Essay 2
The past, present, and future of psychiatric genetics
Introduction: prelude and fugue
The retrospective timescale of half a century, taken throughout this book, is a particularly apt one for psychiatric genetics. In 1959 a landmark event occurred in the United Kingdom that had wider implications for the subject’s development internationally. The Medical Research Council (MRC) Psychiatric Genetics Unit at the Institute of Psychiatry in the University of London was founded under the directorship of Dr Eliot Slater. It was housed on the campus of the Institute of Psychiatry/Maudsley Hospital in a building that became known as the ‘Genetics Hut’. Austere and modest in its scale, it nevertheless became internationally famous, at least among the tiny band of researchers on psychiatric genetics that then existed around the world.
Although it was the first university-embedded unit of its type in the United Kingdom, the Hut’s intellectual roots traced back to Germany where, in 1917, the German Research Institute for Psychiatry (Deutsche Forschungsanstalt für Psychiatrie) in Munich was founded and directed by Emil Kraepelin. He established a genealogical and demographic department in his institute and appointed as the head a pioneer of psychiatric genetics, Ernst Rudin. His department contained a number of talented researchers working on family and twin studies, including Bruno Schulz, Hans Luxenburger, and Franz Kallmann, who shared with Rudin an enthusiasm for the science but not his politics. Rudin was an enthusiastic supporter of the National Socialist (Nazi) party and an influential adviser concerning its policies in eugenics and ‘racial hygiene’, whereas Luxenburger was at one point banned from teaching at the university because of his open criticism of Nazi ideologies. Eliot Slater, who began a fellowship with Rudin in 1934, became outraged by his political views. While in Munich he fraternized with Jewish intellectuals and married one of them, the poet Lydia Pasternak, sister of Boris the celebrated writer. (Their daughter, Catherine Oppenheimer, is a contributor to this volume.) Slater returned to London in 1936 and began the task of founding psychiatric genetics as a new discipline in the United Kingdom. In the same year Kallmann, who was half Jewish, fled to the United States where he was offered a position as the first head of a new medical genetics department at the New York State Psychiatric Institute, essentially founding psychiatric genetics in the United States. Other visitors to the Munich department in the mid-1930s included a young Dane, Erik Strömgren, who went on to become the pre-eminent Scandinavian psychiatric researcher of his generation.
In the wake of World War II, the discrediting of Nazi eugenics policy also tainted psychiatric genetics, which died out in Germany. Thus the Munich genealogy department provided both the prelude to psychiatric genetics in Europe and North America and a flight from the subject in Germany that was to last for over 30 years.
The era of twin and adoption studies
The idea that mental illness has a tendency to run in families is almost certainly an ancient one. There are patient case records dating back to the early 19th century in the museum of the hospital where I do my clinical work, London’s Bethlem Royal Hospital, showing that doctors then were already attempting to record systematically whether their patients’ disorders were hereditary. Of course diseases can run in families because of a shared environment that might include anything from exposure to toxins or infectious agents to shared upbringing and lifestyle. Normal traits, behaviours and characteristics run in families too. For example, a study that I carried out 20 years ago in Cardiff with my then registrar (1) showed that the ‘risk’ of attending medical school among the first-degree relatives of Cardiff medical students was 80 times that of the general population. We further showed that the pattern of inheritance was compatible with a Mendelian recessive gene. We published our results not to convince colleagues that there was a gene to clone, but rather to point out that it is dangerous to make simple genetic assumptions about a complicated trait.
Fortunately there are two types of ‘natural experiment’ that can come to our rescue in teasing out the effects of genes, ‘nature’ and within-family ‘nurture’. These are twin studies and adoption studies. The English polymath Sir Francis Galton first suggested studying twins ‘as a criterion of the relative powers of nature and nurture’ in 1876 (2), but this was before much was known about the biology of twinning. It was Wilhelm Weinberg, a German obstetrician and pioneer of statistical genetics, who, at the beginning of the 20th century, made the first inference that there are two main types of twin: monozygotic (MZ) or identical twins who result from the splitting of a single egg and who have 100% of their genes in common, and dizygotic (DZ) or fraternal twins who result from the simultaneous fertilization of two eggs and have the same proportion of genes in common as siblings, i.e. 50%. In the 1920s twin studies began first by Siemens, a dermatologist (and again a German) investigating the inheritance of skin moles. This was followed by Luxenburger’s study of schizophrenia, the first of its kind.
The basic idea of the classic twin method is that greater resemblance for a trait or disorder in MZ than DZ pairs reflects the influence of genes. However, this depends on an additional assumption about twins, that MZ and DZ pairs share the environment to roughly the same extent. This ‘equal environments assumption’ is open to criticism, particularly because half of DZ pairs are of opposite sex. For this reason many early twin studies were restricted to same-sex pairs; however, even in same-sex pairs MZ twins might be treated more alike by parents and be dressed alike, placed in the same class at school, have more friends in common than DZ pairs. As it turns out, studies have found that these sorts of environmental sharing do not much bias the results. Indeed, a repeated surprise finding emerging from twin studies of common traits is that the role of shared environment is often small.
A different potential source of bias in twin studies depends on how twins are selected for inclusion in the research. Very early twin studies of schizophrenia tended to focus on twins where at least one of the pair had been chronically hospitalized. They also often relied on the subjects being referred to the twin researcher by the doctor in charge of their case. Together these factors meant that early twin studies tended to include the most severe cases and the most conspicuous or memorable ones, and it is likely that this led to a bias in favour of concordant pairs (i.e. pairs where both had the disorder) and identical pairs, tending to inflate MZ–DZ differences and giving a ‘more genetic’ appearance. To overcome this problem Slater, in 1948, established a systematic way of ascertaining twins from the hospital-based register at the Maudsley and Bethlem Royal Hospital where every inpatient and outpatient, at their point of registration, was asked the question, ‘Are you a twin?’ Elsewhere, (e.g. the Scandinavian countries) it was possible to ascertain twins systematically via national registers and match these data with hospital case registers.
The 1960s saw the first flourishing systematic twin studies being carried out on a variety of disorders, but there was a particular focus of attention on schizophrenia which, at the time, was one of psychiatry’s ideological battlegrounds. Earlier twin studies created suspicion in the minds of some that their authors had a hereditarian bias. There was even the idea advanced by Jackson (3), a psychoanalyst, that twins, particularly identical twins, may suffer from ‘ego identity’ problems more than singletons and would thus be particularly prone to psychotic disorder. In the later 1960s, a disparate group of critics often referred to collectively as ‘anti-psychiatrists’, such as the followers of R.D. Laing on the existentialist left and of Thomas Szasz on the radical right, questioned the very existence of schizophrenia as a ‘real’ disorder. Like other mental illness schizophrenia, according to Szasz, was just a myth.
In the United Kingdom social psychiatry had a powerful influence in the 1960s and 1970s and many viewed genetics as dismally deterministic. Indeed, Slater had the experience of a social psychiatrist colleague bursting into his office and ‘whooping for joy’ following the publication of a paper by the Finnish psychiatrist Pekka Tienari which purported to find zero concordance for schizophrenia in identical twins. However, Tienari’s findings were in contrast to those of Slater’s unit. A postdoctoral psychologist from the United States, Irving Gottesman, worked with Slater’s long-time assistant, James Shields, to study schizophrenia using the Maudsley twin register. Their research (4) was notable for its careful methodology, including rating of twin data ‘blindfolded’ to the identity or zygosity of the subject by multiple diagnostic experts. They found over 50% concordance in monozygotic twins compared with just over 10% concordance in dizygotic twins. These figures were much in line with several other studies published at around the same time, including a follow-up study from Tienari who found that several co-twins from his earlier negative study had become ill, making the total pattern of results wholly consistent with a genetic effect in schizophrenia.
For those who remained unconvinced clinching evidence came from adoption studies. The first of these was by Leonard Heston (5) who was still a trainee psychiatrist in the United States when he obtained blindfolded diagnoses on two groups of adults who had been fostered or adopted away from their natural mothers in infancy. The first group consisted of 47 offspring of mothers with a diagnosis of schizophrenia who had given birth when they were inpatients in Oregon state mental hospitals at a time when state law dictated that such offspring be separated from their mothers within 72 hours. The second group of adoptees consisted of 50 age- and sex-matched controls whose mothers were free from psychiatric illness. The results were quite striking in that five of the offspring of schizophrenic mothers became schizophrenic whereas none of those with healthy biological mothers were so affected.
Soon after, a series of publications began appearing as a result of a collaboration between American and Danish psychiatrists, led by Seymour Kety, using Danish adoption and psychiatric registers. These were larger in scale and had the advantage of allowing a variety of study designs. The first was, like Heston’s, an adoptees study looking at the biological offspring of schizophrenics compared with control adoptees, but this time a substantial number of the schizophrenic parents, about one third, were fathers. This meant that criticisms of Heston’s study concerning the possible role of prenatal or perinatal maternal influences could be addressed. The second design was called an adoptee’s family study; the starting point was adoptees who had become schizophrenic and whose biological and adoptive relatives were compared. The finding of a small sample of adoptees with biological parents but who had been raised by adopting parents one of whom became schizophrenic, permitted the third type of study design, the cross fostering study; these adoptees were compared with those in the schizophrenic biological parent group who were raised by normal parents. All three types of study pointed clearly in the same direction: What mattered in creating an increased risk of schizophrenia was sharing genes rather than sharing environment. This led Kety to remark, ‘If schizophrenia is a myth, it is a myth with a strong genetic basis’.
Adoption and, even more importantly, twin studies continue to play a role in psychiatric genetics up to the present time, and have recently provided more sophisticated insights using modern statistical modelling techniques that capitalize on the availability of high-speed computers. However, in the 1960s and 1970s they were not able to show which genes were involved, or where they lay on the genome, the 23 pairs of chromosomes that carry our genetic material. Therefore, for many, studies that actually visualized chromosomes were a more exciting route to follow.
Cytogenetics and the joy of seeing chromosomes
The existence of chromosomes had been known since the mid-19th century. However, an understanding of their roles only began in the 1950s, as technology for visualizing them became developed. Most of the time chromosomes are extremely elongated structures contained in the cell nucleus invisible under a light microscope. However, when cells divide the chromosomes become shorter and more compact, aligning themselves in pairs. If cells are stimulated to divide, the cell division then chemically arrested and the cell broken down, it is possible to stain the chromosomes with a dye and visualize them under a microscope. Early cytogenetics depended on taking a photograph of stained chromosomes, cutting out the images of individual chromosomes with scissors, arranging the 22 pairs of ordinary chromosomes (or autosomes) in order of size by hand, picking out the sex chromosomes (X and Y) and re-photographing them to give a final image of the 23 pairs—a so-called karyotype.
This incredibly crude-sounding approach actually yielded astonishing breakthroughs, the first of which was the discovery by Lejeune, in 1957, that Down’s syndrome is caused by an abnormal karyotype most commonly involving an extra chromosome 21 (6). Discoveries of other so-called trisomies followed, including Klinefelter’s syndrome (men with an extra X chromosome, hence XXY rather than the normal male XY), as well as discovery of deletion syndromes where a chromosome or part of a chromosome goes missing. A good example is Turner’s syndrome where the appearance is female though there is only one X chromosome (X0) rather than the normal two (XX).
Both Klinefelter’s syndrome and Turner’s syndrome are of psychiatric interest. Men with Klinefelter’s usually have lower IQ and, by some reports, an increased rate of schizophrenia. Women with Turner’s syndrome have normal intelligence but tend to test poorly measuring performance rather than verbal reasoning. More recently they have been reported to have increased rates of autistic features, particularly when their single X chromosome is inherited from their mother (7). The cytogenetic discovery of greatest interest in the early years of cytogenetics concerned men with an extra Y chromosome (XYY). Jacobs reported finding that 3% of men in Carstairs, a Scottish secure psychiatric hospital for offenders, had XYY karyotypes (8). They were also unusually tall and had below average intelligence. But was the XYY syndrome, as it came to be called, really a newly discovered genetic cause of antisocial behaviour? A key question was how common was the XYY karyotype in the general population. Further research revealed its presence was somewhere between 1/1000 and 1/2000, which meant at least 1000 adult XYY men in Scotland (the total population at that time being about 5 million) were not in Carstairs Hospital. A subsequent study (9) remarkably screened nearly all non-institutionalized men in Denmark who were over 1.84 m in height, approximately 4000. Twelve had XYY karyotypes, five of whom (42%) had criminal records compared with 9% of non-XYY men. However, the records of the XYY men involved mainly minor offences, effectively refuting the idea the syndrome predisposes to violent criminality.
Cytogenetics continues to be refined in its methodologies and is now a vital branch of laboratory diagnostics in learning disability and medical genetics; however, the karyotypes that one can see using a light microscope proved to be less informative in psychiatic genetics than many had hoped for. Interestingly, there has been a resurgence of interest in cytogenetics as methods have been developed to detect much smaller, subtler, submicroscopic changes in chromosomes.
Statistical models and muddles
While early cytogenetics did not offer much explanatory power for the bulk of psychiatric disorder, a different set of technologies were beginning to be employed. Disorders such as schizophrenia and bipolar disorder were clearly not Mendelian, where genes are either dominant (when a single copy is sufficient to cause the disorder) or recessive (when two copies, one from each parent, are required). Family studies did not show clear-cut patterns of transmission as predicted by Mendel’s first law, the law of segregation, where half the offspring of an affected parent show the disease in the case of dominant genes, and a quarter of the offspring of unaffected ‘carrier’ parents show the disease due to recessive inheritance. Twin studies revealed incomplete concordance in identical twins, indicating that the non-genetic factors played a role. Therefore the only way that such disorders could result from the effect of single genes would be through incomplete penetrance, when an individual carries a disease genotype without manifesting the disorder. Slater (10) was the first to put forward a plausible single-gene model of schizophrenia by invoking this notion. He showed that, assuming the disorder is a lifetime risk in approximately 1% of the general population, a dominant gene with a specified incomplete penetrance and a specified frequency and population could explain the risks in various categories of relatives of individuals with schizophrenia. He studied the hypothesis empirically, using a graphical method to solve the parameters of his model.
In contrast, Gottesman and Shields (11), working in Slater’s unit, put forward a more radical ‘polygenic’ model based on the work of Falconer (12), a statistical geneticist. He postulated that what is inherited in common disorders is a liability contributed by multiple genes, each of small effect, so that in the general population the liability tends to have a bell-shaped (or normal) distribution and only those individuals who live beyond the threshold at the upper tail of the curve actually become affected. Relatives of affected individuals have an augmented liability so that more of them lie beyond the threshold then do members of the general population. This model allows calculation of a measure called ‘correlation in liability’ for various categories of relatives which, in turn, can be used to estimate the heritability, the proportion of variation in liability to the disorder that can be attributed to genes. There subsequently ensued a debate that lasted for almost three decades as to whether the single-gene or polygenic model of schizophrenia was correct. (Similar debates existed about other disorders, but again it was schizophrenia that was the most hotly contested battleground.)
One of the problems about differentiating between single-gene and polygenic models is that both are comparatively ‘elastic’ and therefore difficult to refute. More sophisticated types of modelling were developed in the 1980s as high-speed computers became widely available. In the meantime an influential piece of evidence that argued against the single-gene model was another graphical method demonstrating that many of the available data points from family and twin studies of schizophrenia plotted outside the envelope compatible with the biologically plausible range of single-gene parameters (13). The same result was later found for bipolar disorder (14). When speaking to clinical audiences on the topic, I have often been asked about my confidence in dismissing single-gene models when Slater’s solution seemed so plausible. This led me to re-examine Slater’s original work. Interestingly, he never actually formally tested whether his schizophrenia single-gene model was a satisfactory statistical explanation of the data, and if one applies a simple chi-squared goodness-of-fit test the model can be unequivocally rejected (1,15).
Biometrics and behaviour genetics
The debate over whether schizophrenia could be best explained by a single or polygenic inheritance model provides a specific example of a type of controversy present since the beginnings of genetics as a new branch of biology in the early 20th century—the so-called ‘Galton versus Mendel’ controversy. Francis Galton was an English polymath born in 1822, the same year as Gregor Mendel, and far more celebrated than Mendel during his lifetime. Galton was the first to suggest the study of twins as a means of assessing the relative contributions of nature and nurture and in one of his most famous works, Hereditary Genius, he laid the foundation of behavioural genetics. Galton was interested in the resemblance between relatives for continuous traits, which he saw as being predominantly normally distributed (i.e. following a bell-shaped curve). He was the first to describe the phenomenon of ‘regression to the mean’ whereby parents with extreme characteristics (e.g. great height or mental ability) tend to have offspring who are closer to the average. Here he provided the beginning of a fundamental method in statistics, regression analysis, subsequently developed by his disciple Karl Pearson, who first devised the correlation coefficient as a measure of similarity between pairs of observations (e.g. continuous traits in pairs of relatives).
In contrast to the influential writings of Galton and his followers, Mendel’s laws, originally published in 1866, remained almost completely ignored for the next 35 years. When ‘rediscovered’ they had a revolutionary effect, explaining transmission of present/absent characteristics, but were seen by Galton’s followers, the biometric school, as largely irrelevant to the majority of human characteristics that are continuous. It was not until 1918 when Ronald Fisher showed that many genes of small effect, behaving in Mendelian fashion, could add up to produce a continuous trait that has a normal distribution (16). Fisher’s rapprochement between Mendelian and Galtonian ideas, however, was contained within a mathematically dense and difficult piece of work that only really impressed the statistical cognoscenti who were rare among biologists. Subsequently Mendelian and biometric genetics progressed in parallel, often in a non-overlapping fashion, through much of the 20th century.
The mainstream of behavioural genetics was definitely in the biometric camp and, with regard to human behaviours, heavily influenced by interpretation of twin studies. The subject matter included both continuous traits, such as personality and cognitive ability as reflected in IQ tests, and present/absent traits, such as psychiatric disorder which could be considered as having an underlying continuous liability as in the Falconer-type model. As examples, Figure 2.1 shows concordances for a number of traits of psychiatric relevance. These data provide two important messages. The first is that all of the disorders shown have greater similarity in MZ than DZ twins, indicating genetic effects. The second is that no disorder shows complete concordance in MZ twins even though these are genetically identical, indicating that all the disorders have an environmental component. The environment can be conceptualized as having two main components: shared environment, that causes family members to resemble each other, and non-shared environment, that operates at the individual level and therefore contributes to differences between members of the same family. Thus it becomes possible with twin data to estimate the size of the contribution to the variation within a trait according to the additive genetic component, the heritability, and the variation attributable to shared and non-shared environment.
Such concepts about a partitioning variance had been around for several decades and were imported into human genetics originally from agricultural genetics, where knowing the heritability of milk yield in cattle or the size of tomato fruits has practical implications for selective breeding. A graphical method called path analysis, put forward by the American geneticist Sewall Wright in the 1920s, lent itself perfectly to deriving the necessary equations to estimate the variance components. However, the technical advance that eventually allowed statistical model fitting based on twins to advance rapidly was the advent and widespread availability of high-speed computers in the 1980s. Some results using this approach, which is now known as structural equation modelling, are illustrated in Figure 2.2.
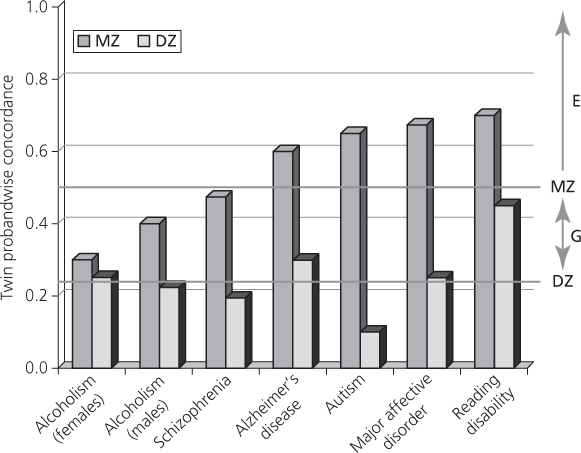
Fig. 2.1 Common behavioural disorders.
From R. Plomin, M. J. Owen, P. McGuffin, the genetic basis of complex human behaviors, in Science. 17 June 1994: Vol. 264 no. 5166 pp. 1733–1739. Reprinted with permission from AAAS.
All of the traits and disorders shown here are heritable, that is they show significant additive genetic effects. ‘Significance’ is formally tested in structural equation modelling by eliminating, in turn, components from the model and seeing whether any make a difference that is significantly different from chance. By contrast, for most of the traits it is possible to drop the shared environmental component without worsening the fit, the exceptions being reading disability and IQ in childhood. It is surprising that the environment shared within families contributes little or nothing to family resemblance, but it is a finding that is completely consistent across the behavioural genetics literature, largely supported by studies of MZ twins reared apart. Twins separated soon after birth are rare, and the studies of them inevitably rely on unsystematic recruitment methods, including advertisements in the media, that could result in sampling biases. Nevertheless they tend to show that for measures such as personality tests twins reared apart are no less similar than twins reared together. Indeed, one early study actually found that twins reared apart had slightly more similar personalities than twins reared together (17). Various explanations of this phenomenon have been put forward, including the idea that twins in particular, but perhaps siblings in general, tend to consciously or unconsciously differentiate themselves from each other in their behaviours. Similarly it may be that parents’ treatment of offspring, however hard they try to deal with them equally, inevitably includes different approaches to different members of a sibship, what behaviour geneticists have sometimes called contrast effects. It is also sometimes suggested that peers outside the family actually have greater effect on the behavioural development of children than their brothers or sisters. These hypotheses are all difficult to test, but what is clear is that, for characteristics such as IQ, the effects of shared environment are substantial in childhood (and cannot be dropped from the explanatory models), but diminish in adulthood and are overwhelmed by additive genetic effects in later life (18).
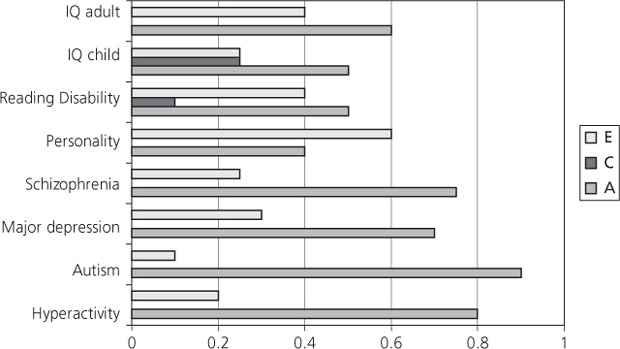
Fig. 2.2 Additive genes (A), common environment (C), and non-shared environment (E).
Data from P. McGuffin, B. Riley, and R. Plomin. (2001). Genomics and behavior. Toward behavioral genomics. Science 291(5507), 1232–1233.
In addition to examining the genetic architecture of single traits, structural equation modelling provides ways of exploring the overlap between traits and the extent to which they share genetic or environmental underpinnings. This is of particular interest in psychiatry where there is frequently an admixture of symptoms from two or more disorders that, at least according to conventional classification schemes, are considered to be more or less distinct entities. A good example is depressive disorder and generalized anxiety. Here analysis of twin data from a population-based study in the United States showed that the genes contributing to anxiety and depression overlap entirely, and that phenotypic differences are contributed by environmental effects (19). Subsequent analyses (20) largely confirmed this finding, although suggested the existence of additional genetic effects specific to depression. A study that was more controversial when it was first published a decade ago took a similar approach to analysing twin data based on the Maudsley Hospital register (21). The authors addressed the issue of whether schizophrenia and bipolar disorder show overlapping causes. The results pointed to separate specific, as well as overlapping, genetic effects contributing to both disorders. These findings have been borne out by molecular genetic studies and a genetic overlap between schizophrenia and major affective disorders is now the accepted orthodoxy even though such proposition was almost heretical a decade ago.
Locating and identifying genes
Our ability to map genes depends upon the phenomenon called genetic linkage, whereby genes close together on the same chromosome disobey Mendel’s second law, the so-called law of independent assortment. In his experiments on pea plants Mendel observed that pairs of characteristics are inherited independently of each other. He of course had no knowledge of chromosomes or of the physical basis of inheritance. A few years after Mendel’s laws were rediscovered, the English biologists Reginald Punnett and William Bateson observed that there were sometimes departures from Mendel’s independent assortment, and coined the term ‘linkage’ to describe them. Subsequently Thomas Hunt Morgan, an American who worked largely on the genetics of fruit flies, proposed that genes are carried on chromosomes. He further proposed that genes that are close together on the same chromosome tend not to be separated during meiosis, the type of cell division that results in the formation of sperms and eggs.
Meiosis is a rather complicated process during which pairs of chromosomes, one set derived from the paternal line and the other from the maternal line, come to lie alongside each other and exchange parts of their material. This process, called ‘crossing over’ or recombination, is essentially like shuffling the genetic deck of cards. It thus ensures that sexual reproduction optimizes genetic diversity with attendant evolutionary advantages. Morgan correctly inferred that recombination between pairs of genes will occur 50% of the time if they are carried on different chromosomes or are widely separated on the same chromosome. For genes closely aligned on the same chromosome, recombination rates of less than 50%, that is linkage, may be observed, with the rate of recombination approximately proportional to the distance between pairs of genes. Today we still measure genetic distance in gene mapping studies using units named after Morgan; the human genome is approximately 35 morgans (M) or 3,500 centimorgans (cM) in length. (Strictly this is the ‘sex averaged’ length of the genome since there is more crossing over in female meiosis than in male meiosis.)
Organisms that have a very short life cycle and where mating patterns can be manipulated by researchers (e.g. fruit flies) are ideal for studying linkage between pairs of genetically determined characteristics. Human beings, who have long life cycles and a preference for selecting their own patterns of mating, are much less suitable. Special statistical methods are required to help assess whether the rate of crossing over, or recombination, is significantly less than the expected rate of 50% that occurs in independent assortment. One of the first tests of linkage in humans was devised by Lionel Penrose, a psychiatrist and pioneer statistical geneticist (22). It was based on the rate of sharing of alternative forms (‘alleles’) of genes in pairs of siblings. Around this time geneticists were beginning to explore the relationship between diseases and genetic markers, that is Mendelian characteristics that are reliably measured, such as blood groups. The most useful genetic markers are ‘polymorphic’, with two or more alleles that are common in the population.
Tests such as the one devised by Penrose were useful for detecting the presence of linkage but not for estimating how much recombination is present; that latter provides a means of estimating the distance between the two points on a chromosome (‘loci’) where the two genes are located. Modern methods for estimating recombination, and hence genetic distance, are essentially derived from an approach put forward by Newton Morton in 1955 called the LOD (log of odds) score method. This is based upon plotting out a series of odds ratios (odds on linkage versus no linkage) between two characteristics over a range of possible recombination rates from zero (complete linkage) to 0.5 (independent assortment). The method uses the common log of the odds which gives LOD scores the useful property of simply being summed (rather than multiplied) over a collection of families to give a combined score, where the LOD score peaks provide the best estimate of recombination. Hence if one trait is a genetic marker of known location on a chromosome and the other is a disease it is possible to infer that the gene conferring susceptibility to the disease maps close to the marker.
The main problem about applying this approach to identify the genes involved in any disease, even ones that show simple Mendelian inheritance within families, is that until the mid-1980s the genetic markers that were available to researchers were few in number. They included ABO and various other blood groups, some enzymes that could be measured in red cells, various polymorphic plasma proteins and polymorphisms within the HLA system, the components of the major histocompatibility complex (MHC) that determines the success or rejection of tissue transplants. Despite this, optimistic researchers began collecting families multiply affected by disorders such as schizophrenia and bipolar disorder, with a view to employing these pre-DNA ‘classical’ markers to carry out linkage studies.
In my own study commencing in 1979 I was fortunate to obtain guidance and collaboration from world-class London-based laboratories directed by Peter Cook and Ruth Sanger at University College London and by Hilliard Festenstein at the Royal London Hospital. Nevertheless I was only able to explore about 30 different marker systems in 12 families. The study proved to be almost entirely negative but, with certain optimistic assumptions, I concluded that I had at least managed to exclude a major schizophrenia susceptibility locus from about 6% of the genome (23).
The advent of the ‘new genetics’
The development that opened up the prospect of mapping disease genes was the discovery of a brand-new type of DNA-based genetic marker. Restriction fragment length polymorphisms (RFLPs) would allow the creation of a set of ‘tags’ to search the entire genome and track down all major genes leading to disease susceptibility. Briefly, RFLPs are detected by cutting up DNA with bacterial enzymes that recognize specific ‘restriction sites’, stretches of DNA that are recognized by specific enzymes and show variability in the population. If a restriction site is present at a particular point the DNA is cleaved. If the site is altered or absent, DNA is not cleaved. This leads to fragments of different sizes that can then be separated by placing them on a gel and passing an electric current through it, causing movement of fragments according to their length. The gel is then blotted onto paper and the DNA fragments identified with specific probes which, in the beginning, were radioactively labelled. The process is called Southern blotting, named after the Edinburgh scientist, Ed Southern, who first described it. This technology was so revolutionary that the editor of the American Journal of Human Genetics coined the term ‘new genetics’ to describe it (24). Researchers conducting linkage studies now had several hundred markers to work with; by the end of the 1980s detailed linkage maps of evenly spaced markers throughout the 23 pairs of chromosomes were available.
Not surprisingly, the first major successes involved less common diseases with simple Mendelian modes of inheritance; the prime psychiatric example was Huntington’s disease, where the gene was located on chromosome 4 (25). Soon researchers were using RFLPs in attempts to track down genes contributing a major role in the risk of schizophrenia and bipolar disorder. Two high-profile studies were published in the journal Nature, each based on rather unusual populations, attractive for gene mappers. The first looked at a large extended family with many members affected by bipolar disorder or depression from a rural Old Order Amish community in Pennsylvania (26). There appeared to be strong evidence of linkage between affective disorder and markers on chromosome 11.
The following year a short report suggested involvement of chromosome 5 with schizophrenia in a family containing a maternal uncle and nephew pair both affected by the condition and showing an abnormality called translocation, where a piece of chromosome is broken off and attached to another. The nephew’s mother was unaffected, but was a ‘carrier’ of the translocation (27). Subsequently Sherrington et al. (28) explored the chromosome 5 region with DNA markers in Icelandic families (like the Amish, from a partial genetic isolate), as well as some British families multiply affected by schizophrenia. They found significant evidence of chromosome 5 markers assorting with the disorder. Others were unable to replicate a schizophrenia finding, but this still left the possibility of heterogeneity of schizophrenia at the molecular level such that some forms were linked and others unlinked to the markers on the chromosome 5 region (15). Consequently enthusiasm for linkage studies in psychiatry persists, and interest in the new genetics of mental disorders continues.
Evidence of increasing interest, even before the publication of the two Nature papers, was reflected in the establishment of the first World Congress of Psychiatric Genetics in 1988. It was organized by a committee under the chairmanship of Tim Crow, arguably the most prominent biological psychiatrist in the United Kingdom at that time and a recent convert to the idea that schizophrenia was largely a genetic disorder. I was pessimistic that the conference would attract more than a handful of delegates, given the scepticism and hostility to genetics I experienced during my years as a trainee psychiatrist. My doubts were dispelled when more than 300 researchers attended with well over 100 offers of presentations.
Around the same time another agreeable surprise, funding of a network to explore the molecular neurobiology of mental illness (MNMI) by the European Science Foundation (ESF), occurred. This arose from a proposal by Roger Marchbanks, a biochemist in the United Kingdom, and myself. Psychiatric departments and genetics laboratories across Europe joined forces to collect data and analyse the DNA from large numbers of multiply affected families with schizophrenia and bipolar disorder. Previously, linkage studies in psychiatry were too small to detect anything other than fully penetrant Mendelian genes. As previously mentioned, such genes were probably rare. The MNMI network was coordinated by a steering committee chaired by a leading French neurobiologist, Jacques Mallet, and included several rising figures in human genetics such as Leena Peltonen from Finland, Francois Clerget-Darpoux from France, and Kay Davies from the United Kingdom. Launched with a workshop that subsequently became an annual event, it can be viewed as a kick-start for molecular psychiatric genetics across Europe.
Soon after, the National Institute of Mental health (NIMH) funded a similar network in the United States. The ESF and NIMH began a collaboration, reaching agreement on basic issues such as a common diagnostic approach and an attempt to establish US–European diagnostic reliability (29).
Many linkage studies progressed in North America, Europe, Asia, and Australasia, made more feasible by a shift from RFLPs to a newer generation of DNA markers called microsatellites. They result from variation in the number of repeats of DNA bases (e.g. dinucleotides, or repeated runs of two bases) that occur throughout the genome. They are generally more polymorphic (or variable) than RFLPs and therefore provide more useful tags by which to track down genes within families. The discovery of microsatellites and methods of genotyping resulted in more useful, finer-grained linkage maps with which to search the genome.
The promise of ‘positional cloning’
The justification for all the effort that was being put into linkage studies, not just in psychiatry but across the whole of medicine, was that ‘positional cloning’ provided a method of discovering the aetiology of disorders where knowledge of the underlying pathogenesis was scant or absent. The broad principle is that linkage allows a method of pinpointing the genes involved in disorders to specific chromosomal locations. It is then possible to move from location to identifying the gene itself, studying the sequence and structure and eventually looking at the variation in the protein that the gene encodes to discover how a miscoding contributes to the disease. With much of the ‘new genetics’ positional cloning was often trickier than the optimists had anticipated, even with single-gene disorders, but it eventually worked. In 1993, 10 years after the first report of linkage on chromosome 4, a paper announcing the cloning of the Huntington’s disease gene was published (30). It immediately provided new insights into the biology of the disorder and a new mechanism of mutation that turned out to be particularly relevant for neuropsychiatric and neurological disorders. The Huntington’s gene contained a repeated sequence of three DNA bases, an unstable trinucleotide repeat. Once expanded beyond a critical length of 40 repeats it becomes pathogenic and causes the protein that the gene encodes to have an abnormal structure.
The other advances of psychiatric relevance were in studies of families containing multiple early-onset cases of Alzheimer’s disease (AD) where the disorder appeared to follow a Mendelian dominant pattern of inheritance. Three genes were identified, the amyloid precursor protein gene on chromosome 21 and two others that had similar structures to one another. They were presenilin 1 and presenilin 2. Carrying the relevant mutations in any one of these three genes was demonstrated to be enough to carry a virtual certainty of developing presenile dementia (31). However, early-onset forms of AD are very rare, whereas late-onset forms, particularly beyond the age of about 80 years, are extremely common. What then might be the relevance of genetics of the early rare forms to later common forms? The answer is probably very little (32). However, the discovery of another linkage signal on chromosome 19 (33) eventually led to discovery of an association between the apolipoprotein E gene (ApoE) and AD that proved to be an important risk factor for the common type of the disorder. Association is a related phenomenon to linkage but it is is observed in populations rather than in families. This occurs either because a marker itself has a direct effect on the risk of disease or because the marker is so extremely close to a disease susceptibility gene that the relationship between the marker and the gene remains undisturbed over many generations of recombination. In the case of ApoE the association with AD turned out to be with the alternative form of the gene, or allele, called E4.
Although it took much longer, the same general approach of searching for associations with genes within regions of the genome where there appeared to be a linkage signal resulted in the discovery of several novel genes associated with schizophrenia. Interestingly, all of them, which included neuregulin, dysbindin, and D amino acid oxygenase activator (DAOA), encoded proteins that are involved in the type of signalling in the brain that occurs via the chemical messenger glutamine (34).
Another gene involved in the liability to schizophrenia was discovered in a slightly different way. A large Scottish family with many members affected by schizophrenia, as well as other severe disorders, was discovered. Mental illness appeared to coincide with carrying a chromosome abnormality called a translocation in which there were breakages resulting in most of a chromosome 1 having a chromosome 11 fragment attached (35). The Edinburgh group studying this family went on to clone the breakpoint on chromosome 1 and identified a gene which they named ‘disrupted in schizophrenia-1’ (DISC1). At first many thought that this was a one-off family and that DISC1 was unlikely to be generally relevant to schizophrenia. However, it turned out that when association studies were carried out comparing schizophrenic patients with healthy controls, the former group more often had a particular set of variants within the gene. It was subsequently shown that DISC1 is not just relevant to schizophrenia but, as in the original Scottish family, variants in the gene are also associated with depression and bipolar disorder. Indeed, when researchers began to explore the new ‘schizophrenia genes’ in other conditions, associations with bipolar disorder again emerged, in line with the findings discussed earlier from the Maudsley twin study (21) that suggested a genetic overlap between the two conditions.
Gene–environment interplay and the nature of nurture
Until about a decade ago molecular genetic studies in psychiatry paid comparatively little heed to the environment. This was surprising, given earlier evidence from twin studies suggesting a sizeable environmental component in all common psychiatric disorders. On the other hand, the twin analyses make an assumption, likely an oversimplification, concerning gene–environment interplay; namely, that genes and environment simply add up to produce a clinical picture, the phenotype. However, behavioural genetic studies on animals have demonstrated that genes and their environment can interact in a multiplicative way, as genes may have an effect on sensitivity to the environment. A further complication is that genetic and environmental effects may be correlated. For example, parents usually provide their offspring with the environment in which they are reared as well as their genes, so that alcoholic parents may provide their offspring with early exposure to alcohol as well as a genetic predisposition to heavy drinking. Alternatively, some types of behaviour that have some genetic basis may influence the type of environment an individual selects. For example, a genetic predisposition to antisocial behaviour may elicit a response in others that encourages aggression or predisposes an individual to consort with peers who indulge in antisocial behaviours. Thus, once we take into account gene–environment interaction and covariation, aptly termed by my colleague Robert Plomin, ‘the nature of nurture’, ‘pure’ genetic or environmental effects are more difficult to distinguish than has traditionally been acknowledged.
Among the examples of studies using purely statistical ‘premolecular’ methods are those looking at adversity and depression in families and twins. A study during the 1980s revealed that not only did depression cluster in families, but so did adverse life events. Measuring life events in a relatively objective way, using a semi-structured interview, demonstrated that familial clustering was due to the same events affecting multiple members of a family. However, twin studies have shown that less objectively measured life events, collected by self-report questionnaires, are moderately heritable. Furthermore, the tendency to report life events and to report depressive symptoms appear to have overlapping genetic effects. In sum, the data, at least for subjective reports of adversity, suggest that the relationship with depression involves both gene–environment interactions and gene–environment correlations.
The first successful attempt to take this further by looking at a specific gene was published in 2003 in a landmark paper by Caspi et al. (36). They studied a cohort of nearly 1,000 individuals who had been followed from birth in Dunedin, New Zealand. When Caspi and colleagues looked at depressive symptoms in relation to objectively measured life events there was the expected clear relationship between the two. The novel aspect of the study was that the subjects were also genotyped for a common variation in the gene that encodes a protein called the serotonin transporter. This particular molecule is of great interest in neurobiology and neuropharmacology because it is the site of action of the most widely used type of antidepressants, the selective serotonin reuptake inhibitors (SSRIs). The variant that Caspi and colleagues studied was in a region, the promoter, that controls how active the gene is. The variant exists in two forms—a long (L) and a short (S) form—the L form resulting in higher gene activity than the S form. Subjects who had inherited the short form from both parents (SS) were significantly more reactive to life events than subjects who had inherited two copies of the long form (LL), while the so-called heterozygous individuals (LS) were intermediate in their response to life events.
One reason this paper is so often cited is because some researchers have been sceptical of the findings or failed to replicate them. However, if one reviews all the available data, the negative studies are entirely based on subjective self-report measures of life events which, as mentioned, are almost certainly ‘contaminated’ by genetic factors that overlap with those predisposing subjects to report depressive symptoms. There are also several lines of biological data suggesting that this serotonin transporter promoter variant has an effect on environmental reactivity (37). These include studies on rhesus macaque monkeys which have a similar variant and show a similar gene–environment interaction when exposed to the stress of separation from their mothers. In summary, we should not conclude that the serotonin transporter gene is ‘the gene for’ depression, but rather that it is one of many genes that contribute to the disorder. And, more specifically, the promoter variant is an example of a gene variant affecting sensitivity to the environment.
From candidate genes to genome-wide association
Studies of the serotonin transporter, and other genes in the same chemical pathway, are examples of candidate gene studies. Candidate genes are so called because they encode a substance thought to have relevance to a disease, for example, based on knowledge about the mode of action of the drug used to treat the disease. In candidate gene association studies researchers simply compare the frequency of variants in that gene in affected individuals versus a control group. Countless such studies were performed in depression, bipolar disorder, and schizophrenia during the 1990s, as RFLPs, microsatellites, and variants based upon a single base change (single nucleotide polymorphisms or SNPs) were discovered in potential candidate genes or their regulatory regions. Sadly, no consistent pattern emerged in any of these disorders until researchers honed in on ‘positional candidates’, such as neuregulin or dysbindin mentioned earlier, within regions identified by linkage studies of schizophrenia. Studies of gene–environment interactions also provided some consistency. However, what many genetic researchers really yearn to do is a genome–wide association study (GWAS) searching the entire genome of cases and comparing them with unaffected controls.
It has long been recognized that association studies have greater power than linkage to track down genes of small effect. They are therefore potentially ideal in complicated polygenic disorders. The downside of association studies is that in order for a genetic marker to tag a disease susceptibility gene it has to be within the gene itself or extremely close to it. Linkage can be used to track genes in families over distances as great as 10–15 cM and hence, if we remember that the genome is about 3500 cM long, only a few hundred markers are needed to span the genome. By contrast, association can only be detected over a distance of a small fraction of a centimorgan, so that hundreds of thousands of roughly evenly spaced markers are needed for a GWAS. The first big milestone on the path to being able to perform a GWAS was the completion of the sequencing of the human genome.
The nearly complete annotated human genome sequence was announced in 2000, with most of the gaps being filled in over the next 3 years. This was the culmination of the Human Genome Project, an international governmental project charitably complemented by the parallel project of a commercial enterprise called Celera that sought to patent the genome. In Janary 2001 Science and Nature devoted whole issues to the descriptions of the new and revolutionary findings, with accompanying articles discussing the likely uses and consequences. One of the uses was the subsequent delivery of a SNP map and the demonstration that SNPs are plentiful enough to conduct a GWAS. A subsequent milestone was a major project called HAPMAP which explored the way in which pairs of SNPs are inherited together on the same chromosomes in differing human populations. The final milestone was the development of a technology enabling many SNPs to be typed in a single experiment. This involves SNP micro-arrays (‘SNP chips’) on which thousands of probes that detect SNPs within DNA are placed on either a glass slide or an array of tiny beads. The first micro-arrays produced contained around 10,000 SNP probes and current commercially available versions are able to detect over 1 million SNPs.
There was an early win with the GWAS approach. A important new gene was discovered by comparing individuals with macular degeneration, a common eye disease, to healthy controls. The first full-scale GWAS was the Wellcome Trust Case Control Consortium (WTCCC) study published in Nature in 2007. Groups of 2,000 cases each of seven diseases, one of which was bipolar disorder, were compared to 3,000 controls. The large samples were needed for two reasons. First, the disorders were common complex ones likely to involve many genes of small effect. Secondly, the micro-arrays used in the study detected half a million SNPs, leading to a huge multiple testing problem. The multiple testing problem is as follows. Typically in a case-control comparison a ‘significant’ result is one observed by chance one time in 20 or less, a 0.05 or less probability of a false positive. A researcher conducting half a million simultaneous experiments would therefore find 25,000 appearing to be significant using the 0.05 criterion, even if no difference between cases and controls exists. Hence, a highly stringent criterion of significance is required in GWAS which conventionally is taken as a probability of 5 × 10–8 or less.
The WTCCC study was a methodological landmark and produced important new findings in some diseases, including Crohn’s disease and type I and type II diabetes. A parallel GWAS had been performed in the United States on bipolar disorder, and data were also emerging from studies in the United Kingdom and Ireland. One of the hazards of any association study is that different ethnic groups have different frequencies of genetic marker alleles and may have different frequencies of diseases. Therefore mixing together different populations can yield spurious results. Mindful of this problem, the American, British, and Irish studies focused only on white subjects of European origin. Nick Craddock from Cardiff University and Pamela Sklar from Harvard formed a consortium that combined the data on nearly 5,000 cases of bipolar disorder and an even larger set of controls (38). This study, stupendous in scale compared with almost anything in psychiatry preceding it, resulted in the discovery of two completely novel genes that had never previously been implicated in the aetiology of bipolar disorder, ANK3 and CACNA1C. Each is an important ion channel gene, associated with the structures that sit on the surface of nerve cells and are involved in rapid transmission of messages involving passage of ions (e.g. sodium or calcium) across the cell membrane. This was a particularly compelling finding in bipolar disorder given that many of the mood stabilizer medications used in long-term treatment were originally developed as anti-epileptic drugs and have the common property of stabilizing nerve cell membranes.
CACNA1C has subsequently been implicated in schizophrenia and unipolar depression and GWASs involving many thousands of subjects have identified new genes conferring susceptibility to schizophrenia, autism, AD, and bipolar disorder. Regrettably, although the sample sizes amassed for unipolar depression are even bigger than for other psychiatric diseases, no consistent ‘hits’ have emerged that can be replicated across different datasets. This perhaps suggests that depression, the commonest of the major psychiatric disorders, is actually more heterogenous than the rest, and collaborative efforts involving huge samples collected across the United States and Europe are currently taking place to attempt to discover more homogenous subgroups that might give more consistent signals. Meanwhile a recent positive and tantalizing finding emerging in unipolar depression is a possibility that a larger number of tiny pieces of genome go missing in people who developed depression than in those not depressed.
Copy number variants
Earlier on I described the excitement that occurred in the 1950s and 1960s when geneticists discovered that psychiatric syndromes were sometimes associated with having extra chromosomes, parts of chromosomes, or missing chromosomes that could be seen with the light microscope. Later, using more sophisticated techniques such as fluorescent in situ hybridization (FISH), smaller bits of missing chromosome could be detected. Half a century later much excitement has arisen with the discovery, largely as a by-product of GWAS technology, that much smaller submicroscopic stretches of DNA are detected as missing (deleted) or having more than one copy (duplicated). This type of copy number variation (CNV) is responsible for a large amount of the differences between individuals’ genomes. Large CNVs, consisting of tens of thousands or even a few million DNA bases, are individually rare with frequencies in the general population of less than 100. However, many of them are dotted across the genome, so that the probability of anyone carrying at least one large CNV is high. A study in our laboratory estimated that about one in three healthy controls has at least one large deletion CNV, suggesting that the majority of large, rare CNVs are ‘silent’. However, it does appear that the CNV ‘burden’, defined as the number of large CNVs or the number of CNVs that disrupt genes, is increased in a variety of psychiatric disorders, particularly schizophrenia and autism. Indeed, some rare CNVs have repeatedly been found in both disorders and confer a much higher risk (although never a certainty) of the disorder than the SNP variants in genes that have been uncovered by association studies. Thus it appears that the early advocates of major gene theories of schizophrenia were not entirely wrong and it is likely that the both schizophrenia and autism are contributed to both by common genetic variants of small effect and rarer variants of large effect, and that there is some genetic overlap between the two conditions.
The story is just beginning to unfold with depression. In a sample of around 3,000 cases of mainly recurrent and severe depression, our research suggests an excess of CNVs consisting almost entirely of deletions that disrupt coding regions of genes (exons). Interestingly, we see the largest difference between cases and controls who have been screened for never having had any psychiatric disturbance in their lifetime. Unscreened controls (who are ostensibly well but may have had psychiatric symptoms in the past) also have significantly fewer large CNVs than cases, but more than the ‘super healthy’ screened controls. This might suggest that having a lower CNV burden has something to do with a genetic basis for well-being or resilience.
Pharmacogenetics and pharmacogenomics: personalized pharmacotherapy?
One hope of the GWAS approach was that it would lead to the discovery of genes useful in predicting response to treatment and development of side effects. The general idea is a much older one and goes back more than half a century, before genome-wide studies were possible. Speculations began in the 1950s that rare and idiosyncratic adverse reactions to certain drugs had a genetic basis. The first discovery relevant to psychiatric patients was that one in 3,000 individuals receiving electroconvulsive therapy (ECT) were ‘slow acetylaters’. They were slow to metabolize drugs like the muscle relaxant suxamethonium, and hence had a prolonged, potentially hazardous recovery from general anaesthesia. The medical geneticist Arno Motulsky subsequently suggested that a new branch of genetics dealing with drug response should have a bright future and coined the term pharmacogenetics. Since then many genetic variants involved in the breakdown of medications have been discovered, the biggest family involving the cytochrome P450 enzyme system in the liver. These show a large degree of variation, the extent varying between ethnic groups, which is thought to have evolved because of advantages possibly conferred by resistance to naturally occurring toxins. Many of the drugs used in psychiatry are broken down via cytochrome P-450 enzymes, such as the one encoded by a gene called CYP2D6, that shows a high degree of variation. Slow CYP2D6 metabolizers are much commoner that slow acetylaters, which may have relevance for the occurrence of side effects in people taking certain antidepressants.
Pharmacokinetics refers to the rate at which drugs are metabolized. Another source of potential genetic variability has to do with pharmacodynamics. This involves genes that produce variation in the site at which drugs act, such as the promoter variation in the serotonin transporter gene in relation to stress response. A number of studies show that patients who carried the double dose of the less active SS form responded less well to selective serotonin reuptake inhibitor antidepressants than patients who had at least one (more active) L form of this genetic variant.
Several other tantalizing findings have emerged from taking a candidate gene approach to pharmacodynamics; for example, some genetic variants seem to predict emergence or worsening of suicidal ideation during antidepressant treatment. Such findings require replication and more research before conclusions about clinical benefit can be drawn. Potentially more readily translatable results from elsewhere involve rare, more idiosyncratic types of effects. For example, carbamazepine—a drug used to treat epilepsy and sometimes as a mood stabilizer in bipolar disorder—causes a rash, joint pains, fever, and possibly death in a small proportion of patients. This reaction, the Stevens–Johnson syndrome, is associated in East Asians with a variant in the HLA genetic complex. The genetic finding is currently being explored as a possible basis of routine clinical genetic testing.
There have now been three genome-wide studies looking at response to antidepressants. Each produced a positive finding, but unfortunately no agreement exists across studies. This suggests that, contrary to expectations, no genes on their own will provide a clinically useful test. However, calculations based on genome-wide data allow an estimate of the heritability of antidepressant response, probably in the region of 30–40%, suggesting that a combination of genes might provide the basis of clinically useful tests. In summary, many areas in psychiatric genetics are turning out to be more complex than originally hoped or foreseen and, consequently, it is still too early to know whether personalized pharmacotherapy informed by genetic tests will become a reality.
The future and postgenomic psychiatry
The history of genetics over the past 50 years has been full of surprises accompanying the discoveries about the molecular basis of inheritance and the structure of the genome. Therefore I should be circumspect and conservative in rounding off of this essay by sticking to predictions concerning current research.
Beginning with pharmacogenetics and genomics, analysis that has occurred thus far has been fairly straightforward (e.g. looking across the genome for signals that predict therapeutic response or common side effects). In the near future I foresee researchers turning to integrative analysis that takes into account the complexity of what goes on when a disorder, such as depression, is treated. This would involve monitoring the blood level of drugs, as well as looking at what other markers reveal. This concerns not only whether certain gene variants are present or absent, but also the level of activity of genes. Such studies comprise transcriptomics (studies of the level of messages contained in RNA which convey information from the nucleus to the cell) and proteomics (studies of the levels of all proteins that can be detected in the blood). They would also consider the role of epigenetic mechanisms.
Epigenetics is likely to prove the next ‘big thing’ in pharmacogenetics, as well as across psychiatric genetics generally. An explanation of epigenetics is complicated, although the concept is relatively straightforward. My colleague Jonathan Mill has produced an excellent metaphor. When we go to hear an orchestra we see the conductor reading from a score, but more often than not the conductor’s working score will not only contain what was written by the composer, but also marks the conductor has made to remind him of how he thinks the music should be interpreted. What we hear corresponds to the phenotype, and what the composer has written is the genotype—the conductor’s marks correspond to the epigenotype. Epigenetics, then, is concerned with the marks, some of them reversible, that are placed on the genome by events that can include where a gene comes from (father or mother), exposure to early adversity, or exposure to a certain diet or drug. Broadly speaking, the marks involve the chemical processes of methylation and acetylation, and modification of histones, structures within chromosomes that enable DNA to be tightly coiled and packed into the nucleus of the cell. Methylation has the effect of switching off the message from a gene, as does tight coiling of DNA around histones. Removing methylation or uncoiling histones has switching-on effects.
To date, the study of epigenetics in psychiatry has just begun, but already there are strong hints that it reveal much—for example, why genetically identical, monozygotic twins differ in many respects such as being discordant for illnesses like schizophrenia or depression. And this is another area where technology is moving rapidly. For example, an ingenious adaptation of the bead array technology mentioned earlier in connection with GWAS now enables the methylome (i.e. methylation patterns across the entire genome) to be scanned in a single experiment, so we can expect results soon to come thick and fast.
Another set of methods that will soon advance our knowledge is referred to as next-generation sequencing. This enables very rapid reads of the genome sequence at comparatively small cost. Whereas the original sequencing of the complete human genome took more than a decade and cost billions of dollars, it is now possible to sequence anyone’s genome for a few thousand dollars in about 3 weeks. It is already feasible to sequence all the coding regions within the genome that are expressed as proteins (the exome), and soon results of a study of 10,000 exomes, comparing cases and controls that include 3,000 patients with either schizophrenia or autism, will be available.
Perhaps large-scale sequencing studies should provide answers to several conundrums that are relevant to human genetics generally, not just to psychiatric disorders. For example, the exome is only a small fraction of the genome, carrying codes for proteins. This discovery that the coding region of the genome was so small, together with the finding of the Human Genome Project that there are only about 25,000 human genes, surprised many. It led to questions as to whether most of the genome was ‘junk’, and if so why? It soon became clear that the junk or non-coding DNA within genes in the so-called introns, does have a function in determining how different variants of the same proteins are made up in different tissues (called splice variants), and affecting stretches of DNA that are important in controlling levels of gene expression. In between genes there are elements of DNA that have been conserved through evolution, regions likely to have some function that may affect expression of genes at a distance. One particular type of non-protein-coding DNA is now known to code for short forms of RNA responsible for a phenomenon called RNA interference, a mechanism by which the signals sent by genes can be controlled.
In conclusion, many mysteries concerning the human genome remain, as do many clues concerning the role of non-coding DNA in controlling those parts of the genome that code for proteins. Such clues will need to be pursued, as will the part played by epigenetic phenomena in psychiatric disorder. Progress up to and since the sequencing of the human genome has generated huge amounts of data and information so that computational biology and the whole new science of bioinformatics is assuming an ever-increasing importance. Meanwhile, one of the earliest technological innovations of human genetics, the twin study, will continue to have a key place in psychiatric genetics. This will include continuing refinement of disease boundaries and overlaps, as well as use of concordant and discordant twins to explore new territories such as epigenetics.
References
1 McGuffin, P. and Huckle, P. (1990). Simulation of Mendelism revisited: the recessive gene for attending medical school. American Journal of Human Genetics 46, 994–9.
2 Galton, F. (1876). The history of twins, as a criterion of the relative powers of nature and nurture. Journal of the Anthropological Institute 5, 391–406.
3 Jackson, D. (1960). The etiology of schizophrenia. New York: Basic Books.
4 Gottesman, I. and Shields, J. (1972). Schizophrenia and genetics: a twin study vantage point. New York: Academic Press.
5 Heston, L. (1966). Psychiatric disorders in foster home reared children of schizophrenic mothers. British Journal of Psychiatry 112, 819–25.
6 Lejeune, J., Gautier, M., and Turpin, R. (1959). Etude des chromosomes somatiques de neuf enfants mongoliens. Comptes Rendus Hebdomadaires des Séances de l’Académie des Sciences 248, 1721–2.
7 Skuse, D. H., James, R. S., Bishop, D. V. et al. (1997). Evidence from Turner’s syndrome of an imprinted X-linked locus affecting cognitive function. Nature 387, 705–8.
8 Jacobs, P. A., Brunton, M., Melville, M. M., Brittain, R. P., and McClemont, W. F. (1965). Aggressive behavior, mental sub-normality and the XYY male. Nature 208, 1351–2.
9 Witkin, H. A., Mednick, S. A., Schulsinger, F. et al. (1976). Criminality in XYY and XXY men. Science 193, 547–55.
10 Slater, E. (1958). The monogenic theory of schizophrenia. Acta Genetica et Statistica Medica 8, 50–6.
11 Gottesman, I. and Shields, J. (1967). A polygenic theory of schizophrenia. Proceedings of the National Academy of Sciences of the U S A 58, 199–205.
12 Falconer, D. (1965). The inheritance of liability to certain diseases, estimated from the incidence among relatives. Annals of Human Genetics 29, 51–76.
13 O’Rourke, D. H., Gottesman, II, Suarez, B. K., Rice, J., and Reich, T. (1982). Refutation of the general single-locus model for the etiology of schizophrenia. American Journal of Human Genetics 34, 630–49.
14 Craddock, N. (1995). Genetic linkage and association studies of bipolar disorder. PhD thesis, University of Wales College of Medicine, Cardiff.
15 McGuffin, P. (1991). Models of heritability and genetic transmission, in: Häfner, H. and Gattaz, W. (eds.) Search for the causes of schizophrenia, pp. 111–25. Berlin: Springer-Verlag.
16 Fisher, R. (1918). The correlation between relatives on the supposition of mendelian inheritance. Philosophical Transactions of the Royal Society of Edinburgh 52, 399–433.
17 Shields, J. (1962). Monozygotic twins, brought up apart and brought up together. London: Oxford University Press.
18 McClearn, G. E., Johansson, B., Berg, S. et al. (1997). Substantial genetic influence on cognitive abilities in twins 80 or more years old. Science 276, 1560–3.
19 Kendler, K. S., Neale, M. C., Kessler, R. C., Heath, A. C., and Eaves, L. J. (1992). Major depression and generalized anxiety disorder. Same genes, (partly) different environments? Archives of General Psychiatry 49, 716–22.
20 Thapar, A. and McGuffin, P. (1997). Anxiety and depressive symptoms in childhood—a genetic study of comorbidity. Journal of Child Psychology and Psychiatry 38, 651–6.
21 Cardno, A. G., Rijsdijk, F. V., Sham, P. C., Murray, R. M., and McGuffin, P. (2002). A twin study of genetic relationships between psychotic symptoms. American Journal of Psychiatry 159, 539–45.
22 Penrose, L. (1952). The general purpose sib-pair linkage test. Annals of Human Genetics 17, 120–4.
23 McGuffin, P., Festenstein, H., and Murray, R. (1983). A family study of HLA antigens and other genetic markers in schizophrenia. Psychological Medicine 13, 31–43.
24 Comings, D. E. (1980). Prenatal diagnosis and the ‘new genetics’. American Journal of Human Genetics 32, 453–4.
25 Gusella, J. F., Wexler, N. S., Conneally, P. M. et al. (1983). A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 306, 234–8.
26 Egeland, J. A., Gerhard, D. S., Pauls, D. L. et al. (1987). Bipolar affective disorders linked to DNA markers on chromosome 11. Nature 325, 783–7.
27 Bassett, A. S., McGillivray, B. C., Jones, B. D., and Pantzar, J. T. (1988). Partial trisomy chromosome 5 cosegregating with schizophrenia. Lancet 1, 799–801.
28 Sherrington, R., Brynjolfsson, J., Petursson, H. et al. (1988). Localization of a susceptibility locus for schizophrenia on chromosome 5. Nature 336, 164–7.
29 Williams, J., Farmer, A. E., Ackenheil, M., Kaufmann, C. A., and McGuffin, P. (1996). A multicentre inter-rater reliability study using the OPCRIT computerized diagnostic system. Psychological Medicine 26, 775–83.
30 The Huntington’s Disease Collaborative Research Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–83.
31 Liddell, M. B., Lovestone, S., and Owen, M. J. (2001). Genetic risk of Alzheimer’s disease: advising relatives. British Journal of Psychiatry 178, 7–11.
32 Gerrish, A., Russo, G., Richards, A. et al. (2011). The role of variation at AbetaPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer’s disease. Journal of Alzheimers Disease 28, 377–87.
33 Pericak-Vance, M. A., Bebout, J. L., Gaskell, P. C., Jr et al. (1991). Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. American Journal of Human Genetics 48, 1034–50.
34 Harrison, P. J. and Owen, M. J. (2003). Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet 361, 417–19.
35 St Clair, D., Blackwood, D., Muir, W. et al. (1990). Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16.
36 Caspi, A., Sugden, K., Moffitt, T. et al. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–9.
37 McGuffin, P., Alsabban, S., and Uher, R. (2011). The truth about genetic variation in the serotonin transporter gene and response to stress and medication. British Journal of Psychiatry 198, 424–7.
38 Ferreira, M. A., O’Donovan, M. C., Meng, Y. A. et al. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nature Genetics 40, 1056–8.