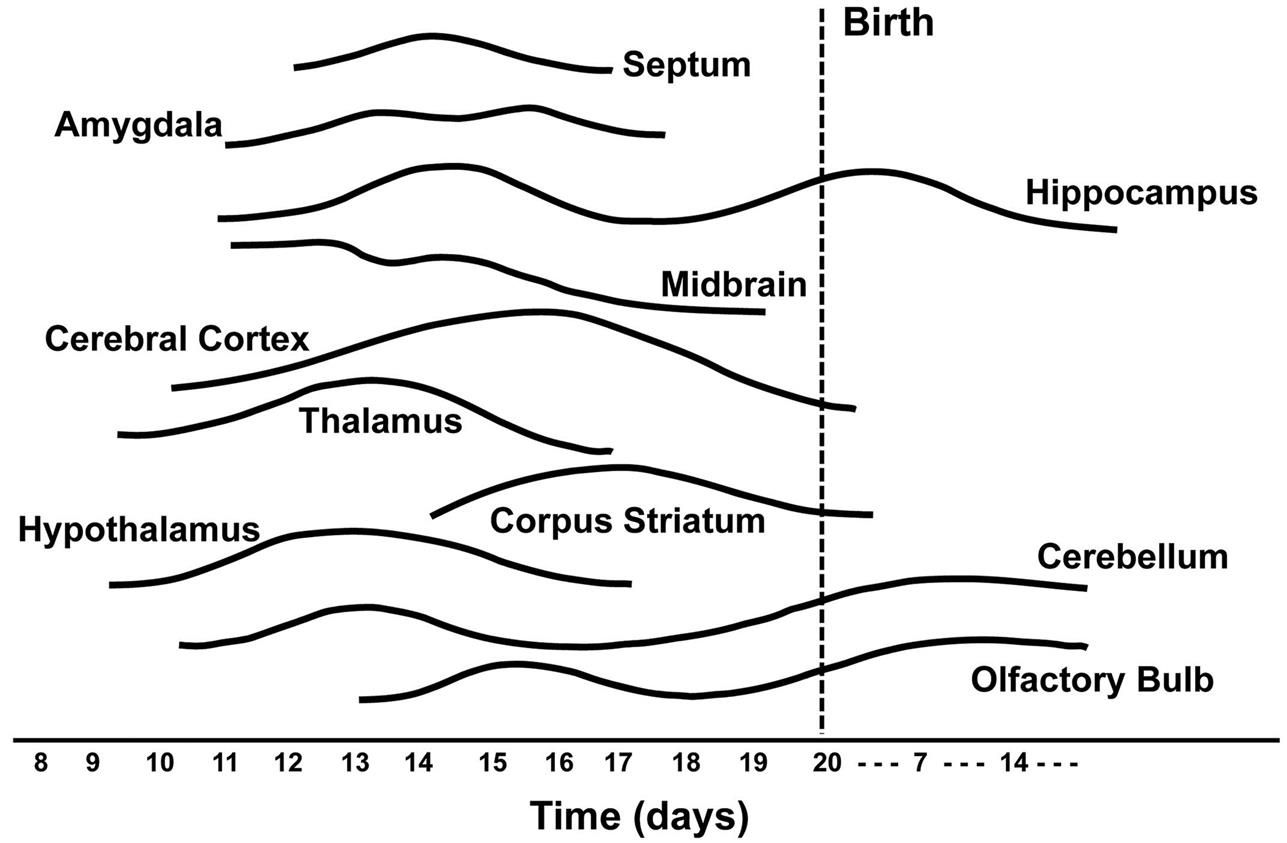
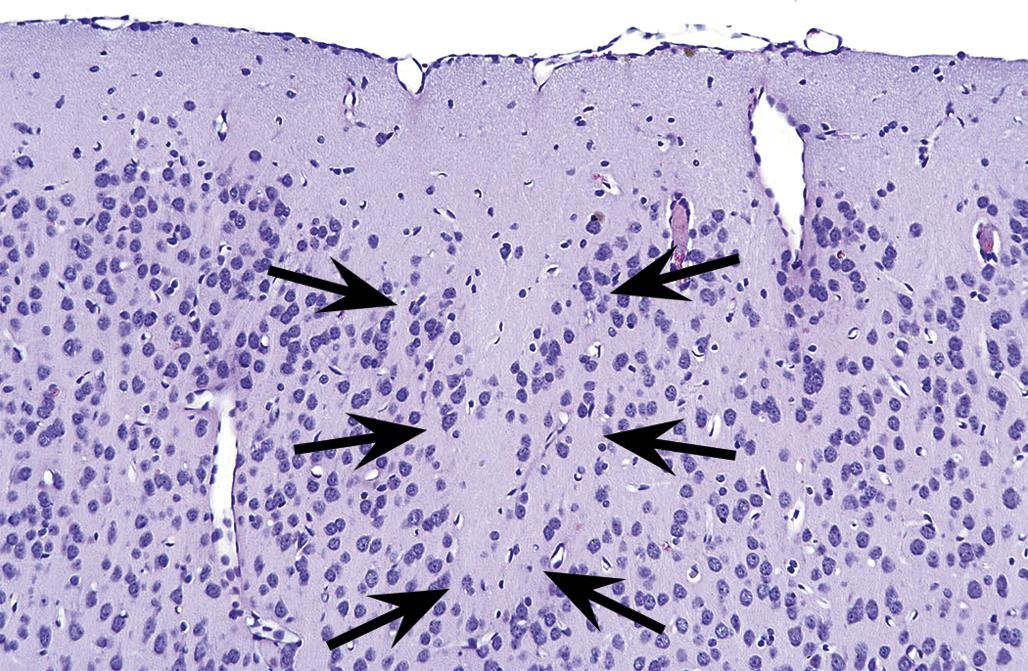
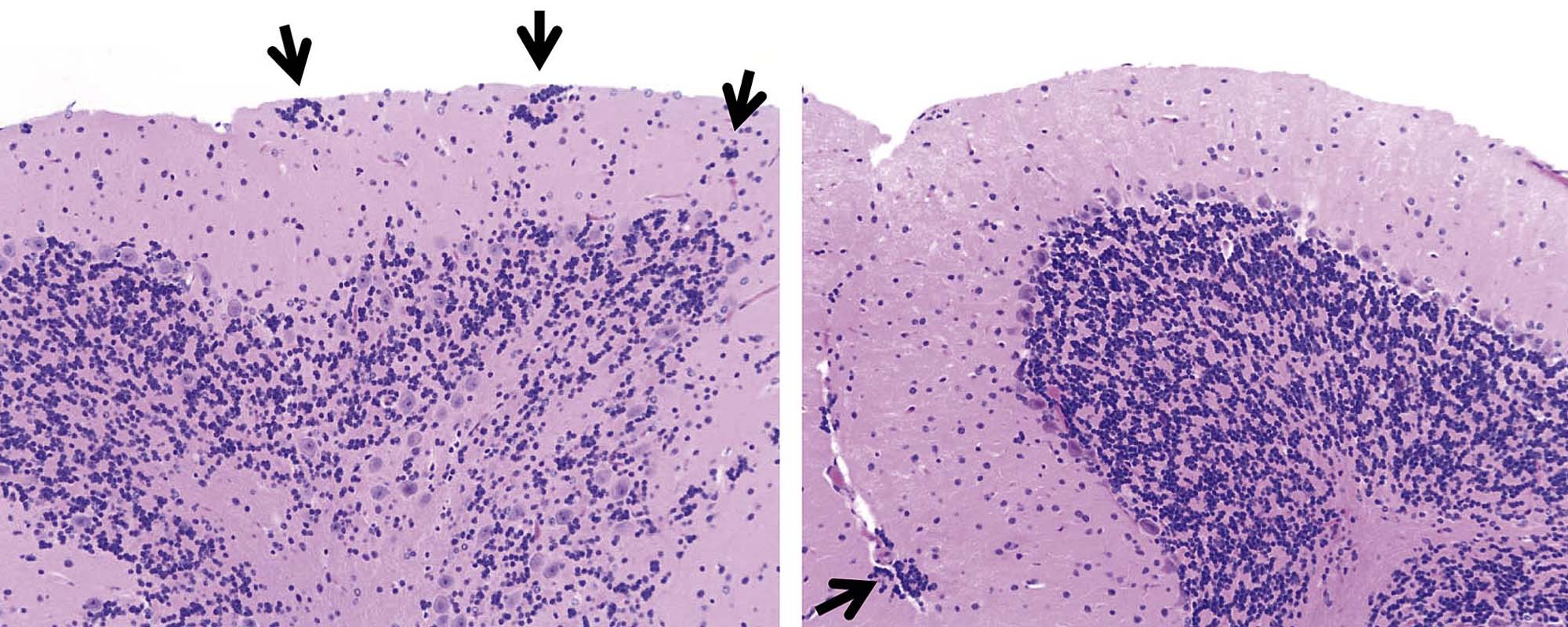
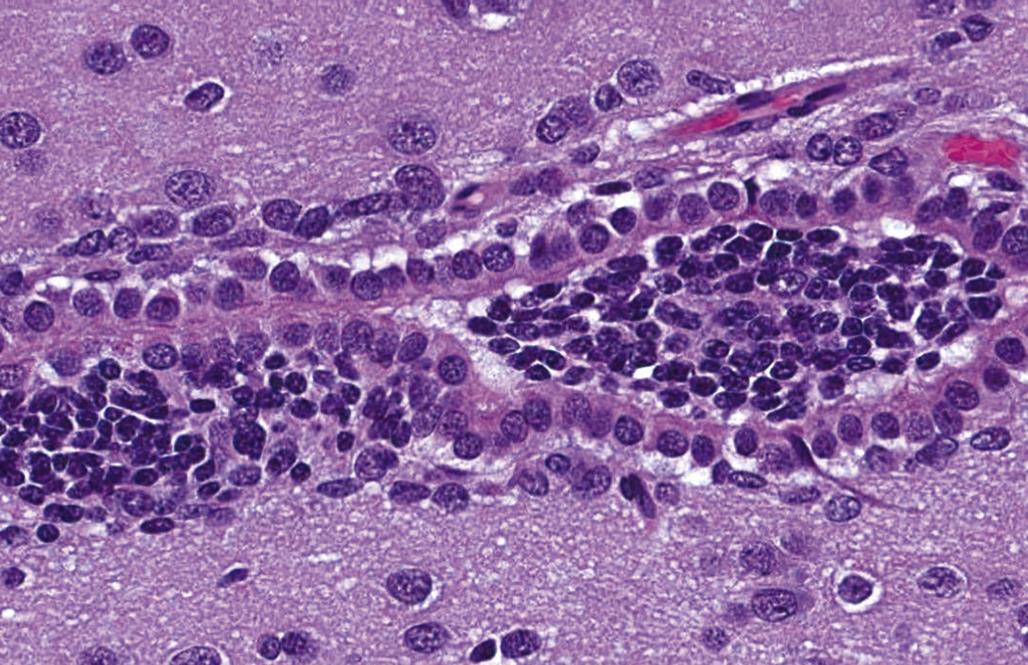
The time during life when exposure occurs will influence both the sensitive cell populations and the potential range of lesions. Different brain regions have unique windows of vulnerability based on when they undergo their peaks of neuronal (Figure 21.10) and glial production and differentiation during development, so transient exposures during different periods of life will elicit divergent outcomes. Adult exposures to neurotoxic agents typically yield necrotic neurons, axonal or myelin degeneration, neuropil vacuolation, and/or reactive gliosis. In contrast, changes usually seen in developing individuals include abnormal cell numbers (termed “dysplasia”; Figure 21.11), displaced (or unmigrated) cells (termed “ectopia” or “heterotopia”; Figure 21.12), and/or aberrant differentiation (indicated by defective myelination or synaptogenesis), but rarely glial reactions; this difference from the adult lesion pattern results from the constant remodeling that occurs during formation of the nervous system. In general, dysplastic and ectopic neurons persist into adulthood. However, some ectopias appear to be transient in nature as indicated by the ability to find aggregates of displaced neural stem cells in brain ventricles of young animals (Figure 21.13) but not adults.
The distribution of lesions among various neural cell populations often depends on cell type–specific architectural or functional factors. For instance the rich synaptic beds in the cerebral and cerebellar cortices as well as the hippocampus make these three regions common targets for neurotoxicants. The cell type–specific factors that direct neurotoxic effects to a particular area also help to explain species differences in responsiveness to various agents. The classic example of this principle is the heightened sensitivity of dopaminergic neurons in the Primate substantia nigra to the prodrug 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Conversion of MPTP into the toxic metabolite 1-methyl-4-phenylpyridinium (MPP+) leads to widespread acute neuronal death, which manifests as acute-onset parkinsonism. The ability of neuromelanin, a dark pigment found specifically in the dopaminergic neurons of the Primate substantia nigra, to preferentially bind both MPTP and MPP+ accounts in large part for the higher susceptibility of primates relative to rodents to MPTP-induced neurotoxicity (although some mouse strains are also quite sensitive to MPTP administration).
Study Design
Toxicant-induced structural lesions in the CNS and PNS may be assessed as a stand-alone problem (the usual case in a diagnostic laboratory setting) or as an organized, prospective study of neurotoxic potential (the typical case in the regulated setting for developing new agricultural chemicals or drugs). In general, diagnostic neuropathology is undertaken in a clinical practice (e.g., medical or veterinary medical office) to ascertain whether or not a xenobiotic can be linked to neural damage. The objective of such diagnostic procedures is to facilitate the return of the patient to full (or reasonable) neurological function, so morphological assessment of the nervous system is confined to nonintrusive imaging modalities like computed tomography (CT) or magnetic resonance imaging (MRI), electrophysiology to assess nerve conduction velocities, or to minimally invasive pathology methods like peripheral nerve or skin biopsies (for assessment of intraepidermal nerve fiber density). These techniques are increasingly used in assessing the nervous system in regulatory studies. However, neural evaluation in the majority of regulatory-type nonclinical studies is done either as one aspect of a general screen for toxicity to all organ systems (i.e., “general toxicity studies”) or to provide a comprehensive examination of structural damage in many nervous system sites (i.e., “dedicated neurotoxicity studies”). The differing aims of these studies warrant a smaller list of neural tissues for general studies relative to the larger battery of nervous system domains used for studies devoted mainly to a detailed neuropathology analysis.
It is impossible to define a single duration of exposure that is suitable for investigating all neurotoxicants, as different agents elicit a fluctuating spectrum of lesions that peak in incidence and degree at various times after exposure. When known in advance, the timing of necropsies should be arranged to occur when toxicant-induced neural lesions are at their peak, keeping in mind that many agents exhibit multiple peaks characterized by distinct kinds of lesions. This principle is well illustrated by the excitotoxic agent kainic acid, in which neuronal necrosis occurs at 1–4 days following exposure but synaptic terminal disintegration develops at 4–14 days. In many cases, neurotoxicity studies are designed to recapitulate specific requirements listed in regulatory guidelines. Testing requirements are relatively specific for dedicated neurotoxicity studies but permit a fair degree of discretion for general toxicity studies.
Tissue Fixation
Neural tissues for general toxicity studies typically are fixed by immersion in neutral buffered 10% formalin (NBF, a 3.7% formaldehyde solution, approximately pH 7.4). This choice is dictated by historical and practical considerations. NBF is available in bulk from many vendors. The small size of the formaldehyde molecule permits it to easily and quickly penetrate dense neural tissues, while the reactive aldehyde group promotes rapid tissue fixation. Nervous system samples fixed in this manner may be processed routinely along with samples from other organs, thus reducing the cost and labor required to prepare tissue blocks for histological sectioning.
For dedicated neurotoxicity studies, the preferred choice for preserving neural tissues for microscopic examination is intravascular perfusion because it offers the best preservation of neural morphology in all CNS and PNS structures (Figure 21.14). Perfusion introduces fixative deep into the nervous system, thus speeding fixation, and it prevents the induction of many artifacts that tend to arise when unfixed CNS tissues are manipulated during necropsy. This method permits the use of fixative mixtures that include methanol-free formaldehyde (often freshly prepared from paraformaldehyde powder) and/or glutaraldehyde. Combinations of formaldehyde and glutaraldehyde are sometimes referred to as McDowell-Trump’s fixative (usual composition: 4% formaldehyde and 1% glutaraldehyde) or various modifications of Karnovsky’s fixatives (example: modified Karnovsky’s fixative, which is typically 2% formaldehyde and 2.5% glutaraldehyde).
Glutaraldehyde is commonly used for preserving cell organelles for ultrastructural analysis. This molecule is a more effective cross-linking agent since it has two aldehyde moieties, but its length greatly slows tissue penetration. Furthermore, glutaraldehyde alone or in combination with formaldehyde enhances the stabilization of lipid-rich membranes, especially myelin. Perfusion fixation typically is conducted at pressures ranging from 70 to 120 mm Hg (i.e., the peak intravascular pressure during systole). Other critical aspects of perfusion fixation—buffering capacity, osmolarity, pH, and temperature—have been discussed in greater detail in other publications (see Further Reading for details). After a suitable period of time following perfusion fixation, neural tissues are removed from the carcass. In general, they are postfixed by immersion for an additional period in NBF. Specimens slated for detailed analysis of myelin and/or electron microscopy analysis are often postfixed a second time in 1% osmium tetroxide (OsO4) to further stabilize lipid-rich membranes prior to embedding in hard plastic. Osmium postfixation even enhances a pathologist’s ability to evaluate myelin sheaths in paraffin-embedded cross-sections of nerve as osmium metal forms dark (and electron-dense) deposits in lipid-rich myelin laminae. Fixation in OsO4 must be performed in a chemical hood as osmium vapors are highly irritating and also readily bind to and discolor the corneal surface and skin.
Immersion-fixed neural tissues often exhibit artifacts associated with handling at necropsy and/or suboptimal processing. Examples include dark neurons (Figure 21.14), myelin bubbling, and neuropil vacuolation (all of which are discussed in more detail later). Their presence does not preclude evaluation of neural tissue, but greater care must be taken to avoid false-positive results (i.e., interpretation of artifactual changes as genuine lesions) and false-negative outcomes (i.e., the inability to detect or properly construe modest lesions that may be camouflaged by more widespread artifactual changes).
Many molecular pathology procedures may be performed in NBF-fixed tissues, but certain IHC techniques and polymerase chain reaction (PCR) often require harvesting of unfixed tissue. Intravascular perfusion with ice-cold, buffered physiological saline may decrease the core temperature enough to lessen postmortem autolysis until the specimens can be removed and frozen. In some instances, reduced-strength fixatives (e.g., 1% or 2% formaldehyde) may be used to provide better tissue preservation while retaining the ability to retrieve labile antigens, although a pilot study is recommended to ensure that the altered fixation protocol will support the desired endpoints. In terms of fixative choice, it is best to start with prior knowledge of the required endpoints, then preserve the necessary tissues accordingly.
Brain Weights
Brain weight offers two major advantages as an endpoint for detecting neurotoxicity. First, brain weight is inherently quantitative and objective, which removes a potentially major source of bias in its acquisition and interpretation. Second, weights may be acquired at necropsy to provide a fast and simple clue that a neurotoxic event has occurred. Brain size typically is evaluated both as a measured absolute brain weight and as a calculated "relative weight" (most often reported as a ratio of brain weight to total body weight).
Brain weights may be taken at necropsy on fresh (i.e., unfixed) or perfusion-fixed organs or after some period of fixation. Either option is considered acceptable as long as weights for all subjects in a given study are taken from organs treated in the same fashion. The handling needed for weighing can induce several artifacts in some brain regions, so in dedicated neuropathology studies separate cohorts of animals for each treatment group are needed to obtain brain weights and perform histopathological assessments.
The key to acquiring reproducible brain weight data sets is to ensure that all organs are harvested in a consistent manner. For example, the olfactory bulb of rodents comprises 6%–7% of the brain weight, so this region should always be included or excluded when building a study data set. Similarly the brain should be separated from the spinal cord at a standard position. Typically a visible external landmark, such as the obex (the point on the dorsal medulla oblongata at which the fourth ventricle closes to become the central canal of the spinal cord), is used as a visual marker in order to provide long-term consistency in tissue harvesting.
Tissue Collection and Trimming
Neural tissues are prone to artifacts if handled roughly or excessively during necropsy. For instance, prompt removal of the brain from the cranial vault, even shortly after intravascular perfusion with fixative, can increase the propensity for artifactual cell shrinkage and increased amphophilia (i.e., able to bind both acidic [e.g., eosin] and basic [e.g., hematoxylin] dyes) that are the hallmark features of “dark neurons” (Figure 21.14). A better technique for reducing brain microanatomy artifacts is to perfuse the subject with fixative followed by careful removal of the skullcap and submersion of the entire head (with brain left in place) into fresh fixative for at least 24 hours before removal. If the brain is removed rather than fixed in situ, immediate immersion into a suitable fixative is preferred relative to delays associated with additional handling (e.g., for weighing).
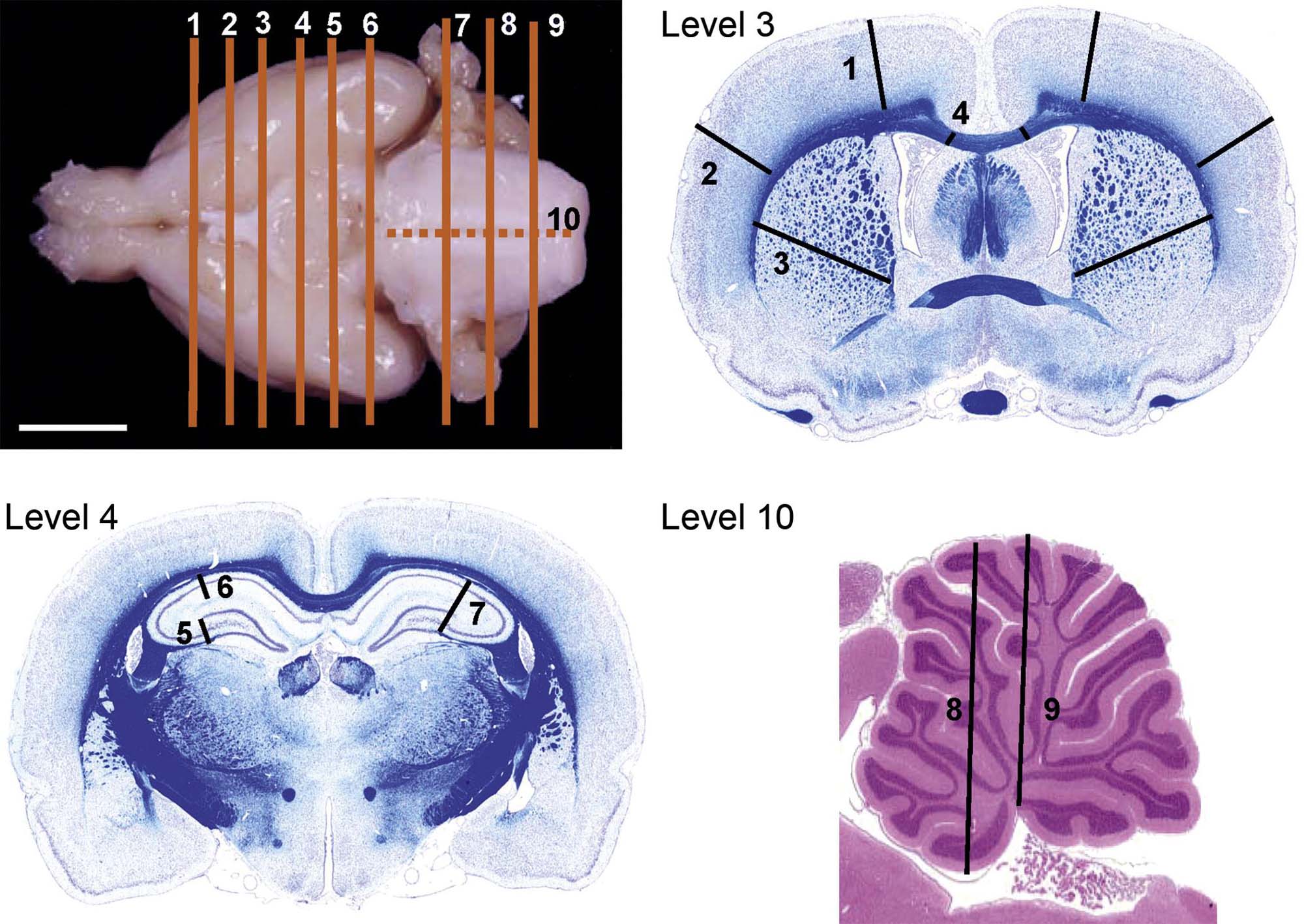
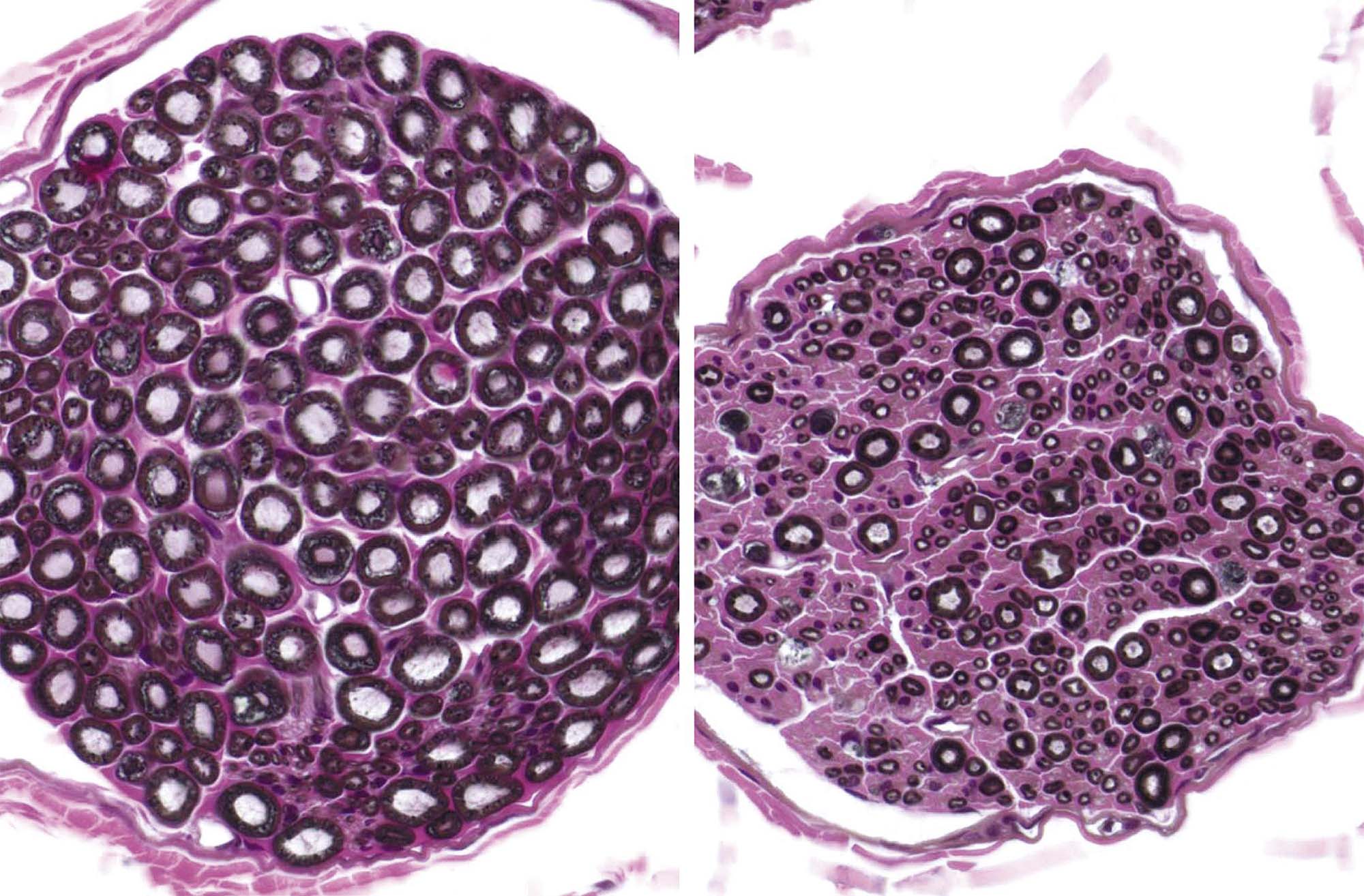
In general, toxicologic neuropathology requires sampling of the CNS and PNS at multiple sites. The minimal approach to sampling often is a matter of institutional preference. For example, common brain sampling schemes for general toxicity studies are 3–7 full coronal (“cross” or “transverse”) brain sections or one mid- or parasagittal (longitudinal) section and 3–5 coronal hemisections. Dedicated neurotoxicity studies typically collect double to triple this number of sections. Trimming the brain and spinal cord using freehand coronal sections, in which planes for sample acquisition are defined using reproducible external or internal anatomic landmarks alone, is a fast and reliable approach for screening studies. Brain molds (i.e., acrylic or metal matrices with multiple parallel slots) may be used if desired when all organs from the test group are of suitable size to fit the apparatus (typically true for adults but not juveniles). The advantage of this latter approach, especially when using fixed brains, is that the slabs can be made quite thin by placing blades in adjacent slots of the mold. However, one caution is that slicing tissue, especially brain, too thin may make it difficult to obtain a full section at microtomy; this is especially true when producing full transverse sections of the brain from primates, canines, and other large animals. The spinal cord and nerves should be examined in both longitudinal (either parasagittal or oblique) and transverse sections as damage to axons in these tissues is often best showcased in the longitudinal orientation.
The keys to proper nervous system sampling are to make sure that critical sites serving major functions and that are targeted by known toxicants are taken. These sites include (as listed alphabetically) the basal nuclei (specifically the caudate/putamen region), cerebellum, cerebral cortex (multiple regions), hippocampus, hypothalamus, medulla oblongata, midbrain, olfactory bulb (especially in rodents), pons, and thalamus in the brain; three segments of spinal cord (cervical, thoracic, and lumbar); dorsal root ganglia; peripheral nerves; and the eye (for retina) with attached optic nerve (i.e., cranial nerve II). Additional sites may be taken if warranted by other factors (e.g., the presence of neurological signs or a known tendency for agents with a given structure or mechanism to target a specific neural domain). Organs should be sampled so that the resulting tissue sections are fairly homologous (i.e., exhibit the same structural landmarks) among animals both within and across studies. The tissue dissection strategy, both at necropsy and during trimming, should progress in a similar manner between animals and across studies to enhance consistency and to avoid missing tissue regions.
Neurohistological Procedures
In general, CNS and PNS samples are embedded in paraffin. The advantage of this approach is that neural tissues can be processed along with other organs. Some regulatory guidelines require that PNS tissues instead be embedded in plastic (usually glycol methacrylate (GMA), i.e. “soft plastic”) or hard plastic resin (e.g., araldite, epon, Spurr’s). Soft plastic or resin embedding allows tissues to be sectioned at thicknesses of 1–2 μm rather than the standard 4–8 μm used for paraffin. If properly fixed and prepared, thinner sections may yield better resolution of fine architectural detail during light microscopic evaluations, especially for nerves (Figure 21.15), but in our experience significantly improved resolution is achieved only using hard plastic. The increase in resolution provided by osmium postfixation and resin embedding is of great benefit whenever there is a need to visualize myelin sheaths (and compare sheath thickness to axon size) or to detect myelin or axon degeneration or regeneration. Many studies rely on paraffin embedding and H&E stains, which at least in longitudinal sections generally provide a suitable screen for the presence of axonal degeneration.
The histological analysis of neural tissues typically is performed in a tiered manner. The examination starts with sections stained using hematoxylin and eosin (H&E), which permits the general evaluation of cytoarchitectural features. Where needed, pathologists will request serial sections stained using special neurohistological methods to reveal additional detail. Such special stains illuminate particular cell populations or probe potential processes that have been shown to be vulnerable to toxicant-induced disruption. Nissl stains (e.g., cresyl violet (Figure 21.16), thionine) used to detect RNA bound to the rough endoplasmic reticulum (Nissl substance) show the fine cellular details of more neuronal populations than does a simple H&E stain although the standard H&E stain also highlights (to a lesser degree) the Nissl substance. Reduced Nissl staining indicates that neuronal function has been impaired.
The health of myelinating cells is examined using methods that label intact myelin, such as Luxol fast blue (LFB, a histochemical method) or myelin basic protein (MBP, a marker protein localized using an IHC method). Neurons and myelin can be visualized in the same section by combining stains (e.g., cresyl violet with LFB (Figure 21.16)). Toxicant-induced neuronal death and/or axonal degeneration can be revealed using fluorescent (e.g., Fluoro-Jade) and metallic enhancement (e.g., amino cupric silver) procedures that collect preferentially in damaged cells (Figure 21.7). Sites of neural injury can also be defined using special techniques to reveal activated glia (e.g., enhanced GFAP (Figure 21.8) or Iba1 (Figure 21.9) expression) collecting at sites of prior neuronal injury.
Contrary to conventional wisdom, labor, money, and time may actually be saved by designing neurotoxicity experiments so that a battery of neurohistological stains are produced automatically as the first tier of the neuropathology examination (e.g., H&E, Fluoro-Jade, and anti-GFAP), if only for the high-dose treatment group and the negative control cohort to start. The reason for this choice is that potential neurotoxic lesions of a subtle character can be more readily identified and rapidly evaluated if sections have already been produced and stained in advance. While the other stains often reveal changes that were not readily seen on the H&E-stained sections, the most important aspect of any evaluation of the nervous system is the preparation and evaluation of an adequate number of sections from multiple CNS and PNS regions to allow for hazard identification and risk assessment. In blunter words, regardless of the special stains used, it is not possible to visualize an abnormality in a portion of the brain, spinal cord, ganglion, or nerve that is not prepared and examined.
Morphometry
Quantification of neural features in samples that are acceptable for performing measurements in two dimensions (2D)—for instance, fiber diameters in nerve cross sections or thicknesses of brain regions as performed in developmental neurotoxicity studies—is a conceptually simple but moderately laborious means for obtaining objective data during a neuropathology evaluation. Common 2D means for quantification include linear (e.g., lengths, widths, heights, perimeters) and area assessments, typically taken at multiple levels (Figure 21.17). Examples include lengths (e.g., greatest mid-axial length of the dorsal cerebral cortex) and areas (e.g., dimensions of the dorsal profile of the cerebellar hemispheres) of major regions in intact brains; thicknesses or areas of particular domains on histologic sections (e.g., height of the cerebral cortex, width of the basal nuclei); and counts of small structures (e.g., cells in a given nucleus, axonal density). To be valid, such measurements must be gathered on highly homologous samples (e.g., tissue sections with comparable internal landmarks), and ideally using a coded (“blinded” or “masked”) analysis in which the investigator does not know the treatment history for the specimens.
Quantification of objects in three-dimensional (3D) structures, such as neuron counts in dorsal root ganglia or in specific brain nuclei, requires a stereological investigation. Investigation design is exceedingly important for this category of specialized analysis. Particular emphasis must be placed on randomly but systematically examining the entire structure, with a sampling parameter that ensures that each and every object of interest has, at the beginning of the study, an equal opportunity of being counted/measured. Stereology generally is employed to answer specific questions rather than as a screening tool for potential neurotoxicity due to the slow and labor-intensive methodology (as described below).
Teased Fiber Preparations
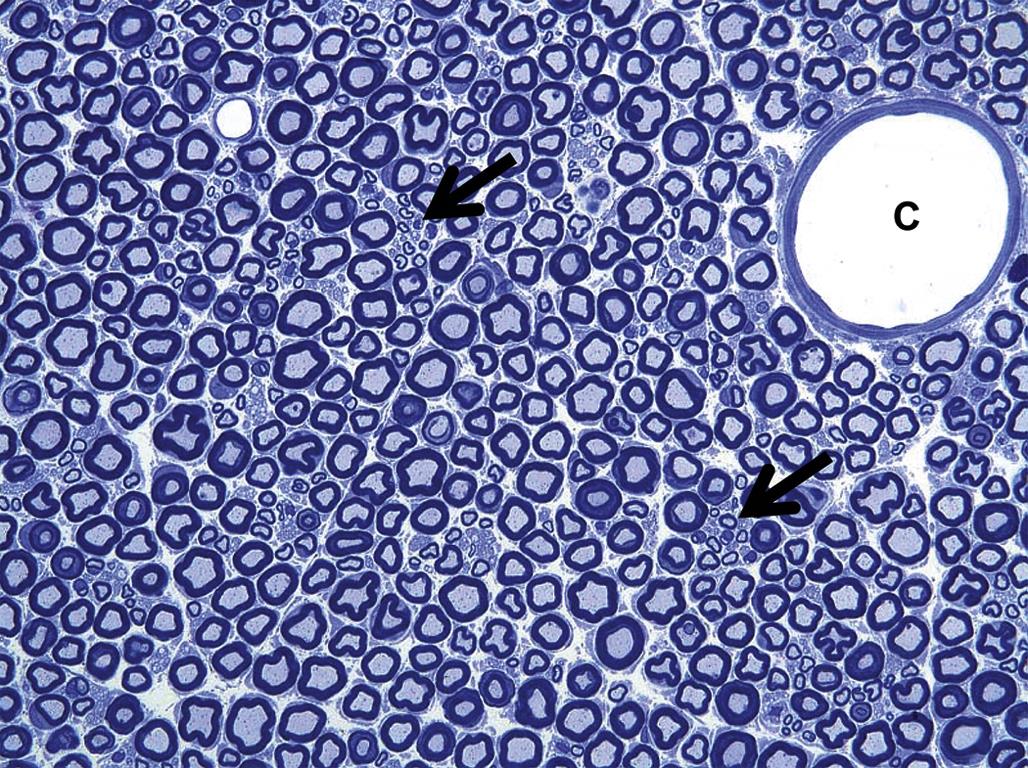
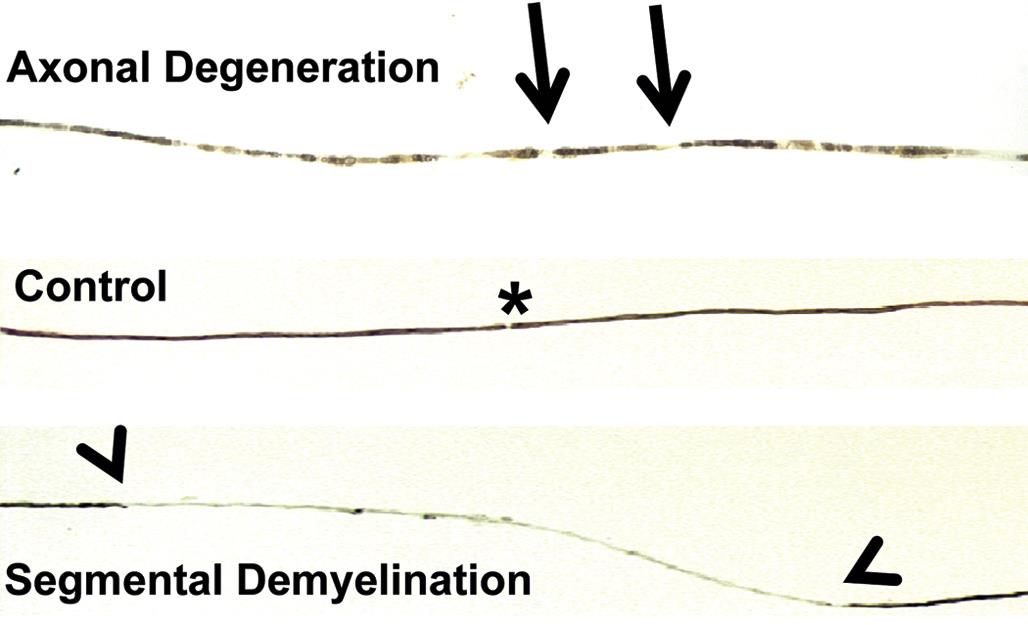
Isolated nerve fibers, consisting of a single axon and its myelin sheath obtained from a nerve, can be evaluated for lesions over long distances (typically 0.5–1 cm). Details of the nerve fiber teasing method are beyond the scope of this chapter, but in brief nerves are fixed by immersion in an aldehyde, separated into individual nerve bundles using forceps, and then postfixed in 1% OsO4 to stabilize and darken the lipid-rich membranes. Individual myelinated axons are teased apart in cedar wood oil under a stereomicroscope using forceps, mounted on slides, dried, and coverslipped. Features evident for inspection include the myelin integrity, the appearance of the nodes of Ranvier, and the intermodal distance, which can be used to discriminate between degenerative and regenerative processes affecting the axon and its myelin.
Special Anatomy-Based Techniques for Neurotoxicity Assessment
Given the importance of neurotoxicity as a health hazard, considerable effort is ongoing to further refine the battery of tests that are available to diagnose neurotoxic conditions in the clinical setting and/or to identify potential neurotoxicants among newly developed and existing agents to which humans and animals are exposed. Morphological changes again represent the “gold standard” for optimizing these evolving procedures.
Noninvasive Imaging
The multiple whole animal imaging modalities that have been developed in the last two decades exhibit great promise as tools for toxicologic neuropathology. Major advantages include their ability to be used repeatedly during life (albeit at low resolution), thus permitting observations of lesion progression over time, and the capacity to translate both the technology and the findings in test animals directly to humans. The main disadvantage of noninvasive imaging as applied to animal studies is insufficient structural resolution at the microscopic level, but recent technical advances permit assembly of 3D images from postmortem specimens at fairly high resolutions (down to 20 μm, i.e., magnetic resonance microscopy (MRM)), albeit using very expensive equipment.
Imaging modalities may be used to examine anatomic features or functional attributes, or to correlate functional changes to structural lesions. Technologies that provide anatomic data—computerized tomagraphy (CT), magnetic resonance imaging (MRI), MRM, and ultrasound (US)—form images using endogenous properties of cells and tissues to define the margins between structures. Platforms that yield functional data—optical imaging, positron emission tomography (PET), and single-photon emission computed tomography (SPECT)—reveal features of a specific biochemical or metabolic pathway within a given structure. In particular, PET and SPECT are powerful means of probing neural responses as they depend on selective accumulation of appropriate radiolabeled tracers (e.g., [18F]-fluorodeoxyglucose, a glucose analog) in various CNS structures. A series of 2D images is gathered and assembled into a “z-stack” using an automated algorithm to produce a 3D low-resolution representation showing where the tracers have localized. A more precise anatomic localization may be gained by aligning functional data sets from PET and SPECT in register with CT or MRI images.
Stereology
Precise and unbiased quantitative assessment of the distribution, orientation, number, shape, or size (i.e., volume) of objects in 3D is an important approach for investigating neurotoxic agents. The attraction of this strategy is that stereological methods are quite sensitive to abnormalities in the number of a given class of structures (e.g., neurons, neuronal processes), which generally is not the case for macroscopic and microscopic (including 2D morphometric) procedures that form the basis of conventional neuroanatomic examinations for toxicant-induced neuropathology. The primary disadvantage of stereology is that processing and analysis may be time-consuming. Because one requirement of stereology is the ability to specifically define the limits of the region of interest, structures that are selected most commonly for stereological analysis are domains that are common sites for neurotoxic damage, such as the pars compacta portion of the substantia nigra, basal forebrain nuclear regions (which can be identified by selective staining for choline acetyltransferase), other brain nuclei/neuronal areas whose limits can be defined, PNS ganglia, and nerve cross sections.
In general, stereology for evaluating toxicologic neuropathology focuses on two tasks. The first is defining the region of interest, and determining the size (usually the volume) of that area by measuring area (Cavalieri-based method) and average section thickness. Once the region of interest is defined, the objects of interest can be counted and/or measured using unbiased sampling techniques like the optical disector/fractionator or a physical disector. The goal is typically to count/measure 100–200 objects of a particular kind (example: neurons) throughout the specimen (example: a dorsal root ganglion) in a systematic random manner within 8–10 or more levels (example: serial or step sections of tissue) taken through the region of interest. Optical disector methods use sections with a thickness preferably in excess of 20 microns. Physical disector methods use thinner sections but rely on the use of “look up” sections for comparison to a reference section. Stereology may reveal changes not apparent during standard morphologic evaluations, such as prior neuron loss (detectable as decreased neuron counts), changes in neuron size, and loss of subsets of axons. While slow relative to standard neuropathology evaluation, the discriminative power of quantitative neuroanatomical analysis increases as one moves from gross assessments (e.g., brain weight measurement) to simple microscopic endpoints (e.g., acquisition of areal or linear dimensions using morphometric measurements) to enumeration of specific objects (stereology).
Biomarker Evaluation in Fluids and Tissues
In many cases, the initial neurological changes that might be detected following xenobiotic exposure will not be macroscopic and microscopic structural lesions or functional abnormalities but rather reflect molecular, neurochemical, and/or ultrastructural (i.e., submicroscopic) effects. Recent neurotoxicological research has focused on finding more rapid, less invasive, and less laborious means for identifying neurotoxic risks. Discovery and validation of novel biomarkers has been a particularly active area of inquiry in this regard.
Clinical Pathology
Changes in peripheral blood samples (e.g., whole blood, plasma, serum) generally are uninformative in terms of characterizing neurotoxic changes, but CSF analysis may be of use in certain instances. The utility of CSF as a substrate stems from its link to interstitial fluid (ISF), which is the principal liquid constituent in the brain and spinal cord immediately surrounding neurons and glia. Collection of CSF is less invasive and injurious to the CNS parenchyma than acquiring a neural tissue sample. Common assays include biochemical tests (e.g., protein levels, concentrations of neurotransmitters and their metabolites) and the evaluation of cell numbers and morphology. In general, CSF collection in rodents is confined to satellite groups of animals as the anesthesia protocols required for this evaluation may impact other endpoints.
Analysis of CSF has reasonable sensitivity but low specificity as an indicator of CNS health. The reason is that the range of aberrant changes in this fluid is narrow relative to the many possible neurological diseases, particularly if the analysis is limited to assessing total and differential cell counts and/or protein concentrations. Diseases in the meninges and neuropil adjacent to the ventricular system (the “CSF compartment”) usually produce greater variance in CSF parameters than do diseases in the deep parenchyma of the brain and spinal cord (the “ISF compartment”). Since most neurotoxic lesions affect the deep neuropil, properties of CSF samples from individuals with neurotoxic conditions generally are within normal limits.
Neurochemistry
Neurochemical measurements are common endpoints in neurotoxicological studies. Such tests provide not only evidence that an agent has altered one or more neurobiological parameters but also point toward specific mechanisms of neurotoxic action.
Neurotransmitter systems are classic biomarkers. Their importance during development (as morphogens) and adulthood (as signaling molecules) stems directly from the need to properly maintain their synthesis, release, and removal. Neurotransmitters may be assessed by many means. Chemical assays (e.g., chromatography or microdialysis to define transmitter levels) and molecular biological techniques (e.g., northern analysis of RNA levels, western analysis or enzyme-linked immunosorbent assays (ELISA) for peptide transmitters, receptors, and signaling cascade proteins) can directly detect specific elements contributing to the function of a given transmitter system. The measurements reveal the quality (presence or absence) and quantity of particular elements within a given pathway but do not directly assess the functional integrity of the system. Thus increases or decreases in neurotransmitter levels induced by neuroactive agents are not necessarily indicative of a neurotoxic effect unless they occur with other functional (e.g., behavioral, electrophysiological) or structural (i.e., neuropathological) changes.
Expression of GFAP is another frequent biomarker for CNS damage, including that caused by neurotoxicants. Accumulation of GFAP results from astrocyte hypertrophy; cell expansion requires enhanced production of all cell constituents, and GFAP is key among them as the major intermediate filament protein. Biochemical assays (e.g., radioimmunoassay, sandwich ELISA, and western analysis) or IHC stains (see below) indicating increased GFAP levels are a sensitive, simple approach for confirming the presence of a CNS response to prior damage, although the cause of the change usually cannot be defined without other tests (e.g., histopathology), and sometimes not even then as GFAP expression may rise in the absence of histopathological changes. Therefore stand-alone increases in GFAP above control levels may be indicative of neurotoxicity if a history of exposure is known. An important consideration in interpreting such increases is that GFAP levels will also be higher following injurious events other than neurotoxic damage (e.g., increased corticosteroid levels, normal aging). Interpretation of decreased GFAP levels in adults or any alteration of GFAP expression in developing animals remains uncertain.
Enzyme histochemistry is another common means for detecting changes in biochemical and metabolic pathways that might serve as biomarkers for neurotoxic lesions and their mechanisms. An advantage of these methods is that they detect the presence of a functional protein. Enzyme histochemical tests that are commonly used in neurotoxicologic studies include visualization of extravasated horseradish peroxidase as a probe for xenobiotic-induced damage to the BBB and assessment of acetylcholinesterase (AChE) inhibition in fluids and tissues to indicate exposure to organophosphate (OP) or carbamate insecticides.
Toxicogenomics
The application of various “omics” disciplines including assessments of DNA (genomics), messenger RNA (transcriptomics), or protein (proteomics) responses is increasingly used to define early steps in the pathogenesis of neurotoxicity. The basic principle is that the expression pattern of a molecule can be probed qualitatively or quantitatively in homogenized tissue to determine a molecular signature that may be used in diagnosing the presence and progression of a given disease. These methods provide high-throughput, high-resolution molecular analysis.
The “omics” platforms serve two primary roles in neurotoxicological research. The first is to assess variations in the molecular signature among different cell populations (e.g., neurons vs astrocytes, granule vs pyramidal neurons). The second aim is to examine molecular pathways that play roles in xenobiotic responses that might be used to diagnose or treat particular health conditions (e.g., receptors capable of interacting with antidepressant drugs, signaling cascades impacted by exposure to drugs of abuse). Other roles will be added as this technology matures as a means for neurotoxicological exploration.
“Omics” techniques are quite sensitive to the quality of the starting material. Tissues must be collected as rapidly as possible after death to prevent artifactual molecular degradation in neural cells (especially in the deep CNS parenchyma) that arise due to postmortem acidosis, dehydration, hypoglycemia, and hypoxia. In general, samples should be collected fresh and frozen at −80°C until analysis, but molecular data can often be obtained from fixed or fixed and previously embedded samples as long as the fixation length was relatively short and the blocks were not subjected to extreme temperature fluctuations during their time in storage. Where known in advance, the experimental design can accommodate parallel collection of fresh tissue for “omic” (and other neurochemical) endpoints and fixed tissues for routine neuropathology evaluation.
Success in toxicogenomic studies generally depends on careful experimental design. One critical aspect is that such studies must be built with a clear question in mind. In most cases, this requirement means that experimental subjects should be as nearly identical as possible except for a single variable, such as a manipulation (e.g., genotype or treatment) or physiological state (e.g., age, genetic background, sex). An additional factor that is critical for success is an appropriate sampling strategy, which typically rests on isolating the correct cell population for analysis. Cell harvesting may be done by gross dissection of large tissue blocks where regional homogeneity at the macroscopic level is evident. However, the differential expression of various genes at the cell level invariably results in a readout in which expression data represents an “average” from many neural cell types. The alternative approach is to use laser capture microdissection (LCM), which is more laborious but permits sampling of a defined cell population.
Mammalian Models for Neurotoxicity Research
Neurotoxicity testing is based on the premise that any anatomic, biochemical, or functional effects that develop in the nervous system of animals after exposure to a particular agent would also be likely to occur in exposed humans, in a qualitative sense if not in a quantitative fashion. Accordingly, neurotoxicity bioassays are designed to discover target sites (i.e., cell populations or organ subregions), elucidate dose-response relationships, catalog similarities and differences in sensitivity among species, and determine mechanisms of toxicant action. Another important aspect of neurotoxicity testing is to define any special attributes of certain classes of individuals who are likely to have nervous systems that are particularly vulnerable to neurotoxic agents. Examples of such classes are developing animals (as neural cells and circuits are established over an extended period (Figure 21.10)) and senescent organisms (who theoretically have less functional reserve available to compensate for neurotoxic episodes).
The bulwark of conventional hazard identification and risk assessment in vivo involves toxicity studies in young adult rats. This choice is dictated by practical factors (particularly cost), but is permissible because the Rodent nervous system is qualitatively similar to that of other mammals. Rats typically are enrolled on studies at 2–3 months of age, and exposures occur over standard lengths of time (usually 4, 13, or 26 weeks). Neural evaluation occurs as a survey of selected CNS and PNS functions and structures in conjunction with surveys of other major body systems. Such general toxicity studies screen for major effects in key neural regions using a battery of endpoints: in-life observations, collection of gross findings and the brain weight at necropsy, and histopathological assessment of selected critical CNS and PNS regions (Table 21.2). If adverse neural events are not observed using these measures, additional neurotoxicity testing generally is not performed. However, enhanced neurotoxicity testing is undertaken if an agent either impacts a neurological parameter (anatomic especially, but also functional) or has a molecular form that suggests a structure–activity relationship to a known neurotoxicant. Such enhanced testing adds more endpoints (e.g., expanded behavioral testing, electrophysiology, neurochemical measurements, and additional sections for microscopic evaluation) to probe the operational integrity of the nervous system more fully.
Developmental neurotoxicity testing (DNT) is a related but separate strategy that examines the special susceptibility of the immature nervous system. Again, rats are the preferred species for testing due to their fecundity and short gestational length (approximately 20 days). Pregnant animals usually are exposed to a test article from gestational day 6 (the approximate time at which the blastocyst implants in the uterine wall) through postnatal day 11 (the approximate end of major neuronal production in the rat) or postnatal day 21 (the conclusion of neuronal and glial expansion as well as initial circuit generation and myelination). The battery of endpoints is comparable to that used for enhanced neurotoxicity testing in adult rats. A special feature that must be remembered when designing DNT studies is that the appropriate experimental unit is the litter and not the individual offspring, since the dam is the entity that has been exposed to the agent.
Although rats are the most common test species for evaluations of neurotoxicity, they may not always be the most suitable species for predicting specific effects in humans. Different species likely react in diverse ways to the same agent due to species-specific variations in neural anatomy (especially barriers), developmental timing, pharmacokinetics, and/or inherent pharmacodynamic differences. Since the importance of these interspecies discrepancies often is incompletely or even totally unknown, any neurotoxic effect seen in animals is presumed to foreshadow a risk to humans even though effects seen in animals may not be the same as those produced in humans. Consequently a clear understanding of the pathogenesis and the mechanism(s) of neurotoxicant action that lead to neural damage are critical to produce the best risk assessment decisions.
Nervous System Responses to Neurotoxic Injury
The ability of toxic agents to disrupt neural activities depends on many parameters. These factors typically are specific to given entities, such as particular CNS regions or cell populations or subcellular structures. This section describes the most common responses of the nervous system to toxic injury. Frequent artifactual changes are defined where relevant so that they will not be misinterpreted as evidence of neurotoxicity.
Anatomic Lesions
In general, structural damage that manifests in the nervous system may be divided using one of two fundamental approaches. The first is to catalog anatomic changes resulting from injury to a specific cell population (e.g., neuron, myelinating element) or cell part (e.g., axon, synapse). The terms applied to such conditions are commonly designated in broad terms using the name of the targeted cell or cell part followed by the suffix “-opathy.” The second option is to diagnose alterations based on the class of lesion that they represent (inflammation, neoplasia, etc.). Basic classes of neural lesions (Table 21.1) are briefly described below.
Cell type–specific lesions
Neuronopathies
Certain toxicants attack specific groups of neurons, usually residing within the CNS. The lesions have been characterized chiefly using rats, but humans typically are vulnerable as well. For instance, 3-acetylpyridine irreversibly destroys the inferior olivary nuclei, thus inducing cerebellar ataxia by decimating the climbing fibers leading to the cerebellum. The neurotoxic effects of MPTP are largely confined to the dopaminergic neurons of the substantia nigra, resulting in cell death in humans, other primates, and some strains of mice (with humans representing the most sensitive species). A mix of cycad toxins have been linked to the induction of amyotrophic lateral sclerosis/Parkinson-dementia complex (ALS-PDC) in natives of Guam with the expanded range of symptoms being attributed to variable blends of toxicants that act individually to target neurons in the cerebral cortex, substantia nigra, and spinal cord. In many instances, the sensitivity of particular neuronal groups to toxic agents first is discerned via their distinct patterns of functional alterations and/or neurochemical changes rather than by morphological analysis.
The basic neuropathology findings associated with a neuronopathy depend on the length of time during which the toxic injury has been taking place. Degeneration (alteration of a cell or tissue to a less functional form) may be an acute or chronic condition characterized by a variety of subcellular microscopic changes. For example, a subset of neurons in layers III/IV of the cingulate/retrosplenial cortex targeted by MK-801, a N-methyl-D-aspartate (NMDA) antagonist, develop fine cytoplasmic vacuoles within hours of exposure (Figure 21.18), but their nuclei and cell membranes are intact. Neuronal vacuolation is caused by expansion of intraneuronal cytoplasm or membrane-bound organelles as a consequence of retained fluid or metabolic byproducts. In principle, degenerative changes may be reversed if exposure to the toxicant ceases in time and/or the dose was below the lethal threshold.
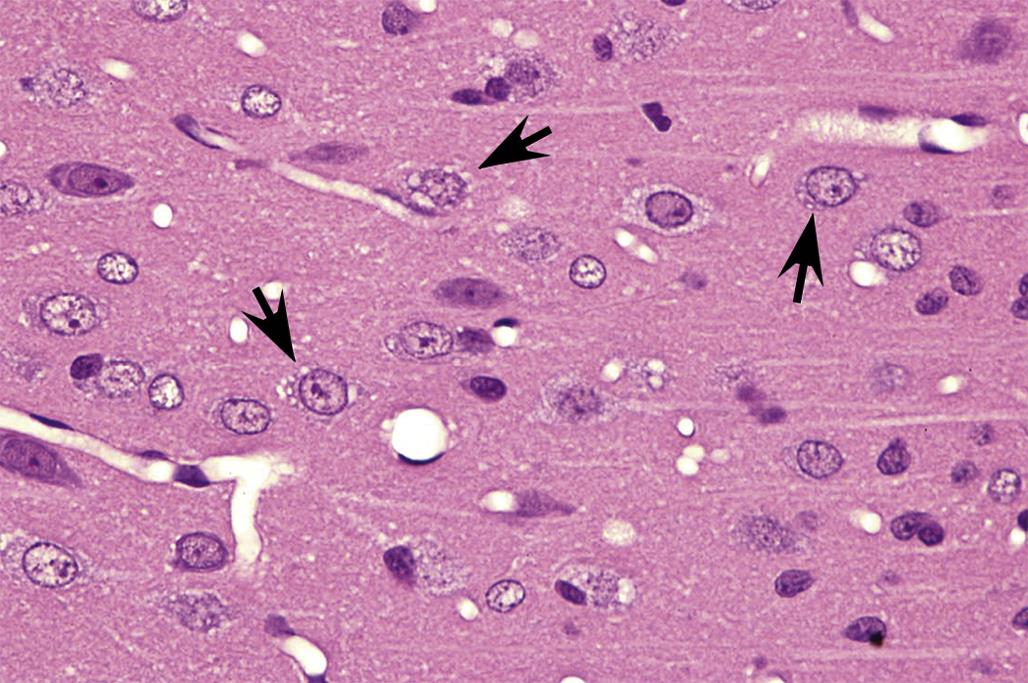
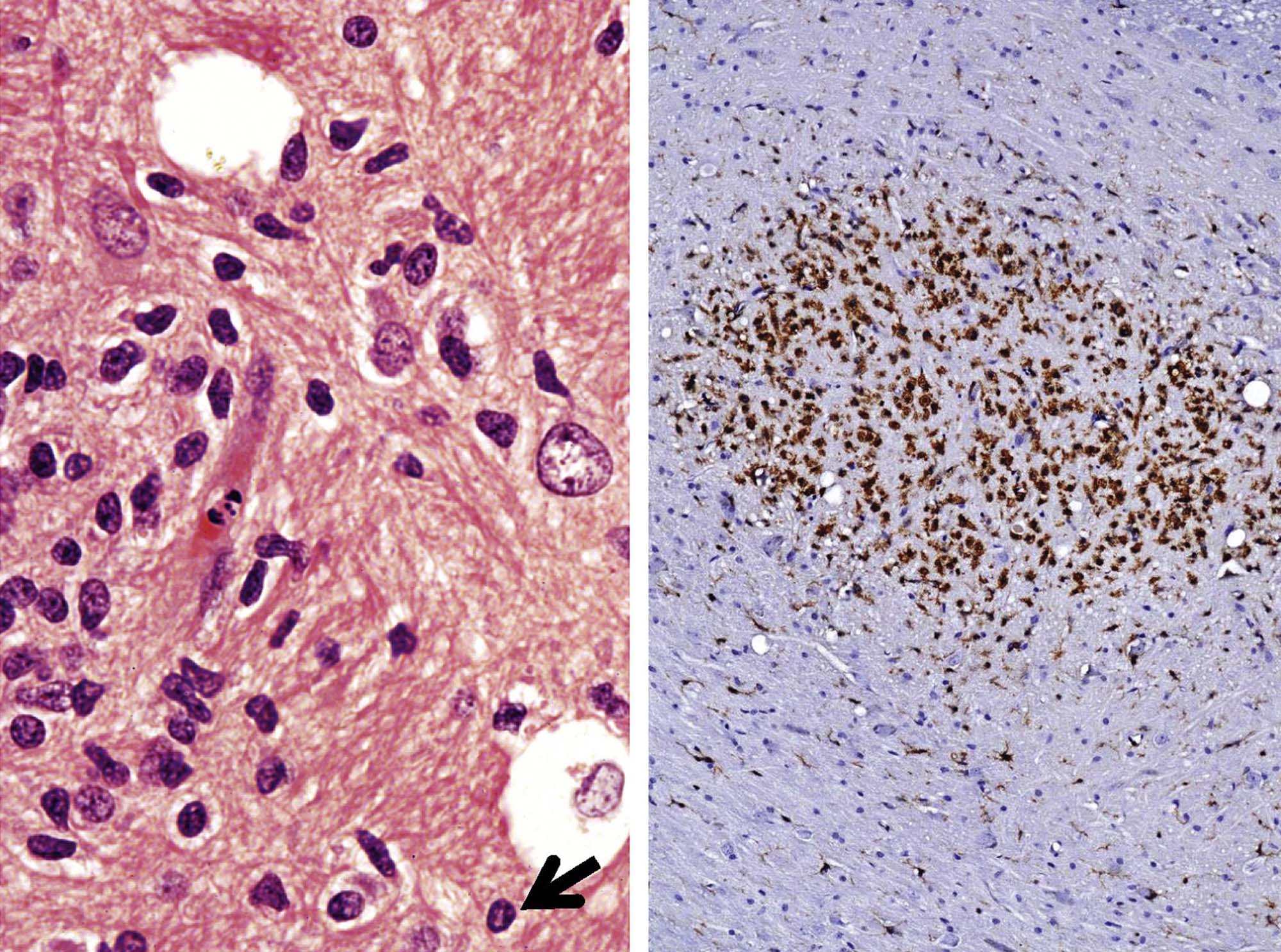
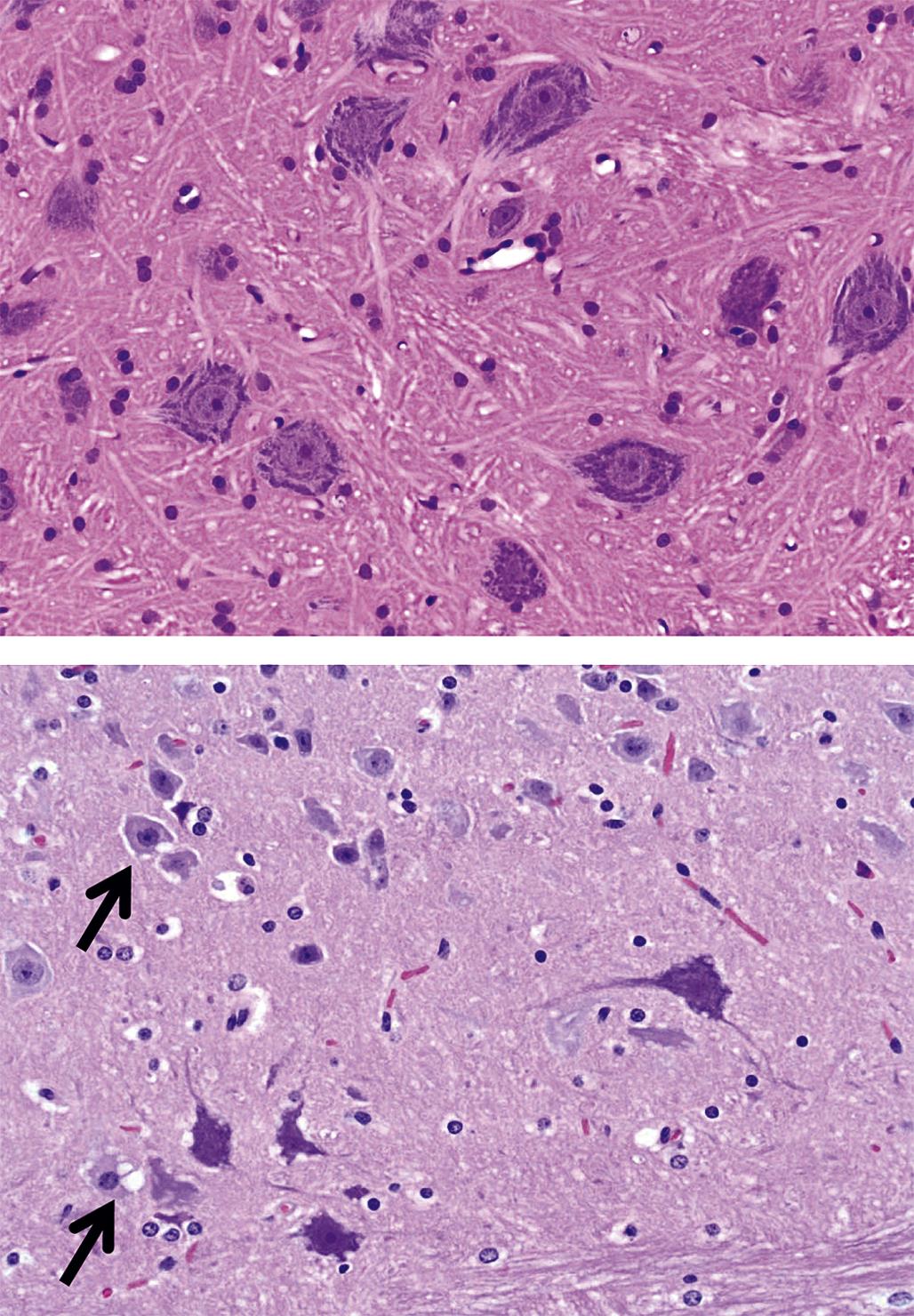
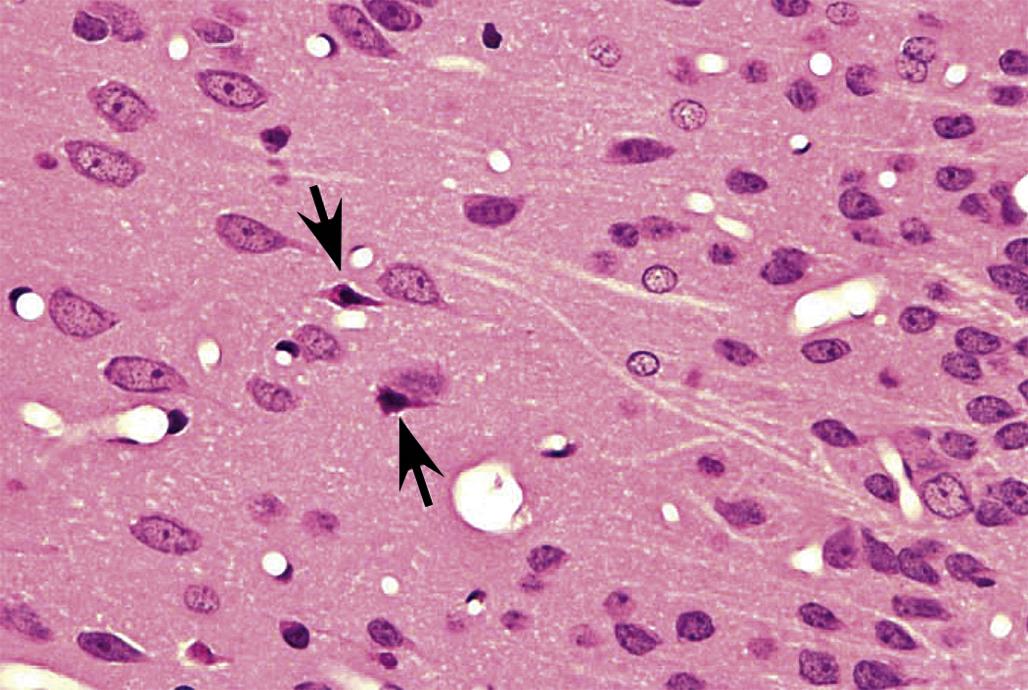
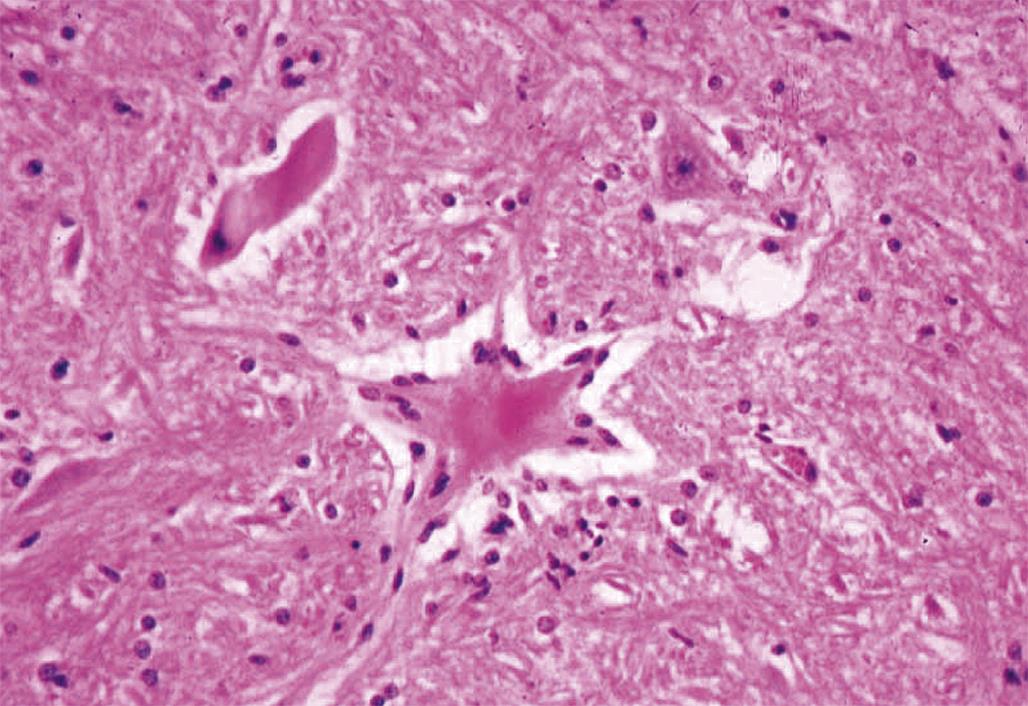
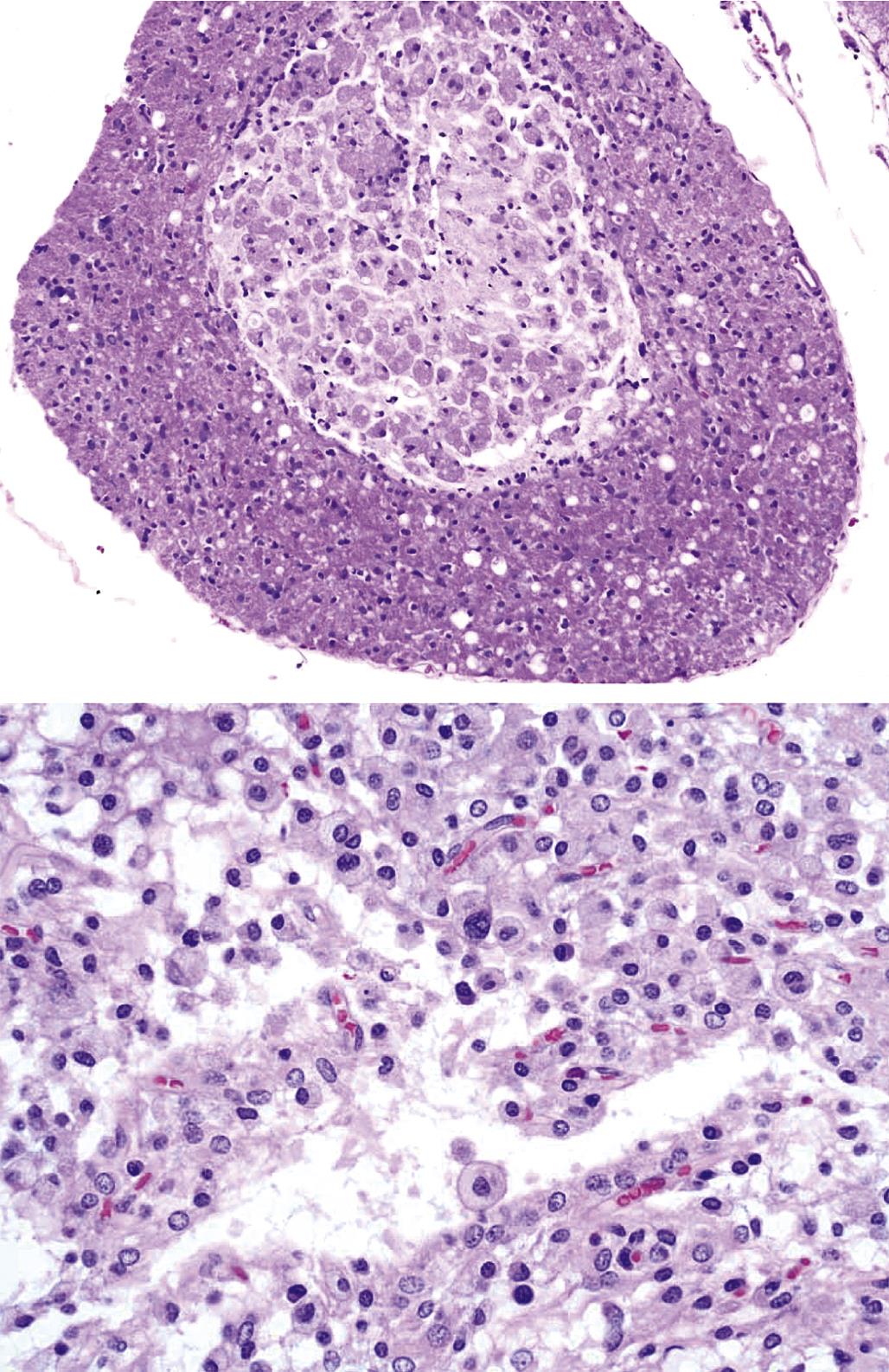
Degeneration may, especially within in neurons, progress to necrosis (Figures 21.19 and 21.20). The hallmark features of toxicant-induced neuronal necrosis in the CNS are contraction of the cell (leaving a clear, peri-cellular halo); condensation and/or fragmentation of the nucleus; and loss of the Nissl substance, leading to hypereosinophilic cytoplasm; the latter feature is the classic trait of “red dead” necrotic neuron (Figures 21.5, 21.19, and 21.20). Genuine neurotoxic lesions often harbor an array of affected cells, with responses ranging from acutely injured to dead and disintegrating. Neuronal necrosis may be detected using standard H&E staining, but increased sensitivity is provided by Fluoro-Jade (performed on frozen or paraffin-embedded tissue sections) and cupric silver stains (performed on frozen sections) (Figure 21.7). With time, dead neurons may attract complements of microglia that help to remove the cellular debris in a process termed “neuronophagia” (Figure 21.21). The remnants of necrotic neurons are cleared rapidly. Unless evaluated in the proper (typically early) time frame, quantitative investigations may be required to detect neuron loss. Exceptions to this include areas where neurons are arranged in an orderly manner, including the various CA regions of the hippocampus and the Purkinje cell layers. Even subtle neuron losses in these well-defined brain regions may be detectable as “gaps” in the neuron layer. Stains to detect astrocyte reactions may be particularly helpful to determine if the “gaps” indicate prior neuron loss or are normal anatomic variations.
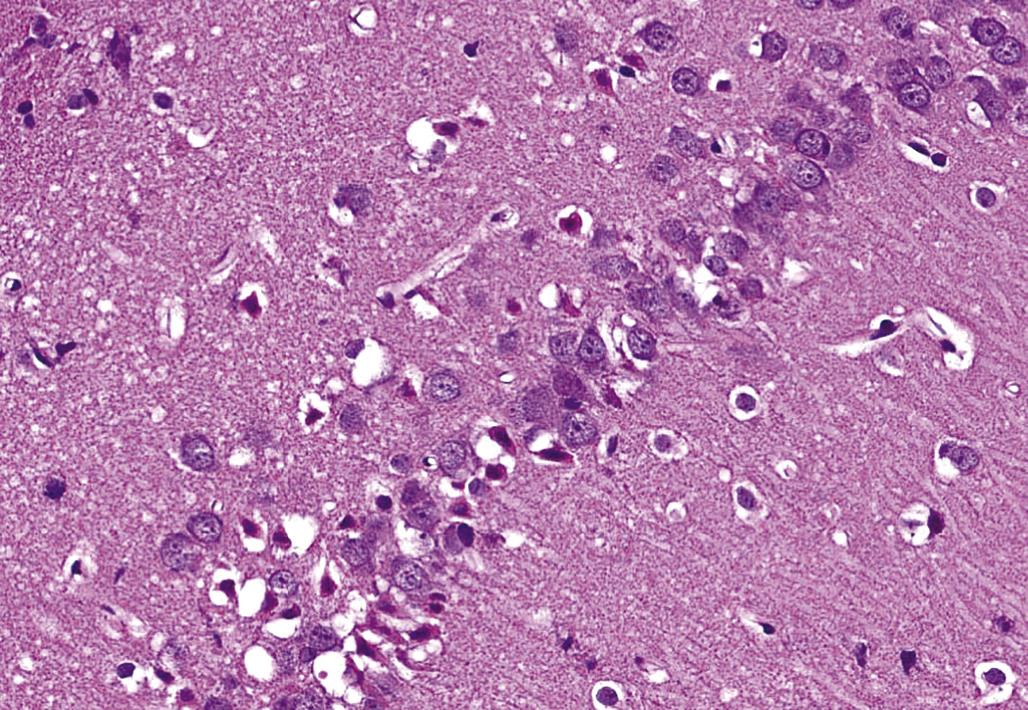
Neuronal heterotopia (or ectopia) is a distinct category of neuronal lesion. Instead of cell loss, the fundamental change is a developmental disturbance leading to abnormal migration and/or terminal differentiation. A common presentation for ectopic neurons is to see clusters of cells located in atypical sites, leading to disrupted neuronal organization. The misplaced cells exhibit the typical features of the neural site at which they normally would have been found. The most frequent locations for these changes are the cerebral cortex, hippocampus, and cerebellum (Figure 21.12); any layer or multiple layers may be affected. Minor heterotopiae often are seen as isolated cell clusters in grossly unaffected brains, while major lesions frequently occur together with brain malformations like hydrocephalus or microcephaly. Aberrant anatomic development often but not always is accompanied by functional alterations. Alcohols like ethanol, methanol, and methylazoxymethanol (MAM) are potent disruptors of neural cell migration.
Neuronal storage diseases result from accumulation of materials in affected cells. Many storage diseases arise from inborn genetic defects that lessen or prevent production of an enzyme needed for normal metabolism. In the absence of functional enzyme, its substrates accumulate within organelles (e.g., Golgi apparatuses, lysosomes) or the cytoplasm. Toxic causes of neuronal storage diseases include cationic amphophilic drugs, which lead to phospholipidosis, and chronic ingestion of swainsonine (the toxic principle in locoweed). Lesions in mild (usually acute) cases of neurotoxicant-induced neuronal storage disease may be reversible if the exposure is halted, but severe (chronic) lesions usually progress to full-blown cell engorgement (Figure 21.22) and eventually to neuronal degeneration.
Axonopathies
Other toxicants do not attack neurons, but rather damage their processes. These lesions may develop within the CNS, where they are typically confined to specific tracts, or in the PNS. In many instances, neurotoxicants with this capability first were discovered following occupational exposures in humans. Three basic lesion patterns can develop.
The first is the primary axonopathy, which is produced when the axon itself is the major site of damage. Examples include agents that cross-link macromolecules in distal axons, like acrylamide, carbon disulfide, and n-hexane. The fiber proximal to the xenobiotic-induced lesion remains intact while the fiber distal to the lesion disintegrates since it has lost connection to the neuron. The primary changes affecting the distal axon are fragmentation followed by formation of elliptical digestion chambers that hold cellular debris (Figure 21.23). This appearance should be referred to as Wallerian-like degeneration as it resembles genuine Wallerian degeneration, which is a sequel to physical (e.g., trauma) rather than chemical nerve transection. Fragmentation may begin within hours of axonal injury, but in practice axon degeneration may require a day or more to appreciate with the light microscope. Over time, macrophages of hematogenous origin (often termed “gitter” cells) are drawn to the site to phagocytize the axonal debris (Figure 21.24).
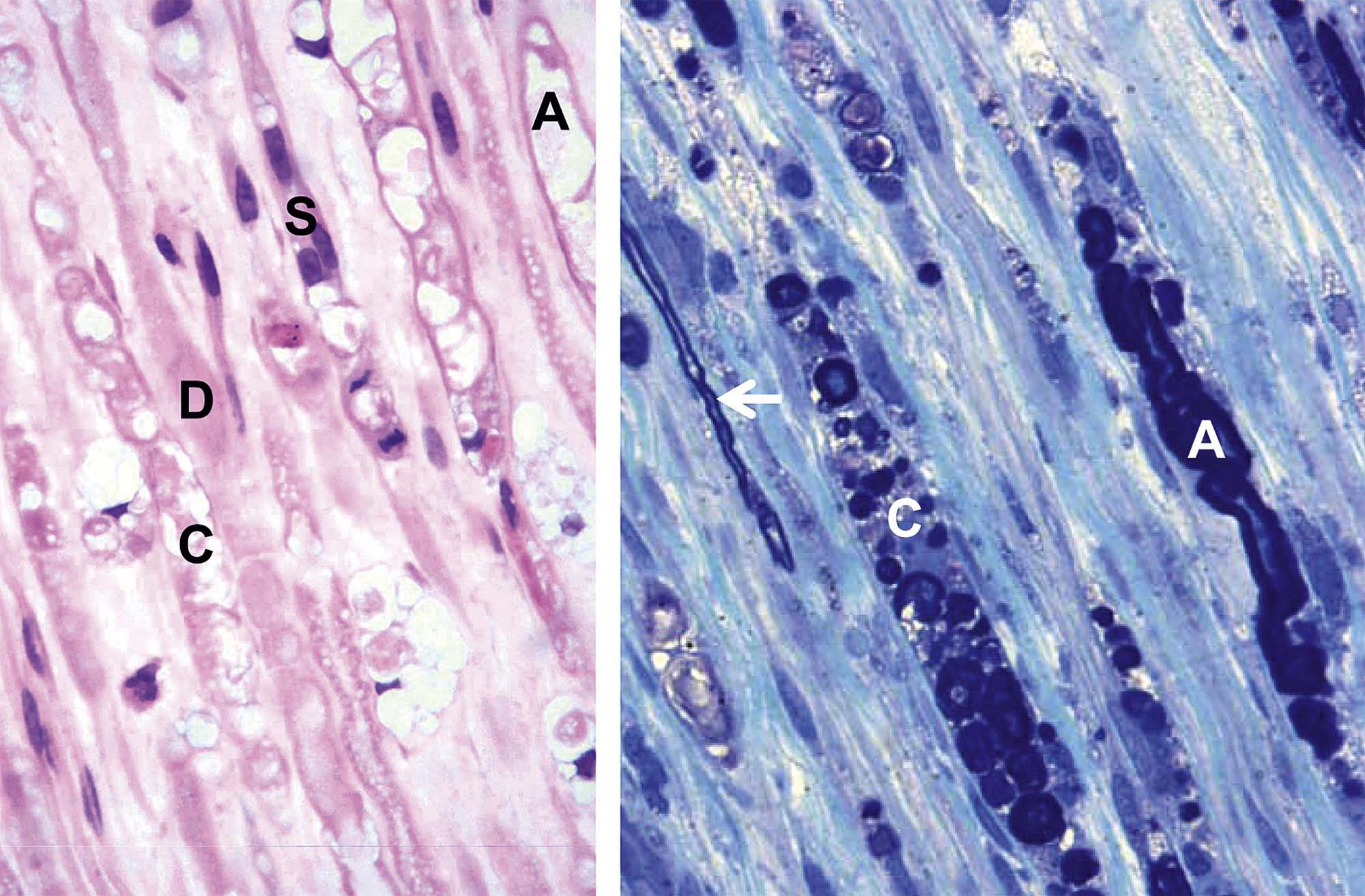
The next pattern is the secondary axonopathy, which occurs when the capacity of a neuron to export materials is compromised below the minimal level needed to sustain its axon and/or nerve terminal. For instance, β, β′-iminodipropionitrile (IDPN) exposure induces a proximal axonopathy because it interferes with slow axonal transport. Continued inability by the neuronal cell body to supply its processes will result in gradual progression of the degenerative changes to more proximal portions of the axon (termed “dying back”); bipolar primary sensory neurons with both central and peripheral axons may exhibit this change in the two processes simultaneously. Morphologic features of the neuronal body and proximal axon may be morphologically normal even as the changes in the distal axon grow in their extent and severity.
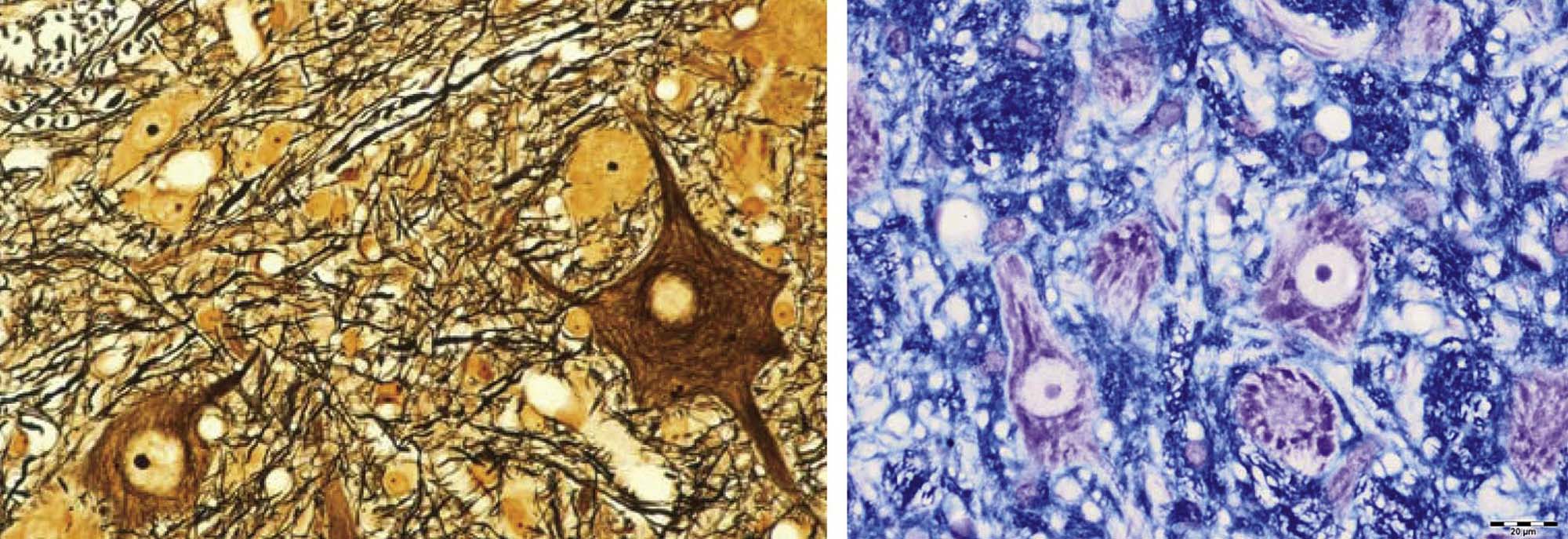
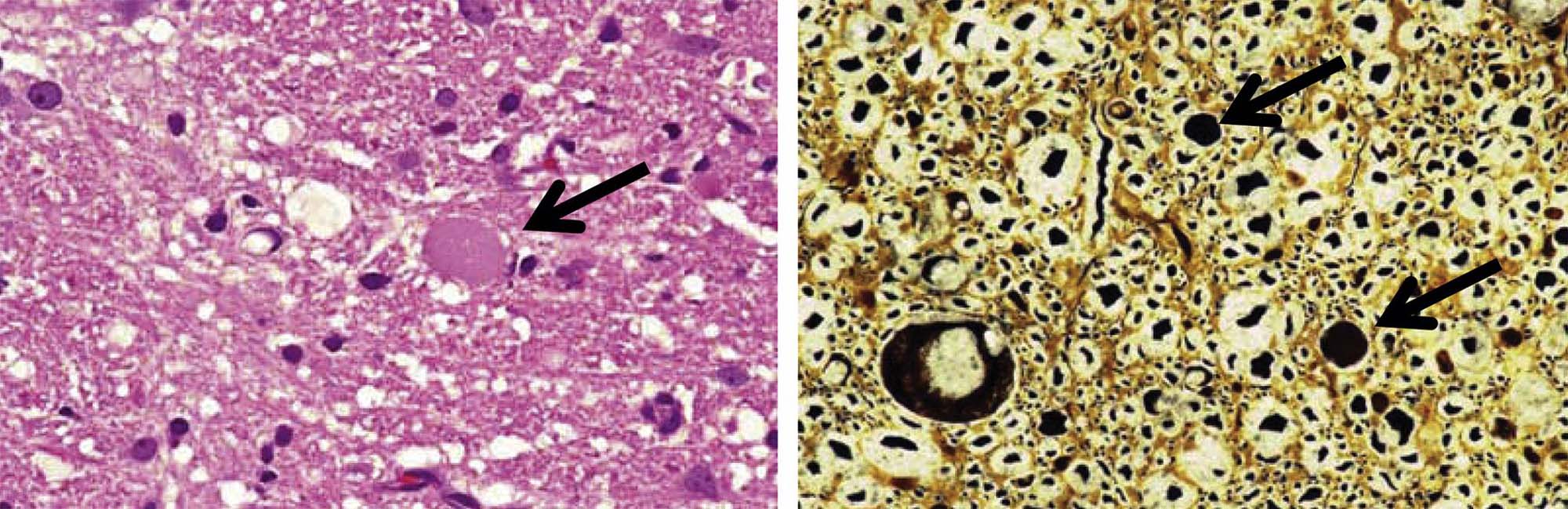
The third pattern is neuroaxonal dystrophy, which is characterized by axonal expansion (often termed “spheroids”). In general, widespread neuroaxonal dystrophy is more common as an inherited or acquired neurodegenerative disease in humans and animals than as a consequence of toxicant-induced damage, although a few agents (e.g., aluminum, methylmercury) can produce spheroids as one element of their pathological presentation. These enlarged fibers appear as large, fairly homogeneous, elliptical swellings by light microscopy, which are shown to harbor a tangled web of cytoskeletal elements and cellular organelles by electron microscopy. The swellings are eosinophilic when stained with H&E and black when viewed in silver-impregnated sections (Figure 21.25), and they are much larger in diameter than adjacent “normal” axons. The proposed pathogenesis is a disturbance in retrograde axonal transport that results in progressive accumulation of less flexible materials at points of axonal constriction (i.e., the nodes of Ranvier, which are the gaps formed between the edges of adjacent myelinating cells).
Regeneration of PNS axons can occur once the axonal damage has been cleared provided that the endoneurial tube is intact. The Schwann cells proliferate to form solid columns of cells (termed “bands of Büngner”) that reach from the site of the axonal lesion to its distal terminus. At the same time, the proximal axon grows distally into these Schwann cell columns, which guide the axon as it travels to reestablish its link with the site that it originally innervated. Regeneration of the axon requires a transient boost in macromolecular synthesis (mainly proteins) in the neuronal body. This need is accommodated by dispersion of the Nissl substance and translocation of the nucleus to the cell periphery, a change termed “central chromatolysis.” This repair response in the neuronal cell body is termed the “axonal reaction,” although it is very common to observe axonal degeneration without seeing any associated changes in the neuronal body. Regeneration of axons proceeds at the same velocity as slow axonal transport, and thus is limited to several millimeters a day; full functional recovery takes weeks to months for distant effector organs, especially in larger species. In general, axonal regeneration of differentiated CNS axons is ineffective.
These axonal changes may be recognized in sections stained routinely with H&E, but in many instances special procedures are undertaken to provide additional contrast. The techniques may highlight the axon itself, either by silver impregnation (e.g., Bielschowsky’s (Figure 21.25) or Bodian’s stains) or IHC labeling to detect an axonal marker (e.g., neurofilament protein (NFP), which is one cytoskeletal constituent). Alternatively the procedure may label the myelin sheath using a histochemical stain (e.g., LFB) that stains lipid, an IHC method to localize a myelin marker (e.g., MBP), or fixation enhancement to increase the contrast between the axon and its sheath (e.g., postfixation in OsO4) (Figure 21.26). These myelin-detecting procedures may serve as effective means to indirectly highlight primary axonal damage. A definitive means of differentiating between the axon and myelin as the primary target site is to isolate individual fibers in teased preparations. Axonal lesions are characterized by the absence of the central axon with multifocal attenuation of the Schwann cell profile over long distances, while primary myelin damage leads to segmental loss of the myelinating cell with retention of the denuded but intact axon (Figure 21.27).
Synaptopathies
The human nervous system houses trillions of synapses, with each neuron playing host to many thousands. Ultrastructural evaluation may be required to investigate synapses. Their minute size prevents synapses from being observed in standard light microscopy sections except that disintegrating synaptic terminals may be visualized using the cupric silver stain.
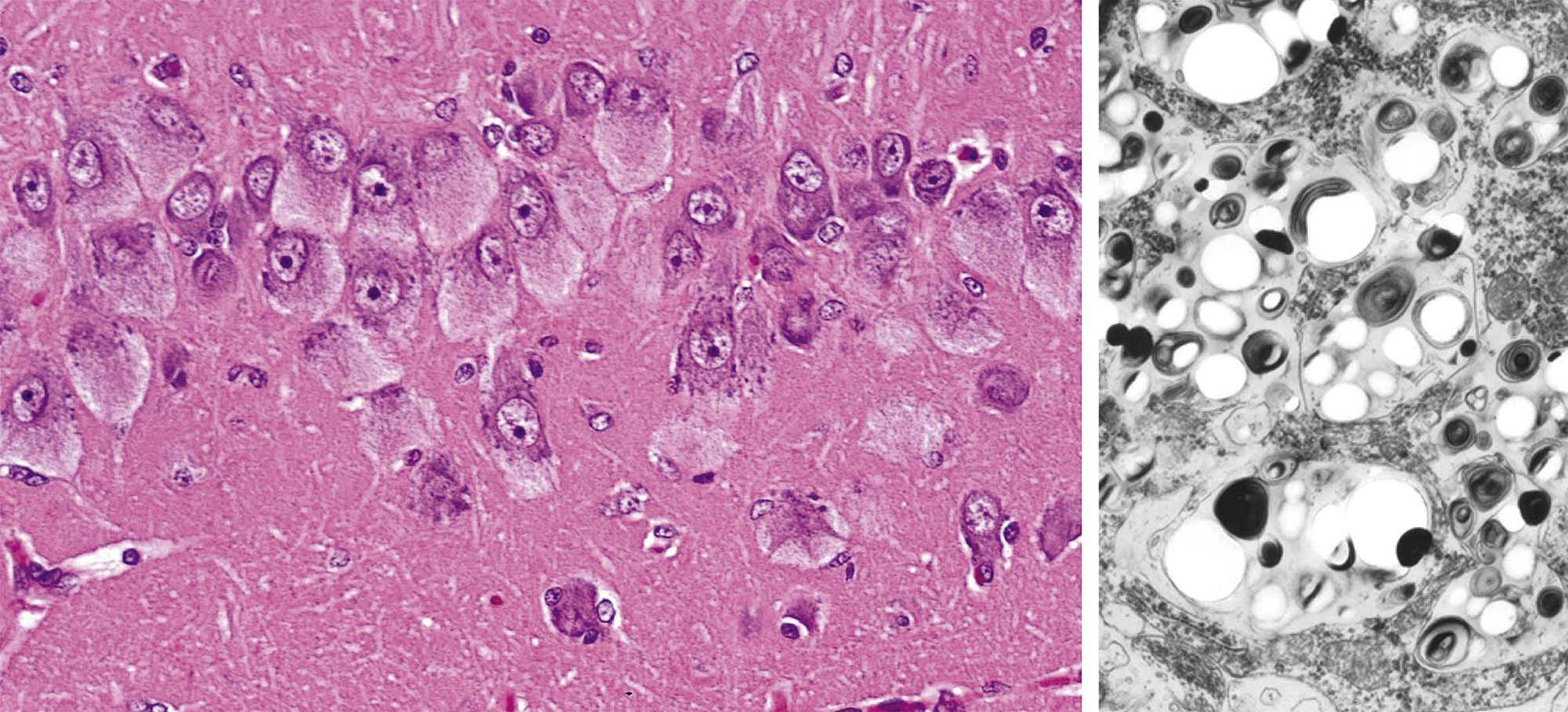
One major change in experimental neurotoxicity reports is altered membrane maintenance and turnover in axonal terminals (Figure 21.28). Lesions may affect either the presynaptic or the postsynaptic elements. Agents capable of altering membrane structure include chloromycetin, which targets cerebrocortical motor neurons; diphenylhydantoin (DPH), which impacts cerebellar neurons; and 3,4-methylenedioxymethamphetamine (MDMA), which transforms monoaminergic neurons in the striatum. Common anomalies include thickening of the synaptic membranes or formation of membranous inclusions in the axon terminals. The progressive nature of the inclusions that characterize the DPH-induced lesion suggests that their pathogenesis begins with accretion of interconnected tubules in gradually expanding axon terminals, followed by the buildup of inclusions and the eventual formation of axonal spheroids. Ultimately the affected neurons undergo coagulation necrosis, seemingly as a consequence of excessive synaptic degeneration. The membranous inclusions in the MDMA-injured cells also contain ubiquitin, which provides added confirmation that this finding is an outcome of significant damage to cell membranes.
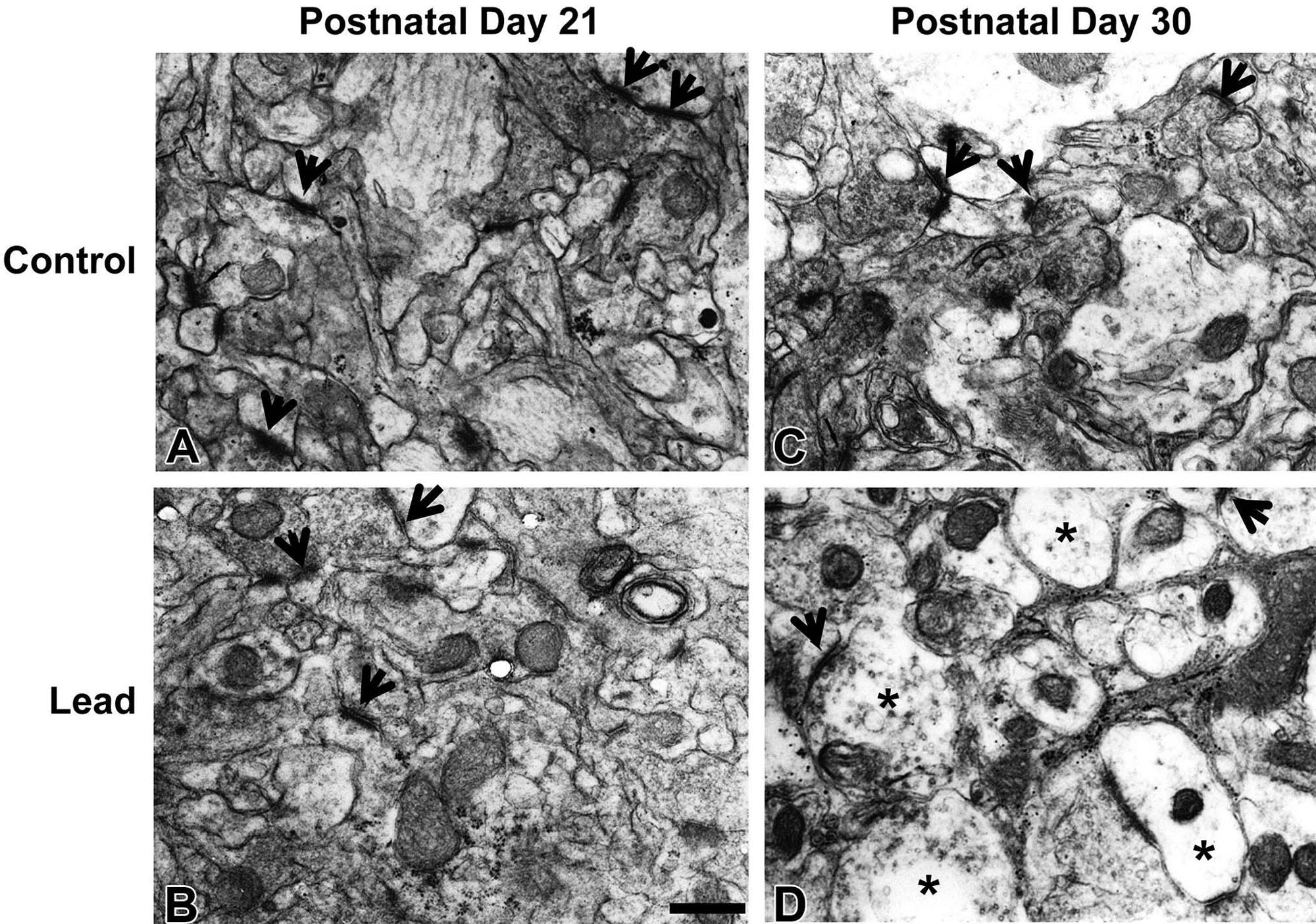
Other ultrastructural lesions induced by neurotoxicants have also been observed as precursors of degenerative changes in CNS synapses. For instance, simultaneous incubation with amyloid-beta (Aβ), a protein fragment of amyloid precursor protein (APP) thought to be responsible for neural lesions in advanced Alzheimer’s disease, and ryanodine induces mitochondrial swelling and a severe reduction in the numbers of small synaptic vesicles and synapse-anchored proteins like synaptophysin and actin in rat cerebrocortical synaptosomes. Depletion of synaptic actin can be reversed by incubation with caspase inhibitors, suggesting that extended abnormalities of synaptic structure can invoke the apoptotic pathway in affected neurons.
Myelinopathies
Toxicant-induced damage to myelin can develop within the CNS or PNS, depending on whether or not the population of injured cells is the oligodendrocyte or the Schwann cell. From a practical standpoint, however, the more important consideration usually is to define the manner in which the lesion is incurred.
Production of myelin to surround axons does not occur at the same time in all parts of the nervous system, but instead develops progressively. In the cerebrum, sizeable white matter tracts and commissures are the first to be insulated, followed in sequence by the major subcortical centers (e.g., thalamic and basal nuclei) and finally the cerebral cortex. Myelination in the cerebral cortex occurs first in projection zones before spreading later to the association areas. In the spinal cord, cervical tracts are myelinated prior to those in the lumbar region. The myelination process is not fully completed for weeks after birth in rodents, and for years in human infants.
Myelin is particularly sensitive to neurotoxicants during two stages of development. The first is a period of rapid glial cell proliferation (i.e., the first two postnatal weeks in rodents). The second is the time of active myelination, the peak of which occurs on approximately postnatal day 20 in rodents. Therefore the location and extent of myelinotoxic insults will vary with the timing of the exposure.
Primary demyelination is produced when myelin sheaths are the main target of the insult. In such instances the axons are not harmed, and accordingly are preserved intact within the disintegrated myelin sheath. The classic agent responsible for inducing this effect is tellurium. The typical appearance of white matter tracts or nerves with primary demyelination is swelling of the affected sheath segments (where each segment is the product of a single cell) with formation of myelin bubbles (also termed balloons or blebs). These bubbles may be discerned from digestion chambers produced in axonopathies because the bubbles surround continuous (i.e., viable) albeit shrunken axons in close association with myelin debris and macrophages. Primary demyelinating insults generally result in a fairly diffuse distribution of white matter damage, which is readily detected in conventional tissue sections that have been processed to demonstrate myelin in white matter tracts (Figure 21.29).
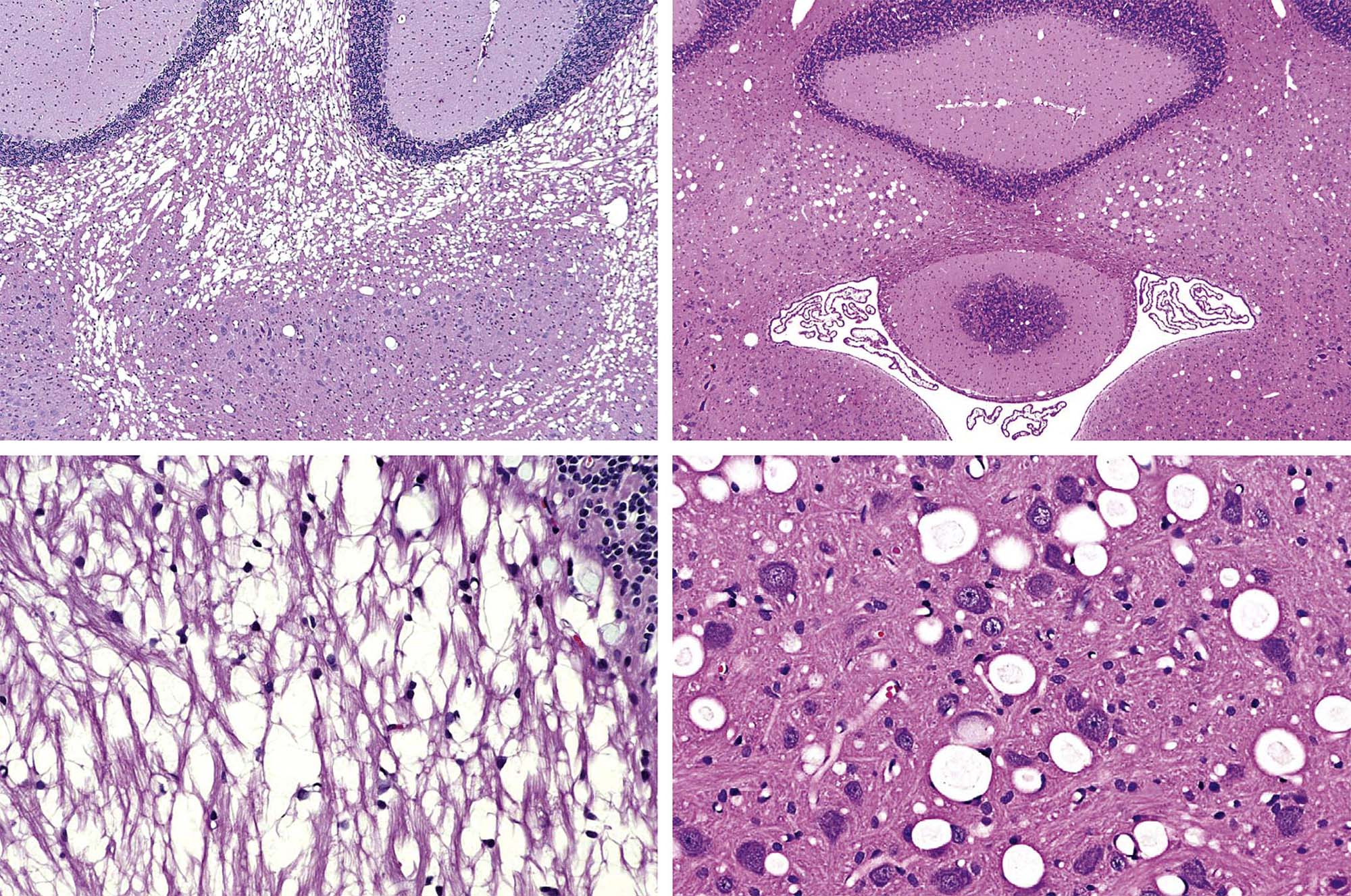
A variant of primary myelinopathy is myelin edema, resulting from fluid accumulation within the myelin sheaths. Hexachlorophene is the prototypic neurotoxicant that induces this lesion. Myelin edema can occur in both the CNS and PNS and usually appears as vacuoles within myelin-rich regions in the absence of myelin degeneration. The volume expansion caused by the increased fluid leads to splitting of the myelin lamellae. Mild cases may be reversible to some extent, but pronounced or chronic lesions typically will lead to secondary axonal damage.
Secondary demyelination results from irreversible degradation of a myelin sheath following the initial loss of its axon. The microscopic appearance of myelin bubbles in this scenario includes loss of the axon, showing that the myelin disintegration follows the prior axonal damage. Conversion of myelin lipoprotein to fully degraded fat typically takes approximately 2 weeks.
Regeneration of myelin sheaths generally is confined to the PNS, and can occur fairly rapidly since it arises from cell precursors already in place at the lesion site. In the PNS, each Schwann cell envelops a single axon, so damage to the Schwann cell will lead to segmental loss of myelin (Figure 21.27) that is more severe at the level of the spinal nerve roots (i.e., where the axons are of larger caliber and thus are encompassed by longer and thicker myelin sheaths). As remyelination proceeds, each gap is filled by a greater number of Schwann cells producing a series of shorter segments with thinner sheaths than were present in the original.
Gliopathies
In many respects, non-myelinating glia are more resistant to neurotoxic insult than their neuronal and myelin-producing neighbors. Injured glial cells typically respond by swelling. In most instances the affected elements are astrocytes. Lesions of minimal to moderate extent are often reversible if the cause of the change is removed.
The usual appearance of swollen astrocytes is that of cell expansion and/or vacuolation (Figure 21.30), the outcome of fluid or material accumulating in the cytoplasm or a membrane-bound organelle. The change occurs most frequently in the brain, and usually presents most prominently in the gray matter. Microscopically the parenchyma of this region is riddled with small holes, some of which are perivascular in location or appear to compress nearby neurons. In general the effect exhibits bilateral symmetry.
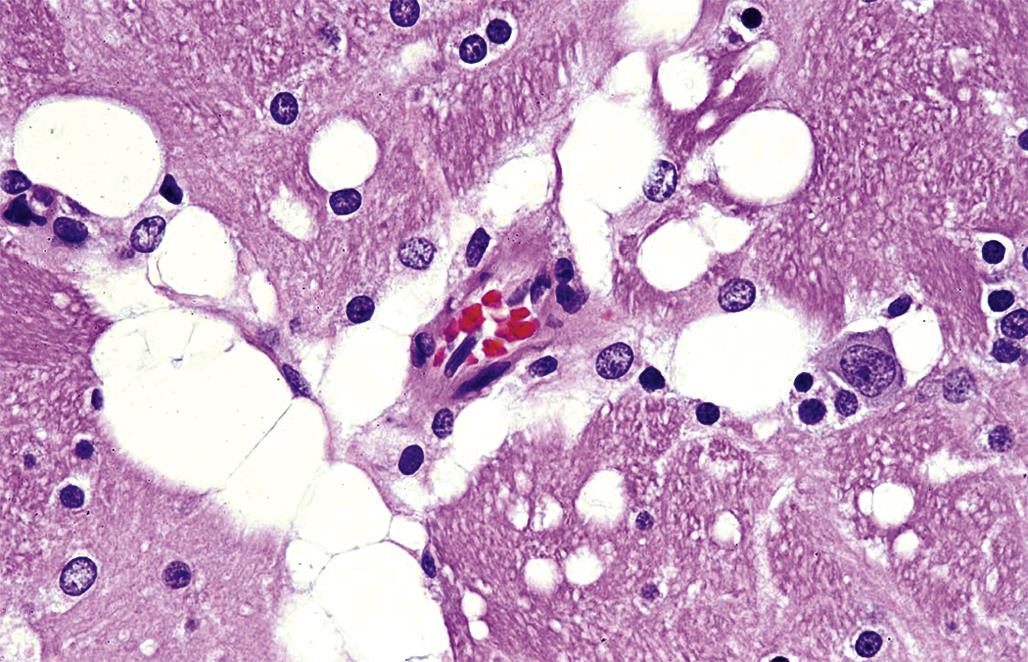
The formation of Type II astrocytes embodies an unusual variant of astrocyte swelling. This cytotoxic response results from nuclear expansion following exposure to elevated levels of nitrogenous waste products (the chief of which is thought to be ammonia); thus, these elements represent a secondary toxic response to primary hepatic disease. Affected astrocytes have swollen clear nuclei with very thin rims of marginated heterochromatin, sometimes with enlarged nucleoli, and indistinct cytoplasm (Figure 21.31). These cells tend to accumulate in the neocortex, basal nuclei, and hippocampus.
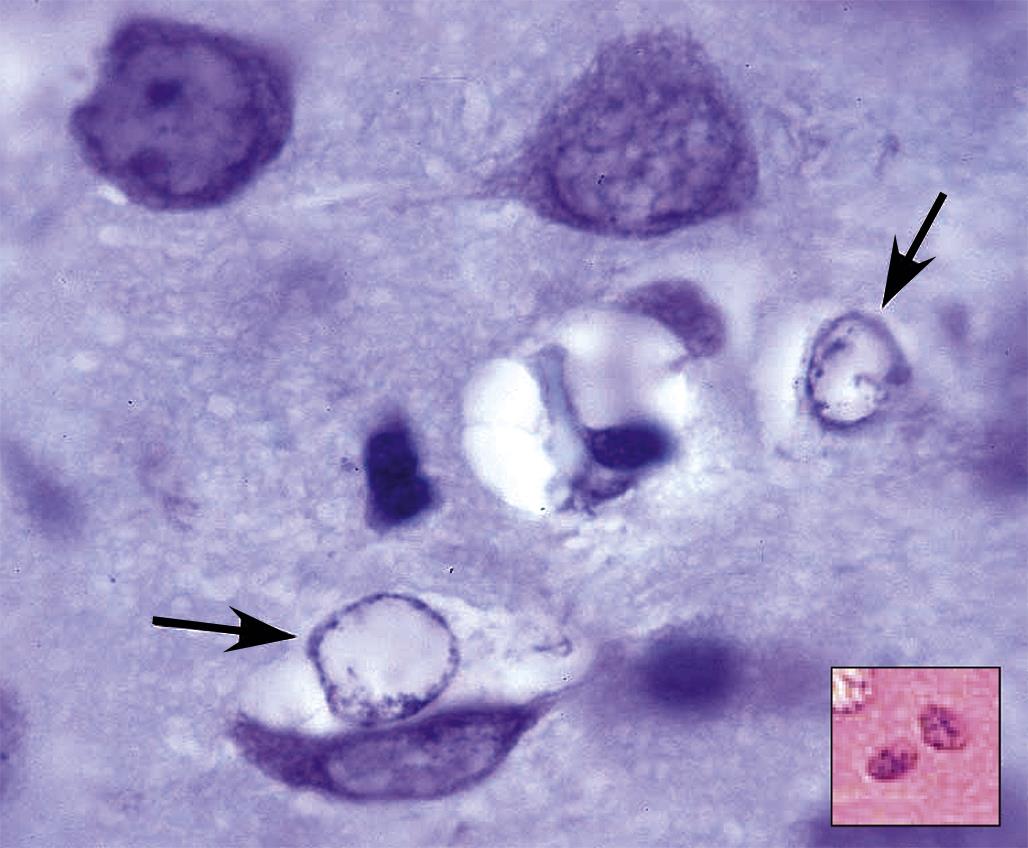
Glial Reactions to Neural Injury
Glia play prominent roles in the repair response following toxicant- also induced damage to other populations of neural cells. The main responders are the non-myelinating cells of the CNS—astrocytes (i.e., true glia) and microglia (i.e., the bone marrow–derived resident histiocytes of the CNS)—but oligodendrocytes and Schwann cells also participate in such efforts.
Hypertrophy and hyperplasia are the main reactions by activated CNS glia to neural damage. The generic term for this change is “gliosis,” which implies enhanced production of multiple cell lineages, but cell type–specific increases (e.g., astrocytosis, microgliosis) are also possible. The presentations and functions served by these reactions are quite distinct. Astrocytosis occurs to fill or encompass damaged regions in the CNS, usually produced as a consequence of neuronal or pan-cellular necrosis. Reactive astrocytes are seen in H&E-stained sections as large, stellate cells with large, pale nuclei and scant to modest amounts of pale eosinophilic cytoplasm (Figures 21.8 and 21.24); they may also be seen in IHC-stained material via their increased expression of cytoskeletal proteins like GFAP (Figure 21.8) and vimentin. A variant of reactive cell termed the “gemistocytic” astrocyte may be observed in some CNS lesions as many plump, brightly eosinophilic cells supporting numerous processes (Figure 21.32). The functional basis for the gemistocytic transformation is not clear.
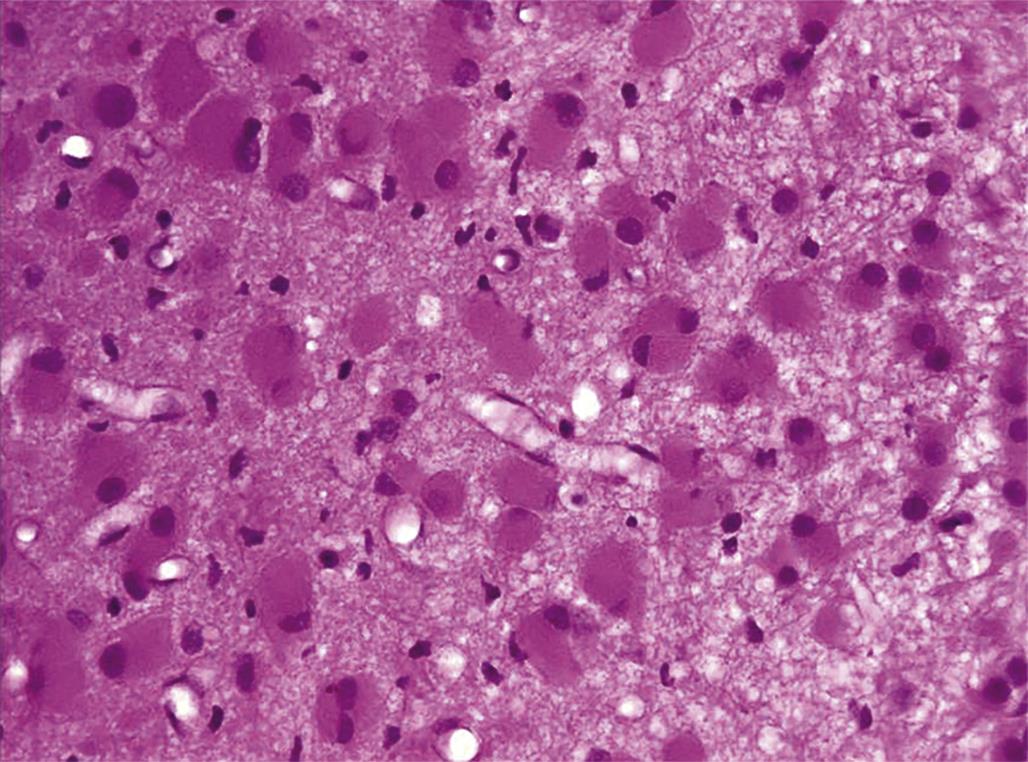
Microgliosis results from proliferation of the resident “immune” elements in the CNS, usually in response to more localized neuronal insults. Microglia can perform both surveillance (e.g., antigen presentation) and effector (e.g., phagocytosis) functions. Reactive microglia are identified in H&E-stained tissue as small, elongate, sometimes twisted nuclei near damaged CNS neuropil (Figure 21.9). Special stains may be used to raise their visibility, such as IHC labeling for Iba1 (Figure 21.9) or histochemistry to detect the lectin Griffonia simplicifolia.
The response by reactive oligodendrocytes is termed “satellitosis” as the myelinating cells aggregate around degenerating neurons, presumably in an effort to support their survival and repair. Reactive oligodendrocytes are evident in H&E-stained sections as partial to complete rings of cells encircling abnormal neurons (Figure 21.33). In general, oligodendrocytes are the least reactive population of glia. Pathologists must be careful to avoid undue haste in interpreting the relevance of satellite cells near neurons as neoplastic lymphocytes in a peri-neuronal location may mimic the appearance of oligodendrocytes (Figure 21.33).
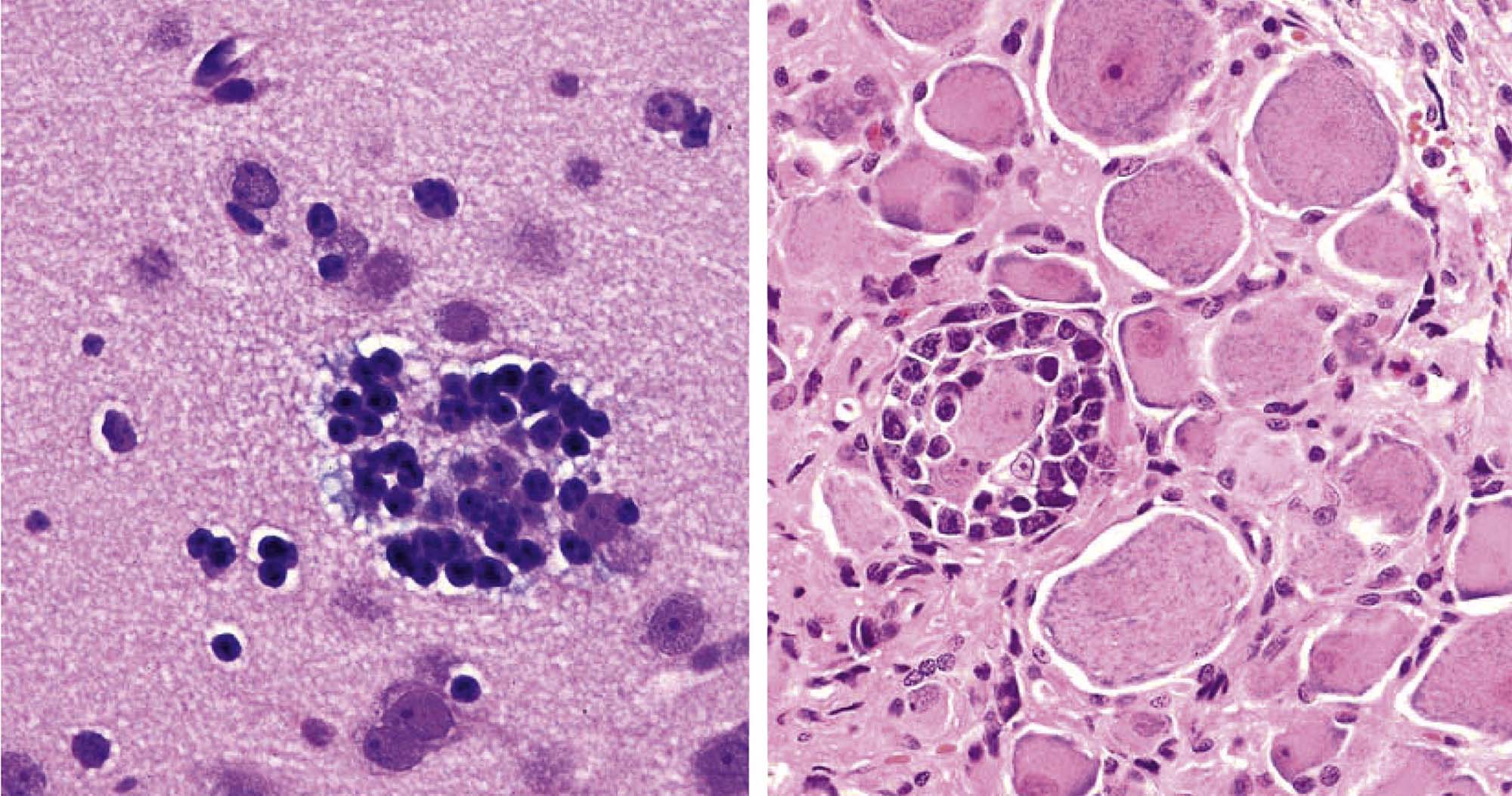
Global Classifications of Toxicant-Induced Neural Lesions
Several kinds of toxicant-induced abnormalities that damage the nervous system recapitulate similar processes that occur in non-neural tissues elsewhere in the body. In general, these lesions are categorized as nonproliferative or proliferative based on the prominence of cell division as a mechanism for enhancing the degree of neural damage. The remainder of this section reviews the basic features of the major lesion types in both these categories.
Nonproliferative Lesions
Developmental disturbances as a class are an important outcome following exposure to many neurotoxicants (Table 21.1). The most common macroscopic abnormalities are neural tube defects (NTDs). The NTDs usually result from enhanced cytotoxicity during the period of neurulation, a phase in which the neural folds of the planar embryo progressively elevate and then fuse to form the neural tube (the precursor to the CNS). The spectrum of NTDs ranges from partial to complete nonfusion of the neural tube (see Figure 25.3 for examples) Figure citation. The correct chapter is "Embryo, Fetus and Placenta" by Rousseaux and Bolon. The correct figure is no. 3, which shows 7 yeloow-hued mouse fetuses against a dark blue background. Severe variants include anencephaly (absence of the brain due to complete nonfusion), exencephaly (exposure of the brain due to failure of the cranial vault to close around or over the brain), and/or spina bifida (failure of the spinal cord and/or vertebral arches fuse). Partial closure leads to encephalocele or myelocele (smaller foci of localized nonfusion in which brain or spinal cord tissue, respectively, protrudes through a fissure in the skeleton) or meningocele (only the meninges protrude through the defect). Anencephaly and exencephaly are lethal malformations, while individuals with one of the other NTDs may survive (though often with some degree of lifelong neurological impairment).
Other classes of toxicant-induced developmental defects include microcephaly (presenting as a reduction in cerebrocortical size; Figure 21.4), cerebellar hypoplasia (affecting the entire organ or just the hemispheres), and neuronal heterotopiae (Figure 21.11). These lesions result from toxicant exposure sometime after neural tube closure. The former two changes reflect excessive neuron death and reduced formation of neural circuits, while the heterotopiae generally stem from aberrant migration of newly made neurons and/or defective differentiation of the radial glia that serve to guide migrating neurons to their appropriate positions.
Hydrocephalus results from dilation of one or more channels within the cerebroventricular system. The change is observed most commonly in the lateral ventricles. The presentation often includes reduced thickness of the overlying brain parenchyma due to increased pressure from the accumulated CSF; the reduction arises by hypoplasia of neural cells if the lesion begins early during development but usually represents atrophy if the change starts after birth. Hydrocephalus is an end-stage lesion that serves as the final common outcome for many different etiologies. In the toxicologic neuropathology setting, hydrocephalus in immature subjects frequently occurs as a compensatory mechanism to fill space within the cranial vault left vacant by xenobiotic-induced reductions in neural cell numbers in the cerebrum. In contrast, in adults the usual cause is CSF blockade by occlusion of the mesencephalic aqueduct (of Sylvius) by a mass such as an abscess (a possible sequel to immunosuppressive therapy or placement of an intrathecal catheter for direct xenobiotic delivery into the CNS) or neoplasm (an occasional finding in rodent carcinogenicity bioassays).
Infarcts (colloquially termed “strokes” in human patients) are characterized by regional necrosis of a neural domain, usually within the brain or spinal cord. The cause is interruption of the blood flow to a particular area as a consequence of blockage or rupture in a larger artery or vein. The absence of blood flow leads to ischemia of all cells within the region supplied by the affected vascular arcade; neurons are most sensitive to ischemia due to their very high basal metabolic rates. A transient disruption in flow typically culminates in neuronal necrosis, which will appear as a reduction or absence of these cells in an otherwise intact region of parenchyma (Figure 21.34). In many instances the numbers of astrocytes and microglia (Figure 21.34) will be enhanced in regions where the neurons used to reside, serving as a lasting marker of previous neuronal damage.
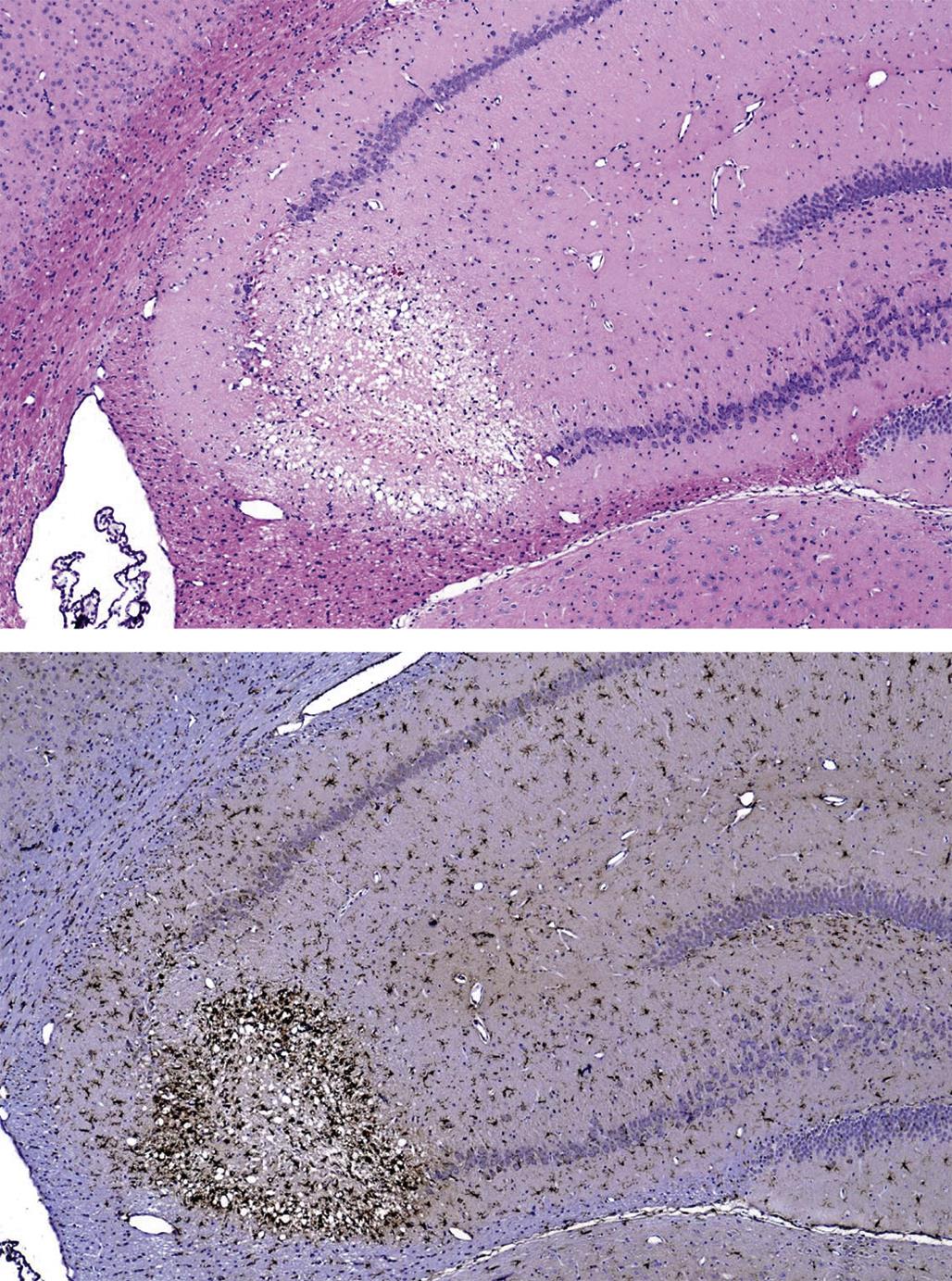
Persistent interference with the blood flow will decimate all cells within the domain, which will finally lead to liquefactive necrosis of the affected parenchyma (Figure 21.24). Eventual removal of the necrotic debris by activated microglia and macrophages recruited from the blood (gitter cells) will leave a cyst that typically has a marginal zone enriched with fibrous (intensely GFAP-positive) astrocytes (i.e., a glial scar). The site and cause of the infarct rarely are seen on gross or histopathologic examination. Potential toxic causes of infarcts usually are confined to intravascular thrombosis secondary to endothelial cell damage and/or activation of the clotting cascade. Agents that can produce these circulatory injuries include bacterial endotoxins or certain chemicals.
Inflammatory lesions result from damage to the neural parenchyma in the CNS and/or PNS by accumulations of astrocytes and/or microglia in conjunction with circulating leukocytes (of one, a few, or all classes). Because leukocyte infiltrates can occur in the absence of tissue damage (Figure 21.35), the concept of “inflammation” should only be invoked when other evidence of neural injury is evident along with the leukocytes; such features may include neural-specific changes like axonal and/or myelin degeneration or gliosis, or they may be more general findings like edema, hemorrhage, necrosis, and vascular congestion. Neural inflammation as a primary toxic response typically results from direct placement of a drug delivery apparatus into the CNS (Figure 21.35), especially if coupled with delivery of a potentially irritating test article. It may also occur as a secondary consequence of agents that incite another disease processes (e.g., immune dysfunction, neural damage) in which the barriers protecting the nervous system are breeched and the injured cells and newly exposed neural antigens become targets. Examples of this latter class include experimental allergic encephalitis (EAE) in rodents, and perhaps some cases of multiple sclerosis in humans.
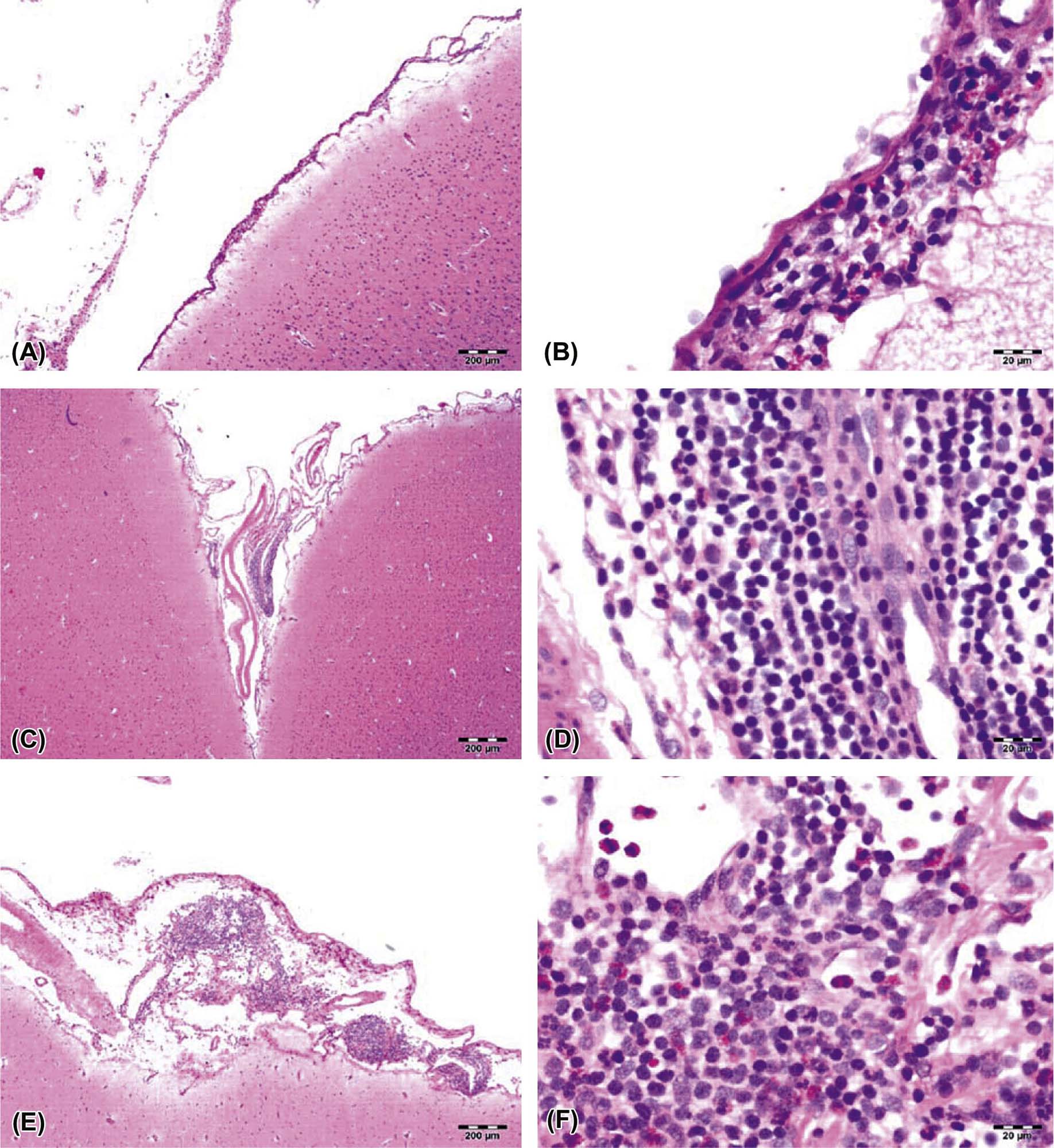
Several pigments may be observed within the CNS, either as reactions to administration of a neurotoxic agent or as an incidental finding. Lipofuscin accumulates in the cytoplasm of large CNS neurons (mainly pyramidal cells), astrocytes, and oligodendrocytes via gradual accretion of cell degradation byproducts. This autofluorescent material represents the phospholipid-rich residue of autophagosomal lysosomes produced in cells undergoing substantial lipid peroxidation of their membranes. The microscopic appearance of the granules generally is faint yellow-brown in H&E-stained sections, pink in periodic acid Schiff (PAS)–stained sections, and dark blue to purple in LFB-stained sections (Figure 21.36). In many cases, lipofuscin is stored spontaneously in aged animals, more so for nonhuman primates and humans, and to a lesser extent in carnivores, than for short-lived rodents. These “natural” deposits elicit little if any cytotoxicity in affected cells. In contrast, lipofuscin has been reported to accumulate in rodents given some neurotoxic agents, including alcohol and lead, where its extensive accumulation may be associated with cytotoxicity.
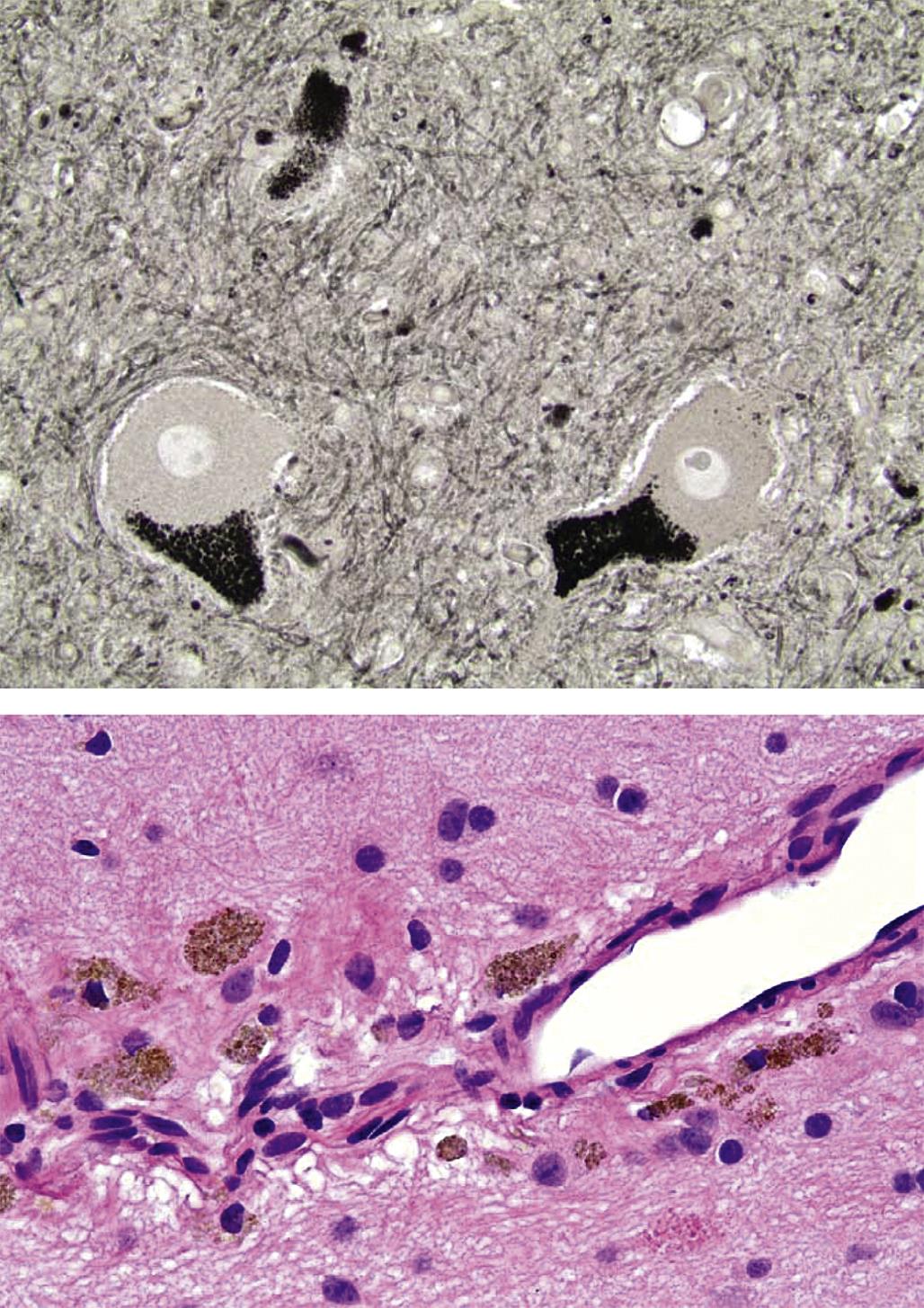
Deposition of hemosiderin within the CNS represents another form of pigment associated with certain neurotoxic treatments. The pattern of deposition depends on the nature of the exposure. For example, Aβ, the endogenous toxicant linked to cerebral amyloid angiopathy (CAA) in Alzheimer’s disease, has been associated with microscopic perivascular hemorrhages (Figure 21.36) in affected brain regions of amyloid precursor protein (APP)-transgenic animal models as well as human patients. In contrast, larger foci of hemorrhage and hemosiderin accumulation may be evident at sites where implantation of a direct delivery device has produced microtrauma to the blood vessels coursing through the neuropil. Very small deposits of pigment, presumably hemosiderin, in the Purkinje cell layer can be a hint that there was a prior loss of cells from this neuronal population.
Storage diseases may occur as a consequence of neurotoxicant exposure, when the active agent reduces or halts the activity of an enzyme required for normal metabolism. The prototypic toxicant-induced neural storage disease is produced by swainsonine, an alkaloid toxin fabricated by plants of the genera Astragalus, Oxytropis, and Swainsona (e.g., locoweed) that inhibits α-mannosidase, a protein involved in the hydrolysis of N-linked glycosides. Affected neurons are observed to have many cytoplasmic vacuoles (representing swollen Golgi bodies) by light microscopy. A variant of xenobiotic-induced storage disease is drug-induced phospholipidosis (DIPL), in which phospholipids accumulate in multilamellar (myeloid) bodies within the cytoplasm (Figure 21.22). Small molecules associated with this lesion are classed chemically as cationic amphophilic drugs (CAD), and they represent essential therapies for treating many indications: angina, depression, hypercholesterolemia, and malaria, to name a few. The usual neural cells targeted by these drugs are large neurons, such as those in dorsal root ganglia and the spinal cord gray matter. In contrast to traditional storage diseases, DIPL results from selective lysosomal uptake and/or retention of the undigested xenobiotic substrate rather than from enzyme inhibition. Confirmation that a molecule has produced lysosomal alterations requires transmission electron microscopy, so in nearly all cases analysis for this finding is limited to experimental animals.
Proliferative Lesions
An infrequent but confirmed consequence of xenobiotic exposure is neurocarcinogenesis, or the induction of neoplasms in the CNS and/or PNS. Most neural neoplasms, including the bulk of xenobiotic-induced lesions, in animals and humans are glial tumors arising in the CNS. These glial neoplasms may evolve from a single cell lineage (e.g., astrocytoma, oligodendroglioma) or from multiple lines (e.g., mixed gliomas, which contain both astrocytic and oligodendroglial elements). In general, chemically induced glial tumors in experimental animals do not evolve into the aggressive glial malignancies (e.g., glioblastoma multiforme) characteristic of lesions observed in human patients. Neural cancers originating from other CNS cell types have been described after exposure to xenobiotics, albeit rarely. These entities include medulloblastoma, a primitive neuroectodermal tumor (PNET) of the cerebellum; granular cell tumors (in rodents) and meningiomas; and ependymomas (typically occurring in the spinal cord central canal).
In the PNS, neural tumors most commonly arise from Schwann cells only or from Schwann cells in combination with endoneurial and perineurial fibroblasts that form the connective tissue sheaths bundling axons within nerves. Traditionally, these neoplasms have been designated as Schwannomas (benign or malignant) and neurofibromas (for benign lesions) or neurofibrosarcomas (for malignant), respectively, but recently some workers have preferred to combine them into a single class as "nerve sheath tumors" (benign or malignant). The PNS axons themselves cannot give rise to neural tumors since they merely represent cell processes, and not cells. However, PNS cell bodies in various ganglia (including the adrenal medulla) can serve as the source of peripheral tumors of neural origin. Chemically induced PNS tumors usually will possess features of Schwannoma (i.e., originating only from Schwann cells).
Differential diagnosis of neural tumors is complicated by two main factors. The first is the difficulty in discriminating between reactive and neoplastic neural cells, particularly in the PNS. Schwann cells proliferate extensively as a regenerative response to any form of damage, so any connective tissue neoplasm that encompasses a nerve is likely to include a substantial proportion of activated Schwann cells intermingled with the neoplastic tissue. A second factor is that tumor cells often express unusual complements of proteins. Such altered signatures are particularly evident for malignant neoplasms, which may produce greatly reduced amounts of typical marker proteins or manufacture multiple markers that are characteristic of an immature pluripotent cell. From a practical perspective an increase in the number of neoplasms following exposure to a xenobiotic is an adverse event regardless of the cell of origin, so the main focus should be on discriminating between reactive and neoplastic lesions.
Neurocarcinogens include alkylating chemicals, radiation, and certain viruses. In general, exposures leading to carcinogenicity occur during development (e.g., by transplacental or neonatal exposure) or during young adulthood. The same agent (e.g., N-ethyl-N-nitrosourea (ENU)) can produce increases in the incidences of both glial and non-glial neoplasms in the CNS. Some neural tumors only develop following direct introduction of the toxicant into the CNS (e.g., choroid plexus carcinomas following intracerebroventricular injection), indicating the importance of barrier systems (e.g., BBB) in protecting neural tissues from blood-borne toxic agents.
In the PNS, tumors of peripheral nerve origin must be distinguished from neuromas, which are focal, nonneoplastic lesions that form at the site of a local traumatic injury (commonly transection with displacement or removal of the distal trunk). Neuromas represent the disordered proliferation of elongating proximal axons that have failed to find their former pathways and thus are unable to reestablish connectivity with their effector organ. The presence of numerous axons is a diagnostic feature of neuromas as Schwannomas contain few if any axons. Toxicants cannot induce neuromas as axons cannot become neoplastic. However, toxicants may produce extensive axonal sprouting which collectively may form masses that mimic the effects of tumors.
Background Neuroanatomic Findings and Their Implications
Certain incidental changes are frequently misidentified as neuropathological lesions by inexperienced researchers. The alterations described here are common artifactual findings in vertebrate brains. Identified artifacts typically should not be reported in the pathology data set. However, systematic distribution of a background finding, where one dose group only is involved or the relationship between the lesion incidence and dose is uncertain, may be recognized and noted in the microscopic data or included as a comment to the organ/tissue at the discretion of the study pathologist. The key for such incidental changes is to properly discuss their significance, or more often their lack of significance, with respect to assessing the risk of neurotoxicity.
“Dark neuron” artifact in the brain is the most problematic spontaneous change, as it is misinterpreted as neuronal degeneration with distressing frequency. This artifact occurs more regularly in certain brain regions, particularly the cerebral cortex (Figure 21.14), hippocampus, Purkinje cell layer of the cerebellum, and large pyramidal neurons of many brainstem nuclei and the spinal cord gray matter. Affected neurons typically have darkly amphophilic nuclei and cytoplasm, with larger-sized neurons often having more readably visible cytoplasmic borders. Some dark neurons may exhibit twisted, corkscrew-shaped extensions. Dark neurons have been shown to result from rapid transfer of water out of the neuronal cytoplasm into adjacent glial cells or into the peri-neuronal neuropil. Within an affected region, all dark neurons have similar features, in contrast to genuine foci of neuronal degeneration in which injured cells exhibit a range (i.e., early to advanced) of neurodegenerative stages (Figure 21.20).
A common presentation for dark neuron artifact is as an asymmetric, focal column of affected cells extending from the deep layers of the cerebral cortex down to the hippocampus. This appearance is consistent with application of superficial pressure to inadequately fixed CNS tissue, leading to localized glutamate release or to other perturbations of neuronal homeostasis. Induction of excitotoxicity postmortem may be involved in the pathogenesis of “dark neurons,” as administration of glutamate antagonists (MK-801, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), etc.) at necropsy has been suggested to reduce their numbers. The usual method for mitigating this artifact is to fix by perfusion and, if time permits, to follow this by an additional period of in situ postfixation (by immersion) following removal of the calvarium.
Neuronal autophagy in ganglia (predominantly dorsal root ganglia, but also occasionally sympathetic ganglia) is the most common form of neuronal death in the PNS and occurs with some frequency in untreated control animals. This change usually is characterized by cell swelling, pale/granular cytoplasm, and the presence of irregular, dark eosinophilic, globular inclusions (lytic chromatin); infrequently, the affected neuron is encircled by satellite glial cells (Figure 21.37) or lymphocytes. Ganglionic neurons may undergo a more typical necrosis reaction—indeed, this finding typically has been termed "neuronal necrosis" historically—but these PNS cells seldom if ever acquire the classic “red/dead” appearance that develops in necrotic brain neurons (Figures 21.19 and 21.20).
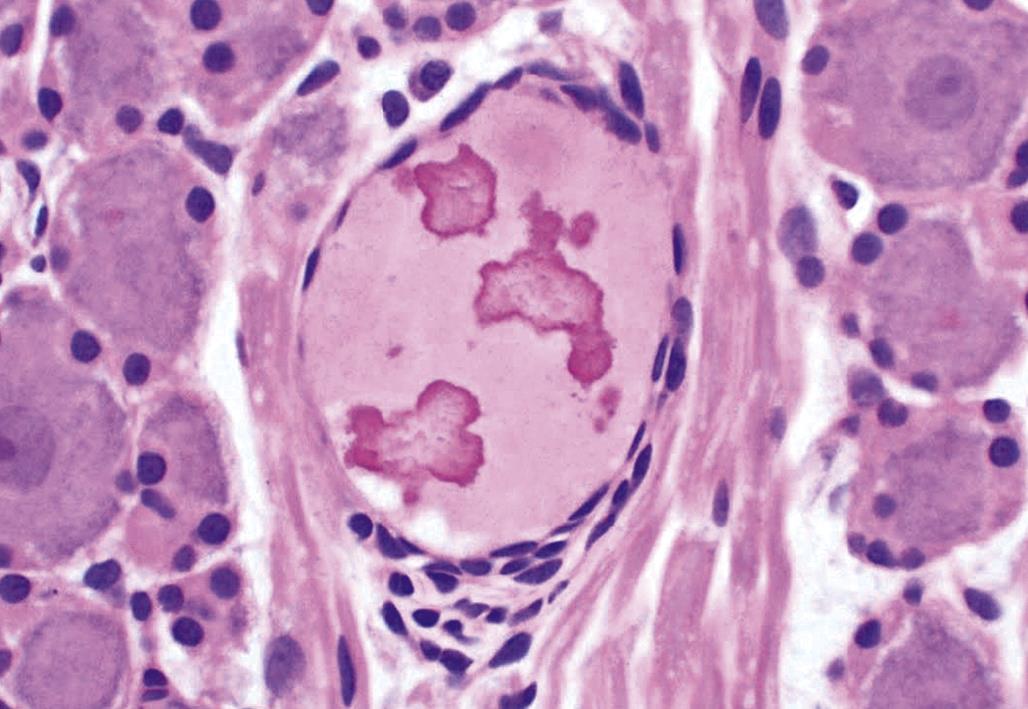
“Myelin bubble” artifact generally involves large myelinated fibers of the spinal cord white matter tracts and PNS major somatic nerve trunks. The finding presents as clear, localized, round to elliptical spaces surrounding an intact axon; the spaces may occur in isolation or in chains (Figure 21.38). The change often is confined to one or a few axons, so it is more readily recognized in longitudinal sections than transverse ones. The main differential diagnosis for this change is axonal degeneration, a true neurotoxic lesion that presents as a string of digestion chambers that contain fragmented axonal and/or myelin debris (sometimes along with phagocytic “gitter” cells). Myelin bubble artifacts tend to develop in immersion-fixed, paraffin-embedded specimens. The mechanism is unknown, but the suggested pathogenesis is manipulation of unfixed tissue leading to tissue fluid accumulation within the traumatized myelin before the fixative solution penetrates the sample.
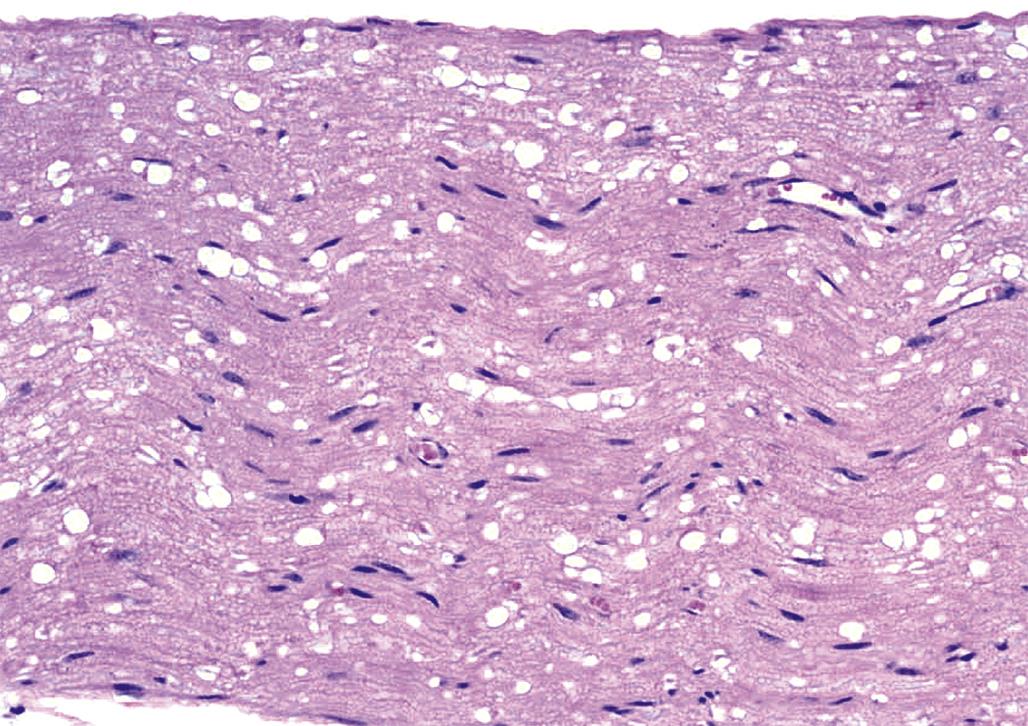
“Vacuolation” artifact can take several forms in the CNS and PNS (Figure 21.39). Diffuse vacuolation may be observed if fixation is delayed. However, this circumstance is unlikely in experimental studies, and can be managed by careful scheduling of tissue harvesting for diagnostic cases. A more frequent variant in CNS white matter is focally extensive collections of large vacuoles in densely myelinated tracts of the cerebrum, cerebellum, and brainstem. The vacuoles may have irregular borders and be filled with pale, homogeneous material (termed “Buscaino bodies”) or have smooth margins and be empty. These vacuoles are more extensive if tissues are retained in alcohol for an extended period—such as holding in the alcohol bath stage on an automated tissue processor over the weekend—possibly as a consequence of excessive lipid extraction. Vacuoles of various sizes are commonly observed in dorsal root ganglia neurons (and less often in sympathetic ganglia neurons) of control animals from many species (Figure 21.40), although they also have been attributed to organophosphate expsoure.
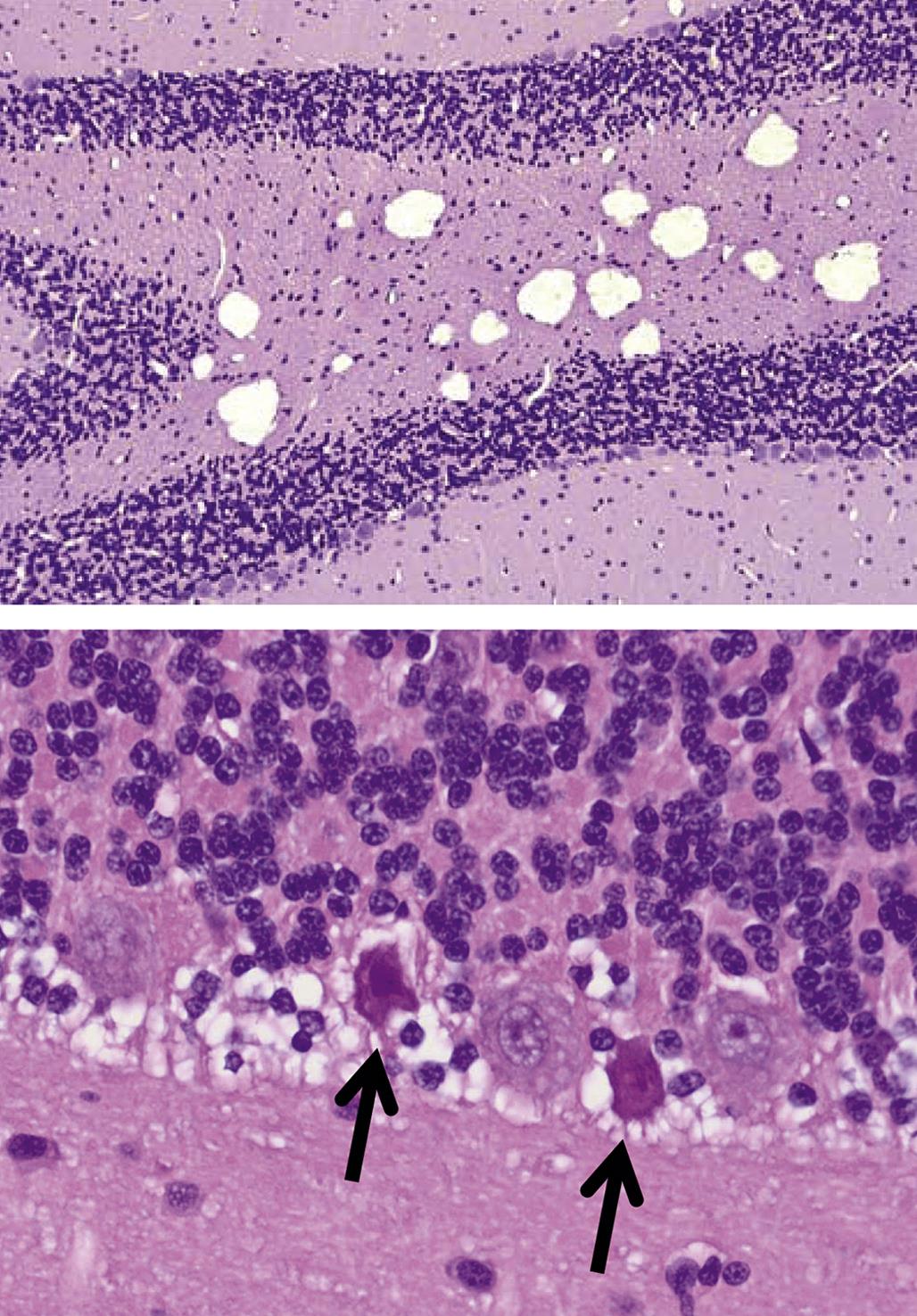
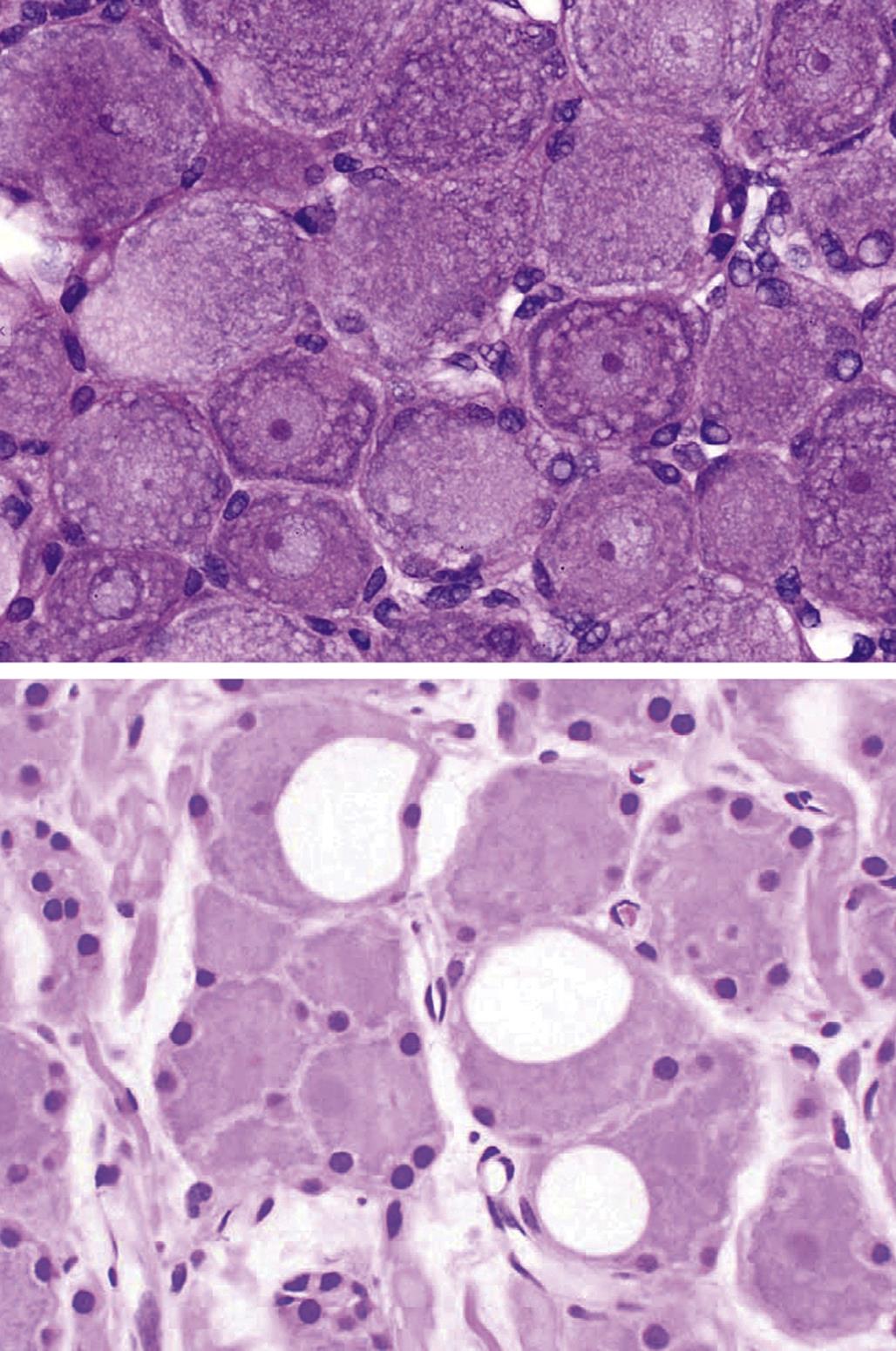
Another form of vacuolar artifact in the CNS results from swollen organelles in particular cell types, especially astrocytes. These may occur as small clear holes in the cytoplasm or as larger, clear vacuoles in the astrocytic processes adjacent to shrunken neurons or blood vessels. The main toxicant-induced differential to this artifact is intramyelinic edema, in which the myelin sheaths encircling intact axons are disrupted by small to large vacuoles (Figure 21.29). Genuine neurotoxic vacuolar lesions usually exhibit a bilaterally symmetrical distribution and are fairly widespread, while the artifactual changes are often limited to one or a few locations.
Axonal “spheroids” (i.e., focally enlarged axons) are occasionally encountered in white matter tracts of the brainstem and spinal cord as a spontaneous change in older animals. Animals from all treatment groups may be affected, including the negative control cohort, and no relationship to xenobiotic dose is evident. A common site for this finding is the cranial portion of the dorsal funiculus, which is the terminus for the ascending sensory tracts in the spinal cord. Spheroids also can result from neurotoxicant exposure (Figure 21.25), but in such cases their number is higher and they usually exhibit a dose-response relationship.
Melanin occurs as a normal pigment in certain sites within the CNS. This location should not be surprising since melanocytes are derived from precursors of neural crest origin. Typical areas for deposition are the meninges in pigmented mice (Figure 21.41) and certain sheep breeds or the neurons of the substantia nigra, the locus coeruleus, and some other basal nuclei in primates, including humans. The neuronal form, termed neuromelanin, is minimal at birth but accumulates over time. The meningeal melanin has not been linked to a specific function, nor is its extent altered by exposure to neurotoxicants. The proposed tasks for neuromelanin are protective, such as storage depots for oxyradicals resulting from metabolism of monoamine neurotransmitters (e.g., dopamine (DA) and norepinephrine (NE)) or residual products of autophagy. This role is supported by the ability of neuromelanin to selectively sequester the neurotoxic metabolite MPP+, a monoamine analog. A more active function for neuromelanin is suggested by the loss of pigmented neurons in basal nuclei in concordance with the onset of certain neurodegenerative diseases.
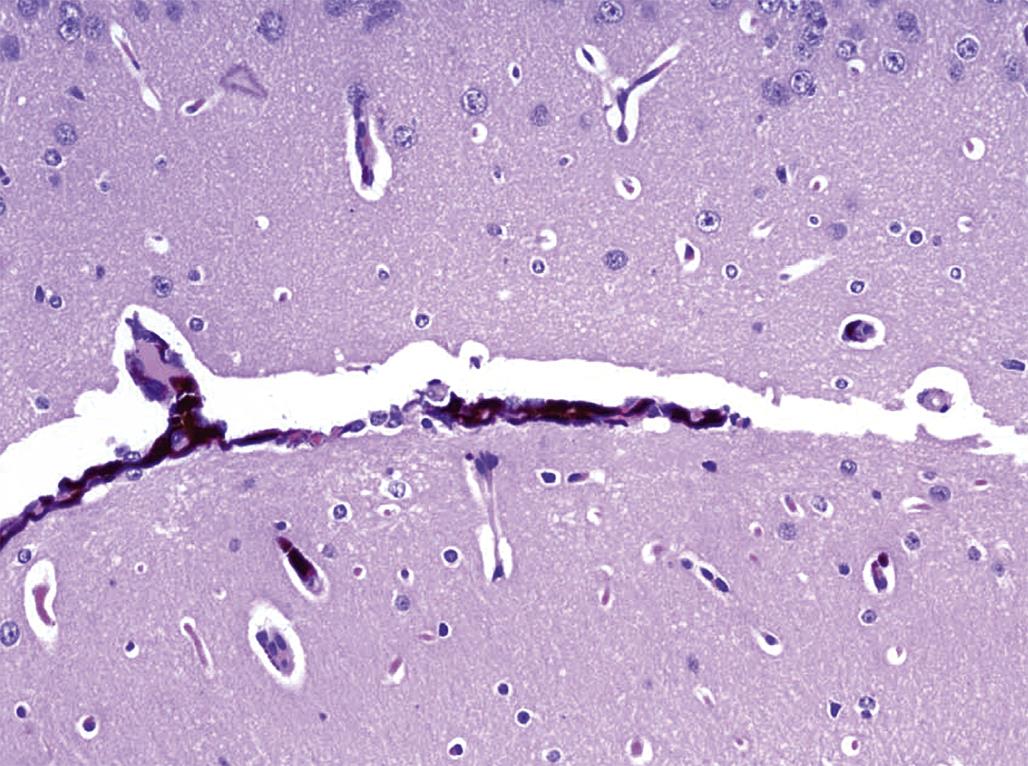
The ancillary tissues within the CNS are host to two common incidental changes. The first is focal fibrosis of the meninges, which appears as a localized accumulation of fibroblasts and collagen. The second is occasional small aggregates of leukocytes in the choroid plexus, and to a lesser extent near meningeal blood vessels. These foci usually contain mature lymphocytes, mainly of the T-cell lineage, and they are seemingly present to fulfill a surveillance function for the acquired arm of the immune system. Mast cells scattered within the perineurium are a frequent finding in large nerve trunks of the PNS, especially in rodents. In general, none of these changes is impacted by neurotoxicant exposures. In contrast, the presence of granulocyte and/or macrophage aggregates in neural tissues is associated with an immediate response by the innate immune arm to a noxious stimulus, though usually one caused by irritation or bacterial contamination associated with implantation of a direct delivery device into or near the CNS rather than as a direct neurotoxic effect induced by the test article.
One key factor required for success in the practice of toxicologic neuropathology is a solid understanding of normal neuroanatomy. In particular, normal features must be recognized as such and not misdiagnosed as lesions. The typical danger of this sort encountered in the nervous system are the circumventricular organs (CVOs), six small CNS domains (Figure 21.42) containing many small, fenestrated capillary loops located adjacent to the ventricular system. Five are found near the third ventricle (listed from rostral to caudal, and dorsal to ventral): the subfornical organ (SFO), vascular organ of the lamina terminalis (OVLT), pineal gland, subcommissural organ (SCO), and median eminence. The sixth, the area postrema (AP), is in the brainstem near the fourth ventricle. Neurons are found in three CVOs (the AP, OVLT, and SFO); the pineal gland is comprised of pinealocytes, while the SCO consists of specialized ependymal cells. Common misdiagnoses applied to the CVOs by inexperienced investigators include congenital cysts, inflammation, and neoplasia. In contrast to CVOs, genuine cysts (Figure 21.43) have irregular contours, are lined by simple or stratified epithelial linings, and may contain secretory material, while inflammatory and neoplastic lesions appear as disorganized cell aggregates that may penetrate and disrupt the adjacent parenchyma.
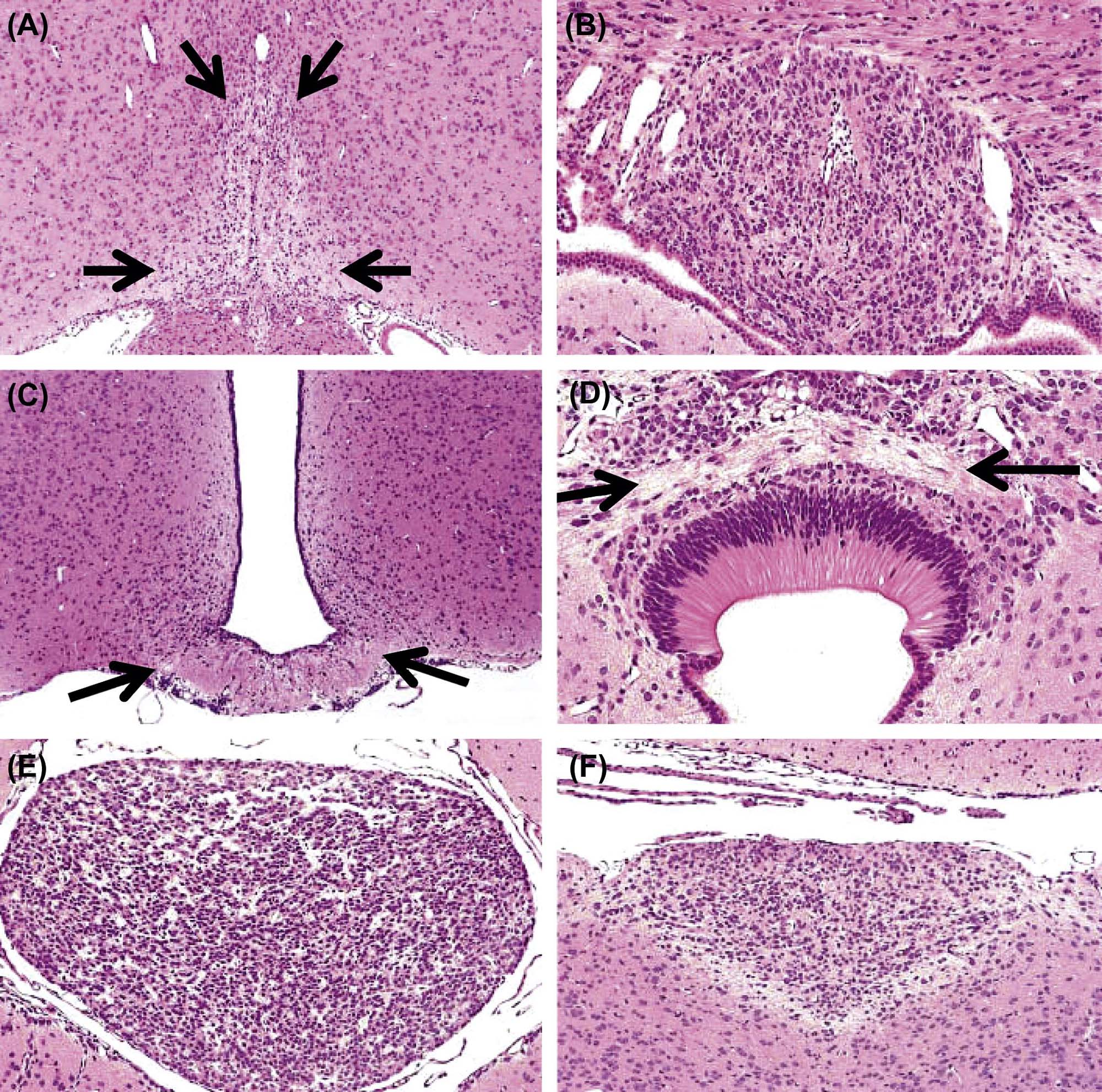
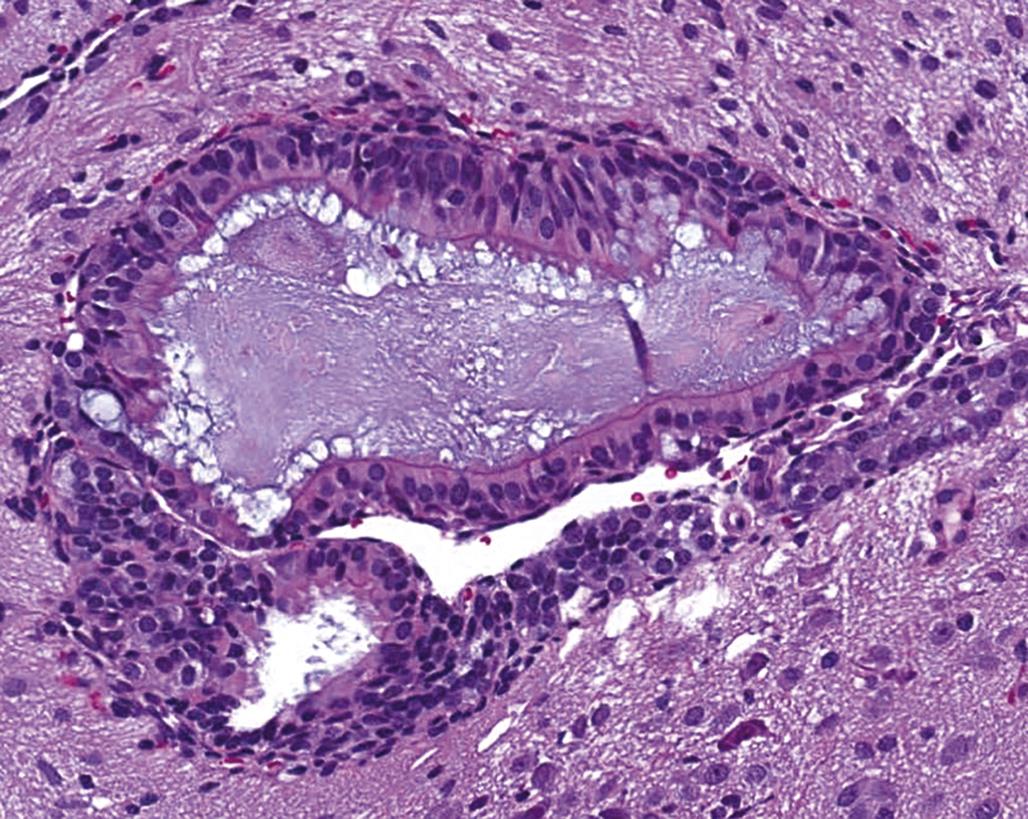
Mechanisms of Nervous System Injury
Neurotoxicants have been shown to produce their effects in many different fashions. Basic mechanisms may target any of several neural elements: particular structures, specific cell types, or certain molecular pathways. The precise nature of the resulting neuropathological lesions will be dictated by the mode of action. Accordingly, the remainder of this section will briefly review basic mechanisms of neurotoxicity using prototypical neurotoxicants (Table 21.1).
Aberrant Cell Migration and/or Differentiation
The typical lesion resulting from abnormal migration and terminal differentiation of neuronal precursors is heterotopia (Figure 21.12), but the mechanisms by which neurotoxicants produce this change are incompletely understood. Experiments with avian and rodent embryos suggest that damage to neuronal stem cells, radial glial (which guide neuronal precursors to their appropriate positions), or both may be factors. Several hypotheses have been advanced to explain the defects.
One alternative is that pluripotent stems cells may be destroyed before they can produce enough daughter cells to populate one or more waves of neuronal migration. This effect may be confined to the neuroepithelium of the neurulating embryo, or it may also impact partially differentiated structures such as ganglia (which are derived from neural crest cells). A second option is that the spatial and/or temporal expression of critical morphogens (growth factors, neurotransmitters, etc.) is disrupted for one or several lineages of neural cell precursors. This consequence may lead to several kinds of functional abnormalities in the affected cells: misdirected migration, reduced movement in the proper direction, and/or incorrectly timed terminal differentiation. In most cases, neurons will seek—and usually succeed, at least in part—to connect with their normal cellular targets. Nonetheless, behavioral or other neurological deficits may be observed since the resulting fiber tracts typically will be too long and insufficiently wired to support normal neural activities.
Altered Intracellular Transport
Toxic agents that disrupt the transfer of essential macromolecules from the cell body to its distant processes will produce degeneration in outlying structures. This finding is typical of axonopathies, in which blocked slow axonal transport results in chemical rather than physical transection of the axon. Axonotoxic chemicals generally induce this effect by promoting the formation of covalent cross-links between macromolecules (e.g., neurofilaments). The resulting disordered filamentous masses will lodge at axonal constriction points like the nodes of Ranvier.
The axon distal to the plug, including the presynaptic terminal, will starve and eventually disintegrate as a primary lesion, to be followed in time by secondary degeneration of the local Schwann cells. The longest axons (e.g., those in long tracts in the spinal cord white matter and in PNS nerve trunks) are affected first. Regeneration can occur in PNS axons if the insult is transient, but affected CNS axons cannot be restored. The neuron generally will survive since the supply of nutrients to its metabolically active body remains intact, although chronic exposure to certain agents (e.g., IDPN) can induce an irreversible loss of CNS motor neurons.
Cell Turnover
Chemical exposure in the rat has been linked to heightened incidences of glial neoplasms (mainly in the deep cerebrum), granular cell tumors (cerebral and cerebellar meninges, chiefly on the dorsal midline), and malignant reticulosis (cerebral and cerebellar meninges). These tumors are thought to evolve from populations of partially committed, oligopotent stem cells that retain the capacity for low-level cell division throughout adulthood. The specific sites are the cerebral subventricular zone adjacent to the lateral ventricles for the glial masses, or fibroblasts or facultative phagocytes in the meninges. These cell populations offer targets for genotoxic carcinogens (discussed later in the “Macromolecular Adducts” section), but cell proliferation is a necessary requirement by which genetic mutations will become fixed in the genome. Cell proliferation occurs in corresponding sites in adult rodents, dogs, monkeys, and humans.
Abnormal cell turnover is also an important factor in neurotoxicity. During development, increased or decreased programmed cell death at an inappropriate time will either reduce or prevent neuronal processes from correctly connecting with their target cells. Such disruptions typically will perturb anatomic and functional maturation. Similarly, toxicants can alter postnatal neuronogenesis in the brains of mature individuals, which can impact behavioral and cognitive abilities (e.g., learning, memory).
Energy Depletion
Reduced availability of energy stores within highly active neural cells, especially neurons, is a common predisposing factor to cell degeneration and eventual cell loss in many brain regions. The mechanism of action involves the capacity of such neurotoxicants to interfere with enzymes in the electron transport chain by which the mitochondria replenish ATP; in the absence of this energy storage molecule, neurons lose their ability to sustain their ionic gradients and thus fail to produce action potentials. The neurotoxic moiety may reach the target neurons by a circuitous route.
For example, MPTP is converted to the toxic metabolite MPP+ by peri-synaptic astrocytes in a two-step reaction that requires the enzyme monoamine oxidase B (MAO-B). Subsequently, dopaminergic neurons in the substantia nigra selectively sequester MPP+ using transporters for the reuptake of monoamine neurotransmitters. Finally, MPP+ enters the mitochondria where it quenches the activity of reduced nicotinamide adenine dinucleotide (NADH) dehydrogenase, the first enzyme in the electron transport chain. Other agents (e.g., bromethalin, hexachlorophene) damage myelinating cells by uncoupling mitochondrial oxidative phosphorylation, which leads to fluid accumulation in the interlamellar space (Figure 21.29).
Excitotoxicity
Excitotoxic neurodegenerative lesions are thought to result from imbalances in reciprocal feedback between neuronal populations that produce the excitatory neurotransmitter glutamate and the inhibitory neurotransmitter γ-aminobutyric acid (GABA). For example, trimethyltin (TMT) increases glutamate release in the hippocampus while reducing the synthesis and reuptake of GABA and glutamate. Enhanced excitation results in persistent influx of ions across the neuronal plasma membrane, including entry of calcium (Ca2+). The rising Ca2+ tide finally floods the neuronal cytoplasm and propels the cell to begin a death spiral. The initial lesion of punctate vacuolation in neurons, consistent with swelling of intracytoplasmic tubular organelles (Figure 21.18), occurs within hours of injury and may progress to full-fledged necrosis (Figures 21.19 and 21.20) over 1 or 2 days.
The vulnerability of various neuronal populations to TMT-induced excitotoxicity is affected by many factors. For example, mice are more sensitive to TMT than rats. However, the lesion pattern diverges in these two species; in the hippocampus, mice preferentially develop dentate gyrus lesions with little involvement of cells in the CA regions, while the converse is true in rats. The sensitivity of the CA neurons is age-dependent in rats with lesions developing only after the hippocampal pyramidal cells mature (i.e., after postnatal day 7). This phenomenon highlights the need to evaluate neurotoxicity under various conditions when seeking to identify and characterize new neurotoxic agents.
Macromolecular Adducts
Known neurocarcinogens in rats (e.g., acrylonitrile, ethylene oxide, nitrosoureas) chiefly produce glial neoplasms following prenatal or prolonged postnatal exposure. These agents are all potent DNA-alkylating chemicals, indicating that the likely mechanism of neural cell initiation is the formation of DNA adducts leading to mutations in critical genes. The typical deep cerebral location of the tumors suggests that the target populations are likely to be retained progenitor cells in the cerebral peri-ventricular zone or subcortical white matter. It is not known whether or not neurotoxicant exposure is responsible for the preponderance of glial tumors in humans, or which chemical(s) or critical developmental periods in people might be most vulnerable to neurocarcinogenic toxicants.
Neurotransmission Disruption
Neurotoxic agents that block synaptic neurotransmission can induce profound neurological dysfunction in the absence of major structural lesions. Classic presentations include altered contraction of skeletal muscles in certain body regions or all major muscle groups, abnormal cognitive abilities, and/or dysfunction of the autonomic nervous system (ANS). Affected subjects can recover if vital functions (e.g., breathing, nutrient intake) can be maintained until the affected synaptic elements are restored. Disruption of synaptic function occurs in several ways.
Decreased Neurotransmitter Release
Attenuated release of neurotransmitters is responsible for botulism and tetanus, both of which are caused by toxins produced by bacteria of the genus Clostridium as metabolic byproducts when grown under anaerobic conditions. Botulism develops in fish, birds, and mammals following bacterial colonization in the digestive tract or deep wounds, or by ingestion of the preformed toxin in contaminated food. Botulinum toxin serves as a protease to degrade docking molecules needed for fusion of synaptic vesicles with the presynaptic membrane of the axon terminal. In the absence of vesicle fusion, acetylcholine is not released and the postsynaptic membrane is not stimulated. Skeletal muscles cannot contract, so flaccid paralysis ensues (e.g., limberneck, the manifestation of botulism in waterfowl).
In contrast, tetanus arises when bacteria within infected puncture wounds generate tetanospasmin, which is taken up at neuromuscular junctions in the body periphery and then transported to the CNS by retrograde axonal transport. Tetanospasmin also functions as a protease that thwarts vesicle docking, but affected synapses fail to discharge the inhibitory transmitters GABA and glycine. The removal of feedback control that modulates the excitatory signals to skeletal muscles results in the characteristic presentation of tetany (i.e., intermittent or constant skeletal muscle contraction).
Persistent Neurotransmitter Activity
Sustained neurotransmission is another recognized route to synaptic neurotoxicity. This effect can be produced in several fashions, and typically occurs in association with CNS neurons. One means is to increase neurotransmitter release from presynaptic terminals. This approach explains the efficacy of mirtazapine (Remeron), a tetracyclic antidepressant that acts as an α2-adrenergic blocker to promote the discharge of serotonin (5HT) and norepinephrine (NE).
A second, more common way for maintaining neurotransmitter levels in the synapse is to reduce the rate at which transmitters are removed. Inhibition of neurotransmitter reuptake is the desired pharmacological response of several selective 5HT reuptake inhibitor (SSRI) antidepressants, such as fluoxetine (Prozac), paroxetine (Paxil), and sertraline (Zoloft). However, self-medication with two such drugs and/or related pro-serotonergic agents (e.g., amphetamines, monoamine oxidase inhibitors (MAOI), some nutraceuticals (e.g., St. John’s wort, Hypericum perforatum), and certain opioids) can over-stimulate organs with high numbers of 5HT receptors (e.g., CNS, gastrointestinal tract ANS), resulting in the potentially lethal “serotonin syndrome” (also called “serotonin toxicity”). Some agents can lessen reuptake of two neurotransmitters simultaneously: dopamine (DA) and NE—the effect of bupropion (Wellbutrin); or 5HT and NE—duloxetine (Cymbalta) and venlafaxine (Effexor). St. John’s wort is thought to inhibit the uptake of DA, 5HT, and NE, although the exact mechanism remains unclear.
Reduced Neurotransmitter Metabolism
A third strategy is to decrease the degradation of neurotransmitters. This effect is produced by treatment with MAOIs, where the two MAO isoforms are bound to the outer mitochondrial membrane of most cells, including neurons and astrocytes; MAOs promote the oxidative deamination of DA, 5HT, and NE. Similarly, inhibition of synaptic acetylcholinesterase (AChE) by exposure to carbamate or organophosphate (OP) insecticides prevents the hydrolytic removal of acetylcholine from synapses in the CNS and PNS as well as at neuromuscular junctions. The OPs irreversibly phosphorylate the serine residue at the active site of AChE, so any recovery must await the synthesis of new enzyme. Administration of pralidoxime (2-pyridine aldoxime methyl chloride (2-PAM)) early after OP exposure can reduce the extent of neurotoxicity by hydrolyzing the OP–serine bond that inactivates AChE. In contrast, the bond between a carbamate and AChE is reversible, with the hydrolytic removal over several hours leading to the gradual restoration of AChE activity.
Termination of Transmembrane Ionic Gradients
Some agents affect neurotransmission by altering the character of the action potential. The maintenance of normal ionic gradients across the charged membranes of neuronal processes is critical for sustaining the ability to conduct impulses. Neurotoxicants may disrupt the flow of a single ion, or they may impact multiple currents. For example, the type I pyrethroid insecticides allethrin and tetramethrin prolong Na+ influx, lessen the peak of the Na+ flow, and decrease the steady-state K+ efflux, apparently by modulating resting (i.e., closed) Na+ channels so that they open more slowly than normal. In contrast, tetrodotoxin blocks the extracellular pores of Na+ channels, thwarting the inward flow of Na+ ions and thus preventing propagation of any nascent action potential. Neurotoxic effects on ion channels often occur in the absence of structural lesions.
Unknown Impact on Neurotransmission
The pathogenesis for the delayed neurotoxicity in organophosphate-induced delayed neurotoxicity (OPIDN) remains incompletely understood. Certain OP classes (e.g., phosphoramidates and phosphonates) incite central-peripheral distal axonopathy (dying back polyneuropathy) in association with phosphorylation of a second enzyme, neuropathy target esterase (NTE). This molecule is an integral membrane protein in all neurons. Structurally, NTE is unrelated to AChE and other major serine esterases. The physiological role for NTE during adulthood is unknown; in development, NTE appears to control interactions between neurons and glia. Agents that cause OPIDN are postulated to bind covalently to the NTE active site. Ultimately OPIDN results in an abnormal propagation of neural impulses.
Oxidative Damage
Gradual accumulation of free radical-mediated damage to cellular macromolecules is a well-recognized pathway by which toxic agents can produce cell lethality. In the nervous system, the usual cellular targets are neurons due to their high metabolic rates and correspondingly outsized exposure to oxygen. Within neurons, oxidative damage has been postulated to disrupt expression of critical genes, damage cell and organelle membranes, and/or distort signaling pathways. The role of oxidative stress has been examined in the neurotoxicity of copper (Cu), iron (Fe), manganese (Mn), rotenone, and other redox-active chemicals.
Certain toxicants can become concentrated in susceptible neurons, thereby extending the period during which oxidative injury can occur. For example, Mn is localized in the basal nuclei of primates, including humans, apparently due to efficient blood-borne delivery by the Fe-carrying protein transferrin and/or the strong affinity that Mn has for neuromelanin deposits in certain primate nuclei (e.g., substantia nigra). One proposed pathogenesis of Mn-induced neurotoxicity is oxidation of DA. The degree of DA modification may be raised synergistically in neurons with elevated intracellular concentrations of Fe, which is a potent oxidizing agent in its own right. Rodents are much less susceptible to Mn neurotoxicity as their basal nuclei neurons do not contain neuromelanin, and thus are less able to selectively sequester Mn.
Loss of antioxidant molecules in the nervous system is an indirect way of promoting damage by oxygen free radicals. For example, reduced glutathione (GSH), an intracellular reducing agent that protects against oxidative damage to cell macromolecules, is concentrated in the neuropil and white matter tracts of the CNS, especially astrocytes, but is expressed in high levels in only a few neuronal populations (e.g., cerebellar granule and Purkinje cells, sensory neurons in dorsal root ganglia). This distribution overlaps with the expression of xanthine oxidase (XO), an important enzyme that generates reactive oxygen species as a byproduct of purine catabolism; XO activity is present at much higher levels in astrocytes relative to neurons. Thus administration of 1,3-dinitrobenzene (DNB), which is metabolized by XO to form free radicals, injures astrocytes to a much greater extent than it does neurons.
Vascular Impairment
Increased vascular permeability (termed dysoria) resulting from damage to the BBB and/or blood–nerve barrier (BNB) is a common mechanism employed by several neurotoxicants. In this setting the microvasculature is the primary target for toxicity. Nonetheless once the barriers have been breached, other CNS cells may experience direct toxic effects of their own. For example, lead (Pb) neurotoxicity is associated with altered vessel diameters (constricted or narrowed); mural changes (endothelial swelling, necrosis); and thrombosis in CNS capillaries, especially those in the cerebrum and cerebellum. Neuropil near damaged vessels is edematous as a consequence of fluid and protein extravasation. Nearby neurons (cerebrocortical pyramidal cells and cerebellar Purkinje cells) become necrotic, and in chronic lesions there are associated with regional accumulation of reactive astrocytes. At least some neuronal death may arise from direct Pb-induced toxicity to neurons, since dead cells have been observed in CNS regions that lacked evidence of vascular damage. The primary changes induced by Pb in PNS trunks are Wallerian-like axonal degeneration and segmental demyelination mainly affecting motor nerves.
Instead of a definitive “barrier,” it may be more useful to view the protection of the brain imparted by the specialized endothelial cells (and to a lesser extent astrocytes) of the BBB and BNB as selective gateways that allow the passage of some essential molecules and attempt to inhibit the passage of others. In fact, due to intended (due to anatomic variation) or unintended disruptions in the “barriers” as well as the numerous areas where the “barriers” are known to be less selective (e.g., some of the CVOs, certain brain nuclei especially hypothalamic nuclei), nearly every molecule, even large biologics, may enter the brain/spinal cord/nerves in some areas.
Multiple Mechanisms
Some neurotoxic agents appear to cause neural damage by several mechanisms at once. Hexachlorophene induces massive intramyelinic edema by uncoupling oxidative phosphorylation from ATP synthesis in oligodendroglia and by altering GABA concentrations in CNS neurons. Methylmercury produces profound neuronal necrosis in many brain regions by disrupting BBB permeability, altering neural cell metabolism, inhibiting synthesis of RNA and protein, and promoting oxidation and denaturation of cell proteins and membranes. Ethanol decreases DNA, RNA, and protein levels in neurons and astroglia and also incites apoptosis of neural cell precursors during development. In our experience, many agents elicit multiple chemical and molecular changes that can be linked to the induction or progression of neurotoxicity, and only careful attention to defining the appropriate study design can permit conclusions to be drawn regarding the primary mechanism of neurotoxic action.
Summary
The regimented arrangement of anatomic and chemical attributes within the CNS and PNS dictate the functional responses by which an individual maintains homeostasis and interacts with his/her external environment. These same stereotypical features are responsible for the patterns of neurotoxic changes produced by numerous xenobiotics. The susceptibility of the CNS is further enhanced by the extraordinary metabolic needs and limited capacity for repair. Taken together, these characteristics render the possibility of neurotoxic damage among the greatest challenges in navigating the world around us, and in bringing new chemical products to market.
The means for identifying new neurotoxic hazards as well as assessing and managing the risk posed by exposure to such agents are evolving as technical advances increase the affordability, speed, and utility of more detailed neuropathology assessments. The current reliance on limited batteries of functional (e.g., behavior, functional observational battery) and structural (e.g., gross lesions, brain weight, histopathology) endpoints for screening is a reasonable first-tier approach for use with general toxicity studies. More extensive investigations are the norm for second-tier instances where neurotoxicity has been observed or is predicted to be likely based on prior observations. Over time, we predict that current third-tier approaches like noninvasive imaging to follow disease progression throughout life and more extensive “omics” evaluations to explore molecular mechanisms responsible for neurotoxicity will be used on a more regular basis. The next decade or two hold considerable promise for a substantially improved understanding of neurobiology in both health and disease, and the knowledge gained using various toxicants to probe the CNS and PNS will be instrumental in this renaissance.