Dysbiosis, Inflammatory Bowel Disease, and Celiac Disease
The immune activity represented in responses to commensal and pathogenic microbes comes at a cost. Even successful immune responses and protective inflammation may cause damage to the epithelium that must be repaired and replaced by ever-active stem cells, after pathogens are successfully cleared. If a pathogen cannot be cleared, inflammation can become chronic, with a lasting effect on the structure and cellular components of the intestine and its immune system.
Inflammatory bowel disease, or IBD, is one of the most common chronic disorders of the intestine (Figure 13-15a). It is really two diseases—Crohn’s disease and ulcerative colitis—characterized by chronic inflammation of the gut mucosa that results in pain, diarrhea, and dramatically reduces quality of life.
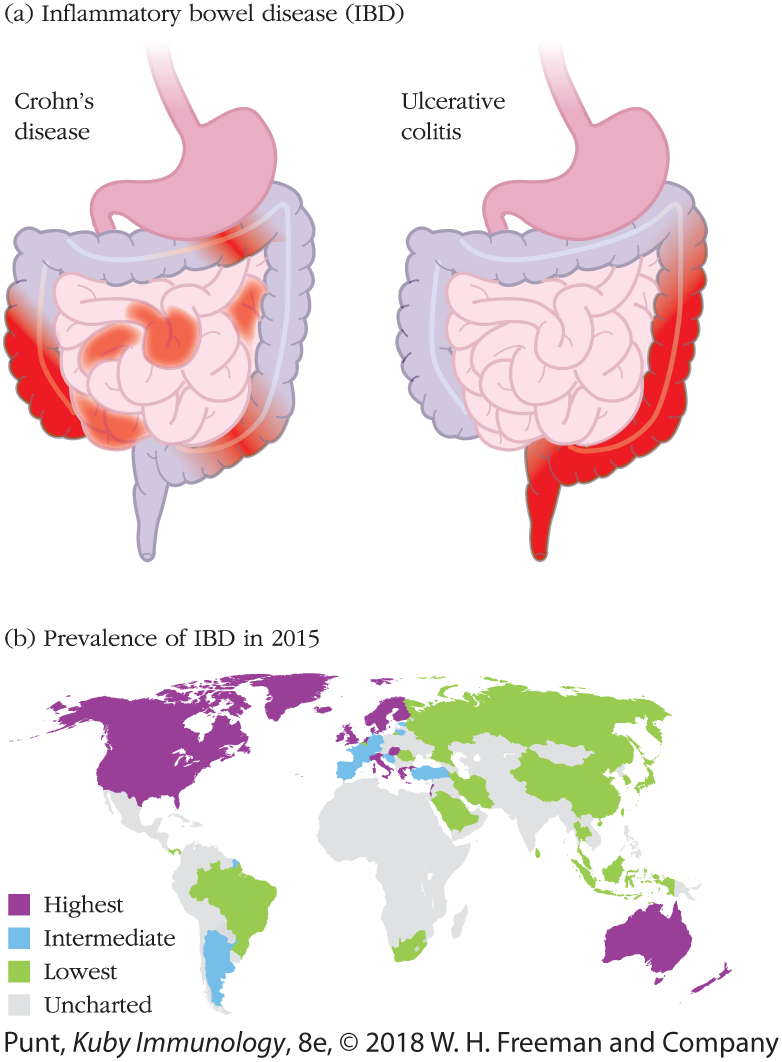
FIGURE 13-15 Inflammatory bowel disease (IBD) distribution in the gut and around the world. (a) The intestinal distribution of the two different variations of IBD. Crohn’s disease can affect any and all areas of the GI tract, whereas ulcerative colitis is found only in the large intestine. (b) The distribution of IBD in world populations. Its prevalence, where measured, is highest in North America, England, Scandinavia, and Australia and lowest in Asia and Brazil. This pattern suggests that diet may have a major influence.
Crohn’s disease is a form of IBD that can affect any part of our digestive tract. It is broadly associated with an inappropriate type 1 response and is characterized by patchy aggregates of inflammatory macrophages in the mucosa. These can form granulomas that are difficult to resolve and distort the microenvironment of the small intestine. Ulcerative colitis is a form of IBD that afflicts only the descending part of the large intestine. It is broadly associated with an inappropriate type 2 response that results in active inflammation of the mucosa with infiltration of neutrophils, reduced mucus boundaries, and direct damage to the epithelium (ulcers).
The causes (etiology) of IBD are not well understood and likely vary from individual to individual. Genetic, environmental and infectious influences all contribute to the development and trajectory of the disorder. Reductions in mucus production, antimicrobial peptide production, tight junction maintenance, regulatory T-cell production, and levels of IL-10 have all been associated with IBD. Alterations in any of these activities can contribute to the loss of tolerance and compromised epithelial integrity featured in these diseases. Interestingly, IBD is also associated with increased levels of IL-23, which activates TH17 and ILC3 cells. As we discussed above, these cell subtypes can contribute to both homeostasis and inflammation, depending on other, yet unclear, environmental influences.
The composition of the commensal intestinal microbiome—and the interplay between microbiome and mucosal immunity—clearly plays a key role in initiating, sustaining and treating IBD. Dysbiosis, the disruption of a healthy microbiome, is now a well-accepted contributing factor to IBD. The rising incidence of IBD in industrialized nations (Figure 13-15b) further suggests that the influence of diet on the microbiome is a root cause.
However, it is difficult to distinguish initiating causes from consequences of chronic intestinal inflammation. Genome-wide association studies (GWAS) have revealed several gene variations associated with increased susceptibility to IBD that may provide better clues. Some of the genetic variations point to defects in pattern recognition receptors, and hence dysfunctional relationships between the gut microbiome and the innate immune system.
For instance, as described in Clinical Focus Box 4-3, individuals who inherit specific variants of the NOD2 gene can be forty times more susceptible to developing IBD. NOD2 is a member of the NLR family of cytosolic PRRs, expressed by multiple innate immune cells in the intestine. It recognizes intracellular bacterial MAMPs and activates inflammatory signaling pathways that generate antimicrobial peptides and enhance the immune activity of antigen-presenting cells (see discussion of our immune response to Salmonella in the previous section).
Mice with defective CARD9 and humans with a variation in the CARD9 gene sequence also are more susceptible to IBD. CARD9 is part of the signaling cascade generated by certain pattern recognition receptors. Interestingly, a defect in CARD9 has an indirect effect on the composition of the gut microbiome, reducing the overall production of intestinal tryptophan. Why is tryptophan important? Its metabolites interact with receptors on gut epithelium, stimulating production of the protective cytokine IL-22. In fact, tryptophan levels—provided only by food and the microbiome because our cells cannot synthesize this amino acid—can partially ameliorate the symptoms caused by CARD9 mutation. This phenomenon is only one of many that illustrate the importance of the dialog between microbiome and host immunity in gut inflammatory disease. There is much more to come, and clarity will certainly inspire new therapies.
Individuals with IBD often suffer from inflammatory disorders of other tissues, including the skin. Canker sores and oral ulcers are common and painful accompaniments. These extra-intestinal manifestations of IBD are thought to arise from a systemic defect in immune tolerance. As we have discussed above, the commensal intestinal microbiome helps tune the immune system and encourages the generation of anti-inflammatory cells, including regulatory T cells. Not all of these cells remain in the intestinal mucosa; many populate other organs and tissues and are thought to contribute to a systemic tolerizing affect. Dysbiosis may not only impair the development of tolerizing influences but can result in the activation of proinflammatory TH17 cell subsets that travel to other organs and wreak havoc.
Celiac disease (CD) is an autoimmune disorder that also affects our intestinal mucosa. People with CD share some symptoms (diarrhea, gastrointestinal discomfort, bloating) with those affected by IBD. However, the specific cause of CD is known and is distinct from the complex causes of IBD. CD is triggered by an immune response to dietary glutens (and other molecules) associated with several grains, including wheat. The immune response to gluten results in the production of IL-15, which activates IELs resulting in epithelial cell death and physical damage to the barrier. Gluten peptides gain access to the lamina propria and trigger predominantly TH1 (versus TH17) responses, as well as NK-cell and B-cell activity.
Gastrointestinal viral infection often precedes CD symptoms, and genetic variations in MHC molecules (as well as many other gene loci) may enhance susceptibility to CD. Finally, microbiomes of people with CD differ from those of healthy individuals, but it is unclear whether this is a cause or consequence of the associated inflammation.
Treatments for IBD and CD differ. A life-long gluten-free dietary regimen helps most, but not all, patients with CD. Immunosuppression and immunotherapy with antibodies that block inflammatory molecules (such as anti–TNF-α) help many, but not all, patients with IBD. The complexity of the disease and the individuality of immune responses contribute to therapeutic challenges that face physicians and patients alike, and there is room for much more research.