General Properties of Immune Receptor-Ligand Interactions
Cells of the immune system must constantly recognize and respond to a myriad of molecular signals from the external environment. Immune system cells use several different types of receptors to recognize ligands. Pattern recognition receptors (PRRs) are found on innate immune cells (e.g., macrophages, dendritic cells, neutrophils, and others), as well as on adaptive immune cells (T and B lymphocytes) and have also been defined on cell types that are not part of the formal immune system, such as neuronal cells, epithelial cells, and others. In contrast, the antigen-specific B-cell receptors (BCRs) and T-cell receptors (TCRs) are found exclusively on the B and T lymphocytes of the adaptive immune system. All of these cells and many others can express cytokines, chemokines, and their receptors in order to communicate with one another as they respond to antigen encounters. Since B and T cells express both adaptive and innate immune receptor molecules, as well as many cytokine and chemokine receptors, they must therefore constantly interpret multiple simultaneous or sequential signals. It is the integration of these many receptor-ligand interactions at the cellular level that will ultimately determine the biological outcome of any immune stimulus.
The interactions of ligands with immune system receptor molecules can be extremely complex. Most students are familiar with the concept of enzyme-substrate binding interactions. Such interactions are usually monovalent (one enzyme molecule binds one molecule of substrate) and of moderate affinity, and only a small number of enzymes will have the capacity to interact with any one substrate. In contrast, interactions between antigens and their receptor molecules may be of extremely high affinity and the diversity of receptors capable of recognizing any given antigen is simply breathtaking. In addition, the affinity of cytokine receptors for their cognate cytokine as well as the level of expression and the placement of receptors on the cell membrane may vary according to the activation status of the cell on which it is expressed (see the chapter opening photograph).
Receptor-Ligand Binding Occurs via Multiple Noncovalent Bonds
A receptor molecule attaches to its ligand by the same types of noncovalent chemical linkages that enzymes use to bind to their substrates. These include hydrogen and ionic bonds, and hydrophobic and van der Waals interactions. The key to a meaningful receptor-ligand interaction is that the sum total of the bonding interactions holds the two interacting surfaces together with sufficient binding energy, and for sufficient time, to allow a molecular signal to be received by the cell signifying that the receptor has bound its matching ligand. Because these noncovalent interactions are individually weak, many such interactions are required to form a biologically relevant receptor-ligand connection. Furthermore, since each of these noncovalent interactions operates only over a very short distance—generally about 1 angstrom (1 Å = 10-10 meters)—a high-affinity receptor-ligand interaction depends on a very close “fit,” or degree of complementarity, between the receptor and the ligand (Figure 3-1).
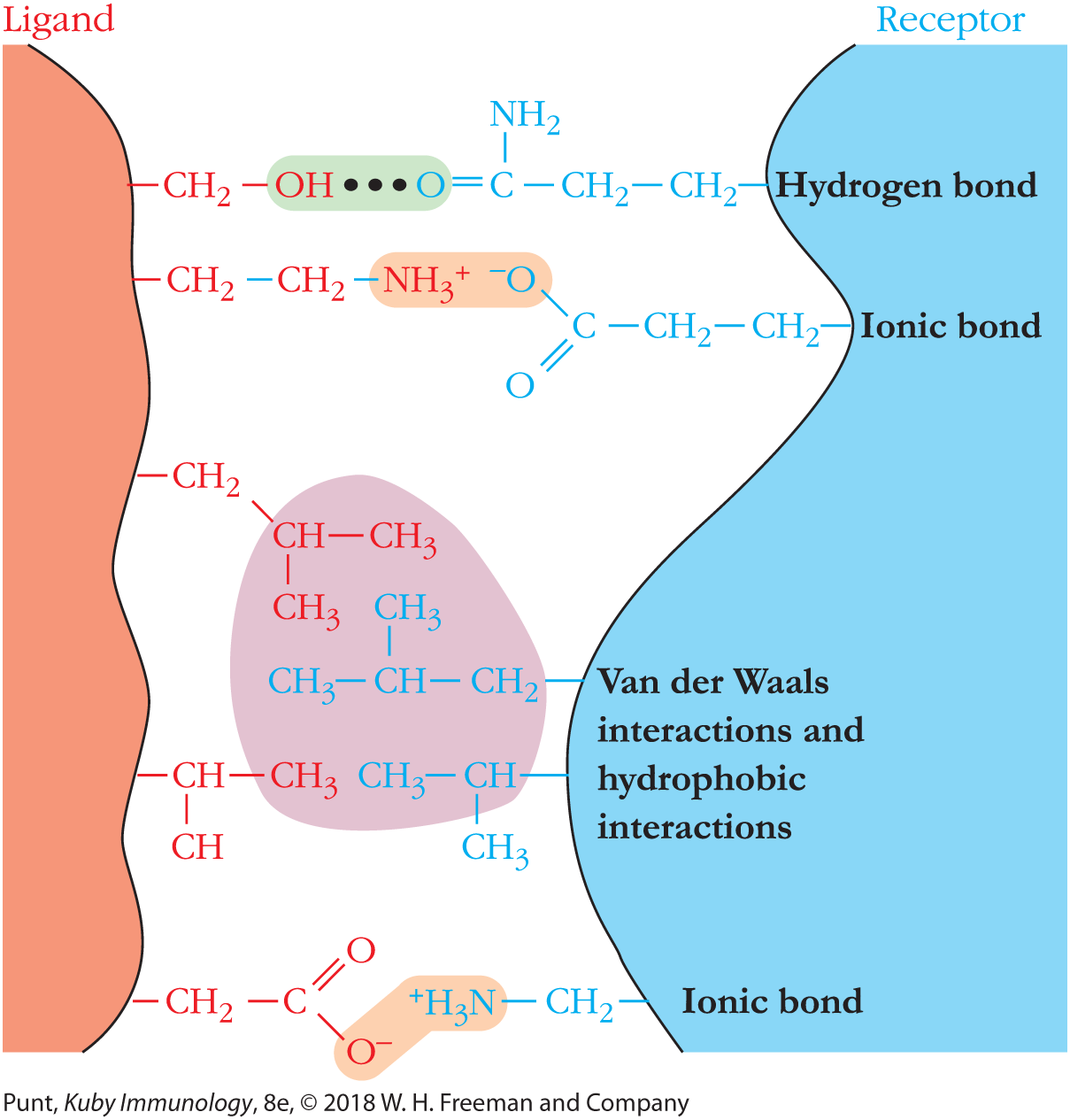
FIGURE 3-1 Receptor-ligand binding obeys the rules of chemistry. Receptors bind to ligands using the full range of noncovalent bonding interactions, including ionic and hydrogen bonds and van der Waals and hydrophobic interactions. For signaling to occur, the bonds must be of sufficient strength to hold the ligand and receptor in close proximity long enough for downstream events to be initiated. In B- and T-cell signaling, activating interactions also require receptor clustering. In an aqueous environment, noncovalent interactions are weak and depend on close complementarity of the shapes of receptor and ligand.
How Do We Describe the Strength of Receptor-Ligand Interactions?
We can describe the strength of the binding interaction between a receptor binding site, S, and a ligand, L, with the following expression:

where Kd, the dissociation constant of the reaction, defines the relationship between the concentration of the receptor-ligand pair, [SL] and the product of the concentration of free receptor sites, [S] and the concentration of free ligand, [L]. The units of the dissociation constant are in molarity (M). The lower the Kd, the higher the affinity of the interaction. Note that when 50% of the binding sites are occupied, [SL] = [S] and the Kd = the concentration of free ligand.
For purposes of comparison, it is useful to consider that the Kd values of many enzyme-substrate interactions lie in the range of 10–3 to 10–5 M, which are analogous to the Kd values of rather low-affinity antigen-antibody interactions at the beginning of an immune response. However, since the antibody genes that are expressed on initial antigen stimulation are mutated and selected over the course of an immune response, antigen-antibody interactions late in an immune response may achieve a Kd as low as 10–12 M. This is an extraordinarily strong interaction; even if the antigen concentration is as low as 10–12 M, fully half of the antigen molecules will be bound to the receptor. Recent work with innate Toll-like receptors places the Kd of interaction of these receptors with their respective ligands in the range of 10–7 to 10–8 M.
The affinity of receptor-ligand interactions can be measured by equilibrium dialysis or surface plasmon resonance (SPR). Both of these methods are described in Chapter 20.
Interactions between Receptors and Ligands Can Be Multivalent
Many biological receptors, including B-cell receptors, have more than one ligand-binding site per molecule and are therefore characterized as multivalent. When both receptors and ligands are multivalent—as occurs when a bivalent B-cell receptor binds to two, identical ligands on a bacterial surface—the overall binding interaction is markedly stronger than that between identical, but univalent receptors and ligands. (Note, however, that the strength of binding through two identical receptor sites on one molecule to two identical ligands on the same cell may be somewhat less than twice the strength of binding through a single receptor site. This is because the bivalent binding may somewhat strain the geometry of the receptor or ligand and therefore slightly interfere with the “fit” of the individual interactions.)
Much of the benefit of multivalency results from the fact that noncovalent binding interactions are inherently reversible; the ligand spends some of its time binding to the receptor, and some of its time in an unbound, or “off” state. When more than one binding site is involved, it is less likely that all of the receptor sites will simultaneously be in the “off” state, and therefore that the receptor will release the ligand. Compare the univalent interaction in Figure 3-2a with the bivalent interaction in Figure 3-2b. The term avidity is used to describe the overall strength of the collective binding interactions that occur during multivalent binding.
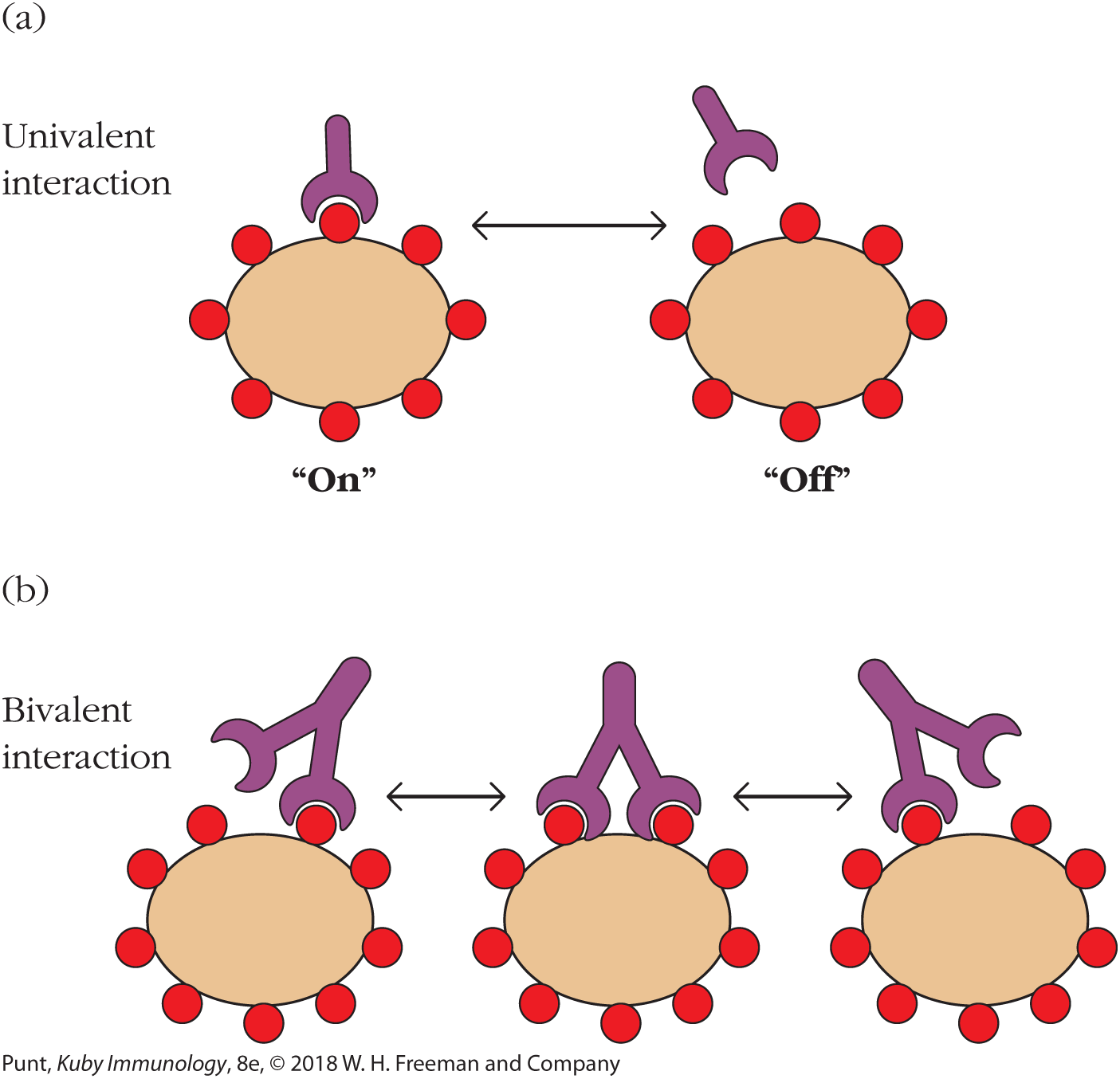
FIGURE 3-2 Univalent and bivalent (or multivalent) binding. (a) A univalent receptor approaches a multivalent antigen. The receptor exists in equilibrium with its ligand, represented here as a red circle. Part of the time it is bound (binding is in the “on” state), and part of the time it is unbound (binding is in the “off” state). The ratio of the time spent in the “on” versus the “off” state determines the affinity of the receptor-ligand interaction and is related to the strength of the sum of the noncovalent binding interactions between the receptor and the ligand. (b) Bivalent or multivalent binding helps to ensure that when one site momentarily releases the ligand, the interaction between the two molecules (or indeed, two cells) is not lost, as it is with monovalent binding.
Because of the fluid nature of the cell membrane, most membrane-bound antigen receptors function in a multivalent fashion, even if individual receptor molecules are monovalent in nature. Figure 3-3 illustrates how a multivalent antigen, interacting with a membrane-bound receptor in a reversible way, incrementally stabilizes increasingly large receptor clusters on the cell membrane. This is because, as one or two stable connections are made, other receptors diffusing randomly in the fluid environment of the cell membrane become caught up into the receptor cluster, which grows accordingly. As we will learn shortly, these receptor clusters facilitate intramolecular interactions on the cytoplasmic side of the cluster that lead to passage of cell activation signals through to the nucleus.
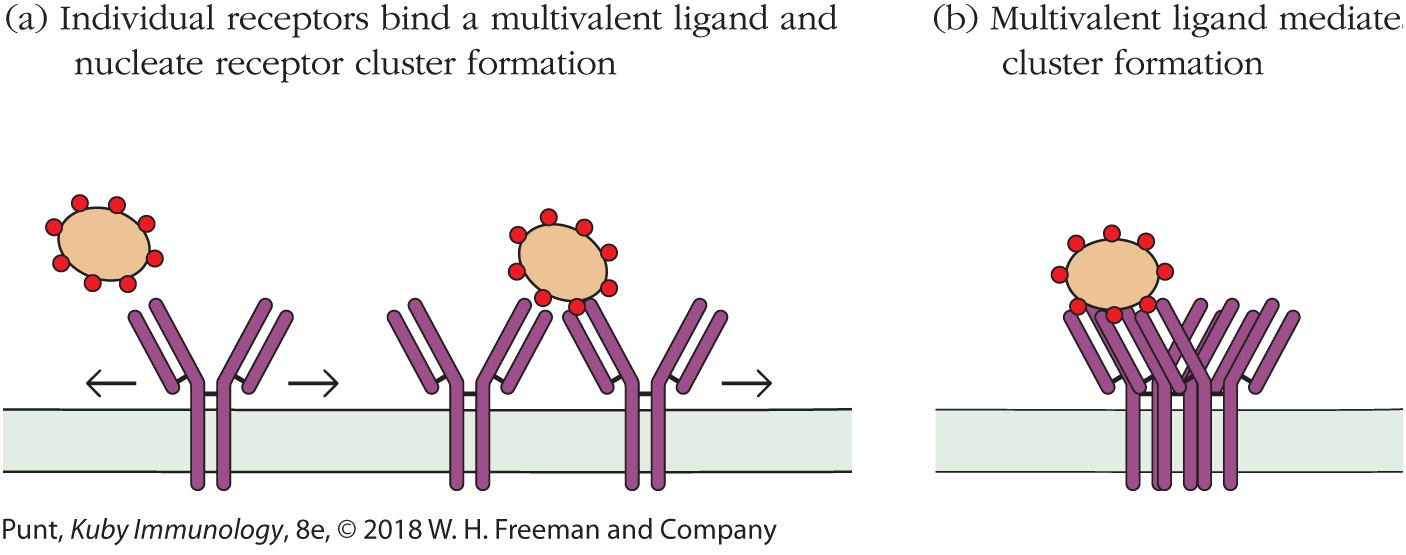
FIGURE 3-3 Cell surface receptors cluster on binding multivalent antigens. (a) Receptors on a cell surface encounter ligands in a multivalent array, for example, on the surface of a bacterial cell. (b) The receptors migrate in the plane of the membrane to form clusters of occupied receptors.
Combinatorial Expression of Protein Chains Can Increase Ligand-Binding Diversity
Immune receptors are required to bind to an incredibly diverse range of antigens and signaling molecules, and the immune system has evolved an impressive number of strategies to do so within the constraints of a finite number of receptor coding sequences in the organism’s DNA. One of those strategies involves re-using a single protein chain in combination with multiple partners to create a variety of different binding sites.
Some cytokine receptors are made up of two protein chains and, in most cases, both of these chains contribute to the ligand-binding site. For example, three different class 1 cytokine receptor alpha (α) chains bind the same beta (β) chain to form receptors for interleukins 3 and 5 (IL-3 and IL-5) and granulocyte-macrophage colony-stimulating factor (GM-CSF), respectively (see Figure 3-4). Since the three-dimensional shape of the ligand-binding site depends on the manner in which the two chains interact with one another, this means that the β chain can bind to multiple cytokines, depending on the α chain with which it is paired. The IL-17 cytokine receptor family uses a similar strategy to bind multiple ligands, using a limited number of gene products.
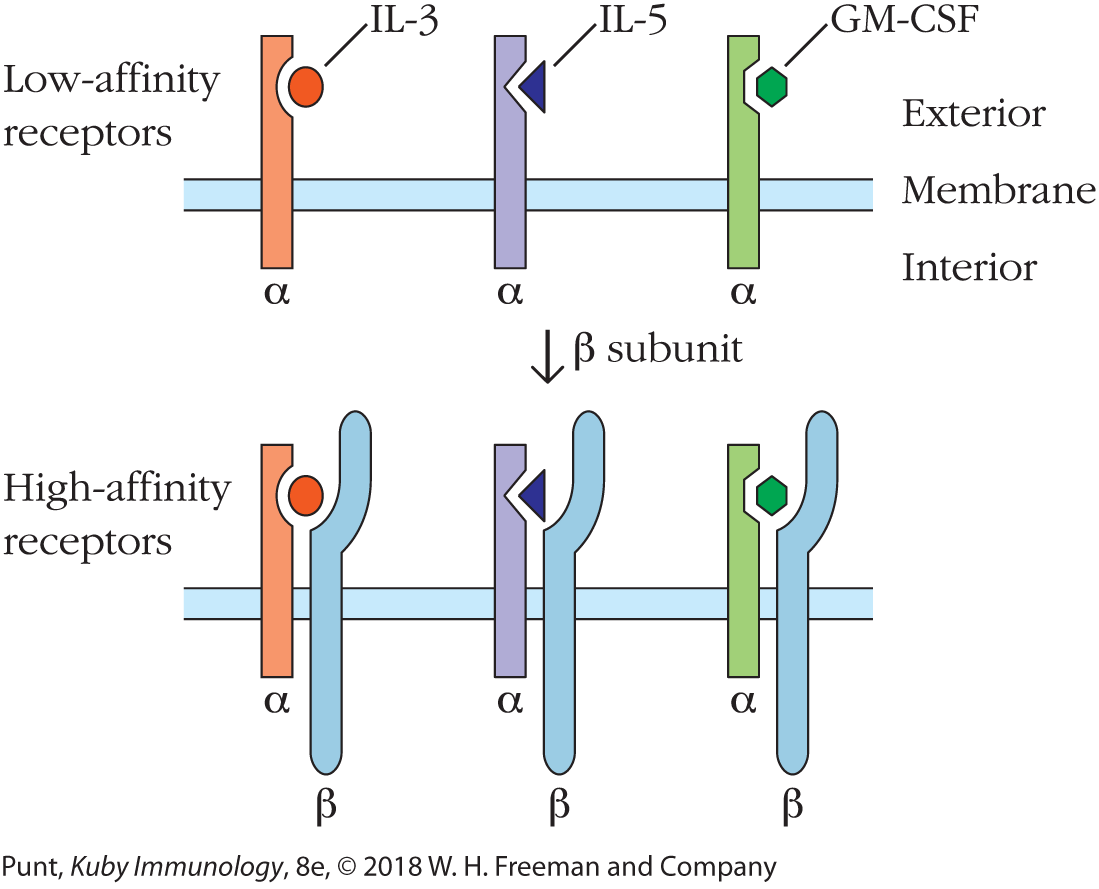
FIGURE 3-4 Combining one receptor chain with different partners allows increased receptor diversity and affinity while minimizing the need for new genetic information. A schematic diagram of the low-affinity and high-affinity receptors for the class 1 cytokines, interleukins 3 and 5 (IL-3 and IL-5), and granulocyte-macrophage colony-stimulating factor (GM-CSF) is shown. The cytokine receptor α subunits exhibit low-affinity binding and cannot transduce an activation signal from the cytokine to the interior of the cell. Noncovalent association of each subunit with a common β subunit yields a high-affinity dimeric receptor that can transduce a signal across the membrane on cytokine binding.
An analogous strategy is adopted by the B- and T-cell receptors (BCRs and TCRs) of the adaptive immune response as well as the Toll-like receptors (TLRs) of the innate immune response when binding antigens. Members of all three of these receptor families are made up of two protein chains per receptor molecule. In the case of the BCR, the two chains are called the heavy (H) chain and the light (L) chain. Both chains contact the antigen, and therefore contribute to antigen specificity. A similar diversity of antigen-combining sites can be generated by pairs of TCR chains or of TLR monomers.
Adaptive Immune Receptor Genes Undergo Rearrangement in Individual Lymphocytes
So far, our discussions have not addressed the striking differences in the extents of diversity among innate versus adaptive immune receptor systems. We will describe these two types of receptor systems in detail in Chapters 4 and 6, respectively. However, we should note here that whereas the number of innate immune receptors of all types totals around 100 or less, the numbers of each of the two classes of adaptive immune receptors, BCRs and TCRs, are measured in units of billions.
The vast diversity of antigen-binding sites expressed by adaptive immune receptors is enabled by the unique ways in which T- and B-cell receptors are encoded in the genome. The DNA sequences that specify the antigen-binding sites in BCR and TCR protein chains are coded in short fragments in the germline DNA. These fragments are then stitched together in random combinations in each different B or T cell. A useful metaphor for thinking about the creation of the gene that encodes the entire antigen-binding site is that of creating a whole meal by selecting one item from each section of a menu and linking them together.
Adaptive immune receptor diversity is therefore generated by the random combinations of protein chains (heavy and light chains), and the random combinations of gene segments encoding each chain. These DNA segments are stitched together by DNA recombination in different combinations in each cell to create unique coding sequences for each heavy or light chain. Additional diversity at the junctions is generated during the DNA recombination process. A very similar process generates mature TCR and BCR genes and this mechanism is fully described in Chapter 6. For now, the point to grasp is that the extraordinary degree of the receptor diversity in the adaptive immune system is generated in part by recombination events at the DNA level (note—this is not RNA splicing!), the details of which are different in every adaptive immune cell!
Levels of Receptor and Ligand Expression Can Vary during an Immune Response
One of the most striking features of the molecular logic of immune responses is that the level of cell surface expression of many immune receptors is coupled to the activation state of the cell. One very clear example of this phenomenon is the receptor for the cytokine interleukin 2 (IL-2), which was one of the earliest cytokines to be discovered and which provides a critical signal to lymphocytes to initiate proliferation and differentiation.
Most resting (i.e., non-antigen-activated) lymphocytes express a heterodimeric (two-chain), intermediate-affinity form of the IL-2 receptor, IL-2Rβγ. The affinity of the IL-2Rβγ form of the receptor is too low to enable it to bind IL-2 at physiological cytokine concentrations. However, once a lymphocyte has been activated by binding antigen through its BCR or TCR, the signal from the antigen-binding receptor causes an increase in the cell surface expression of a third chain of the IL-2 receptor, IL-2Rα, which then combines with the other two receptor chains on the cell surface (Figure 3-5 and chapter opening photo).
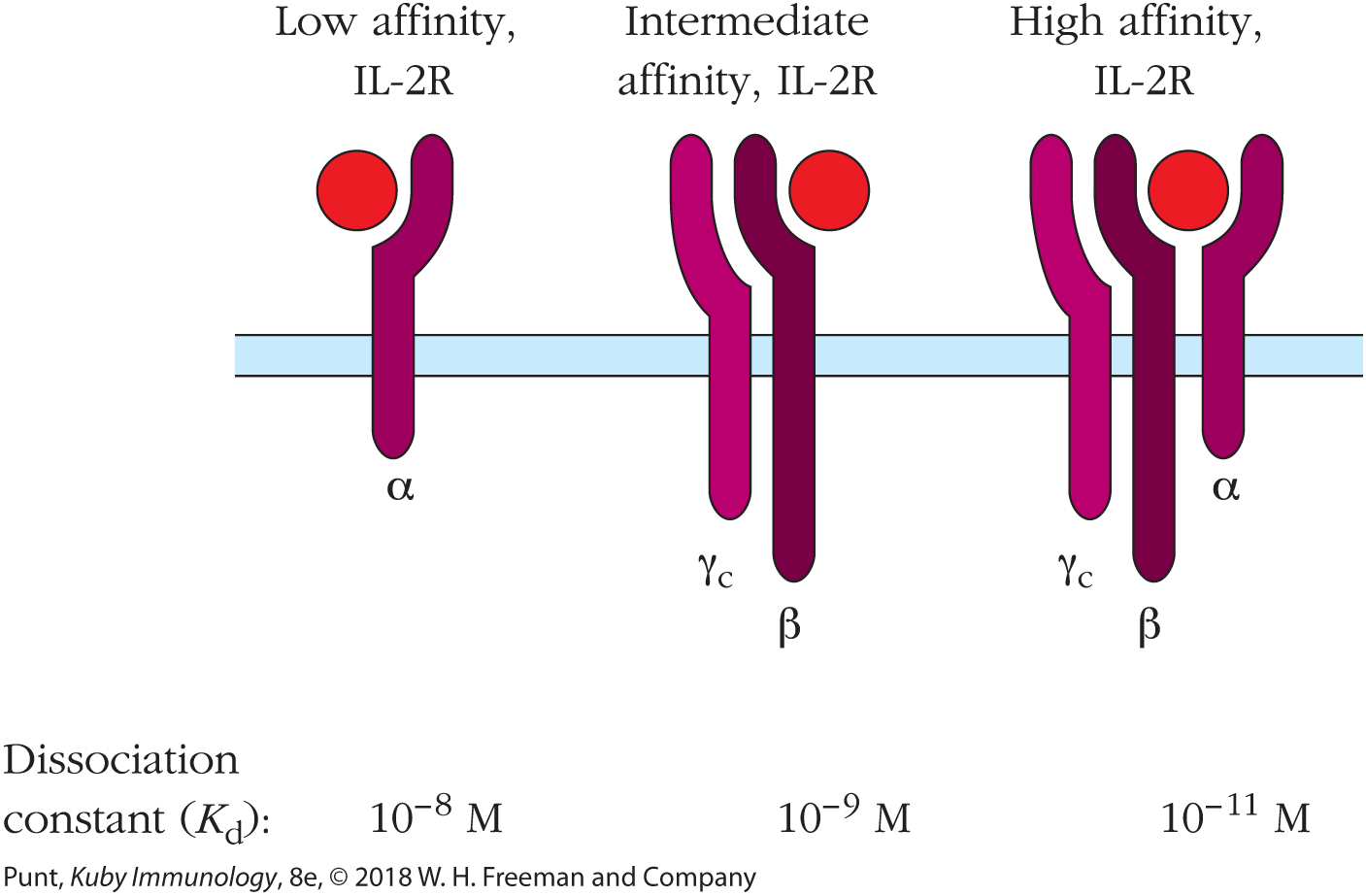
FIGURE 3-5 Comparison of the three forms of the IL-2 receptor. Signal transduction is mediated by the β and γc chains, but all three chains are required for high-affinity binding of IL-2. Dissociation constants reflect increased affinity of the three-chain IL-2R for IL-2.
Addition of this third chain converts the intermediate-affinity form of the IL-2 receptor to a high-affinity form, capable of responding to the cytokine levels found in the lymphoid organs. The functional corollary of this is that only those lymphocytes that have already been activated by antigen binding have cytokine receptors of sufficiently high affinity to respond to physiological IL-2 concentrations. In this way, the immune system both conserves energy and prevents the accidental initiation of an immune response to an irrelevant antigen.
Expression of some antigen receptors is also altered on cell activation. We will learn that B cells express two types of immunoglobulin antigen receptors on their cell membranes, IgM and IgD, that differ in the amino acid sequences of their non–antigen-binding regions. On cell activation, the expression of IgD drops significantly, whereas that of IgM remains constant. Some innate immune receptors also increase their expression patterns on cell activation.
Local Concentrations of Ligands May Be Extremely High during Cell-Cell Interactions
When considering the strength of the interactions between receptors and their ligands, it is important to consider the anatomical environment in which these interactions occur. This consideration becomes particularly important in the context of the adaptive immune system, where cells alter their locations and their binding partners multiple times during the induction and expression of an immune response. In particular, during the activation of a helper T (TH) cell, the TH cell and an antigen-presenting dendritic cell may remain in a complex with one another for 12 hours or more (see Figure 14-17). During this time, the two cells exchange cytokine signals.
Binding of the T cell to the dendritic cell induces redistribution of the microtubule-organizing center of the dendritic cell. That, in turn, causes redistribution of the cytokine-containing secretory organelles (the Golgi body and secretory vesicles) within the dendritic cell cytoplasm, such that the cytokine is released directly into the interface between the two cells before diffusing into the surrounding tissue fluid (Figure 3-6). In the T-cell/dendritic cell interaction, the activation is mutual, and over the course of the 12 hours of contact, the T cell will also redistribute its secretory apparatus and release cytokines directly into this tight intercellular interface that further activate the dendritic cell. An activated TH cell might then dissociate from the dendritic cell and engage in a similar interaction with a B cell or a cytotoxic T cell, again redistributing its secretory apparatus so as to release cytokines directly into the new intercellular junction.

FIGURE 3-6 Polarized secretion of IL-12 (pink) by dendritic cells (blue) in the direction of a bound T cell (green). The higher magnification micrograph shows the secretion of packets of IL-12 through the dendritic cell membrane.
These cell-cell associations are common in the immune response, where the local concentration of cytokines at the cellular interface can be extremely high, much higher than in the tissue fluids generally. This concentration of activating cytokines at the junction between cells is an important mechanism for ensuring their efficient and accurate delivery.
Many Immune Receptors Include Immunoglobulin Domains
There are few macromolecular structures in biology for which the relationship between structure and function is as clearly apparent as in the immunoglobulin domain. First described as a repeating unit in secreted antibody (immunoglobulin) molecules, this domain has since been characterized in a legion of molecules involved in recognition and adhesion functions (Figure 3-7).

FIGURE 3-7 Some examples of proteins bearing immunoglobulin domains. Each immunoglobulin domain is depicted by a blue loop. Note the presence of the characteristic spacing of the disulfide bonds—about 67 amino acids separate the two cysteine residues—shown at the base of each amino acid domain loop.
Within each immunoglobulin (Ig) domain, several parallel β strands are arranged to form a pair of β sheets. Along the amino acid sequence of each β strand, hydrophobic and hydrophilic amino acids alternate so that the hydrophobic amino acids on one sheet are oriented toward those on the opposite sheet and the hydrophilic residues interact with the environment. The two b sheets thus form an extremely stable “hydrophobic sandwich” (see Figure 3-8) in which each domain is stabilized by hydrophobic interactions between the sheets and the protein as a whole is notably soluble.

FIGURE 3-8 The immunoglobulin domain is made up of amino acid residues arranged in β sheets that are connected by variable loops. The two β pleated sheets are shown in two shades of blue. They are held together by hydrophobic interactions and by a conserved disulfide bond (not shown). The three loops of each variable domain, shown in red, vary considerably in length and amino acid sequence and make up the antigen-binding site. They are therefore referred to as complementarity-determining regions (CDRs).
Most immunoglobulin domains contain approximately 110 amino acids, and each β sheet contains three to six strands. The pairing of β sheets within each domain is stabilized by intrachain disulfide bonds. Neighboring domains are connected to one another by a stretch of relatively unstructured polypeptide chain.
So how does this domain structure facilitate the recognition functions of the many proteins that incorporate it? At the ends of each of the β sheets, more loosely folded polypeptide regions (loops) link one β strand to the next. These loosely folded regions can accommodate a variety of protein sequence lengths and structures without disrupting the overall backbone of the molecule. For example, in the BCR, those parts of the molecule that make contact with the antigen, the complementarity-determining regions (CDRs), are located in these loosely folded regions and are highlighted in red in Figure 3-8. It is clear that the CDRs can adopt a multitude of conformations without disrupting the essential β-sandwich framework structure of the molecule.
In other proteins, such as those illustrated in Figure 3-7, the same structural framework can also be used to support loops that contain amino acids important in cell adhesion, or that operate as coreceptors. These properties explain why the immunoglobulin domain is found in so many proteins with recognition or adhesion functions. Together, these structurally related proteins comprise the immunoglobulin superfamily, a term that is used to denote proteins derived from a common primordial gene encoding the basic immunoglobulin domain structure.
Note that different types of recognition proteins may contain different numbers of immunoglobulin domains. For example, antibody heavy chains contain four or five domains, whereas the light chains contain only two. In each case, the antigen-binding function is located within the amino (N)-terminal domain of the protein.
Immune Antigen Receptors Can Be Transmembrane, Cytosolic, or Secreted
For most biologists, the word “receptor” conjures up images of a transmembrane protein, dutifully awaiting the diffusion of a soluble ligand into its vicinity so that it can respond to the ligand’s signal. And indeed, the antigen receptors of the adaptive immune system, the BCR and TCR, are transmembrane proteins, as are many of the innate and cytokine receptors. However, this is not always the case.
First, it should be noted that the B-cell receptor exists in both membrane-bound and secreted forms, which will be described in more detail in the next section. The soluble form of the BCR is called an antibody, and is synthesized only after antigen stimulation of the relevant B cell. Both soluble antibodies and membrane-bound BCRs belong to the immunoglobulin family of proteins, and consist of two identical heavy (H) chains and two identical light (L) chains. In the membrane-bound form, hydrophobic residues at the carboxyl (C) terminus of the heavy chain anchor the receptor into the plasma membrane (Figure 3-9a). After antigen stimulation, the daughter cells of the original B cell begin to secrete soluble antibodies in which the hydrophobic residues at the C-terminal end of the antibody heavy chain are exchanged for more hydrophilic amino acid residues (Figure 3-9b).
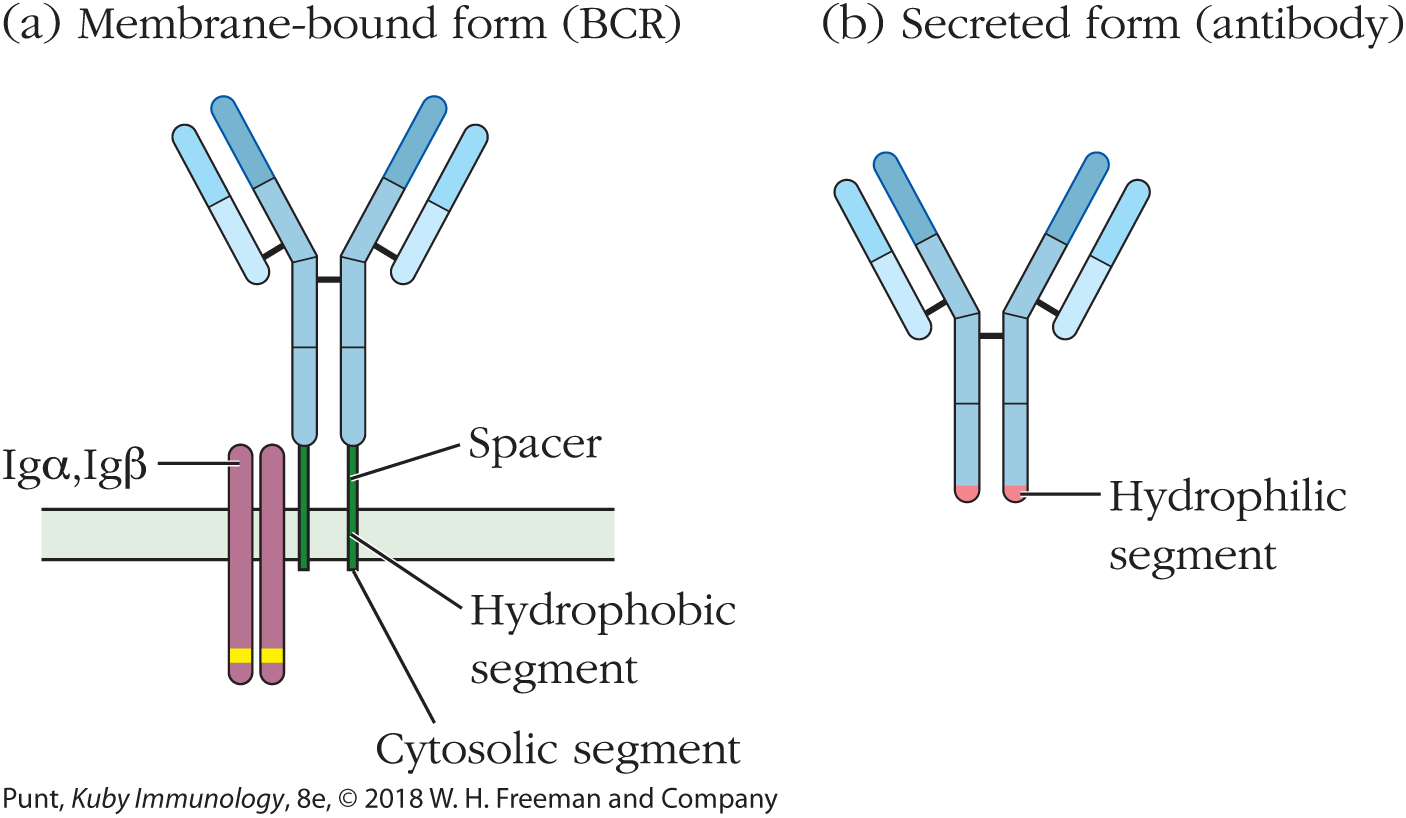
FIGURE 3-9 The BCR exists in both membrane-bound (a) and soluble (b) forms. When a B cell is stimulated, it secretes a soluble form of its receptor that differs in sequence from the membrane-bound BCR at the C terminus but has the same antigen binding site as the membrane receptor. The hydrophobic region that anchors the membrane-bound form onto the surface of the B cell is replaced, by differential mRNA splicing, with a soluble (hydrophilic) amino acid sequence in the secreted antibody.
Unlike their B-cell counterparts, TCRs are always found in membrane-bound form and are never secreted in soluble form. Moreover, as we will learn, the antigens that bind to TCRs are not normally soluble, but are usually located on the surface of antigen-presenting cells in the form of a complex with immune system molecules.
The locations of the antigen receptors of the innate immune system are significantly more variable than those of their adaptive immune system cousins. Some innate receptors, such as mannose-binding lectin (MBL), are entirely soluble, circulating in the tissue fluids. Others, such as members of the TLR family of innate receptors, may either be attached to the plasma membrane, or found in association with the cytosol-facing membrane of intracellular endosomes and lysosomes. Yet others are found within the cytosol, not associated with any intracellular membrane structures. These locations match the functions of the corresponding innate receptors: plasma membrane–bound TLRs bind antigens such as the lipopolysaccharides of gram-negative bacteria, whereas those located on the endosomal membranes are specific for molecules found only within the cytosol, such as CpG-rich bacterial DNA or viral RNA (see Chapter 4).