Embryo, Fetus, and Placenta
Colin G. Rousseaux1 and Brad Bolon2, 1University of Ottawa, Ottawa, ON, Canada, 2GEMpath Inc., Longmont, CO, United States
Abstract
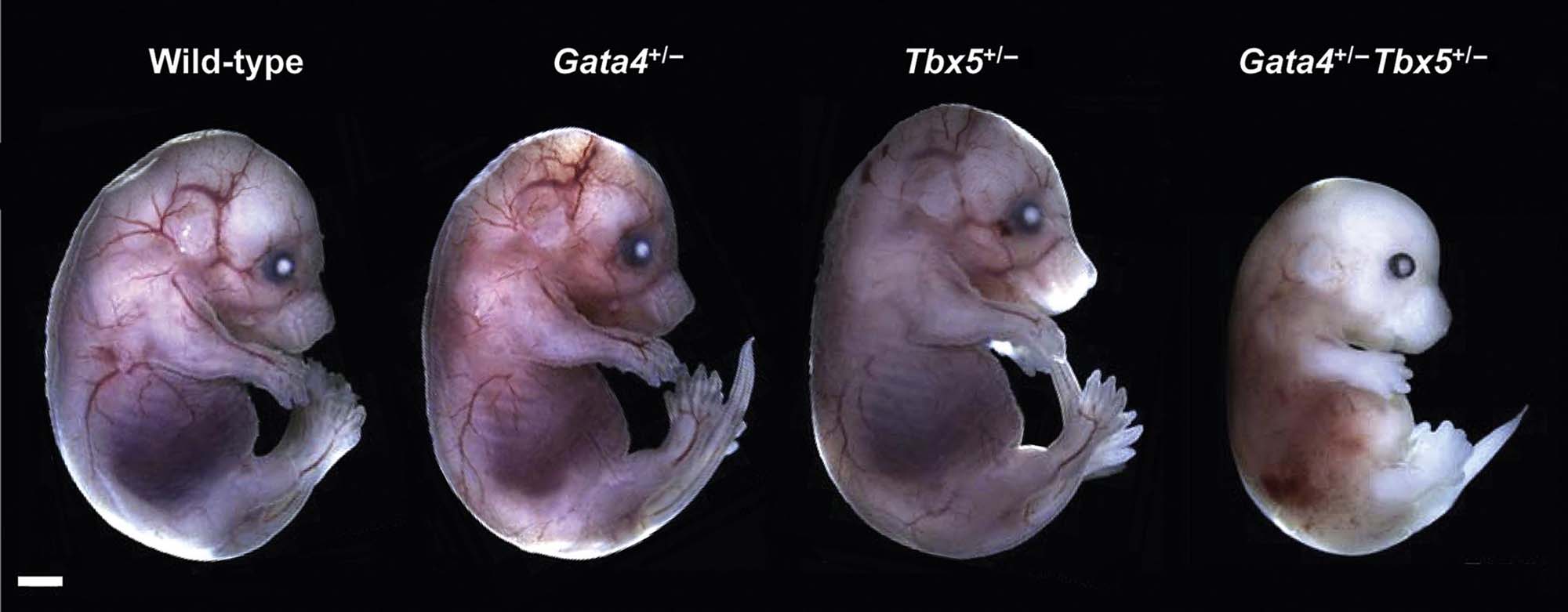
Until the latter half of the 20th century, congenital malformations were believed to arise solely from genetic or infectious causes. The thalidomide tragedy of the late 1950s and early 1960s showed that xenobiotics also could cause birth defects, and as such sensitized the scientific community and the public to the potential hazards of exposing the developing conceptus to foreign substances. Epidemiological and experimental data indicate that in utero exposure to many xenobiotics can result in structural defects and/or functional deficits within progeny. Humans and animals may be exposed to xenobiotics (e.g., drugs, environmental contaminants, metabolic byproducts, metals) during critical period of development, and at least some of these agents can act as teratogens. Accordingly a more thorough understanding of the processes that drive both normal and abnormal development has assumed increasing importance in toxicologic pathology. This chapter will describe normal and abnormal developmental events in selected vertebrate species, detail methods for determining if and how much such processes may have been disrupted by teratogens, and describe common mechanisms of abnormal development along with factors that may influence the extent and severity of such damage.
Keywords
Embryo; fetus; placenta; mechanisms; developmental toxicology; malformations; risk assessment
Introduction
Until the latter half of the 20th century, congenital malformations were believed to arise solely from genetic or infectious causes. The thalidomide tragedy of the late 1950s and early 1960s showed that xenobiotics also could cause birth defects, and as such sensitized the scientific community and the public to the potential hazards of exposing the developing conceptus to foreign substances. Epidemiological and experimental data indicate that in utero exposure to many xenobiotics can result in structural defects and/or functional deficits within progeny. Humans and animals may be exposed to xenobiotics (e.g., drugs, environmental contaminants, metabolic byproducts, metals) during critical period of development, and at least some of these agents can act as teratogens. Accordingly a more thorough understanding of the processes that drive both normal and abnormal development has assumed increasing importance in toxicologic pathology. This chapter will describe normal and abnormal developmental events in selected vertebrate species, detail methods for determining if and how much such processes may have been disrupted by teratogens, and describe common mechanisms of abnormal development along with factors that may influence the extent and severity of such damage.
Basic Principles of Developmental Toxicology
Toxicologic pathology of the embryo, fetus, and placenta, termed developmental toxicology, assesses the effects induced by xenobiotics, physical agents (e.g., heat, ionizing radiation), or metabolic byproducts. In general the effects of developmental toxicants are manifested as congenital anatomical defects or biochemical/functional deficits that become evident during gestation or soon after birth. However, some toxicant-induced effects initiated during gestation may not become apparent for extended periods after birth, as is the case for transplacental carcinogens.
Developmental toxicity in vertebrates may involve damage to either of the two portions of the conceptus: the embryo (or fetus), and the placenta. The embryonic and fetal periods typically are defined by their relationship to organogenesis, which is the stage during which the organ primordia (or anlagen) first differentiate. The embryonic period is that span of time that precedes and overlaps with organogenesis, while the fetal period is that portion of gestation following the conclusion of organogenesis in which the organs expand toward their adult forms. The fetal period encompasses most of gestation in species with long gestations (e.g., humans), while the embryonic period occupies the major portion of gestation in species with short gestations (e.g., rodents). Indeed the majority of developmental events corresponding to the third trimester of the human fetus actually occur during the first postnatal week of life in rats and mice.
Developmental toxicology is closely allied with teratology (derived from the Greek “teras” (monster) and “logos” (study)), the investigation of manifestations, causes, and mechanisms of developmental anomalies as well as means for their prevention. Thus developmental toxicologic pathologists must command a clear grasp of the six basic principles of teratology, first proposed by James G. Wilson nearly five decades ago.
1. Interactions between the genotype of an embryo or fetus and many environmental factors will influence the onset and degree of teratogenicity;
2. The stage of development during which an embryo or fetus is exposed determines its susceptibility to teratogenic agents;
3. Teratogens induce abnormal development according to specific mechanisms;
4. Physical properties of developmental toxicants determine their access to the embryo or fetus;
5. Death, malformation, intrauterine growth retardation, and postnatal functional abnormalities are all manifestations of teratogenesis; and
6. Expressions of developmental toxicity tend to be dose-related, ranging from slight defects or weight decrements to major malformations or death.
With the exception of Principle 2, these six tenets are identical to those that dictate how adult animals will respond to toxicant exposures. Accordingly, no great adjustment is required when shifting from assessment of toxicologic pathology findings in adults to examination of toxicant-induced effects in developing organisms. It should be noted that not all species are equally sensitive to exposure, and not all littermates show abnormalities, or the same abnormality, following treatment. Maternal toxicity also should be considered and will be discussed later in this chapter.
Incidence of Congenital Anomalies
In developed countries the incidence of major malformation is 2%–4% of births per year. Approximately 3% of congenital defects are considered to arise from maternal exposure to teratogens in the environment.
The incidence of developmental abnormalities in domestic animals ranges from 2% to 12%, depending on the species. Morphologic anomalies in laboratory animals have been well characterized, especially in those species commonly used for teratology studies (particularly mice and rats). In mice, major spontaneous malformations incompatible with postnatal survival (e.g., cardiac septal defects, neural tube defects (NTDs), organ agenesis) occur at a frequency of less than 1%. Minor structural anomalies that do not preclude postnatal viability range in incidence from 1% to 5% for lesions like cranial displacement of the gonads, hemorrhage, sternebral asymmetry, and unossified phalanges, and up to 35% for common variants such as renal pelvic cavitation and supernumerary or wavy ribs.
Normal Morphologic Development
The general processes involved in the normal development of humans and most animals are well characterized. Beginning with fertilization, normal morphologic development proceeds through a series of cell expansions and ordered rearrangements that proceed in a stereotypical fashion (Figure 25.1). Terms commonly applied to specific periods during normal development include embryogenesis (the progression of steps needed to form an embryo) and organogenesis (the period in which initial primordia of major organs are formed).
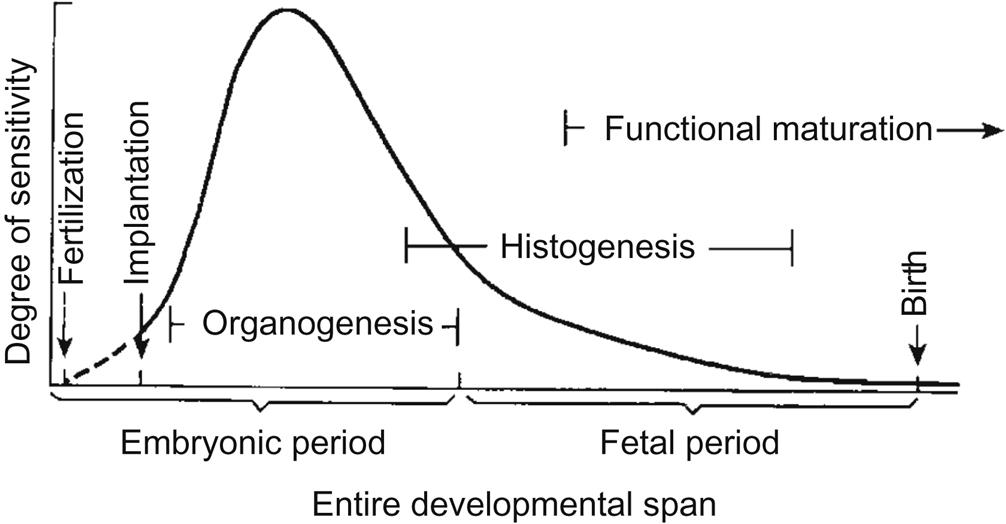
The process of ordered development is guided by multiple factors. Genetic programming, including correct coding and faithful gene transcription and protein translation, is essential to the final differentiation pathway that a cell selects. Accurate intercellular communication is critical to normal development, and occurs via several mechanisms: direct cell contacts, morphogen gradients, and tissue interactions (induction). Cell populations respond to their genetic predispositions as well as their internal and external chemical milieus by choosing one or more functional outcomes—cell shape changes, morphogenetic movements, etc.—that will promote ordered differentiation and growth of the organism. An understanding of these processes is essential before the impact of prenatal exposure to teratogens can be fully comprehended. A more detailed consideration of embryonic, fetal, and placental anatomy and physiology is available through excellent developmental biology and developmental pathology texts (see Further Reading).
The zygote (i.e., a one-cell fertilized embryo) divides repeatedly to produce a morula, a grape-like cluster of fundamentally equal, totipotent embryonic stem (ES) cells. Over time, these cells differentiate into two distinct lineages, the trophoblast (a thin layer that encloses the fluid-filled central cavity, or blastocoele), and the inner cell mass, which will become the embryo. These early developmental stages are relatively resistant to toxic insults because of the totipotent nature of ES cells. Toxicant-induced damage that can severely injure these cells will lead to embryonic death prior to implantation, while less serious injury typically will permit the surviving cells to replenish their full complement with little more than a short developmental delay.
In mammalian embryos the process of gastrulation is initiated shortly after the blastocyst (an embryo with a blastocoele) implants within the uterine wall. During this stage the embryonic germ layers (ectoderm, mesoderm, and endoderm) are formed, and the molecular blueprint for the first organs is established. Toxicant exposures at this developmental stage typically do not produce structural malformations, but rather will cause embryonic death and resorption.
Organogenesis, the most complex stage of development, results in specification of all organs within the embryo. The complexity of this developmental stage means that interference by exogenous toxicants can produce devastating effects on the growing organism, usually in the form of major malformations. Teratogenic effects often are produced by interfering with intercellular communication (which guides appropriate connectivity), cell movement, generation of morphogen gradients (where morphogens are molecules that direct structural differentiation when present at the correct concentration), and programmed cell death (needed to sculpt organ primordia into their final form).
As development progresses in a given organ primordium, totipotent stem cells (which express all genes and so can become any cell at need) evolve to pluripotent stem cells (which can develop into many but not all cell types), then to oligopotent stem cells (which can form only a few cell types), and thence to fully differentiated cells (Figure 25.2). The progressive increase in specialization (termed histogenesis) renders the cells more capable of performing their specific functions but typically renders them more vulnerable to development toxicants.
{kind=link}
Developmental Toxicity Testing and Risk Assessment
The toxicologic pathology aspects of developmental toxicity testing are usually performed using various in vivo test systems. Fundamental aspects of developmental biology at the cellular and molecular levels have commonly employed both invertebrates, such as fruit flies (Drosophila melanogaster) and worms (Caenorhabditis elegans), as well as many vertebrates, including zebrafish (Brachydanio rerio), avians (e.g., chick (Gallus gallus) and quail (Coturnix sp.)), and many mammals. The test species used in conventional developmental toxicity testing are generally mammals (mainly rodents and rabbits). This section describes the main principles of such tests and delineates factors that make various mammalian species the subjects of choice for such analyses.
Hazard Identification and Dose-Response Analysis
The first two steps in the risk assessment process for the developmental toxicity potential of any test article are hazard identification and the dose-response evaluation. These steps are difficult to separate, as hazard should always be evaluated in the context of the exposure route, dose, timing, and duration.
Default Assumptions
Extrapolation to humans of developmental toxicity data derived from animals is a complex process. A number of general default assumptions can be made to guide the risk assignment process.
The first assumption is that an agent, which produces an adverse developmental effect in experimental animal studies, will pose a potential hazard to humans following sufficient exposure during a susceptible stage of development. This supposition has been confirmed for many developmental toxicants but is not true in all cases. The most glaring example of this discordance is the thalidomide catastrophe, in which the mammalian test species used in developmental toxicity testing (rodents) were insensitive to in utero toxicity while human fetuses were exquisitely sensitive. Rabbits, whose embryos are sensitive to thalidomide, were not used in its safety assessment.
The second assumption is that all four manifestations of developmental toxicity—death, structural malformations, growth alterations, and functional deficits—are of concern in defining whether or not an agent is a potential hazard. The notion is easily confirmed for compounds that significantly reduce the number of viable implantation sites, increase the incidence of perinatal death, or lessen the birth weight or postnatal weight gain. These endpoints are ideal for this purpose as they offer objective (easily quantified) measurements of an effect, with the range of interindividual variation being relatively narrow under normal circumstances.
The third assumption is that the types of developmental effects seen in animal studies are not necessarily the same as those that may be produced in humans. This assumption is made because it is impossible to determine a priori which species will be the most appropriate in terms of predicting the specific types of developmental toxicity that might occur in humans, the chief of which are the divergent timing of critical developmental periods (Table 25.1) and fundamental differences in the anatomic, biochemical/molecular, and physiological characteristics of the conceptus (embryo/fetus, placenta, or both). Additional factors such as variations in dose, route, and timing of exposure; unique kinetic and/or metabolic disposition of the test article and its metabolites; and species-specific differences in the mechanisms of action/toxicity also may play a role.
Table 25.1
Timing of Developmental Milestones in Vertebratesa
| Species | Length of gestationb | 2-Cell | 8-Cell | Morula | Blastocyst | Implantation | Gastrula | Neurula | 10-Somite | Embryogenesis completed | |
| Zebrafish | Brachydanio rerio | 60 | 0.75 | 1.25 | 2 | 5 | N/Ac | 8 | 10 | 12 | 48 |
| Chick | Gallus gallus | 21 | 3 | 4 | 5 | 10 | N/Ac | 18 | 24 | 36 | 10 |
| Mouse | Mus musculus | 19 | 1 | 2 | 3 | 3.5 | 5 | 6.5 | 7.5 | 8.5 | 15 |
| Rat | Rattus rattus | 21 | 1.5 | 2.5 | 4 | 5 | 6 | 8 | 9.5 | 11 | 16 |
| Hamster (golden) | Mesocricetus auratus | 16 | 0.75 | 1 | 2.75 | 3.5 | 5 | 6 | 7 | 8 | 13 |
| Guinea Pig | Cavia porcellus | 67 | 2 | 3.5 | 4.5 | 5.5 | 6.5 | 9 | 12 | 15 | 29 |
| Rabbit | Oryctolagus cuniculus | 32 | 2 | 2.5 | 3 | 3.5 | 7 | 7 | 8 | 9 | 17 |
| Ferret | Mustela putorius | 42 | 2 | 4 | 5 | 6 | 13 | 13 | 14 | 15 | 20 |
| Cat | Felis domesticus | 63 | 3 | 3.5 | 4 | 5 | 12 | 12 | 13 | 14 | 28 |
| Dog | Canis domesticus | 63 | 4 | 6 | 7 | 8 | 16 | 16 | 17 | 18 | 40 |
| Pig | Sus scrofa | 115 | 1 | 2.5 | 4 | 5 | 14 | 9 | 14 | 15 | 33 |
| Sheep | Ovis aries | 145 | 1 | 2.5 | 4 | 6 | 16 | 10 | 15 | 16 | 35 |
| Cow | Bos taurus | 282 | 1 | 3 | 6 | 8 | 30 | 14 | 19 | 21 | 40 |
| Horse | Equus caballus | 336 | 1 | 3 | 4 | 6 | 35 | 14 | 18 | 19 | 45 |
| PRIMATES | |||||||||||
| Rhesus | Macaca mulatta | 165 | 1 | 2 | 4 | 5 | 8 | 16 | 20 | 23 | 47 |
| Human | Homo sapiens | 267 | 1 | 2 | 3 | 5 | 9 | 13 | 16 | 25 | 58 |

aTimes are estimated mean values in days postconception except for the zebrafish and chick, where values for all columns (except length of gestation and completion of embryogenesis for chick) are in hours. The day of confirmed mating is designated as the day of conception (gestational day 0).
bFor zebrafish and chicken, the values in this column refer to length of incubation before hatching.
cN/A denotes not applicable in this species.
Table reproduced from Haschek, W.M., Rousseaux, C.G., Wallig, M.A. (Eds.), 2013. Handbook of Toxicologic Pathology, third ed., Academic Press, Table 62.1, p. 2706 with permission.
The fourth assumption is that the “most appropriate species” will be used to acquire data for estimating human risk. Ideally translation of animal data to assess human risk is based on evidence permitting a direct cross-species comparison, such as an equivalent pharmacokinetic (PK) profile or similar reactivity of the putative target in animal and human tissues. In the absence of such data, it is presumed that the most sensitive animal species is the most appropriate for use. This supposition is based on epidemiological (in human) and experimental (in animal) observations showing that, relative to the most sensitive animal species, humans are as sensitive or more so to the great majority of human developmental toxicants.
The fifth and final assumption is that developmental toxicants will generally follow a dose-response curve that includes a distinct threshold. This idea is based on the known capacity of the developing organism to either repair or compensate for a certain amount of damage at the cellular, tissue, or organ level.
How Dose Relates to Developmental Defects
The stage of embryonic or fetal development must be considered before attempting to define the correlation between dose and the consequences of abnormal development. Low doses do not necessarily cause functional deficits, and high doses do not always lead to embryonic or fetal death. The outcome also depends on the stage of development at which the agent is administered and the mechanism by which it acts.
During early development, relatively high exposures to xenobiotics may induce death or may elicit few visible changes. These divergent outcomes result from the wide range of fates open to most embryonic cells prior to the start of organogenesis. Once organogenesis is in progress, lesser concentrations of a substance may produce prominent morphological defects. This change in the pattern of anomalies arises because the death of partially committed stem cells tends to produce cell losses and/or functional deficits, which can no longer be fully reversed by the surviving remnants of the targeted cell population or compensated for by other partly differentiated stem cell collectives.
Toward the end of organogenesis, malformation is less likely and tends to require very large doses. Instead late in embryogenesis xenobiotic exposures are more likely to cause functional deficits and intrauterine growth retardation.
Transplacental carcinogenesis depends on the nature of the carcinogen. Current thinking is that neoplasms can be induced by exposure to very small concentrations during organogenesis or to moderate concentrations after organogenesis. The proposed mechanism of initiation usually is a genotoxic (i.e., mutating) event.
Laboratory Animal Studies
The conventional strategy for developmental toxicity testing programs assesses the ability of the test article to adversely impact development following prenatal exposure during the entire period of organogenesis (i.e., a period spanning the time during which all organs will initially be formed). Program designs commonly test the product in one rodent and one nonrodent species, most often the rat and the rabbit, unless other species (e.g., nonhuman primates) are deemed more relevant (often the case for biomolecules).
The conceptus is exposed to the compound by treating the dam, and the evaluation of toxicity endpoints is performed shortly before parturition. More recent regulatory guidelines recommend longer treatment periods with dosing extending from before or shortly after conception until well after birth (often weaning). Certain study designs have evolved to assess both developmental and reproductive toxicity (DART) endpoints in multiple generations within a single large experiment.
Maternal Endpoints of Developmental Toxicity
Many endpoints for assessing the health status of the dam provide data that also provide information regarding the health status of the progeny. In-life measurements of maternal health include clinical signs, food and water consumption, and maternal weight gain (both the amount and the rate) during treatment. Essential parameters to quantify at necropsy are terminal body weight and target organ weights of the dam, and in some cases histopathological analysis of selected target organs; an important consideration is to also measure either the total weight of the gravid uterus or the maternal carcass weight after uterine removal to avoid bias due to different litter sizes. In addition, the number of implantation sites should be counted. Care must be taken when assessing the relevance of such maternal data because some endpoints (e.g., weight gain over time in rabbits) can vary greatly for reasons unrelated to xenobiotic exposure.
Progeny Endpoints of Developmental Toxicity
In multiparous species the litter is the experimental unit used for statistical analysis of developmental pathology data sets, rather than each individual offspring. Therefore developmental toxicity studies include 10 litters per group (assuming a standard litter size of 8), and not 80 conceptuses. This convention is employed because progeny in a litter are subject to a common environment, resulting from shared influences arising from the specific dam and/or their particular siblings.
Important parameters to measure in all developmental toxicity studies include several measures of litter size—the total number of conceptuses, the numbers of viable and dead conceptuses, and the number of resorptions (abnormal implantation sites)—and the presence of gross structural malformations and incidental variations. The number of dead fetuses and resorptions show a test article’s ability to induce prenatal mortality, and both counts are increased by exposure to many developmental toxicants. In near-term fetuses and neonates, total body weights may be acquired, and the sex ratio can be determined using the anogenital distance. In studies that include collection of parturition and lactation data, several other measurements may be taken for the neonates (or juveniles). The easiest measurements are the number of perinatal deaths, incidence of major structural malformations, birth weights, and progressive weight gain over time. If desired, additional endpoints to consider may include clinical signs and symptoms (e.g., abnormal skin coloration, inability to suckle); simple behavioral tests (usually to test progressive motor development); and histopathological examination.
Certain endpoints require special postnecropsy processing. Common techniques of this nature include analysis of internal organs (accomplished by making multiple free-hand cross sections of fixed fetuses (Wilson’s technique)) or skeletal anatomy (performed by skeletal double staining with Alcian blue and alizarin red S to highlight cartilage and bone, respectively). These latter techniques are time-consuming and require considerable technical expertise on the part of the personnel.
Careful consideration of the study goal is required when designing developmental toxicity experiments. Major malformations are much more likely to be induced by high-dose, short-term exposures that take place during a particular organ’s critical period of development (e.g., in mice, gestational days (GD) 8–9 for NTDs, or GD 10–11 for cleft palate, where the morning after conception is considered to be GD 0). In contrast the typical design used in regulatory studies to assess the risk of developmental toxicity calls for low-level, longer-term dosing throughout organogenesis, which provides a fairly robust means of evaluating prenatal death and growth retardation arising from substantial damage to any organ system. This latter design is especially desirable when the target organ(s) vulnerable to a test article are not yet known.
Animal Models
Rodents
Mice and rats are particularly useful as the initial mammalian test species for most conventional developmental toxicity screens due to their small size, short gestation, large litter size, ease in breeding, and ready availability. Rats have the advantage of producing larger fetuses, thereby enabling easier evaluation. Data are available (both in online databases and from vendors) for rates of spontaneous anomalies in numerous strains and species of rodents. Many inbred mice have a high incidence of strain-specific structural defects. Other mouse strains have low incidences of spontaneous defects and are quite impervious to developing them under the influence of teratogens. This phenomenon means that pairing sensitive and resistant strains of inbred mice in a single study can provide a powerful platform for investigating the influences of genotype and specific molecular mechanisms on the genesis of certain developmental defects, and particularly the potential actions of toxicants in promoting them.
Some institutions use alternative rodent species for teratology studies. In this regard the golden (Syrian) hamster (Mesocricetus auratus) is a model of choice. Like other rodents, this species has a 4-day estrous cycle and produces litters averaging 8–10 pups. However, the gestation length of the golden hamster is the shortest known for a placental mammal (16 days, vs 18–20 days for various mouse strains and 20–22 days for rat strains).
Rabbits
Rabbits are lagomorphs rather than rodents, and thus represent the nonrodent species of choice for developmental toxicity testing. Factors favoring this choice include their larger size (which provides ample samples for analysis), accuracy in timing the start of conception (since ovulation is induced by copulation, occurring about 10 hours after mating), and large number of progeny (ranging from 4 to 12). The length of gestation is approximately 30 days. Some strains produce a spectrum of malformations similar to those of humans when exposed to thalidomide during gestation, but in general rabbits do not appear to be superior to other animal models in predicting the human response to most developmental toxicants.
Nonhuman Primates
In certain instances the preferred nonrodent species might be a nonhuman primate (NHP). A number of NHP species have been employed for this purpose. Factors dictating this decision include close similarities in maternal kinetics and metabolism of xenobiotics, placental structure, and reproductive physiology—especially anatomic and temporal aspects of early embryogenesis. However, in spite of these resemblances, NHPs are avoided when possible due to their scarcity, high cost, long gestation, and monoparous pregnancy (i.e., tendency to produce a single conceptus per pregnancy).
These factors substantially limit sample sizes in treatment groups (typically 3–5 per cohort), and routinely result in a data set in which interactions between the test article and sex cannot be reliably assessed due to the unequal numbers of male and female progeny. Another complicating aspect of developmental toxicity testing with NHPs is that their sensitivity to xenobiotic-induced teratogenicity often differs from that of humans. An example is methotrexate, a folic acid analog, which has been shown to induce skeletal defects (chiefly in the limbs and skull) in humans but produces little or no embryotoxicity in NHPs. Finally, NHPs are commonly not used due to their cognitive abilities and need for substantial environmental enrichment and socialization activities.
Other Vertebrate Species
Other mammals (ferrets, guinea pigs, cats, dogs, swine) are occasionally employed in developmental toxicity studies. However, characteristics such as size, cost, difficulty in handling, long gestation periods, lack of a historical database, and lack of any obvious predictive superiority as well as social acceptability limit their use.
The use of chicken embryos for developmental toxicity screening has the obvious advantages of a readily available, low-cost model with a recognized historical database and short developmental period (21 days at the optimal temperature). However, in the strictest sense, in ovo testing in embryonic chicks is not equivalent to in vivo testing as maternal PK processes and placental transfer can play no role in this avian system. In addition, chick embryos have a relatively high sensitivity to many exogenous agents, and significant differences exist in the course of embryogenesis among avian and mammalian species. Together, these factors typically limit the use of chicken (and quail) eggs to mechanistic studies while precluding their use as a system for developmental toxicity testing.
Human Studies
Epidemiological studies are used in two fashions to assess the potential that an agent has for inducing developmental toxicity in humans. The first approach uses such data in an attempt to identify new teratogens based on increased incidences of structural and/or functional abnormalities in exposed individuals. The second tactic is to test the relevance of developmental toxicity data derived from animal studies to human hazard identification and risk assessment, which is accomplished by determining whether or not the spectrum of defects seen in animals is recapitulated in humans.
Human epidemiological studies have identified many likely human developmental toxicants, including pharmaceutical agents (e.g., anticonvulsants, antineoplastic agents, thalidomide); environmental pollutants (e.g., maternal alcohol abuse or heavy smoking); heavy metals (e.g., methyl mercury); and viral infections (e.g., rubella, Zika). However, such studies by themselves cannot confirm causality and have relatively poor sensitivity.
Maternal Versus Developmental Toxicity
An important question when trying to classify hazards and manage risk is to define whether or not a toxicant-associated developmental defect results from direct damage to the embryo (a primary effect) or is the indirect sequel to some maternal disease process (a secondary effect). The concern arises because mild embryolethality and nonspecific fetotoxicity commonly occur for doses at which dams exhibit more substantial signs of toxicity. The number of possible mechanisms for indirectly inducing maternally mediated effects is probably much lower than the number of direct-acting (i.e., embryotoxic) mechanisms.
Several hypotheses have been suggested to explain this phenomenon. One prominent possibility is failed maternal support of the conceptus (e.g., by anemia or reduced placental circulation), leading to hypoxia in the conceptus. A second prospect is maternal production of some endogenous toxicant by xenobiotic-damaged maternal tissues (e.g., acidosis leading to a precipitous drop in the pH of embryonic tissues); a variant of this scenario is maternal illness leading to an increase in maternal core body (and therefore uterine) temperature—in other words, hyperthermia (fever). Finally, modification of maternal metabolism can intensify or lessen the effects of an agent (e.g., by activation of a nontoxic pro-drug or production of a more toxic or longer-lasting metabolite). Maternal toxicity can promote developmental toxicity by several means, so it is probable that the spectrum of maternally controlled developmental abnormalities in the offspring may be quite broad.
Short-Term Tests
Abbreviated screens for developmental toxicity are often employed to prioritize chemicals for further testing. These tests are typically classified together as “in vitro” assays. However, this appellation is a misnomer as many are instead “ex vivo” or “in vivo” preparations (i.e., utilizing isolated whole organs or intact, viable, nonmammalian organisms, respectively). These short-term assays are not meant to substitute for “in vivo” testing in pregnant mammals. Instead, they expose invertebrate organisms (e.g., flies (D. melanogaster), hydras (Hydra attenuata), worms (C. elegans)) or simplified vertebrate systems (e.g., cultured cells, detached body parts (micromass organ cultures), or whole embryos) to xenobiotics for limited periods.
The short-term procedures are particularly relevant for identifying and characterizing mechanisms of developmental toxicity due to the ability to observe developmental events over time and the complete exclusion of any confounding maternal influences. These methods are also popular as indicators of developmental toxicant accumulation in polluted aquatic environments.
The short-term screens have several advantages over in vivo teratogenicity testing in mammals. The major benefits include their rapidity, low cost, and reduced utilization of sentient laboratory animals. Unfortunately the abbreviated assays also suffer from many significant disadvantages: lack of specificity, inability to model metabolic and PK events, and uncertain applicability of invertebrate models to mammalian species. The lack of metabolic capability may be addressed by partially restoring maternal metabolic function via the addition of cytosolic or S9 microsomal fractions from homogenized liver.
Other Considerations in Developmental Toxicity Testing
As is the case with other toxic effects, developmental toxicity is strongly impacted by PK parameters. The influence of such factors is complicated by the fact that two different organisms are involved, mother and conceptus, both of which have separate and often distinct PK profiles. Maternal uptake, transplacental passage into and away from the embryo, and maternal elimination are critical parameters that govern the access of xenobiotics to the offspring. Biotransformation (metabolism) also plays an essential role in defining the extent of developmental toxicity. In this regard the fully mature maternal metabolic pathways are usually of more importance in dictating the effect of xenobiotics, as enzyme systems in the embryo and fetus usually are incompletely differentiated and/or incapable of high-throughput chemical conversion. Some toxicants may be sequestered within embryonic tissues due to the altered conditions (e.g., lower oxygen tension, reduced glucose stores, pH gradient) relative to those in the dam.
Epigenetic influences (i.e., intrauterine environmental imbalances rather than embryonic genetic anomalies) also may produce long-lasting developmental effects. The classic examples of this phenomenon are maternal metabolic abnormalities, especially diabetes but also some environmental toxicants (e.g., arsenic, bisphenol A). In humans the incidence of congenital defects in women with preexisting diabetes (about 10%) is higher than that of the general population (approximately 3%), particularly if blood glucose levels are poorly controlled during organogenesis (i.e., the first trimester). The abnormalities occur in multiple systems but are common and severe in the head (e.g., NTDs and craniofacial malformations), heart (often atrial and ventricular septal defects), and caudal trunk (hypoplasia or absence of the caudal trunk and hind limbs). The putative mechanism for functional and structural abnormalities is that nutritional imbalances in cells of the conceptus skew the embryonic programming for many neuroendocrine control systems that regulate energy homeostasis and metabolism. These changes not only alter intrauterine development but may also predispose individuals to developing their own metabolic diseases late in life.
Responses to Injury
In many respects a conceptus represents a unique tissue type that just happens to be a transient passenger in its maternal host. The bases for its unique response, particularly during the earlier stages of gestation, are the lesser degree of cellular differentiation and its tolerance for low-oxygen conditions. Both these factors permit more ready repair of damage that would decimate the more differentiated and oxygen-dependent maternal tissues. Nonetheless the fundamental responses of the embryo/fetus and placenta to toxicant-induced damage are similar to the well-recognized reactions that occur in the tissues of toxicant-treated adult animals.
As seen in other tissues the conceptus (and neonate) responds to toxicant-induced injury by initiating a number of mechanisms that either sustains the damaged cells until they can be repaired or tries to mitigate the damage when severely perturbed cells degenerate and die. The basic manifestation of cytotoxicity in the conceptus is cell death, the ramifications of which range from none or slight (in early embryos with pluripotent stem cells) to major structural malformations, growth retardation, or in utero death. Unlike adult animals the developing conceptus seldom mounts an inflammatory response to necrosis (or other insults) until the latter half of gestation, when the immune system has begun to mature.
The plasticity of the early embryo (based on its large complement of totipotent and pluripotent stem cells) allows compensatory growth to completely restore many tissue defects after nonlethal exposure that would induce lethality or major malformations in older individuals. Thus statistically significant growth retardation in many cases is likely to represent a developmental delay rather than a genuine reflection of teratogenic activity per se. To be sure, attempts at repair following developmental toxicant exposure may lead to deformations, which are congenital anomalies due to ineffective regeneration (as opposed to malformations, which are defects produced by tissue injury). An example of a deformation is intestinal atresia resulting from fibrosis following ischemic injury to the intestine.
Death
Approximately 50%–70% of all human conceptuses are lost during the first 3 weeks of development, and by the end of pregnancy another 78% of the early survivors will have died. Chromosomal anomalies are apparent in 60% of human abortuses occurring at less than 12 weeks of gestation, and have a prevalence of 1 in 160 live births. These aberrations account for many multisystemic malformations in defective fetuses that make it to term. Malformed fetuses are born dead 10 times more often than delivered alive, and many of those that survive birth will die shortly thereafter. Thus a general principle of developmental toxicologic pathology is that major structural defects are usually fatal (assuming no medical intervention to repair them). The most common lethal defects that occur during early gestation are NTDs, cardiovascular malformations, and multisystemic anomalies (that cannot be linked to preexisting chromosomal damage).
Nonviable embryos and fetuses are typically eliminated by spontaneous (natural) causes, a process termed terathanasia. However, mortality due to congenital anomalies does not stop with birth. Approximately 8% of human infants with major malformations die during the neonatal and juvenile periods.
Malformations
Embryonic and fetal malformations provide the most spectacular evidence of developmental disaster, and as such have been a source of curiosity and dread for millennia. The modern science of teratology first facilitated investigations of the biological basis of developmental defects, and then expanded to encompass testing for developmental toxicity. The reason for this expanded focus is that numerous xenobiotics are teratogens (i.e., agents capable of inducing malformations) (Table 25.2).
Table 25.2
Examples of Specific Congenital Defects with Known Etiologic Agents in Mammals
| System | Defect | Etiology | Species |
| Central nervous/axial skeleton | Anencephaly/exencephaly | Colchicine | Mouse |
| Injected inorganic arsenic | Mouse, rat, hamster | ||
| Ethylnitrosourea | Rat | ||
| Methylhydrazine | Rabbit | ||
| Retinoic acid | Hamster | ||
| Thalidomide | Rabbit | ||
| Auditory nerve hypoplasia | Quinine | Human | |
| Cerebellar hypoplasia | Triamcinolone acetonide | Baboon, monkey | |
| Encephalocele | Ionizing radiation | Mouse | |
| Hydroxyurea | Mouse | ||
| Hydrocephalus | Ionizing radiation | Mouse, rat | |
| Vitamin A deficiency | Rat, rabbit, pig | ||
| Iniencephaly | Streptonigran | Rat | |
| Microcephaly | Hyperthermia | Guinea pig, rabbit | |
| Methylnitrosourea | Rat | ||
| X-rays | Rat | ||
| Spina bifida | Actinomycin D | Mouse | |
| 7,12-Dimethyl-benz[a] anthracene | Rat | ||
| Thalidomide | Baboon, monkey | ||
| Craniofacial | Agnathia/micrognathia | Injected inorganic arsenic | Mouse |
| Pyrimethamine | Rat | ||
| Retinoids | Hamster, monkey | ||
| Anophthalmia/microphthalmia | Ethylnitrosourea | Rat | |
| Glycol ethers | Mouse | ||
| Cataracts | Mirex | Rat | |
| Cleft face | Ochratoxin A | Mouse | |
| Cheiloschisis/palatoschisis | Dioxin (TCDD) | Mouse | |
| Diphenylhydantoin | Mouse | ||
| Glucocorticoids | Mouse | ||
| Griseofulvin | Cat | ||
| Cyclopia | Veratrum californicum | Ruminants | |
| Microtia and/or synotia | Veratrum californicum | Mouse | |
| Nasal defects | Griseofulvin | Cat | |
| Open eye | Methyl salicylate | Mouse | |
| Retinal defects | X-rays | Rodents | |
| Cardiovascular | Atrial septal defects | Alcohol | Human |
| Dextroamphetamine sulfate | Mouse | ||
| Dextrocardia | Actinomycin D | Rat | |
| Great vessel and vena cava anomalies | Valproic acid | Rat | |
| Tricuspid valve anomalies | Methyl chloride | Mouse | |
| Various defects | Diethylene glycol dimethyl ether | Mouse | |
| Ventricular septal defects | Alcohol | Human | |
| Dextroamphetamine sulfate | Mouse | ||
| Thalidomide | Rabbit | ||
| Respiratory | Lung agenesis | Vitamin A deficiency | Rat, pig |
| Lung hypoplasia | L-Asparaginase | Rabbit | |
| Gastrointestinal | Absent gallbladder | Nitromifene | Dog |
| Retinoic acid | Hamster | ||
| Anal atresia | Colchicine | Mouse | |
| Diaphragmatic hernia | Nitrofen | Mouse, rat | |
| Esophageal/duodenal atresia | Thalidomide | Human | |
| Gastroschisis | Vincristine | Mouse | |
| 6-Azauridine | Rat | ||
| Omphalocele | Actinomycin D | Mouse, rabbit | |
| Hyperthermia | Rat | ||
| Urogenital | Cryptorchidism | Cadmium | Rat |
| Vitamin A deficiency | Rat, pig | ||
| Hydronephrosis | Bradykinin | Mouse | |
| Hydroureter | Vitamin A excess | Rat | |
| Hypospadias | Chlorambucil | Rat | |
| Intersexuality | Androstenedione | Rat | |
| Prunus serotina | Pig | ||
| Ovarian hypoplasia | Diethylstilbestrol | Mouse, rat | |
| Renal agenesis | Chlorambucil | Rat | |
| Injected sodium arsenate | Rat | ||
| Thalidomide | Human | ||
| Musculoskeletal | Arthrogryposis | Anagyrine | Cow, pig, sheep |
| Nicotiana glauca | Pig | ||
| Sudan grass | Horse | ||
| Digit malformations | Cyclophosphamide | Human | |
| Ethylenethiourea | Rat | ||
| Limb reduction defects | Acetazolamide | Mouse, rat | |
| Caffeine | Mouse | ||
| Hydroxyurea | Rabbit | ||
| N-Methyl-N-nitro-N-nitrosoguanidine | Mouse | ||
| Thalidomide | Rabbit, monkey, human | ||
| Muscular dystrophy | Vitamin E deficiency | Rat, rabbit | |
| Polydactyly | Cytosine arabinoside | Mouse | |
| Rib and/or vertebral defects | Ethylene glycol | Mouse, rat | |
| Injected sodium arsenate | Mouse, rat | ||
| Hydroxyurea | Rabbit | ||
| Tail shortened and/or malformed | Colchicine | Rabbit | |
| T-2 toxin | Mouse |

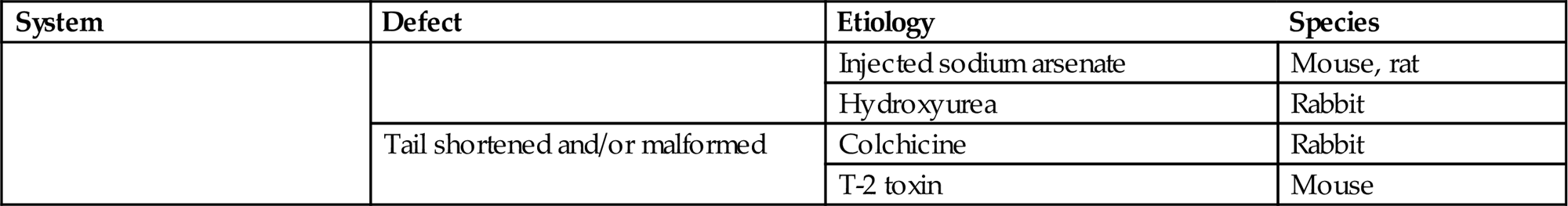
Table reproduced from Haschek, W.M., Rousseaux, C.G., Wallig, M.A., (Eds.) 2002. Handbook of Toxicologic Pathology, second ed., Academic Press, Table IX, p. 917 with permission.
Teratogens often affect multiple species, and induce a similar spectrum of effects across sensitive species. Examples of toxic agents that generate malformations in both humans and laboratory animals include “recreational” (e.g., cocaine, ethanol, nicotine) and therapeutic (e.g., anticonvulsants, antineoplastics, immune suppressants) drugs as well as many environmental contaminants (e.g., heavy metals, plant toxins, solvents). Indeed, some xenobiotics are so potent, and their constellation of defects so reproducible, that they constitute preferred agents for investigating teratogenic mechanisms (e.g., retinoid-induced cleft palate).
Malformations have been reported in essentially all organs and systems of multiple vertebrate species. This section briefly describes some of the more common anomalies observed in human obstetrical practice and animal developmental toxicity experiments. Unless otherwise noted later, these defects have been linked to many pathogens (usually viruses), physical agents (e.g., heat, radiation), and xenobiotics.
Central Nervous System
Dysraphism
Dysraphism is the generic term for incomplete fusion of the neural tube (i.e., a NTD). Variants of this lesion are typically defined based on the affected site. Common forms are cranioschisis (nonfusion of the cranial portion), as exemplified in anencephaly, exencephaly, and encephalocele (Figure 25.3), as well as rachischisis (nonfusion of the caudal region) leading to spina bifida (Figure 25.4) and meningocele. Rarely the entire neural tube may remain open, a condition called craniorachischisis. The pathogenesis for these lesions is failed elevation and/or fusion of the neural folds, or rarely reopening of a thinly sealed fusion site.
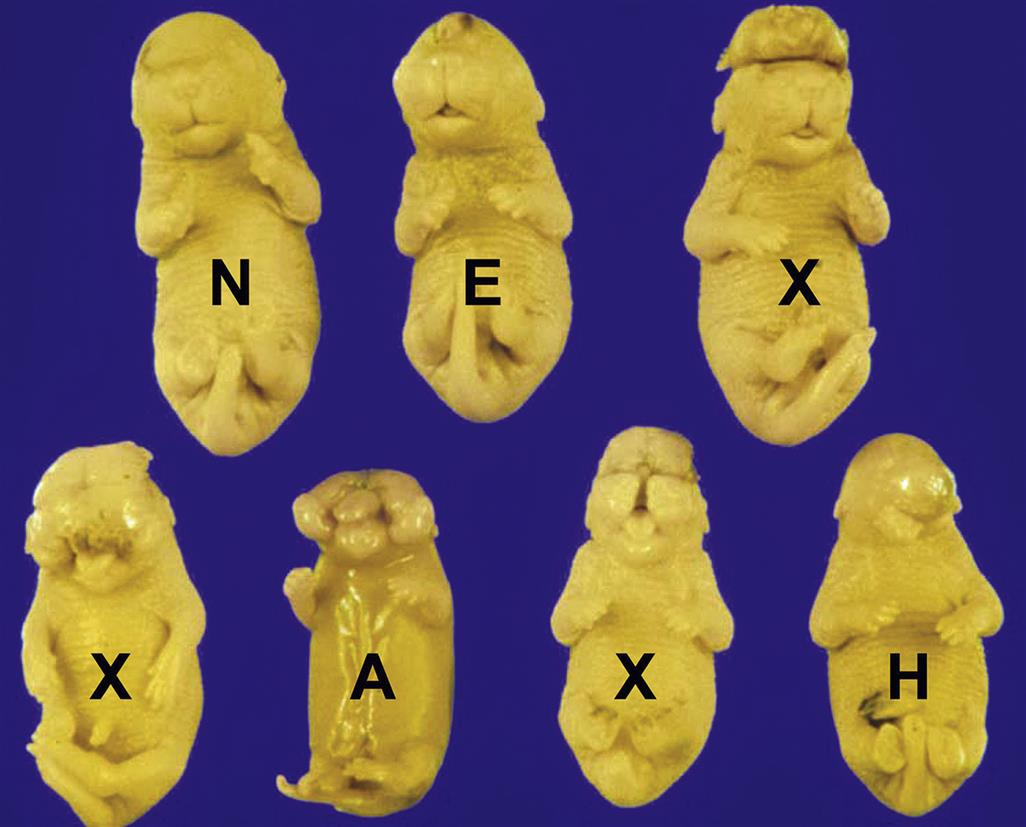
If the head is affected the development of the pituitary gland is often rudimentary. These structural defects are associated with functional abnormalities, which may lead to secondary hypoplasia of endocrine tissues (i.e., adrenal, gonadal, and thyroid) that require pituitary releasing factors for their own maturation.
Anencephaly
Anencephaly (no brain) is more common in humans than in domestic animals. The usual appearance is absence of neural tissue rostral to the brainstem (Figure 25.3). It has a prevalence ranging between 0.8 and 18/10,000 infants, depending on the locale. Areas with heightened prevalence typically have populations with genetic predispositions or who live in regions with substantial environmental contamination. One factor thought to contribute to the severe loss of brain mass is the long gestational period, which permits lengthier exposure to the destructive effects of amniotic fluid.
Exencephaly
Exencephaly (external brain) is seen more frequently in laboratory animals than anencephaly. In exencephaly the entire brain is present but is exposed (Figure 25.3). Mechanisms include disruption of tissue interactions, inhibition of cell proliferation, or general cytotoxicity. The occurrence of exencephaly is greatly increased in certain inbred mouse strains, indicating the importance of genetic factors. Preservation of the exposed brain tissue in animal species with short gestations is proposed to result from a shorter exposure to amniotic fluid, although other factors also must contribute as anencephaly is observed occasionally in mice (Figure 25.3).
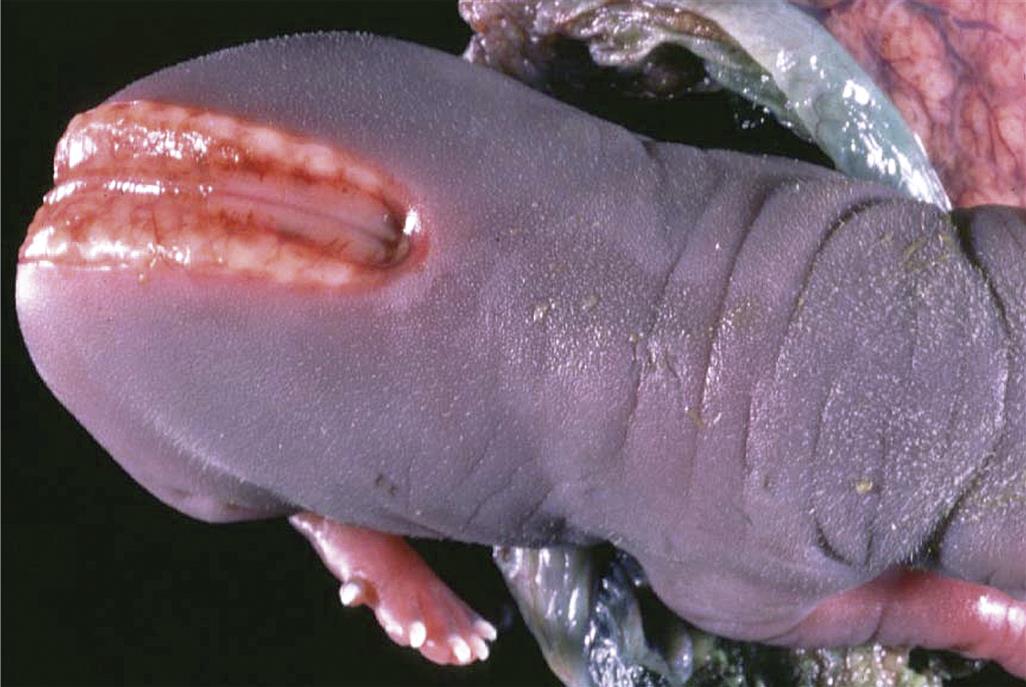
Encephalocele is the extension of brain tissue (usually cerebral cortex and the overlying meninges) through a small opening in the calvarium (Figure 25.3). These lesions typically present as small round nodules on the midline, often in the frontal region. The masses may be covered by skin, or the brain tissue may be exposed. In general the neural tissue is fairly well differentiated. In mice, encephalocele and exencephaly occur together in litters exposed to various xenobiotics, suggesting that they represent different expressions of the basic NTD phenotype. Encephaloceles are much less common than exencephaly in animal models of NTD.
Holoprosencephaly
Holoprosencephaly (formerly termed arhinencephaly) stems from failed division of the prosencephalon (the one primary forebrain vesicle) into the two components of the telencephalon (the two secondary brain vesicles from which the cerebral cortices arise). The resulting single telencephalic ventricle is called a holosphere. The olfactory bulbs and tracts are absent.
The defect typically produces malformations in both the brain and the craniofacial skeleton (Figure 25.3). A common variant is a reduction in the distance between the two eyes, or even their fusion into a single globe located at the midline (i.e., cyclopia). In most instances individuals are so deformed that they expire before birth.
Acrania
In cases of anencephaly and exencephaly, the corresponding skeletal defect is acrania (lack of the calvarium (upper skull)). Associated skeletal reductions or loss affect the petrous temporal bones, sphenoid bone, and internal ear. The base of the skull undergoes normal development. However, the cerebellum, pons, and cranial nerves are usually malformed.
Spina Bifida
Spina bifida is a NTD of the caudal axial skeleton resulting from failure of the neural arches of the vertebra to unite (Figure 25.4). Spina bifida has been reported in all species, with a worldwide incidence in humans of 0.5/1000 live births. However, the frequency varies widely depending on the country (e.g., 0.2/1000 in Japan versus 4.1/1000 in South Wales). This divergence in incidence raises questions regarding potential environmental and genetic influences as an etiology for the lesion in humans.
The extent of spina bifida is variable. Myeloschisis, the most severe form, is characterized by an area of open neural plate with no covering tissues (i.e., the overlying meninges, vertebrae, epaxial muscles, and skin are absent). Pervasive degeneration of the spinal cord results in hind limb paralysis (paraplegia).
The intermediate form is termed spina bifida cystica (due to the cyst-like sac characterizing these defects), which encompasses myelomeningocele and meningocele. Both usually present as a small, fluctuating mass located on or just beneath the skin surface near the base of the vertebral column. The spinal cord is abnormal by definition in myelomeningocele, but may be structurally altered in meningocele.
The least severe form is spina bifida occulta, which is not a NTD in the strict sense and is most commonly asymptomatic. The only indication of its presence may be a sacral skin dimple or abnormally arranged tuft of hair. It is characterized by a gap in one or more vertebral arches, with no protrusion of spinal cord or meninges outside the vertebral canal and no break in the soft tissues or skin covering the area.
Arthrogryposis (multiple joint contractures) is commonly seen in severe forms of spina bifida. The pathogenesis for joint contracture is spinal cord dysgenesis leading to reduced or absent motor activity, which permits soft tissues surrounding joints to become rigid during the course of an extended gestational period.
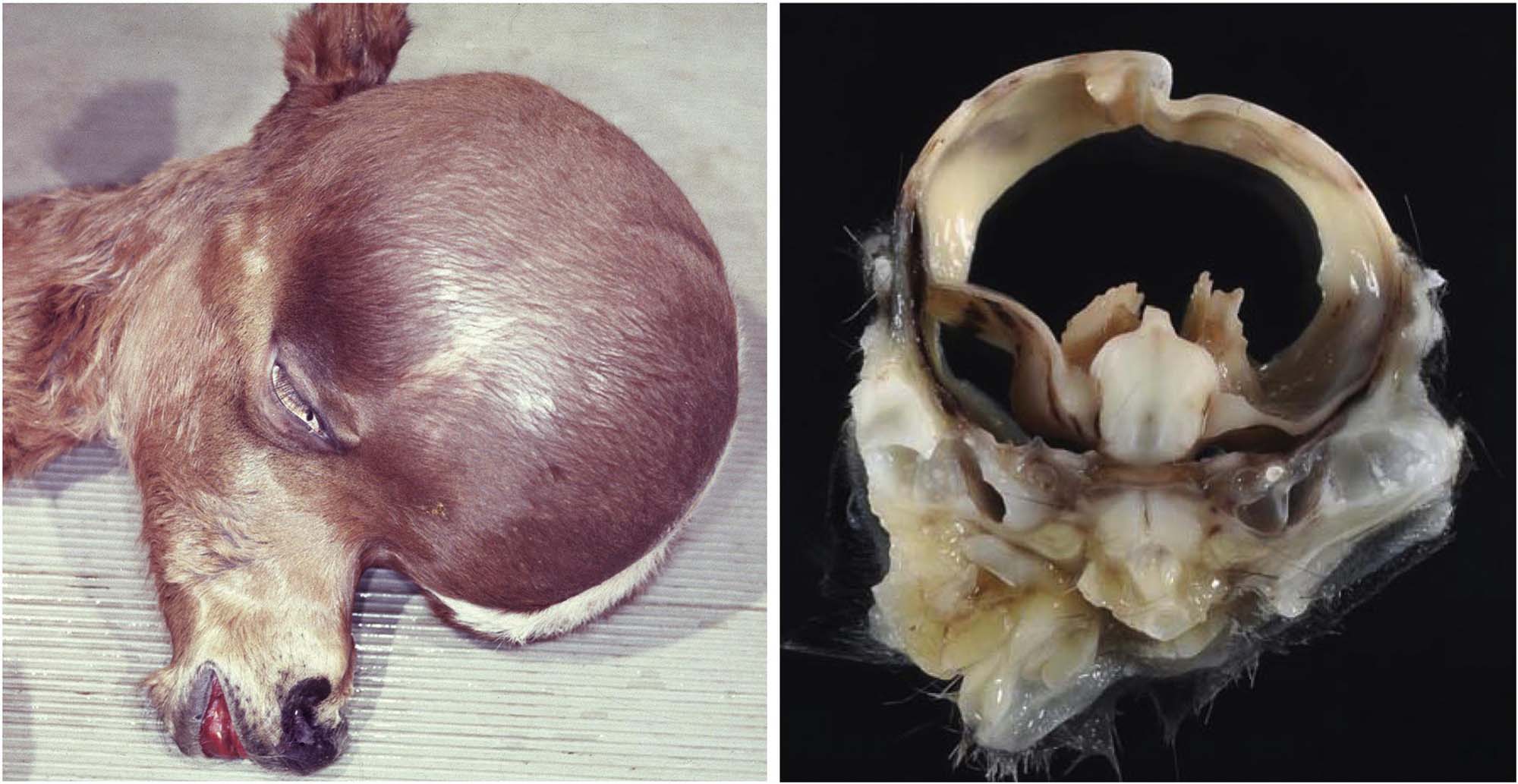
Hydrocephaly (Hydrocephalus)
Hydrocephaly (water brain) results from abnormal accumulation of cerebrospinal fluid (CSF) in the ventricular system, usually in conjunction with pronounced ventricular dilation. The typical presentation at birth is an animal with a markedly domed cranium (Figure 25.5). The overlying brain is thinner than normal but still exhibits its normal traits (distinct cortical layers, white matter tracts, etc.). Extreme cases lead to degeneration and loss of the ependymal epithelium with CSF dissection into the neuropil, especially along white matter tracts. This lesion must be differentiated from hydranencephaly, in which the cerebral hemispheres are absent and their normal location is filled by fluid.
Microcephaly
Microcephaly (small brain) is a primary defect in brain development leading to secondary skull involvement. Thus both the brain (Figure 25.6) and calvarium are diminished in size. It has a reported prevalence of 0.6–1.6/1000 live human births, though new agents likely will lead to higher incidences in some locales (e.g., Zika virus in Brazil). Pure cases of microcephaly, in which brain size is decreased but the calvarium is unaffected, are rare.
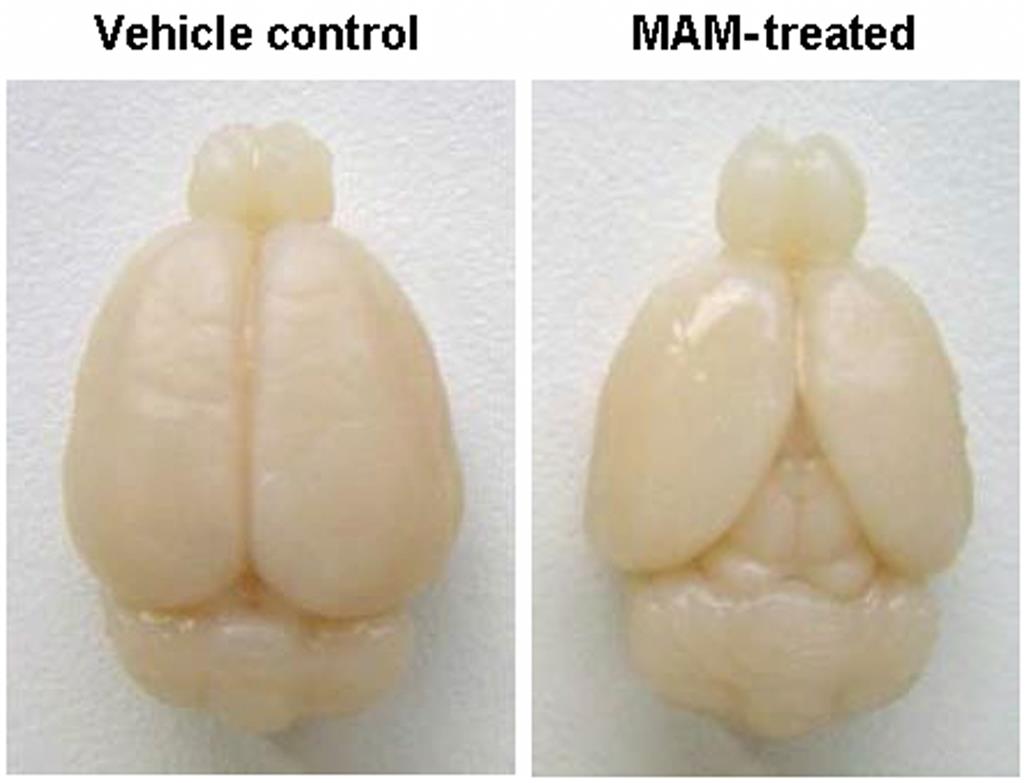
The cerebral hemispheres of microcephalic brains, and particularly the frontal lobes, are reduced in size and exhibit a simplified and sometimes asymmetric pattern of surface convolutions. Microscopic examination reveals the presence of fewer large, differentiated neurons in the cerebral cortex, with corresponding increases in neuroblasts and abnormal spindle-shaped cells.
Craniofacial Structures
Anophthalmia and Microphthalmia
Anophthalmia is the complete absence of all optic structures: the globe, optic nerves, optic foramen, and optic chiasm. Microphthalmia represents hypoplasia. Both conditions may be either unilateral or bilateral. Microphthalmia is often associated with defects of other eye-associated structures, including congenital cataract, coloboma (a gap due to incomplete development) of the iris and choroid, pupillary obstruction, corneal scarring, and ocular muscle imbalance. The correlation of microphthalmia with facial and cardiovascular defects suggests that this is an anomaly of the first branchial arch. These lesions may represent a primary defect (i.e., total failure of optic vesicle formation) or a secondary anomaly (e.g., suppressed forebrain growth with later partial failure of eye development). Another possible mechanism is destruction of a previously formed optic vesicle.
Cyclopia
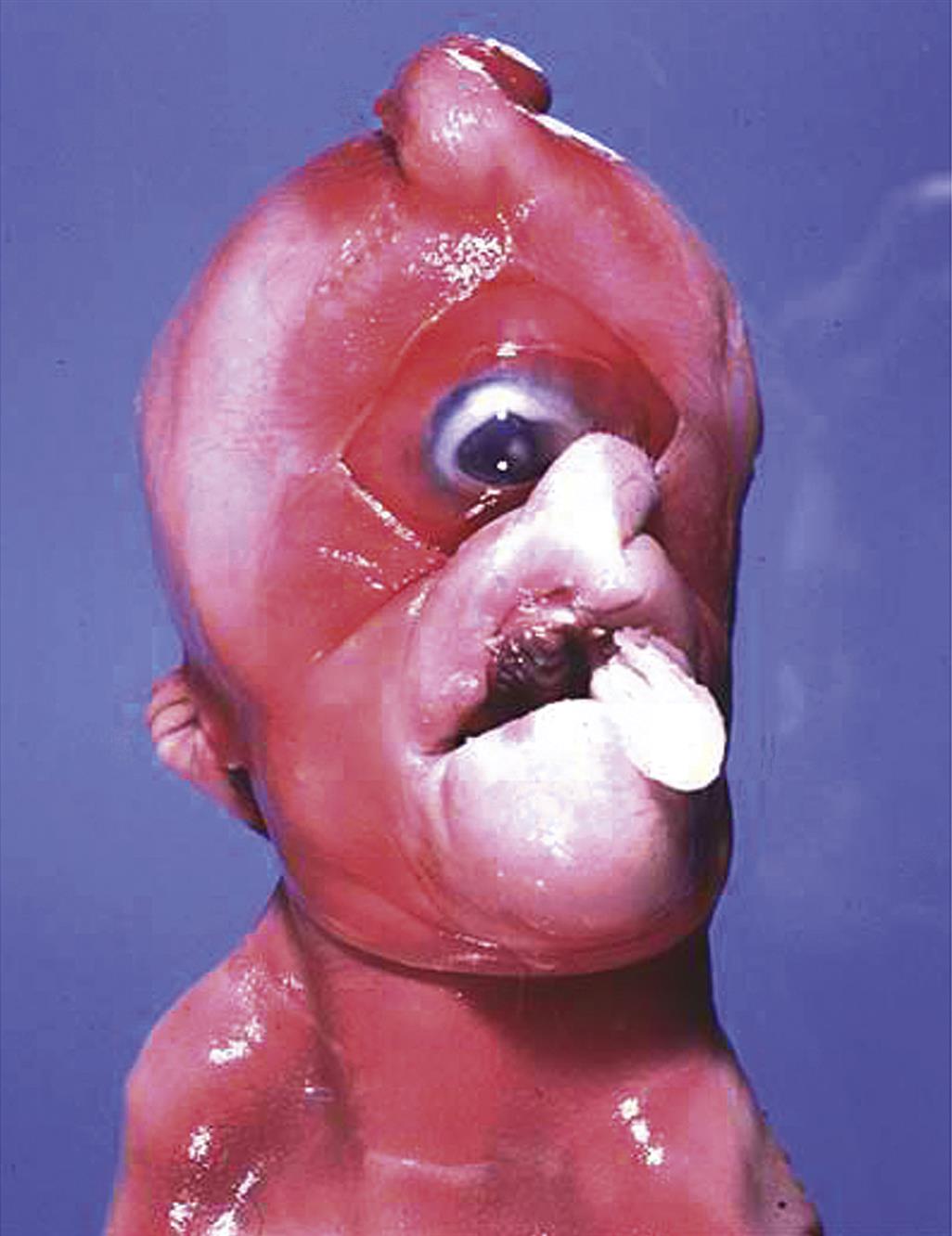
Cyclopia typically coexists with holoprosencephaly. This ocular lesion may present as complete ocular fusion in a single orbit or as two eyes in a single orbit. Eye defects linked with cyclopia include colobomas (gaps) in the iris, retina, and optic nerve; inconstant optic nerve numbers (either one or two is possible), and an absent or abnormal optic chiasm. Cyclopia occurs when the rostral (anterior) portion of the notochord and adjacent mesoderm are deficient in mass. This shortage leads to the aberrant induction of the forebrain tissues followed by severe derangement of midline facial development.
Cyclopia is uncommon in humans. A well-recognized cyclops syndrome occurs as outbreaks in sheep or cattle that grazed on hellebore (Veratrum californicum) at day 13.5 of gestation (Figure 25.7). The responsible toxins are the steroidal alkaloids jervine, cyclopamine, and cycloposine. Several mechanisms of teratogenesis have been proposed: defective craniofacial chondrogenesis, inhibited hedgehog pathway signaling, and altered catecholamine release in the neuroepithelium of the neural tube.
Agnathia and Micrognathia
Total absence of the maxilla or mandible is extremely rare in mammals. Agnathia is a common feature of otocephaly (a congenital head malformation featuring marked mandibular hypoplasia or agenesis in conjunction with fusion or close approach of the ears in the throat region) and may accompany cyclopia; hence, it is seen in ruminant fetuses exposed to Veratrum californicum. Again, this defect appears to originate in the first branchial arch.
Micrognathia (or hypognathia) occurs more often than agnathia. Maxillary micrognathia (Figure 25.3) results from deficient premaxillary tissue during craniofacial development. Mandibular micrognathia may occur with cleft palate, glossoptosis (downward displacement of the tongue), microcephaly, and microphthalmia.
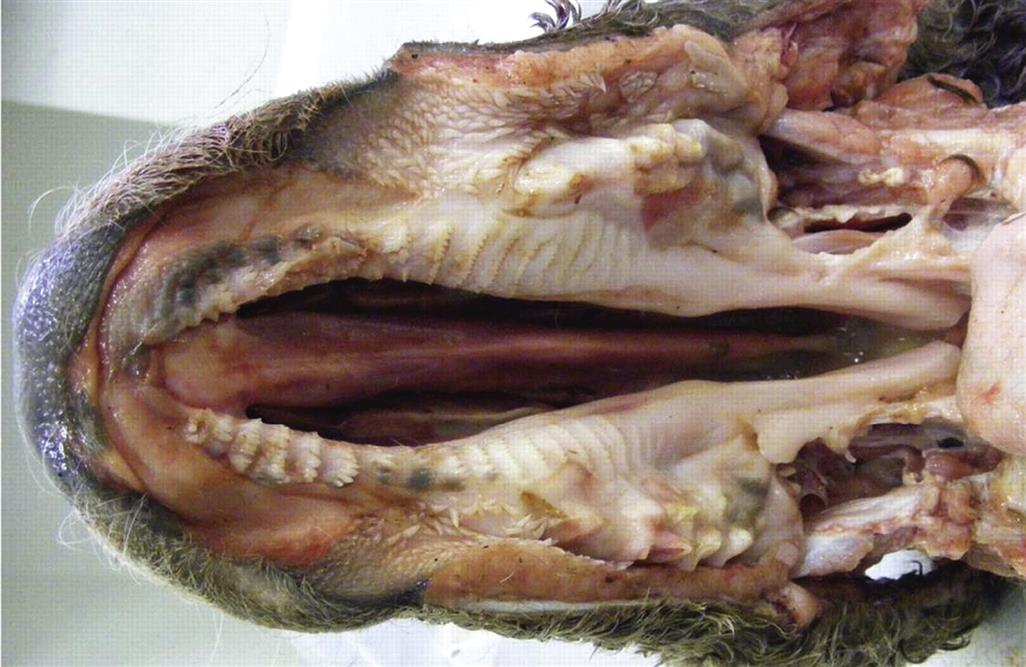
Cleft Lip and Cleft Palate
Palatoschisis, or cleft palate, is an important developmental anomaly of mammals, since the neonate cannot nurse properly without an intact palate (Figure 25.8). A cleft palate usually results in inhalation of milk and aspiration pneumonia. In humans the incidence of cleft palate is 0.5–1/1000 births. The prevalence of cleft palate in animals may be greater than reported, since not all animals that die during or after birth undergo a necropsy (animal autopsy) examination. Cleft palate is commonly bilateral, indicating that neither palatal shelf was elevated.
Cardiovascular System
Atrial Septal Defects
Although not fatal malformations, atrial septal defects (ASD) account for approximately 17% of all human heart defects. The incidence of ASD in domestic animals is presumed to be similar to that in humans. Experimental induction of ASD is not a commonly reported teratogenic outcome.
Four types of clinically significant ASD have been described. First, defects of the oval fossa (the location of the ostium secundum in the interatrial septum) account for approximately 70% of ASD in humans, exhibiting a female:male predilection of 3:1. This lesion is characterized by a patent foramen ovale with a short or fenestrated primary septum, occurring with or without defective development of the secondary septum. The second form is the sinus venosus type, a relatively uncommon condition featuring a high ASD. It results from abnormal absorption of the sinus venosus into the right atrium or from abnormal development of the septum secundum. Third, a persistent ostium primum results from incomplete fusion of the primary septum and the endocardial cushions, with resultant anomalies in the atrioventricular valves. Finally, the least common ASD stems from complete absence of the interatrial septum resulting from a failure of development of both primary and secondary septa.
Many other cardiac malformations have been associated with ASD in humans. Examples include anomalies of the left atrioventricular (AV; also mitral or bicuspid) valve, atresia of the right AV (tricuspid) valve, common ventricle, coarctation (narrowing) of the aorta, noncyanotic patent ductus arteriosus, pulmonary stenosis or atresia, tetrology of Fallot, transposition of the great vessels, and ventricular septal defects (VSD).
Ventricular Septal Defects
These malformations occur more commonly than ASD, accounting for an estimated 30%–50% of human congenital cardiac anomalies depending on the screening method used in the survey. Ventricular septal defects (VSD) are classified according to their location in the septum as either membranous or muscular (Figure 25.9). The muscular type accounts for approximately 10%–15% of all VSDs in humans. In some cases, absence of both parts of the septum coexists with additional cardiovascular anomalies (e.g., aortic hypoplasia, transposition of the great vessels).
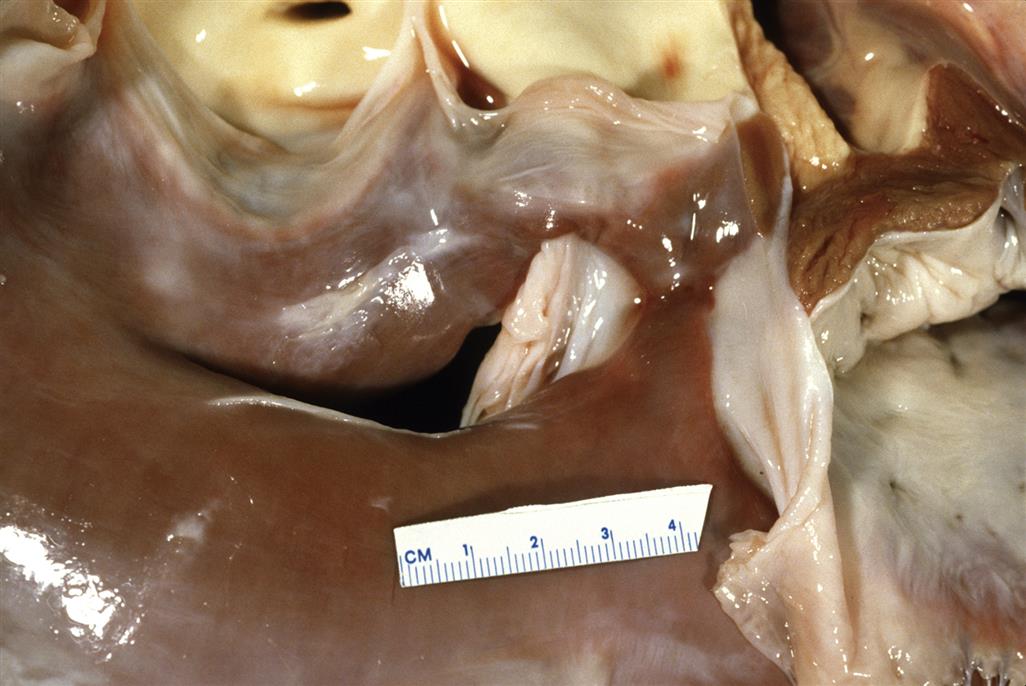
The etiology of VSD is still unclear. In humans, approximately 4% are thought to stem from a chromosomal or genetic defect. Maternal diseases like diabetes mellitus and infections (especially those inducing fever) also may play a role. Experimental treatments that delay closure of the cardiac septae are capable of inducing VSD. Agents include hypoxia; nutritional deficiencies (e.g., folic acid or vitamin A) or excesses (copper or vitamin); and many cytotoxic chemicals.
Transposition of the Great Vessels
This malformation results from an abnormal spatial arrangement of any of the primary blood vessels: aorta, cranial (superior) and/or caudal (inferior) vena cavae, pulmonary artery, or pulmonary vein. The outcomes may range from a change in blood pressure to interruption of the normal right-to-left circulation, depending on the exact location and extent of the mal-positioning. Regardless of the cause, survival requires the presence of additional cardiac anomalies, such as septal defects and persistent ductus arteriosus, which allow mixing of the two parallel circulations. This disorder appears to be more prevalent in male infants, but in animals it is not recognized as a sex-linked trait.
The pathogenesis by which transposition of the aorta and pulmonary trunk takes place is controversial. Three hypotheses include lack of spiral twisting of the great vessels around each other, abnormal division of the truncus arteriosus, and anomalous cardiac looping. The latter possibility has been confirmed in mice exposed to retinoic acid, where altered looping has been linked to hypoplasia of the conotruncal ridges and aorticopulmonary septum, with subsequent delayed fusion of the AV cushions.
Respiratory System
Agenesis or Hypoplasia of the Lungs
Total absence of the lungs, bronchi, and vascular structures–pulmonary agenesis (or aplasia)—is usually unilateral. In rodents, it usually affects the left lung. Aplastic lungs have absent pulmonary and vascular structures with either rudimentary bronchi or abrupt termination of the distal trachea. Agenesis is probably caused by failed interactions between the endodermal components of the tracheobronchial buds and the surrounding mesenchyme.
Pulmonary hypoplasia, commonly associated with renal agenesis, is a more frequent anomaly. The link between these two conditions is explained in part by the ability of fetal urine (an important component of amniotic fluid during late gestation) to promote lung growth as excreted proline directs collagen and mesenchyme formation in the lung. Hypoplastic pulmonary parenchyma resembles fetal lung tissue, having more prominent bronchioles and reduced numbers of underinflated alveoli. Hypoplasia may be primary (e.g., due to genetic defects or a pulmonary viral infection with resulting tissue damage) or secondary (e.g., a consequence of a diaphragmatic hernia following nitrofen administration).
Gastrointestinal System
Aplasia/Hypoplasia of the Caudal Enteric Ganglia
The ganglia of the digestive tract are seeded by migration of neural crest cells. This developmental event is controlled, at least in part, by the glial-derived neurotrophic factor (Gdnf) signaling pathway. Mice engineered to have null mutations of either Gdnf or in one of the two proteins that contribute to its complex receptor, GDNF receptor α-1 (Gfra1) and the RET tyrosine kinase receptor (Ret), do not form enteric ganglia in the caudal portion of their intestines; these animals also lack kidneys. The same condition in humans is termed congenital aganglionic megacolon (Hirschsprung disease) and occurs in approximately 1/5000 births. This lesion has been linked to maternal hyperthermia. To our knowledge, ganglionic aplasia is not associated with gestational exposure to xenobiotics.
Diaphragmatic Hernia
This defect may arise by several mechanisms. These include failed or delayed fusion of the pleuroperitoneal membranes with the septum transversum and the dorsal mesentery of the esophagus, premature return of the intestines from the extraembryonic coelom to the abdominal cavity, and weak or abnormal diaphragmatic musculature. Most herniation sites are located in the dorsolateral portion of the diaphragm, thereby leaving room for part or all of the abdominal viscera to enter the thorax. Diaphragmatic hernias occur in about 1/2200 of human infants and are predominately located on the left side.
Umbilical Hernia, Omphalocele, and Gastroschisis
This trio of related malformations is not rare in humans, and collectively they are common in laboratory animals. Umbilical hernia is relatively minor, as one or several loops of intestine are displaced through the abdominal musculature but remain covered by subcutaneous tissue and skin. Omphalocele is a more marked lesion in which a variable portion of the intestines and liver, and occasionally other organs, remain outside the abdomen in a peritoneal sac due to underdevelopment of the abdominal wall muscles. Over half the time, this defect is accompanied by serious malformations in other organs, particularly affecting the heart but often also the urinary tract and/or vertebral column. Gastroschisis is the most severe form, presenting as eventration of abdominal contents without a sac. Several mechanisms have been proposed as the pathogenesis for this lesion, including aberrant formation of the lateral folds of the ventral embryonic wall, thereby leading to a persistent defect in the abdominal wall; failure of mid-gestational retraction of the extra-abdominal mass of intestines (a normal physiological event during organogenesis) back into the abdominal cavity; and late-term escape of intestinal loops via an imperfectly closed umbilicus.
Urinary System
Renal Agenesis
Bilateral renal agenesis or severe dysplasia is rare in humans, affecting 0.02–0.37 of every 1000 human births. In contrast, unilateral renal agenesis is a fairly common finding at postmortem, with an incidence in humans of 1.0–1.8 per 1000. In such cases the remaining kidney is functional but often is deformed as well; common anomalies include altered orientation (ectopia or rotation), hydronephrosis, polycystic disease, or urolithiasis (calculi in the pelvis). Genital defects are another frequent finding in affected individuals, especially in females. The lesion represents a defect in the earliest stages of organogenesis due to altered morphogen expression in the renal primordium.
Hydronephrosis
Transient closure of the ureters occurs during normal development. If urine secretion by the kidney commences before the reopening of the ureters, the proximal ureters and renal pelvis will become distended (Figure 25.10). Such dilation is often noted in rodents during teratological studies, but this expansion appears to be transient since the incidence in adult animals is much lower than expected based on the number of affected near-term fetuses. Permanent hydronephrosis will result if the obstruction is not reversed. Additional renal defects develop in 40% of individuals with hydronephrosis, including agenesis or hypoplasia of the contralateral kidney, cystic kidney, and hypospadias.
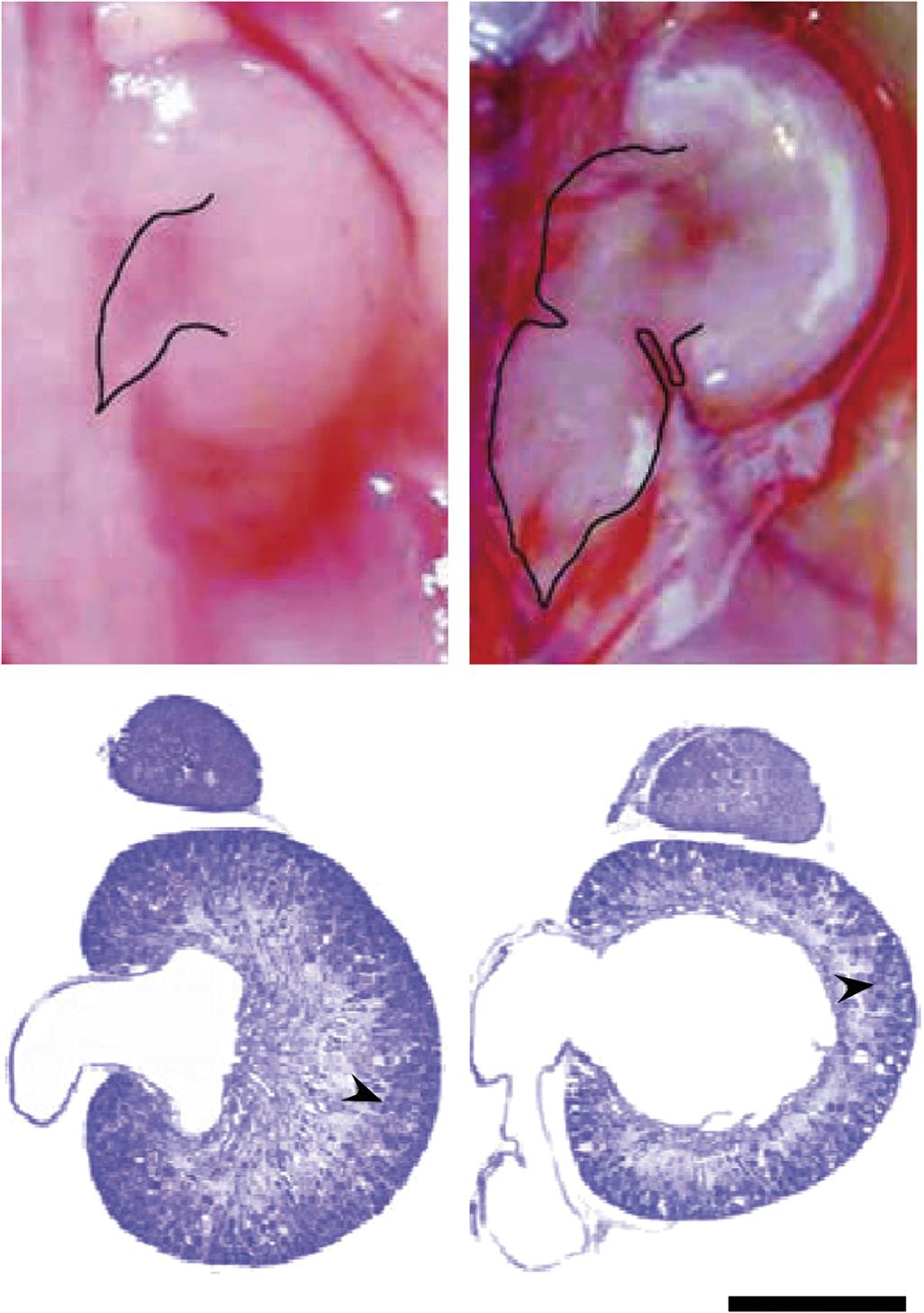
Hydroureter
This lesion is a common anomaly in laboratory rodents (Figure 25.10). Following obstruction the affected ureter dilates, elongates, bends, and becomes tortuous before the development of urinary reflux occurs. Other ureteral abnormalities have been associated with hydroureter, such as duplication, ectopia, ureterocele (i.e., dilation of the distal ureter), and ureterovesicle strictures. Unilateral renal agenesis and renal dysplasia also have been linked to the ureter defect.
Hypospadias
This urethral defect is the most common urogenital malformation. It presents in males as an abnormally placed external urethral orifice, where the opening is located somewhere along the underside of the penis rather than at the tip. In more than 50% of cases the shift in meatus position is confined to the ventral surface of the glans, with the remainder opening on the shaft or in the perineal region. The penis may be reduced in size as well, and often exhibits a ventral curvature (termed chordee).
Hypospadias is thought to arise from the incomplete fusion of the urethral folds and/or failed canalization of the glandular plate. The molecular mechanism is posited to be insufficient production of androgens or androgen insensitivity, thereby resulting in a partially feminized phenotype.
Reproductive System
Cryptorchidism
Failure of one or both testes to descend from their original inguinal position into the scrotum is called cryptorchidism. It is the most common genital defect observed in many species, including humans. The condition is usually unilateral but may be bilateral. An undescended testis is evident at birth in approximately 3% of full term and 30% of premature infants. The testis completes its migration in about 80% of cases during the first year of life, making the genuine incidence of this defect around 1% in humans. The term should not be applied to those laboratory animal species (e.g., rodents) in which the wide inguinal rings permit position-dependent translocation of the testes between the scrotum and the abdominal cavity into adulthood. The proposed molecular etiology is deficiency of gonadotropic hormones.
Intersexuality (Pseudohermaphroditism)
Sexual ambiguity, often referred to as intersex or disorders of sex development (DSD), is a reasonably frequent occurrence in humans and various laboratory animal species. The phenotypic sex of developing mammals is defined by three main factors: the genotype, gonadal development, and the genesis of accessory genital organs (Figure 25.11).
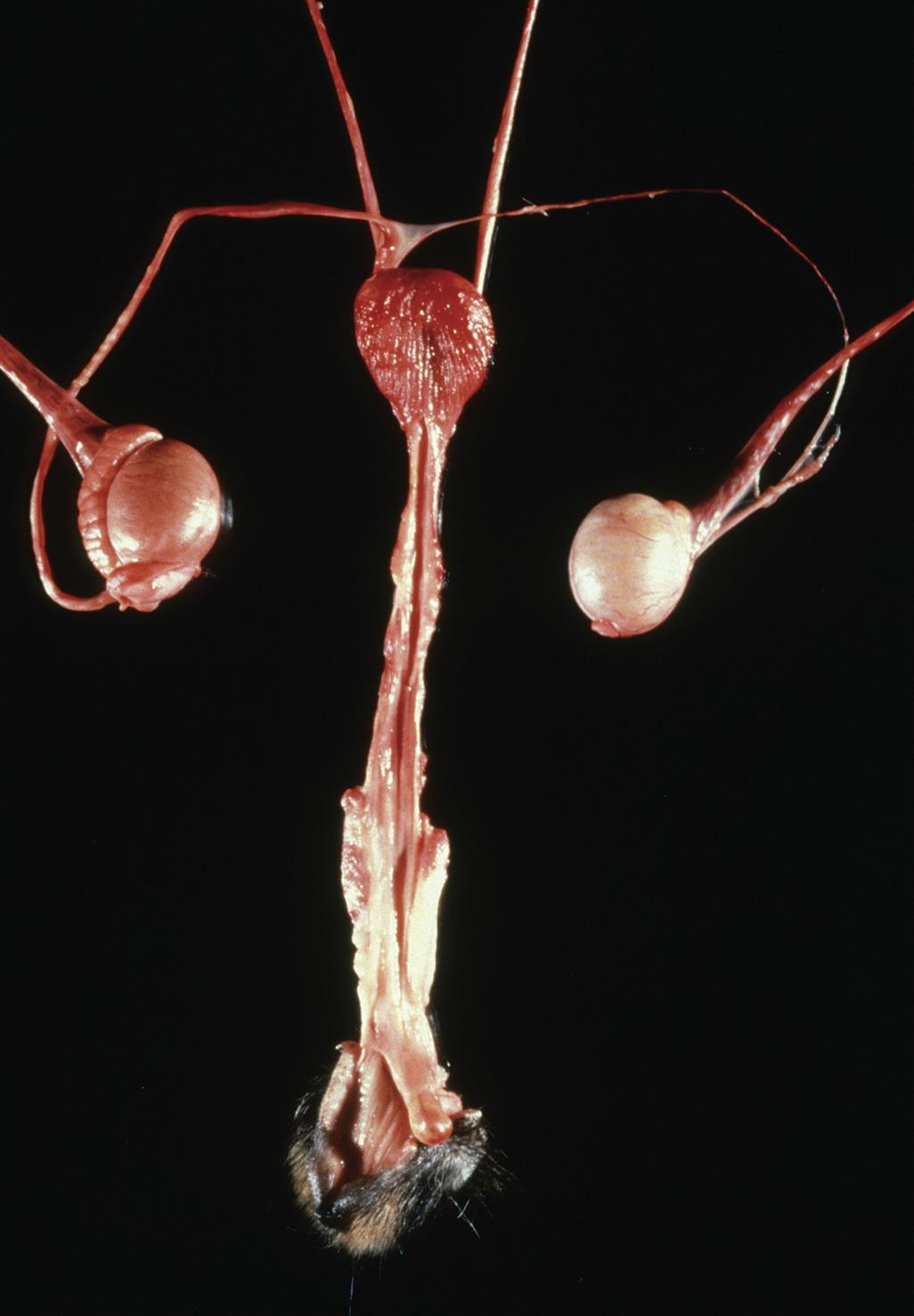
Genotypic sex, the genetic designation of gender depends on the expression of several genes. Genotypic sex is determined at fertilization as either male (XY) or female (XX), except in rare instances where an abnormal number of sex chromosomes are transferred (e.g., XXY, XYY, or XO). In contrast, phenotypic sex depends on the expression of the SRY sex-determining gene; adequate production of the SRY protein results in differentiation as a male, while a lack of SRY leads to a female offspring. If the SRY protein is mutated so that it cannot bind to DNA, sex will be maintained as the default female phenotype.
While numerical and structural aberrations of the sex chromosomes may produce intersex individuals, they are not the only cause of this condition. Gonadal development, and in particular the hormone complement, plays a critical ancillary role in determining the phenotype. Androgens induce male characteristics, while estrogens are responsible for female characteristics. Hyposecretion of appropriate gonadal hormones (androgens for male fetuses, estrogens for females), defective receptors for these hormones, or exposure during gestation to xenobiotics with endocrine-disrupting activities (see later) can produce pseudohermaphrodites, individuals in whom the genotype and gonadal development do not agree with the phenotype as expressed in genital structure and secondary sex traits. This condition arises when animals of a given genotype are exposed in utero to excessive quantities of sex steroid hormones appropriate to the opposite sex.
Skeletal System
Digit Anomalies
Malformations of the digit are relatively common in both humans and laboratory animals. Multiple different abnormalities may occur (Figure 25.12). The common pathogenesis is abnormal remodeling of the apical epidermal ridge (AER) during initial specification of the digital rays early during on limb organogenesis. The type of lesion that develops depends on the manner in which limb differentiation is disrupted.
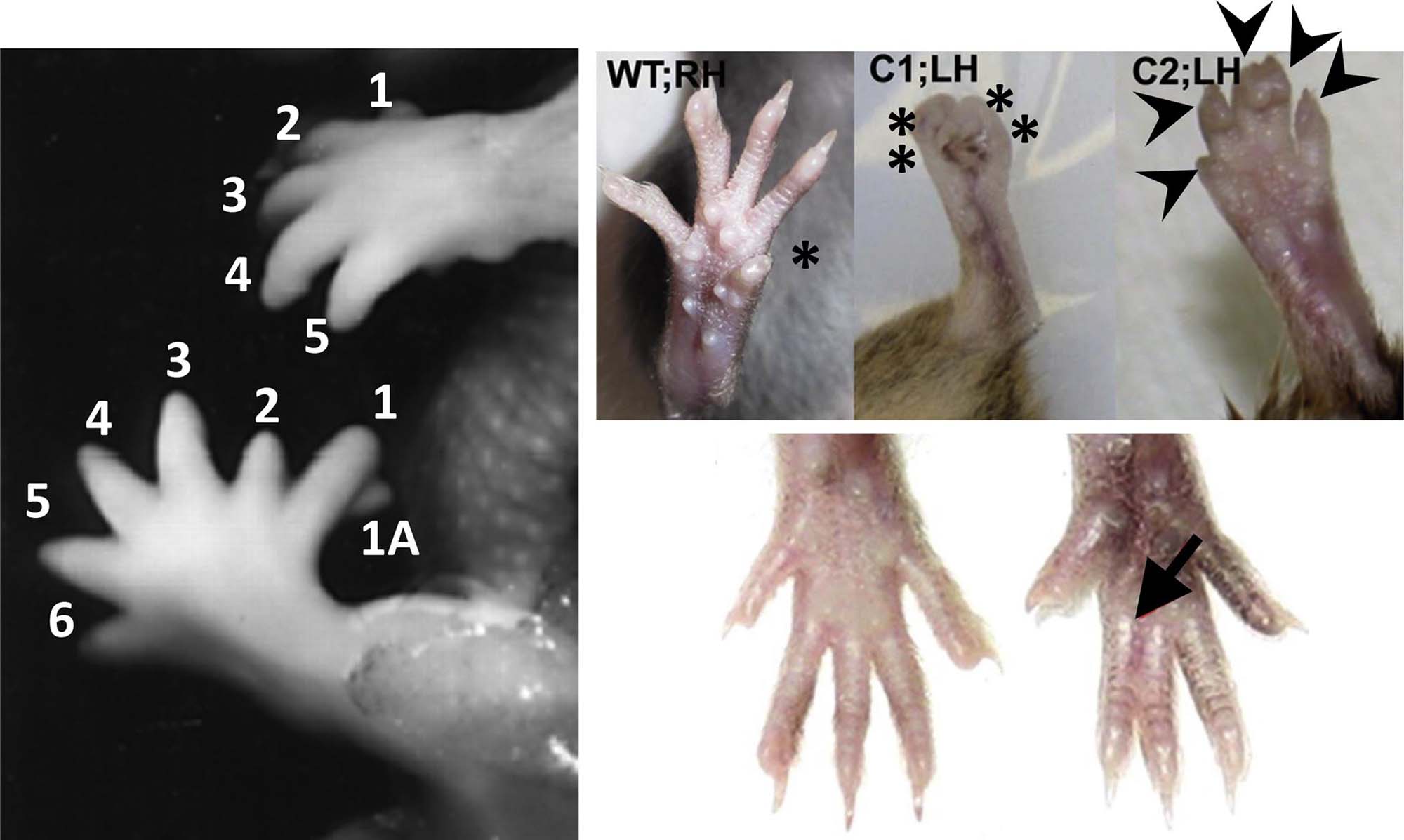
Ectrodactyly, the absence of part or all of at least one digit, results from interference in normal mesenchymal condensation of digital rays. Possible mechanisms include failed intercellular interactions or cytotoxicity of critical stem cells. The most common type of ectrodactyly in humans is the split hand or foot, where the middle digits (II and III) are absent while normal outer digits (I on one side, and IV and V on the other) border the cleft.
Polydactyly, the presence of extra digits is a common anomaly in mammalian species, including humans. The first and fifth digits are most commonly duplicated in humans. In most instances, only a single extra digit is present. Two mechanisms have been proposed to explain this lesion. The first is a failure of programmed cell death, thereby leading to inappropriate pruning of the digital blastema. The second is formation of a supernumerary digital blastema as a consequence of alterations in the composition, configuration, and quantity of extracellular matrix.
Syndactyly, or fusion of adjacent digits, is a moderately common congenital defect. The mechanism is reduced or failed differentiation between two digits, typically due to interference with programmed cell death that is required to separate the digital rays. In most cases the fusion consists of soft tissue, such as cutaneous webbing; osseous union is rare. Syndactyly often occurs with other limb anomalies, such as brachydactyly (digital shortening) and ectrodactyly (especially variants with split extremities). The lesion is often linked to amniotic band syndrome (i.e., ischemia-related defects, especially of the digits, that arise when limbs are ensnared in fibrous bands formed following amniotic damage), and it is a part of many malformation syndromes.
Reduction Deformities of the Limbs
The mass of the limbs may be reduced to a variable degree. In many instances the changes affect only a portion of the affected limb. They may be unilateral or bilateral, depending on the cause. Such defects may occur together with malformations of many other skeletal regions.
Amelia is the absence of an entire limb, while meromelia is the partial absence of a limb. These defects may occur in isolation, and may affect any of the limbs. However, amelia is also associated with defects in the remaining limbs (e.g., talipes equinovarus (club foot)), as well as cleft lip and/or cleft palate and scoliosis (curvature of the spine from side to side).
Hemimelia is absence of one side of the distal half of a limb. This anomaly is one of the most common reduction malformations, typically involving the distal forearm and hand.
Phocomelia (seal limbs) results from differential reduction of the long bones in various limb domains. It is typically characterized by greater foreshortening of the upper extremities with relatively more development of distal regions (hands, feet, and digits) (Figure 25.13). This lesion is extremely rare, but known instances are a result of toxicant exposure. The most famous example is thalidomide, an antinausea agent given during pregnancy to combat morning sickness; this agent induced phocomelia in thousands of human infants following exposure during the critical period of early limb organogenesis (GDs 35–50). The same lesion occurs in some nonhuman primate species and also in rabbits, but not in rodents, following thalidomide exposure.

The pathogenesis for reduction abnormalities involves interference with regional specification of cell populations in the proximal limb bud. Potential mechanisms are disrupted regional interactions between ectoderm and mesoderm, altered morphogen production, failed morphogenetic movements, or disturbed inductive processes. The reduction or absence of proximal structures coupled with the fairly normal appearance of distal features, as in phocomelia, indicates that limb bud damage induced by toxicants does not thwart limb differentiation. Instead, compensatory processes attempt to repair or bypass reductions in cell mass so that surviving structures can attempt to continue their developmental program.
Arthrogryposis
This malformation is characterized by persistent flexure or contracture of one or more joints. Other sequelae include shortened ligaments and fibrous ankylosis of the joints along with reduced amounts of loose connective tissue under the skin. Mechanical compression resulting from increased pressure and/or reduced fetal mobility (e.g., from intrauterine crowding) also can be a cause of this lesion.
Arthrogryposis multiplex congenita, in which multiple joints exhibit ankylosis at birth, occurs in approximately 0.03% of human live births. It is the most common limb deformity in domestic animals, and is often accompanied by defects of the appendicular skeleton (Figure 25.14).

Although usually classified as a skeletal deformity, this malformation typically results secondary to insufficient limb movement rather than from a primary failure in skeletal development. Accordingly, more common causes include gestational diseases of the developing CNS, connective tissue, or skeletal muscles. For example, the absence of neural input (typically due to CNS lesions in the motor neurons or motor tracts in the spinal cord) thwarts skeletal muscle activity, which causes denervation hypoplasia and atrophy, and eventually to arthrogryposis.
Intrauterine Growth Retardation
Individuals affected by intrauterine growth retardation are not born prematurely, but are born with a small size given the length of their gestation (Figure 25.15). Placental insufficiency has often been considered responsible for intrauterine growth retardation, but this is apparently not the usual cause. The placenta’s great physiological reserve capacity means that most placental lesions appear to be functionally unimportant, and do not result in fetal malnutrition.
Intrauterine growth retardation often occurs along with congenital malformations. Common examples include the central nervous system (hydrocephalus, macrocephaly, microcephaly, spina bifida); heart (ventricular septal defects); kidney (renal agenesis); and limbs (arthrogryposis, fractures secondary to osteogenesis imperfecta). The severity of these defects often is inversely correlated with the fetal body weight.
Placental Abnormalities
Gestational or perinatal mortality have been linked to lesions in many placental elements. Early in gestation the most frequent causes are abnormal yolk sac circulation or failed chorioallantoic fusion. These changes typically are attributed to dysgenesis of hemangioblasts (which serve as precursors for both endothelial cells and erythrocytes in the yolk sac) or mesodermal cells (especially trophoblasts comprising the allantois or in fewer cases the chorion). During late gestation the most common finding is disrupted formation of the placental tissue responsible for gas and nutrient exchange (e.g., the labyrinth in rodents, villi in primates). In general, this change presents as insufficient production and/or branching of capillaries (commonly a consequence of an endothelial defect) or abnormal production of trophoblast (Figure 25.16). Placental tissue also may undergo the same changes observed in other tissues, particularly hemorrhage, necrosis, and thrombosis; these findings typically are linked to acute embryonic or fetal loss only if they are extensive enough to disrupt a large portion of the placenta.

Perinatal Toxicology
Developmental events induced by toxicant exposure during gestation may not be manifested until near or shortly after birth, and then as functional deficits rather than overt malformations. Examples of altered function include abnormal cell reactivity (e.g., by immune or reproductive elements), metabolic errors, and neurobehavioral deficits. Although subtle structural defects may be apparent, functional deficiencies are the most common adverse outcomes of this kind. Often these functional changes result from depleted cell numbers due to inadequate repair.
Abnormal cell reactivity in the perinatal period is a common developmental toxicity outcome. The ontogeny (i.e., an individual’s developmental history) of immune system differentiation is commonly impacted by xenobiotic exposures in utero. The main functional deficits resulting from such exposures are inappropriate immunosuppression and immunostimulation, either of which can permit the onset of inflammatory diseases (especially allergic or autoimmune) or neoplasia later in life. Abnormal reproductive functions can follow intrauterine exposure to many agents, including antineoplastic drugs, hormones, pesticides, and polycyclic aromatic hydrocarbon carcinogens.
Developmental dysfunction associated with errors of metabolism commonly arises from genetic mutations or xenobiotic exposures. Such genetically based “inborn” errors are often not expressed until after birth. An environmentally-caused disease, termed a phenocopy because its lesions mimic those of a known genetic ailment, may mirror such congenital genetic conditions. For example, cattle exhibit similar morphologic lesions when afflicted with α-mannosidosis (genetic cause) or locoism (i.e., locoweed toxicity). However, accumulation of α-mannoside in these two conditions results from distinct mechanisms: the inherited disorder arises from the complete lack of functional α-mannosidase protein, while the environmental phenocopy follows α-mannosidase inhibition after ingestion of the alkaloid swainsonine. The existence of phenocopies highlights the difficulty in determining the genuine etiology of congenital defects.
Endocrine Disruption
Xenobiotics classified as endocrine-disrupting compounds (EDC) have received much attention in recent years as potential reproductive and developmental toxicants. These agents are exogenous chemicals that are considered to affect the activity of an endogenous hormone by either acting as an agonist or antagonist in place of the native molecule or alternatively by interfering with the endogenous hormone’s synthesis, secretion, transport, binding, action, or elimination. The main interest in xenobiotics that mimic endogenous hormones, enhance their actions, or act as hormone antagonists has centered on their potential for disrupting reproductive tract development, altering sexual behavior, and impairing fertility as a consequence of aberrant endocrine pathways in the gonads. However, compounds that affect pituitary or thyroid function are also of concern.
To date, endocrine disruption has primarily been a concern raised in animals. Such effects have been demonstrated in wildlife, a notable example being the feminization of male and “super-feminization” of female alligators in central Florida’s Lake Apopka due to accumulation and persistence of pesticide residues in fat. With few exceptions (e.g., diethylstilbestrol (DES) as an etiology of breast and cervical/vaginal carcinomas), causal relationships between exposure to a specific chemical and endocrine disruption have yet to be established definitively in humans. Nonetheless, growing epidemiological evidence suggests that endocrine disruption during development may predispose humans to endocrine-related disorders later in life, as suggested by a global increase in neoplasia of endocrine organs (e.g., adrenal and thyroid glands) and hormone-regulated tissues (e.g., mammary gland, prostate) and metabolic conditions (e.g., noninsulin-dependent (Type II) diabetes, obesity).
Congenital Neoplasia
Tumors in young animals and humans are, by definition, indications of abnormal embryogenesis. Fetal cells are inherently susceptible to carcinogens because of their high rates of proliferation. The incidence of neonatal tumors in humans up to 15 years of age is estimated to be between 98 and 125/million/year. The most common childhood tumors arise from committed stem cells for specific cell lineages, such as leukocytes (leukemia, lymphoma) or mesenchyme (osteosarcoma, rhabdomyosarcoma), although some may be derived from multiple lineages, like nephroblastoma (Wilm’s tumor) and neuroblastoma. Many neonatal tumors result from genetic mutations, but others are consequences of in utero exposure to xenobiotic carcinogens.
Transplacental carcinogens may act directly without prior metabolism or require enzymatic bioactivation to reactive intermediates. If bioactivation is required, it takes place in the embryo or fetus as reactive molecules interact immediately with nearby cellular constituents. The long latent period required for neoplastic transformation has been used to support the argument that many if not all cancers that arise during the period from birth to young adulthood are induced by a prenatal initiation event, usually considered to be a mutation (i.e., DNA-reactive event) that becomes fixed due to rapid cell cycling. For this reason, all stages of prenatal development are susceptible to the action of these agents, and the tendency to develop neoplasia can be passed on to any future offspring.
In most instances developmental toxicants act as either teratogen or transplacental carcinogens, but not both. This division is reasonable as most known teratogens do not act by causing mutations. Indeed the genetic causes of malformations are likely to reflect chromosomal abnormalities rather than chemically-induced mutations, while the genomes of animals exposed to transplacental carcinogens will exhibit many aberrations at the DNA level. It is not uncommon to see childhood cancers in individuals displaying defective development, sometimes but not always with the birth defects and neoplasms occurring in the same organ or system. Although some substances (e.g., heavy metals, mycotoxins, solvents) can cause congenital malformations and cancers, it is not known whether intrauterine exposure to any single agent can induce both conditions in a single individual.
Principles of Developmental Toxicity
The impact of xenobiotic exposure on the developing organism depends on many interrelated factors. Critical parameters inherent in the conceptus (or neonate or juvenile) include the stage of development during which exposure takes place and the individual’s genotype. However, other factors that modify the toxicity of xenobiotics include the parental genotypes, the maternal environment, and the structure and function of placental tissues. These elements are briefly reviewed here. Additional details may be garnered from many supplemental sources (see Further Reading).
Critical Phases of Development
Developmental processes occur in specific locations and at particular times. In many cases the success of subsequent developmental tasks is affected to a greater or lesser degree by the events that have taken place before. The sequence and timing of events generally is consistent for individuals of a given species, and the overall pattern is well conserved across mammalian species. Critical periods for structural development typically occur prior to birth, although certain organs do not fully form until after birth (e.g., the rodent cerebellum). In contrast, both functional and molecular development continues to evolve throughout the neonatal and juvenile periods, not achieving full maturity until puberty. Taken together, these principles indicate that some structure, function, and/or biochemical pathway likely will still be differentiating any time a pregnant individual or her newly born or juvenile offspring are exposed to a xenobiotic agent.
The critical period of intrauterine development is that time during its differentiation and growth during which the conceptus (or any part thereof) has the greatest sensitivity to noxious influences. Critical periods begin during organogenesis, when the structures involved are laying the foundations (i.e., an anlage, or organ primordium) that ultimately determine their final form. Each organ and system has its own critical period or periods. Distinct regions in complex structures (e.g., the CNS) typically have their own critical periods; some (e.g., hippocampus, olfactory bulb) may have more than one such peak of sensitivity (see Figure 21.10 in Chapter 21: Nervous System). Accordingly the same agent may induce different defects depending on when the exposure occurred during gestation.
Modifying Factors
Conceptus
Genetic Background
Strain and Species
Species differences in expression of defective development following xenobiotic exposure have been clearly shown experimentally. As previously mentioned the classic example is thalidomide, which is highly teratogenic in humans and certain nonhuman primates but does not readily cause malformations in rodents and chicken embryos. Glucocorticoids are teratogenic in mice but not in rats. Species differences in teratogenic response are probably a manifestation of many species-specific factors. The most important parameters for dictating the teratogenic potential of an agent in any species are thought to be its PK properties—including the individual rate of transplacental transfer—and differences in the distribution and/or sensitivity of the xenobiotic’s receptors and target cells. The key means for controlling these response factors are the embryo’s complement of genes.
The responses of individuals in outbred species (e.g., humans) are specified chiefly by the person’s own allelic complement. In species with multiple highly inbred strains, individuals for a given strain tend to react in a comparable fashion. In contrast, those of distinct strains may respond in divergent fashions depending on how much concordance exists among their complements of genes.
The divergence among strains can represent intrinsic differences, such as unique patterns of protein expression or timing for developmental events, or they can result from extrinsic differences that are defined by unique maternal biotransformation rates or products. For example, cortisone induces a higher incidence of cleft palate in A/J mice than in either CBA or C57BL/6J mice. The greater vulnerability of the A/J strain appears to reflect both a genetically determined increase in the number of glucocorticoid receptors in the developing maxillary processes as well as the slightly later timing of palatal shelf elevation in this strain. Furthermore, A/J dams metabolize cortisone differently than do CBA dams, thus producing a greater exposure in the A/J embryos.
The importance of the embryonic genotype in determining the response of the conceptus to xenobiotics is readily apparent in the discordant defective development that may occur between dizygotic human twins exposed to a teratogen. This discordance also manifests itself in rodents, since offspring in a single litter are rarely all affected in the same manner or to the same degree following a teratogen exposure. However, such within-litter discordance may arise from other causes. Examples include variations in maternal blood supply to offspring at various locations in the uterine horns and the impact of a sibling’s metabolic profile (e.g., hormone production) on nearby conceptuses.
Multifactorial Threshold
The premise of the multifactorial threshold concept is that the embryo must be genetically predisposed to develop a malformation before the action of a teratogen results in manifestation of the defect. Estimates suggest that approximately 50% of congenital malformations may involve multifactorial inheritance.
Each organ system exhibits a threshold for abnormal organogenesis. Damage below the threshold does not alter the normal developmental program, while damage exceeding the threshold will result in a malformation. The threshold can be impacted by one or more genes as well as by environmental agents. If the genetically determined pathway specifying normal developmental progression is close to the threshold, then only minor noxious stimuli may be necessary to induce an abnormality. A corollary concept is that a higher number of predisposing genes will reduce the severity of the teratogenic insult required to break the threshold. In fact, it is likely that most, if not all, individuals have one or more “weak points” in their genetically programmed developmental plans that are especially vulnerable to influences that could result in abnormal development. The absence of major defects in most individuals speaks to the resiliency of the conceptus.
Developmental Pharmacokinetics
Metabolism
Variability exists among species with respect to the stage in which metabolic systems begin to develop and mature. In general, conceptuses of species with longer gestations (e.g., humans) develop greater metabolic capabilities in utero, while those of species with short gestations (e.g., rodents) typically show little or no enzyme activity until near or after birth, and thus are incapable of extensive in utero metabolism. For a given species, metabolic pathways are much less developed in embryos than they are in fetuses and neonates. Similarly the metabolic proficiency of conceptuses and neonates is less differentiated than is the corresponding set of activities in the dam.
Human hepatic enzyme activities that participate in xenobiotic metabolism begin developing before birth, although the levels vary by enzyme family and subfamily and are well below those of mature adults. For example, hepatic cytochrome P450 (CYP) 3A activity, a critical Phase I enzyme for xenobiotic metabolism accounting for about 30% of fetal CYP content, is about 30%–60% of that found in adults; in contrast, activities of some Phase I enzymes (e.g., CYP2D6 and CYP2E1) are a fraction (10%) of adult levels during gestation and do not begin increasing until adolescence. Thus fetal enzymes can metabolize xenobiotics that have crossed the placenta to make either toxic or nontoxic metabolites. Since these metabolites are water-soluble, they may accumulate in the fetal compartment due to its relatively higher water content. Fortunately, detoxification reactions tend to predominate over activating reactions in fetal tissues. Therefore fetal metabolism usually serves to protect the fetus.
Fetal renal excretion also aids elimination of xenobiotics. During the latter stages of gestation, the glomerular filtration rate increases, resulting in a concomitant increase in the fetal renal drug clearance.
Enzyme Induction
Prenatal exposure to known enzyme-inducing agents does increase enzyme activity in laboratory rodents. Both 3-methylcholanthrene and phenobarbital induce effects in conceptuses that are similar to those seen in mature animals. Again the dichotomous risk and benefit regarding whether enzyme-mediated metabolism will result in detoxification versus bioactivation depends on the xenobiotic in question. The ability of conceptuses to respond to xenobiotics that induce Phase I and Phase II reactions depends on the animal strain.
Tissue Accumulation
Preferential sequestration of some teratogens in embryonic or fetal tissues occurs in some species, resulting in higher toxicant concentrations in the offspring than occur in the maternal blood. Lead accumulates in the fetus by binding to mineralizing fetal bone. Methyl mercury is retained in the fetus because it cannot convert the methylated form to inorganic mercury, which is more readily eliminated. Indeed, fetal sequestration of methyl mercury is so complete that pregnant women are protected from methyl mercury toxicity. Thalidomide is sequestered in the embryos of sensitive species (e.g., rabbit) because embryonic enzymes hydrolyze the parent drug to hydrophilic metabolites that cannot readily cross cell membranes. Many rodent teratogens are weak acids; hence, they tend to concentrate in embryonic and fetal tissues, as the conceptus compartment is more basic than maternal plasma.
Mother
Maternal Physiologic Status
Alterations in maternal homeostasis must be severe to affect the conceptus, since the needs of the growing organism are usually met even at the expense of the mother. Exceptions do exist, however, since approximately 3.5% of all congenital malformations in humans relate to various metabolic and nutritional imbalances. Examples include diabetes (which results in macrosomia or sacral agenesis); malnutrition (spontaneous abortion, stillbirths, NTDs, neonatal deaths); phenylketonuria (microcephaly, mental retardation, cardiac defects); thyroid disorders (e.g., hypothyroidism, which leads to cretinism, deafness, and retardation); and virilizing tumors (pseudohermaphroditism in females).
Maternal Pharmacokinetics
Absorption from the gut is altered during pregnancy because of increased gastric emptying time and reduced intestinal motility. These changes increase the residence times in both the stomach and small intestine, with the effect on the amount of uptake depending upon whether a given xenobiotic is more readily absorbed via the stomach or the intestine. The pulmonary tidal volume also increases during pregnancy, resulting in increased uptake of inhaled xenobiotics. In addition, the volume of distribution markedly increases during pregnancy because of increased total body water and body fat. This rise in volume leads to decreases in the initial blood concentration of absorbed xenobiotics. There is also a more rapid maternal elimination of water-soluble compounds because of increased renal plasma flow and glomerular filtration rate.
A progressive gestational decrease in maternal plasma albumin concentration may affect xenobiotic binding by plasma proteins. For example, decreased serum binding of a number of therapeutic drugs has been seen to occur during pregnancy.
Maternal hepatic enzyme activities escalate during pregnancy, probably because of the growing need to metabolize fetal wastes. Unfortunately, drug metabolism may be reduced, at least for certain xenobiotics, because of competitive enzyme inhibition by steroid hormones. These counterbalancing trends are complicated: estrogen decreases the efficiency of some metabolic pathways, while progesterone induces some enzymes. It is probable that the effect of steroids and other hormones on metabolism will depend on the timing of a gestational exposure as well as the physicochemical characteristics of a given xenobiotic. Metabolic capabilities exist between resistant and sensitive inbred strains of mice, showcasing the importance of maternal metabolism in regulating in utero exposure.
Father
Direct Toxic Effects on Sperm
Teratospermia is the production of morphologically abnormal sperm, which are often functionally deficient as well. This lesion can follow exposure to some toxicants. Direct actions of compounds on sperm may alter their cytoarchitecture and/or their function. The aberrant functional attributes are reported more frequently, and likely are of greater consequence. Examples of functional deficits include altered sperm motility, reduced ribosomal activity (often with decreased RNA content), and impaired protein synthesis. The induction of chromosomal damage is indicative of substantial genetic damage that may be passed to the progeny, and that may result in malformations.
Abnormalities in Seminal Fluid
Xenobiotics dissolved in the seminal fluid can induce secondary morphological abnormalities in sperm, impair sperm motility, and reduce sperm viability. Dissolved toxicants may also have some influence on the uterus, either by directly damaging the endometrial tissue or by impacting more distant maternal functions following systemic uptake. Subsequent adverse effects on the timing of implantation, the number and distribution of implantation sites, and the success of placentation are possible, and some xenobiotics delivered in semen theoretically may be directly toxic to the developing embryo.
Placenta
Placental Biology
Barrier Properties
A major role for the placenta is as the first major physical barrier between the conceptus and any xenobiotics within the surrounding “external” environment (i.e., the maternal circulation). Placental structure evolves over time in mammals. The initial form is comprised of the maternal endometrial (or “decidual”) reaction to the newly implanted conceptus. The placental contribution derived from the embryo’s extraembryonic membranes builds over time until later in gestation the fully functional placenta becomes responsible for many gestational functions, including hormone production (peptides and steroids), immunosurveillance, metabolism of endogenous and xenobiotic materials, and nutrient uptake.
The nature of the barrier layers within the placenta varies among species. A “complete” or epitheliochorial placenta contains a full complement of maternal and embryonic layers (three layers for each: epithelial, mesenchymal, and endothelial) between the two blood supplies. Ruminants have evolved this conformation. The dog possesses an endotheliochorial placenta, having retained maternal endothelium and all three embryonic layers. Humans and animals species commonly used in developmental toxicity testing (e.g., nonhuman primates, rats, mice, and rabbits) exhibit a hemochorial structure, in which the three embryonic derivatives are retained but the maternal layers are absent. The conformation in primates and rabbits is hemo-monochorial (having only one layer of embryo-derived trophoblast), while rodents are hemo-trichorial (having three trophoblast layers). The difference in trophoblast layering appears to be inconsequential in terms of embryonic exposure to xenobiotic agents.
The rates at which xenobiotics are transported across the placenta play a great part in determining whether or not toxic levels reach and accumulate in the conceptus. The major factors affecting xenobiotic passage across any membranous barrier—lipid solubility, molecular size, ionic charge, concentration gradients, potential to form complexes with other molecules, and the availability of carrier molecules—also apply to the placenta. Most drugs are transported by passive diffusion. Therefore as lipid solubility increases, the rate of transfer tends to climb. Materials with sizes greater than 1000 Da do not cross the placenta easily, whereas those less than 600 Da (which includes most drugs) travel with ease. Steric hindrance the process by which the shape and size of a molecule may interfere with molecular binding sites, may occasionally play a part in reducing transport across the placenta. Binding of xenobiotics by plasma proteins is also frequently a major influence, as it decreases the amount of the freely diffusible form that is available for transplacental transport.
Physiological Properties
Since most drugs cross the placenta by passive diffusion, maturational changes in the barrier structure are unlikely to influence xenobiotic exposure. Thus the rate of transfer of small molecules into the fetus is correlated with the rate of placental blood flow. However, if an agent’s permeability is a rate-limiting step in its transfer (e.g., for hydrophilic compounds), physiological changes in the placenta over the full course of gestation may cause significant changes in fetal exposure. Some compounds, mainly vasoactive agents, may alter placental blood flow through their pharmacological activity.
Placental Pharmacokinetics
Metabolism
Placental metabolic capabilities may sometimes play a role in the modulation of embryonic/fetal drug exposure. Enzyme systems in the placenta are subject to induction by xenobiotics. The function of such enzymes is likely to prevent entry of xenobiotics into the embryo or fetus, but the success of this endeavor will depend on the nature of the xenobiotic.
Sequestration
A generalized decrease in nutrient transport has been seen when some xenobiotics bind to the placenta. For example, cortisone exposure decreases glucose transport, and methyl mercury inhibits amino acid transport.
The pH of the conceptus with respect to the dam changes during the course of gestation. During organogenesis the blood and tissues of rodent embryos are more basic than the maternal blood, resulting in accumulation of weak acids in the embryo. Later in gestation, fetal blood is relatively more acidic than maternal blood, leading to “ion-trapping” of basic compounds in the fetal circulation. This trapping can result in a higher concentration of nonionized drug in the maternal circulation and a net transfer to the fetus over a prolonged period.
Mechanisms of Developmental Toxicity
The pathogenesis and etiology of malformations is quite complex, and many mechanisms have been proposed to explain the genesis of toxicant-induced structural defects. Some represent cellular responses to damage, while others actually represent biochemical and molecular elements that are thought to invoke the cellular reactions. The most important current hypotheses are listed later.
It is often difficult to determine the precise cause of a developmental defect. This problem reflects the multiple potential etiologies of specific lesions due to gestational damage. A particular developmental anomaly can be induced via multiple pathways. In other words, different mechanisms of abnormal development often produce a common lesion. This quandary highlights the limited repertoire of pathologic responses to injury (whether genetic, toxic, or otherwise) that can be expressed by the conceptus.
Excessive Cell Death
Cell populations with a high proliferative rate, or those beginning to differentiate, are the most susceptible to cytotoxicity. This sensitivity may result from the greater amount of active (or “exposed”) genome, yielding greater access to a larger number of genes. Such active DNA is highly susceptible to damage by foreign substances that can form adducts via covalent bonding to nucleotides. The exposed DNA can also be subject to nucleotide substitutions in which native elements are replaced by analogs that distort the annealing of the complementary strands. If left in place, such modified bases can engender point mutations during DNA propagation; in many instances, such damage results in gene inactivation, which is often fatal to the affected cell. Another potential point at which toxicants can inflict damage is by altering energy metabolism, as the internal nutritive requirements of rapidly dividing cells in the G1 phase may be higher than those of resting (G0) cells that are common in differentiated tissues of adult animals.
Excessive cytotoxicity is a proven cause of abnormal embryonic development. For example, cell loss localized to a finite region has a dramatic effect on such processes as limb formation and neural tube closure. The pattern of death may encompass large cell groups (i.e., necrosis) or be limited to isolated elements (i.e., single-cell necrosis); importantly, cell loss by necrosis may damage adjacent cells. Early and excessive cell attrition may leave too few cells to initiate critical inductive events at specific stages of development. The lack of positional information associated with this loss will produce incorrect spatiotemporal relationships that can prevent expansion of a given target cell population—and also all future cell collectives that cannot be induced without the help of these affected target cells. Thus small deficits resulting from too much cell death at early points in gestation can cascade into additional deficits affecting many more cell populations later in development. The regenerative capacity of embryonic tissues in compensating for cell death is also vital in defining the extent of toxicant-induced injury.
Interference With Programmed Cell Death (Apoptosis)
Embryonic sculpting by the region- and time-specific activation of programmed cell death (PCD) is a normal phenomenon during development and does not damage adjacent cells. Many structures utilize PCD, including the brain (to remove superfluous neurons), digits (to attain the right degree of separation and appropriate size) (Figure 25.16), and hollow organs (to create cavities in solid primordia). The process involves selective suicide (apoptosis) by cells that do not complete their expected developmental program, and it is an active undertaking designed to prune exuberant cell numbers during histogenesis and organogenesis. Signals that can initiate PCD usually are chemical messages, such as hormones or growth factors. Interference with these signaling pathways may prevent timely cell death and lead to supernumerary structures or cells.
Reduced Cell Proliferation
Diminished rates of cell division can occur following an exposure to a dose of developmental toxicant that is lower than the dose that induces cytotoxicity. The diminished cell numbers may cause growth retardation of one or more organs. Growth retardation of an organ can have dramatic effects that resonate throughout the entire period of development, since a critical mass of one tissue is often necessary to induce many secondary structures. For example, if dental anlagen are not of sufficient mass, teeth will fail to develop and their supporting bony structures will be smaller than normal (Figure 25.17).
The growth of various tissues occurs at dissimilar rates, so reduced proliferation of tissues during histogenesis can result in different degrees of abnormal growth depending on the affected organs. This loss of developmental synchrony is a striking feature of hypovitaminosis A in pigs, where piglets are born blind because premature closure of the cranial sutures leads to smaller optic foramina and compression of the optic tracts as they exit the cranium. Similarly, corticosteroids reduce palatal shelf size, resulting in cleft palate formation in mice.
Failed Cellular Interactions
Genetic factors, exogenous agents, or both can cause abortive connections among various cell populations. For example, abnormal cerebellar development in the weaver mouse mutant (gene symbol: wv) results from a genetically programmed decrease in the numbers of radial glia processes needed to guide the migration of granule cells from the brain surface through the molecular layer to the granule cell layer. Similarly a genetic defect in cattle produces anophthalmia or microphthalmia via reduced or absent contact between the optic cup and overlying ectoderm, thereby preventing induction of the lens placode. Cytotoxic antineoplastic agents as well as ionizing radiation also incite these ocular lesions by the same mechanisms. Hypovitaminosis A disrupts the spatial orientation among cells in mesenchymal condensations through altered cell-adhesion expression, leading to altered shapes of cartilage models and their bony successors.
Impeded Morphogenetic Movements
Cellular migration can be hampered by many mechanisms. Common factors include decreased cell mobility; abnormal quantity or quality of the extracellular matrix (ECM), especially via altered substrate-adhesion molecules (SAMs); aberrant quality of cell-adhesion interactions as mediated through anomalous patterns of cell-adhesion molecules (CAMs), SAMs, or cell-junctional molecules (CJMs); and disruption of cytoskeletal microtubules or microfilaments. Accordingly, altered perception of critical positional information and/or inability to follow the signals seems to be major themes responsible for such defects.
The ECM is composed of glycosaminoglycans (GAGs) such as chondroitin, chondroitin sulfate, heparan, heparan sulfate, hyaluronic acid, and keratin sulfate as well as abundant collagen. These molecules and other SAMs interact with the integrins on cell membranes, allowing adhesion and ultimately directed motion among tissues in defined planes. The ECM content of these constituents varies during different stages of development. High levels of hyaluronic acid in early morphogenesis encourage cell migration by increasing cell proliferation, inhibiting aggregation, and increasing the fluidity of the ECM. During later differentiation, hyaluronic acid levels decrease as cells begin to aggregate and chondroitin sulfate production is increased.
Diminished cell adhesion can also result in deformities. Cortisone disrupts the synthesis of GAGs and collagen, resulting in reduced production and sulfation of ECM constituents. The decreased adhesive qualities of the ECM may contribute to the inability of the palatine shelves to fuse. In addition, cell surface receptors may be altered, thereby thwarting recognition and induction of certain cell collectives.
Microtubules and microfilaments are essential contractile elements of all cells. Functioning of these elements can be affected by disrupting the synthesis and turnover of tubulin, changing the number and arrangement of microtubule organizing centers, altering the phosphorylation of tubulin-associated proteins, and perturbing tubulin polymerization. Certain antineoplastic agents incite developmental toxicity in this fashion: colchicine and vincristine distort microtubule structures, while cytochalasin B inactivates microfilaments. The actions of these cytoskeletal components can also be impacted by indirect means. Calcium (Ca++) is required for appropriate microtubular and microfilamentous function; therefore, treatment with ethylenediaminetetraacetic acid (EDTA) induces heart malformations by chelating Ca++, which paralyzes pioneer cells that must move through the cardiac primordium in order to fulfill normal development.
Reduced Biosynthesis of Essential Components
Altered production of many molecules can have profound effects on normal growth and development. In this regard the usual change is decreased synthesis. All the major molecules required to support rapidly expanding tissues—nucleic acids (DNA and RNA), proteins of all kinds, and energy storage molecules (e.g., the complementary molecules adenosine di- and triphosphate (ADP and ATP, respectively)), as well as the native and phosphorylated forms of nicotinamide adenine dinucleotide (NAD and NADP+, respectively)—are subject to such perturbations following xenobiotic exposure during development.
Inhibition of DNA synthesis is not teratogenic per se. However, cytotoxicity is a common outcome of reduced DNA synthesis, and the resulting cell loss can engender teratogenic effects at future stages of embryogenesis. Important factors in the outcome of such inhibition are the time available for cellular repair, which can be minimal in the rapidly dividing cell collectives that are characteristic of the embryo, and the degree of cell necrosis.
A primary reduction in RNA synthesis may inhibit DNA synthesis since some RNA variants are required for nucleic acid replication. However, decreased RNA synthesis more typically interferes with protein production, thereby depriving metabolically active cells of essential building blocks needed for growth. The degree of RNA depletion correlates well with the cytotoxic potential of many antineoplastic agents.
Inhibition of protein synthesis occurs by several mechanisms. Examples include failure to assemble ribosome complexes, which prevents protein translation; misreading of mRNA and/or premature termination and release of peptide chains, both of which results in the release of defective proteins; and the inability to perform posttranslational modification to produce functional proteins. In general, protein synthesis inhibitors yield little cytotoxicity, but they nonetheless are efficient agents for inducing embryolethality or growth retardation.
Abnormal energy metabolism is generally a critical blow to developing organisms. Inadequate reserves of ATP and NAD/NADP+ can have serious effects on biosynthesis by altering the efficiency of glycolysis and electron transport during critical periods of organogenesis. As adequate energy stores are an absolute requirement for many aspects of cellular function, especially in rapidly dividing cells, it is not surprising that agents which block the synthesis of ATP and/or NADP+ can have disastrous effects on the developing embryo, ranging from decreased cell function to cell death. If the degree of cell death is severe, embryonic or fetal death can follow.
Mechanical Disruption
Damage by agents that physically impact normal cytoarchitectural development can profoundly impact the viability of the conceptus. Such disruptions may sever interactions among neighboring cells, impede morphogenetic movements, and decrease proliferation and growth through pressure-induced necrosis. Changes in the local ionic balance can provoke fluid buildup within cells and fluid-filled organs (e.g., brain, eye) that ruins the integrity of these structures. For example, in the chick embryo, both dimethyl sulfoxide (DMSO) and hypoxia precipitate increases in cerebrospinal fluid (CSF) volume, which increase the pressure within the brain ventricles and spinal cord central canal. Over time, distension can exceed the elastic limits of the ependymal lining, thereby permitting escape of the CSF into the adjacent neuropil. This mechanism is thought to explain such congenital defects as hydrocephalus and syringomyelia.
Altered pressure is also thought to contribute to the genesis of ventricular septal defects (VSD), which are the most common congenital cardiac anomalies. Agents that interfere with aortic arch development can change the blood flow patterns in the heart tube. Over time the distorted flow dynamics increases the intraluminal pressure within the reorganizing heart, thereby preventing normal cardiac partitioning—and in particular, the full closure of the ventricular septum. The chronic pressure overload in the ventricles is commonly accompanied by defects in the atrial septum and/or one or more major cardiac vessels, including patent ductus arteriosus, tetralogy of Fallot (of which VSD is an integral component), or the transposition of the great vessels. In addition to certain xenobiotics, other etiologies capable of inducing these changes include infections (e.g., rubella) and maternal diseases (e.g., diabetes mellitus).
Mechanical constraint has been linked to many instances of gross malformation in external structures. Examples include mal-positioning, reduction, or amputation of the limbs as well as failed closure of the abdomen, neural tube, palate, or philtrum (middle portion of the upper lip). Abnormalities of placental structure (e.g., umbilical cord bands) or function (e.g., oligohydramnios (deficient production of amniotic fluid)) have been linked to this outcome. In like manner, aberrations of uterine structure (e.g., malformations) or function (prolonged severe contractions) have been associated with such lesions. In all these cases, the putative pathogenesis is an extrinsic increase in the mechanical pressure applied to the surface of the developing embryo or fetus.
Heightened pressure may disrupt development by mechanisms other than direct trauma to the conceptus. A common pathway for such changes seems to be insufficient circulation to the rapidly evolving tissues. For example, vascular occlusion by tissue compression (e.g., from intense uterine contractions) will decrease organ perfusion. Alternatively, early embryonic trauma through overzealous manual pregnancy testing of cattle can cause atresia coli in calves. The presumed pathogenesis of such lesions is hypoperfusion, cell hypoxia, eventual endothelial cell loss, necrosis, with eventual repair by fibrosis. Developmental toxicants such as misoprostol (a synthetic prostaglandin E1 analog) and phenytoin as well as certain placental manipulations (e.g., chorionic villous sampling) have been shown to induce leg defects in human infants by vascular disruption in limbs that had first formed normally.
Intracellular pH
The hydrogen (H+) ion concentration in developing cells has been implicated as a common pathogenesis for many developmental toxicants. Decreased intracellular pH interferes with numerous processes, including such critical functions as proliferation, internal signaling, enzyme activity, and cytoskeletal protein polymerization. Thus it is not surprising that toxicant-induced alterations in fetal pH can produce developmental defects. Agents that have been implicated as modifiers of embryonic pH include anticonvulsants (e.g., diphenylhydantoin, sodium valproate), carbonic anhydrase inhibitors (e.g., acetazolamide), and carbon dioxide (which is converted in the body to bicarbonate (HCO3−)). These substances cause many types of malformations, including NTDs and postaxial right forelimb ectrodactyly.
Summary
Developmental toxicology studies, including the toxicologic pathology evaluations that comprise a part of the analysis, are essential contributors to the hazard identification and risk assessment processes for novel xenobiotics. The developing embryo or fetus is often the biological system that is most sensitive to xenobiotic exposure. The complexity of developmental processes makes risk assessment for developmental toxicants fairly difficult. Major factors help produce a constantly changing spectrum of functional and structural manifestations of developmental toxicity, including tissue-specific critical periods of organogenesis; the intricate relationship between the spatial and temporal expression of various genes (especially those for morphogens and their signaling pathways); and interactions between the genotype and various metabolic processes and the toxicant. To date, research in this field has predominantly attempted to identify new hazards and develop new methods for evaluating prenatal toxicity. However, the explosion in our understanding of cellular and molecular mechanisms of teratogenesis that has accompanied the rise of bioengineering, coupled with the advent of powerful new bioinformatic and computing platforms, should permit scientists and regulators to increasingly improve their predictive capabilities, thereby allowing society to avoid, reduce, or even reverse the consequences of developmental toxicity.