Central Nervous System Tumors 32
Emanuela Molinari and Mark R. Gilbert
INTRODUCTION
Tumor of the central nervous system (CNS) can be divided into primary, arising directly in the brain or spinal cord, or secondary, due to metastatic disease. This chapter will discuss both with emphasis on the former to provide a schematic and practical approach to their classification and management. Emphasis has been placed on the new classification of CNS tumors, which combine molecular and histologic features, as this is the way forward in neuro-oncology and it has significant implications in clinical management and clinical decisions.
Brain tumors account for approximately 88% of all primary CNS tumors, and concerns regarding the increasing incidence of brain tumors have been reported over the past three decades, likely due to the increase in life expectancy and increase in brain tumor identification with widespread use of CT and MR imaging. Additionally, this increase in incidence of brain tumor subtypes may partially be attributable to the recent shift in diagnostic categories, thus reflecting changes in diagnostic practice and criteria. Although recent advances in diagnostic and treatment approaches have improved the management, improvement in outcomes for patients with brain tumors has been modest and the survival rate continues to remain poor.
Most brain tumors are sporadic. However, about 5% of primary brain tumors have known hereditary factors, as neurofibromatosis type I and II, tuberous sclerosis, von Hippel-Lindau disease, Turcot’s disease, familial polyposis, and Li-Fraumeni syndrome.
Brain tumors are often morphologically heterogeneous and many of them do progress over time, becoming more malignant due to the accumulation of genetic alterations, resulting in changes in the tumor biology. Although the initial differential diagnosis can be postulated by a combination of radiological findings, location and patient’s age, and occasionally initial behavior of the lesion, the definite diagnosis of CNS tumors requires tissue sampling and is based on an integrated classification combining phenotypic and genotypic characteristics. Although the precision of diagnosis has markedly improved with the integration of histology and genetic markers, further research is needed to establish prognostic and predictive biomarkers thereby guiding individualized treatment and positively affecting the mortality and morbidity outcome of patients.
Metastatic brain tumors are more frequent than primary brain tumors and their incidence is rising due to a combination of factors including advanced screening programs, improved treatments, and consequent increased survival after initial cancer diagnosis, allowing the spread of cancer to the brain to occur as a late complication. It has been estimated that up to one-fourth of patients with a diagnosis of cancer have brain metastases before death with primary sources to be more frequently lung, breast, skin, kidney, and gastrointestinal tract.
Molecular Diagnosis of Primary Brain and CNS Tumors
In 2016 the World Health Organization (WHO) formulated a major reconstruction of CNS tumor diagnoses by combining histological features and molecular parameters. This dynamic pathogenetic classification improves diagnosis by grouping tumors that share similar prognostic markers, and increasingly guides patient management by enabling the use of therapies for entities with similar biological and genetic characteristics. The grading system (WHO) that determines the level of aggressiveness of each type of tumor has been maintained based on histological features with additional criteria for atypical meningioma that includes brain invasion. The malignancy grade divides tumors from 1 to 4, respectively, from less to more aggressive. The grade of malignancy is determined by the area of the most malignant features based on criteria of cellular atypia, mitotic activity, degree of cellularity, degree of necrosis, and/or microvascular proliferation. Low-grade tumors such as the pilocytic astrocytoma, designated as grade 1, are biologically distinct from the grade 2 to 4 as they typically are well circumscribed, rarely undergo malignant transformation and may be cured with surgical resection. Therefore, there are important differences between grade 1 and grade 2 gliomas, the latter are typically not curable with resection and can undergo malignant transformation to grade 3 or 4.
Clinical Diagnosis and Considerations
Brain tumors cause symptoms and signs through a combination of mechanisms. Clinical manifestations depend on location, size, and rapidity of growth. Direct effects of the tumor are related to invasion and compression of the tumor on the brain parenchyma. Secondary effects are mostly related to vasogenic edema. Symptoms may be focal, reflecting the location of the tumor (e.g., hemiparesis); generalized, which are nonlocalizing (e.g., headache); or false localizing, which are caused by raised intracranial pressure (e.g., tinnitus) (see Table 32.1). The most common symptoms are headache, which is usually nonspecific, seizures, mental status changes and behavioral changes, and unilateral weakness (paresis). Between 30% and 90% of patients with brain tumors experience seizures either at presentation or at some time during the disease trajectory, often with progression. Secondary epilepsy is always focal in origin although seizures can secondarily generalize; this is more common in primary tumors than metastases, and is more often associated with slow growing/low grade tumors. The most common signs are paresis, normal examination, and memory impairment. Quite commonly there could be involvement of cranial nerves, optic discs, and visual fields. In children, most frequently symptoms and signs are related to increased intracranial pressure, often associated with ataxia.
Table 32.1Brain Tumor: Symptoms and Signs
Localizing | seizures, hemiparesis, diplopia, aphasia, vertigo, incoordination, sensory abnormalities and dysphagia |
Generalized | headache, nausea, vomiting, dizziness, mental status changes, visual obscurations and seizures |
False-localizing | tinnitus, diplopia, hearing and visual loss |
The time between the onset of symptoms and diagnosis varies and there seems to be a degree of inverse association between apparent delay in diagnosis and poorer outcome. This may be explained by the less specific symptomatology that slower growing tumors are more likely to have in comparison with more severe and localizing symptoms earlier in the disease course of more aggressive tumors.
Acute Complications
The brain and CSF are compressible components contained within a nonexpandable space due to rigid skull in adults. Mass occupying brain tumors cause displacement of the brain from one cranial compartment to another, following the path of least resistance. The most important clinical consequences are increased intracranial pressure and herniation syndromes, which are reflected by a depressed level of consciousness. Five common herniation syndromes can occur, either alone or in combination: subfalcine modification (cingulate), uncal (transtentorial), central, upward and downward cerebellar (tonsillar) herniation (Figure 32.1).
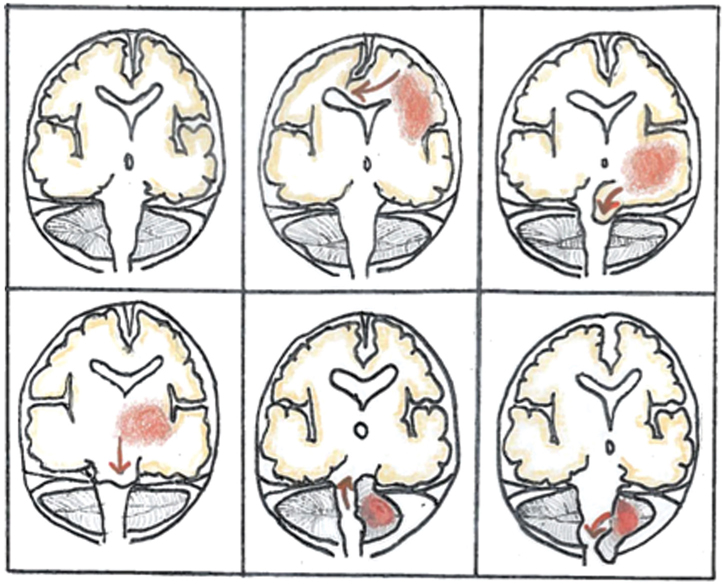
FIGURE 32.1Herniation syndromes.
(edited from a figure courtesy of Heidi Maj)

TREATMENT CONSIDERATIONS
Therapeutic regimens typically encompass multiple treatment modalities. As previously described, patient prognosis remains poor; therefore, treatment should balance the quality of life of patients with the goal of prolonging survival. Experience from long-term follow-up has documented the early and late impact of the disease and treatment on both symptom burden and on the patient’s quality of life (QoL).
Surgery
Surgery is the initial component of the management of CNS tumors. Although occasionally resection can be curative in grade 1 tumors, for higher grade tumors it frequently does not achieve complete tumor removal due to the infiltrative nature of the disease. Surgery is usually aimed at maximal safe debulking of the tumor burden, where the limit is often set by the vicinity of eloquent brain areas and the related surgical risk of neurologic deficits. Surgery helps in the management of acute symptoms, relieving deficits caused by mass effect, and maximizing the benefits of other therapies by providing less tumor burden to be treated and potentially reducing toxicity risks from increased intracranial pressure and improving seizure control in selected patients. In addition to direct damage to the surrounding normal brain tissue, surgery does carry other risks, such as infection and wound breakdown. Stereotactic biopsy is a minimally invasive procedure for diagnostic purposes with very limited morbidity and mortality estimated to be 4% and 0.9%, respectively. However, biopsy may not provide a diagnosis in up to 5% of cases and limited diagnostic accuracy due to the limited sampling. In one study, the diagnosis was changed in 38% of cases when tumor tissue from biopsy was compared to tumor obtained by surgical resection. This discrepancy in pathological diagnosis has significant repercussions in clinical practice, affecting treatment decisions and outcome.
Radiation Toxicity
The tolerance of normal brain parenchyma to radiation treatment is based on the total dose, the dose per fraction, and the volume of brain treated. Neurotoxicity due to radiation therapy (RT) can be acute, subacute, or delayed and late onset. Acute toxicity occurs during or soon after the treatment. It is thought to be due to edema and demyelination causing a clinical syndrome most commonly manifest as excessive somnolence and encephalopathy, and is usually self-limited. Subacute or delayed toxicity manifest up to 3 to 6 months after the completion of radiation but is most commonly seen with the first imaging study after completing radiation treatment, and has been termed pseudo-progression (see below). Late effects may occur month to years after the treatment and are often referred to as radiation necrosis (see below). These chronic complications are thought to be due to damage to the normal cellular component of brain parenchyma and alterations in the function and integrity of the cerebral vasculature. There are a wide range of clinical manifestations, including seizures and cognitive loss potentially leading to dementia. In children with an incompletely developed nervous system, radiation treatment can impair growth and development. A unique delayed radiation toxicity has recently been described as stroke-like migraine attacks after radiation therapy (SMART) syndrome. The disorder is characterized by complex migraine attacks associated with focal neurological deficits, and is associated with marked alterations in imaging that can be misinterpreted as tumor progression. Exposure to ionizing radiation can also increase the rate of other tumor occurrence, as meningiomas, and vascular abnormal formation typically cavernomas.
Chemotherapy Toxicity
Chemotherapy agents used for brain tumor treatment are typically administered systemically either by oral or intravenous routes. Therefore, typical systemic toxicities occur but in addition, patients with CNS cancers are at increased risk of neurologic effects, likely the consequence of disease and treatment-induced brain injury. Seizures, neurocognitive impairment, and worsening of focal neurologic function can follow both systemic as well as locally delivered treatment. For example, chemotherapy-induced cognitive changes may represent the effect of DNA damage, telomere shortening, cytokine deregulation, genetic predisposition to increased chemotherapy vulnerability, as well as potentiating effects on cognitive decline from other concomitant treatments.
Targeted Treatments
Treatments targeting molecular signaling pathways that are essential to tumor growth, such as angiogenesis and growth factors, may also impact neurologic function as well as impair the ability of the brain to repair after injury therefore impacting functional recovery. Most of the long-term side effects of the new treatments are unknown; therefore, it is crucial to systematically assess outcomes aimed to establish the impact of treatments on neurological functions both in the short and long term.
FOLLOW-UP AND MONITORING CHALLENGES
The evaluation and assessment of patients with brain tumors to determine treatment response, progression, and treatment side effects can be very challenging. The notion of treatment-induced changes in imaging characteristics complicate the interpretation of imaging studies. For example, treatment-induced inflammatory changes, often called pseudo-progression, will appear as a contrast enhancing mass with extensive edema. Conversely, some treatments, particularly anti-angiogenic therapies will decrease diffusion of contrast material leading to an improved imaging study but not necessarily indicating a decrease in tumor burden and has been termed pseudoresponse. Radiation can also cause damage to the brain tissue, in particular, the white matter, leading to change in appearances even years after treatment. This damage, referred to as radiation necrosis, can continue to increase further mimicking tumor progression, thus increasing the challenges of disease evaluation for the clinician.
 Postsurgery: MRI should be performed within 48 hours and not later than 72 hours to better detect residual tumor and minimizing postoperative changes that may mimic residual tumor.
Postsurgery: MRI should be performed within 48 hours and not later than 72 hours to better detect residual tumor and minimizing postoperative changes that may mimic residual tumor.
During or soon after radiation treatment: Increases in MRI contrast enhancement can be induced by a variety of processes, such as treatment-related inflammation, postsurgical changes and ischemia, radiation necrosis, and subacute radiation effects. In the first three months (although this may occur later) after radiation treatment (particularly when combined with chemotherapy), pseudoprogression is a recognized event. This pseudoprogression can be seen in up to 25% of patients. Patients with MGMT-methylated tumors are thought to be in higher risks for pseudoprogression. Although the pathogenesis is not known, it is thought to reflect a transient and local reaction characterized by inflammatory response, demyelination and abnormal vessel permeability, due to increased sensitivity to radiation of oligodendrocytes and endothelial cells
The neuroimaging of pseudoprogression is characterized by increased enhancement on MRI caused by abnormal vessel permeability (breakdown of the blood–brain barrier (BBB)) and increased T2 and fluid-attenuated inversion recovery (FLAIR) weighted signal due to edema, and usually, but not always, the absence of increased perfusion. Certain imaging characteristics patterns are also more suggestive for pesudoprogression, as linear pattern of enhancement and periventricular white matter changes, but these findings can also be seen with tumor growth. In most patients, the increase in radiologic abnormalities is clinically asymptomatic. When symptomatic, it usually reflects general cognitive functioning or pre-existing symptoms. As it is partly due to transient demyelination, it may benefit from corticosteroids that decrease the inflammatory response and the BBB leakage.
Later in the disease trajectory, radiation necrosis is a known complication. It is a late delayed reaction, and usually occurs 18 to 24 months after radiation but as early as two months and up to 5 years. The incidence reports vary but has been reported to occur in 3% to 24% of brain tumors. The pathophysiology is thought to be the consequence of vascular change, edema and fibrinoid exudate. On imaging studies, radiation necrosis appears as space-occupying lesion with mass effect therefore very difficult to be distinguished from tumor recurrence. It often affects the area of maximum radiation dose and periventricular white matter appearing with an enhancing “soap bubble” appearance. Metabolic studies may be difficult to interpret due to inflammatory activity and MR spectroscopy may help in differentiating from tumor recurrence by showing increased Lactate/Creatine, decreased Choline/Creatine, and lack of the 2-hydroxyglutarate (2HG) in IDH-mutated tumors, although the sensitivity and specificity of MR spectroscopy remain too low to use this methodology as the exclusive means of differentiating tumor from necrosis.
Radiation necrosis can cause symptoms and decline in neurological function. In clinically symptomatic patients, management options include surgery that will confirm the underlying diagnosis and resolve the mass-related complications and symptoms. Further options include addition of anti-VEGF treatment to reverse the effect of increased VEGF expression in the white matter following radiation therapy (RT) that correlates with BBB breakdown and brain edema. This treatment has been proven in a randomized, placebo-controlled clinical trial. A variety of other treatments have been tried, but most reports are anecdotal. These include hyperbaric oxygen therapy, oral vitamin E administration, and laser interstitial thermal therapy.
Conversely, pseudoresponse is a phenomenon that describes imaging changes of signal reduction that are possibly due to “normalization” of the BBB and are often associated with treatments as anti-VEGF therapies.
PRIMARY BRAIN AND CNS TUMORS
There are more than 130 types of primary brain and CNS tumors. The section will focus on the most common and those of particular scientific interest. Despite classification based on the appearance of the tumor cells, the cellular origin for most brain tumors is unknown and there are no recognized precursor lesions that define a premalignant status. Primary brain tumors are grouped accordingly to their histological appearances that most closely resemble normal CNS cell constituents. Neuroepithelial cells are thought to give rise to gliomas, pineal tumors, and embryonal tumors such as medulloblastoma), meninges, choroid plexus, germ cells, and sellar origin (including pituitary tumors and craniopharyngiomas) (Figure 32.2). Brain tumors are thought to arise from neural stem, progenitors cells, or de-differentiated mature neural cells that undergo malignant transformation. Glial tumors account for approximately two thirds of all intracranial tumors with age-related incidence by defined molecular and histological subtypes. Childhood tumors have different incidence, different localization preference (i.e., posterior fossa with main involvement of the cerebellum) and different molecular profiles; therefore, pediatric CNS cancers warrant consideration of the impact of age and site of origin as well as outcomes and treatment approaches.
Table 32.2Main Groupings of 2016 WHO Classification of Glial CNS Tumors
Glial neoplasms | Main subtypes | Molecular profile | Grade |
Diffuse Astrocytic and Oligodendroglial tumors | Diffuse Astrocytoma Anaplastic Astrocytoma Glioblastoma Oligodendroglioma Anaplastic Oligodendroglioma Diffuse midline glioma, H3 K27 mutant | IDH mutant IDH mutant IDH mutant and wild-type IDH mutant 1p/19q codeleted IDH mutant 1p/19q codeleted H3K27M-mutated | I III IV II III IV |
Other Astrocytic tumors | Pilocytic Astrocytoma Pleomorphic Astrocytoma (PXA) Subependymal Giant cell Astrocytoma (SEGA) | BRAF V600E mutationBRAF fusion BRAF V600E mutation TSC 1/ 2 mutation | I II I |
Ependymal Tumors | Subepensymoma, maxillopapillary, ependymoma, ependymoma RELA fusion positive, anaplastic * | Allelic loss 22q NF2 mutation Combination of DNA methylation and genomic alterations for spine, infratentorial and supratentotial (CIMP, RELA; YAP) | I-III |
* molecular and histopathology details of 9 ependymoma subgroups go beyond the scope of the chapter but the reader is suggested to refer to the 2016 WHO classification for details
Epidemiology
Primary CNS tumors are relatively rare, accounting for 1.8% of all cancers
US incidence rate for brain and CNS tumors in adults (>20 years old) is 22.36 cases per 100,000, and 5.47 cases per 100,000 in the pediatric population
The population subgroups at higher risk for brain cancer are elderly, Caucasians, men and those living in metropolitan counties
Incidence rate follows a bimodal distribution, with a small peak in early childhood and more pronounced peak in late middle age
The higher incidence for older individuals suggests a possible role for bioaccumulation from environmental toxic exposure
Established environmental causal factors for brain tumors are ionizing radiation and possibly prolonged exposure to hydrocarbons. Exogenous hormone use among women is an established risk factors for meningioma.
Possible protective factors for glioma risk are allergy-related immune responses, elevated IgE, and previous history of chickenpox and/or positive VZV IgG
According to the Central Brain Tumor Registry of the United States, 16,947 deaths are estimated to be attributed to primary malignant brain and other CNS tumors in the United States in 2017
Five-year survival rates after diagnosis of primary brain tumor progressively decrease with age
Meningiomas make up 36.4% of all primary brain tumors
One third of tumors are malignant, with the most frequent being glioblastoma (WHO Grade 4)
CNS tumor are the second largest category of cancer in the pediatric population, with more frequent localization in the posterior fossa for age between 4 and 10 years old
Embryonal or primitive neuroectodermal tumors (PNET) as well as astrocytic lineage tumors are the most frequent before the age of 20
In childhood, there is dichotomy distribution for site (infratentorial and supratentorial) as well as predominance of low-grade tumor with age-group peaks at 5 to 9 years and 15 to 19 years, while high-grade tumors often presents before the age of five and their incidence decrease with age.
Gliomas
Gliomas encompass a heterogeneous group of tumors that affect patients of different ages with often substantial differences in molecular profiles and behavior. Most gliomas in adults diffusely infiltrate the adjacent brain tissue and therefore are often referred as “diffuse gliomas” that encompass grade 2 to 4. Per the 2016 WHO classification, there are three main groups of glial neoplasms, categorized as diffuse astrocytic and oligodendroglial tumors, other astrocytic tumors and ependymal tumors (Table 32.2).
Diagnostic Approach and Clinico-genetic Considerations
Genomic alterations driving gliomagenesis pathways have been analyzed. For example, isocitrate dehydrogenase 1 or 2 mutation (IDH1 or IDH2 mutation) have been established as a common initiating event of carcinogenesis in lower-grade gliomas. Progression to more aggressive tumors is associated with additional genetic changes and more complex chromosomal and genetic alterations (Table 32.3).
Table 32.3Schematic Grading of Glioma by Combined Molecular Profile Subtype
No precursor | Diffuse gliomfas | Focal glioma | ||
EGFR amplification PTEN mutation LOH 10q | IDH mutant | IDH wild-type | BRAF | |
1p/19q codeletion | 1p/19 q intact TP53 mutation ATRX loss | EGFR PTEN CKDN2A | ||
OLIGODENDROGLIOMA WHO II-III | ATROCYTOMA WHO II-IV | ASTROCYTOMA WHO II-IV | PILOCYTIC ASTROCITOMA WHO GR I | |
Additional alterations Often LOH 10q, DCC loss expression | ||||
PRIMARY GBM | SECONDARY GBM | |||
MGMT methylation status | MGMT methylation status | |||
Dismal prognosis Better with MGMT methylation (less frequent) | Good prognosis | Intermediate prognosis Better with MGMT methylation (more frequent) | Poor prognosis | Excellent prognosis |
Specific molecular signatures are now recognized as of crucial biological importance and underpin a new diagnostic approach with significant clinical repercussions and practical relevance in patient’s management (Table 32.4). The major distinction for adult gliomas is based on IDH1 or IDH2 mutations compared with “wild-type” IDH status, which characterizes their distinctive biology and clinical behavior, with better outcomes with IDH mutant gliomas in comparison with IDH wild-type. IDH mutations are associated with specific pattern of methylation and DNA hypermethylation profile. Gliomas with IDH mutations, including glioblastomas, have a better prognosis. For lower grade gliomas (LGG), IDH mutated tumors with loss of one copy of chromosome arms 1p and 19q (1p/19q codeletion), are associated with longer median overall survival. The related strong practical clinical impact of such knowledge comes clear, for example, when identifying IDH-mutated gliomas with G-CIMP low profile. Conversely, the absence of IDH mutation (IDH wild-type) in low-grade tumors marks poor GBM-like prognosis. However, there is a very small subgroup of low-grade tumors (6%) that do not harbor an IDH mutation but have some characteristics of pilocytic astrocytoma (BRAF mutation) that have a very low mortality rate.
Table 32.4Practical Approach to Glioma Subgroups in the Adult Population: Clinical and Molecular Correlations
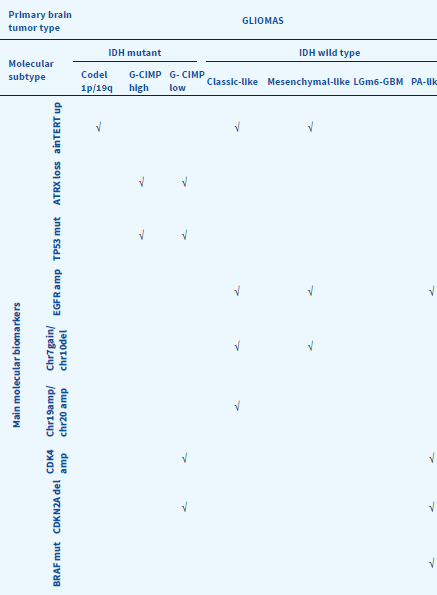
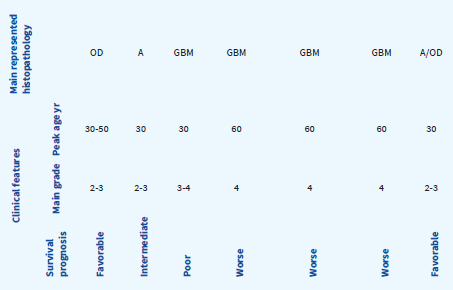
Adapted from Ceccarelli et al., 2016, Cell 164, 500-563
New Classification: Histopathological and Genetic Considerations
The 2016 WHO classification defines both grades 2 and 3 oligodendroglioma by requiring the demonstration of IDH mutation and 1p/19q codeletion. The new classification also sees the delegation of oligoastrocytoma diagnosis to a rare finding of both 1p/19q codeletion and the combination of p53 mutation and ATRX loss. To date, there are only few biomarkers with diagnostic and risk stratification implications and predictive information that guide therapeutic decisions. In particular
O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation
1p/19q codeletion
IDH mutation
BRAF duplication/fusion
Diffuse gliomas in adult and pediatric patients, although with similar histopathologic appearance, are molecularly distinct. Pediatric gliomas rarely have IDH mutations or 1p/19q codeletions. Instead, BRAF and H3K27M mutations are more common, particularly for low grade and midline tumors, respectively. These findings suggest a different pathogenesis and biology for pediatric brain tumors, mandating different treatment considerations, approaches, and eventually different outcomes.
Imaging: Advanced Techniques and Genetic Implications
Neuroimaging is used for diagnostic purposes and to monitor the effect of treatment of brain tumors. Brain computed tomography (CT) and magnetic resonance imaging (MRI), as well as structural and functional techniques, provide information regarding differential diagnoses (abscess, demyelinating plaques, stroke), features helpful in the grading of the tumor, evaluation and treatment planning, and monitoring of treatment response, disease progression and side effects of treatment (radiation necrosis and pseudoprogression). Advanced neuroimaging is increasingly being used to correlate findings with the genetic profile of the tumor, but this remains investigational.
Important aspects of imaging studies include
CT imaging has a role in the detection of hemorrhage (e.g. postoperative), herniation, hydrocephalus.
CT imaging is valuable to detect calcifications within the mass, suggesting brain tumor types as oligodendrogliomas or meningiomas.
Contrast enhancement correlates with local breakdown of the blood brain barrier and is a key feature of high-grade tumor although there are some exceptions, including some low-grade tumor such as pilocytic astrocytomas in children that do enhance.
T2/FLAIR signal abnormalities around the mass lesion correlates with peri-tumoral edema.
Oligodendroglial tumors typically have a heterogeneous image by contrast-enhanced MRI.
MRI spectroscopy is often used to help differentiate tumor from inflammation or radiation-induced injury, although the sensitivity and specificity are limited
•MRI spectroscopy is not specific, detecting peaks of N-acetylaspartate (NAA) decrement because of processes that destroy or replace normal neurons and increased peak of Choline that correlates with increased cell turnover and can be seen with other processes such as demyelinating diseases.
•MRI spectroscopy provides a diagnostic biomarker; detecting accumulation of 2-hydroxyglutarate (2HG) within the tumor as this is associated with IDH-mutated glioma.
Perfusion MRI can help differentiating treatment-related changes from tumor recurrence, and can help plan tumor sampling when contemplating a biopsy.
MRI with diffusion weighted imaging (DWI) can help distinguish a primary CNS lymphoma; an important consideration to avoid profound tumor reduction with corticosteroids leading to a nondiagnostic neurosurgical procedure.
Malignant gliomas (MG) account for more than 75% of newly diagnosed malignant primary brain tumors and carry a disproportionately high rate of morbidity and mortality despite treatment advances. Glioblastoma (GBM), grade 4 by WHO criteria, is the most aggressive tumor subtype and accounts for more than half MG. Anaplastic astrocytoma (AA), WHO grade 3, typically affects a younger adult population and although the prognosis is better than grade 4 tumors, it remains a highly malignant neoplasm. The only established risk factors for MG are exposure to ionizing radiation and rare familial syndromes, such as Lynch syndrome and Li-Fraumeni syndrome. Approximately 5% of patients with MG have a family history of gliomas. Clinically, patients may present with a combination of generalized and localizing symptoms and signs. Patients often complain of headache that does not have specific features, resembling tension-type headache, usually worse in the morning, and presentation with seizure onset is common. Imaging studies, most commonly MRI, reveals an irregular, enhancing mass with associated edema and mass effect. Metabolic imaging using fluorodeoxyglucose positron emission tomography (FDG PET) reveals increased glucose uptake, evidence of hypermetabolism. Evaluation of blood flow and tumor blood volume using either MRI with perfusion sequences or single-photon emission computed tomography (SPECT) demonstrates an increase compared to the contralateral uninvolved brain parenchyma.
Patients with MG present with a variety of neurological complications. These include the following:
Seizures. These are typically focal with secondary generalization. Treatment is required and preferably utilizing the newer antiepileptic drugs (e.g., levetiracetam,lamotrigine, gabapentin) that do not affect the hepatic cytochrome P450 system, thereby avoiding altering the metabolism and clearance of many systemic cancer treatments.
Nonlocalized signs such as confusion and mental status alteration. These are often due to peritumoral edema and increased intracranial pressure, although seizures may precipitate similar findings. Treatment is typically with corticosteroids, although in some situations, hyperosmotic agents such as mannitol or emergency tumor debulking may be required.
Localized signs such as hemiparesis, language dysfunction, and visual field loss. The neurologic dysfunction is typically directly related to the location of the tumor. For example, involvement of the dominant cerebral hemisphere, particularly the posterior frontal lobe and temporal lobe may cause aphasia, whereas involvement of the occipital lobe may result in contralateral hemianopia (visual field loss).
Standard treatment for newly diagnosed MG is maximal surgical resection, despite the infiltrative nature of gliomas. Advantages are as follows:
Defining diagnosis that helps prognostication and drives further treatment options.
Improvement of symptoms resulting from mass effect.
Studies have demonstrated that the extent of resection correlates with outcome. This is particularly important for medulloblastoma and ependymoma.
Specific considerations for glioblastoma and anaplastic gliomas follow.
The median age is 54 years, although GBM can occur at any age. In adults, most current series have reported a median survival of 12 to 18 months even with standard of care treatment.
Glioblastomas may occur de novo, also known as primary or after transformation from a lower-grade glioma. This latter category, called a secondary glioblastoma and which accounts for approximately 10% of all glioblastoma, is characterized by mutation in either the IDH 1 or 2 gene. These patients have a better prognosis than patients harboring the primary GBM.
Primary GBM, defined arising as a de novo glioblastoma, presents with a genetic profile of IDH wild-type. Their characteristic molecular genetic profile includes epidermal growth factor receptor (EGFR) amplifications and mutations, LOH 10q (2/3 of cases), PTEN mutation (1/3 of cases), and p16 deletions.
Secondary GBM, defined as the result of transformation from a lower-grade glioma, presents with IDH1 mutation, TP53 mutations, platelet-derived growth factor (PDGFR) overexpression, LOH 10q.
MGMT methylation state is an epigenetic modification present in 35% to 75% of GBMs that ultimately determines less effective DNA repair and therefore increased chemotherapy sensitivity to alkylating agents such as temozolomide. Additionally, some studies suggest that tumors with MGMT methylation may be more prone to develop “pseudoprogression” after treatment with radiation and chemotherapy.
GBM characteristically enhance after contrast administration on both MRI and CT, often have a central necrotic cavity, and more peritumoral edema, and are more likely to cross the corpus callosum.
Standard of care since 2005 EORTC-NCIC study for newly diagnosed GBM following resection is radiotherapy (RT 60Gy) with concomitant and adjuvant temozolomide (75 mg/m2/d followed by 6 monthly cycles of 150 to 200 mg/m2/d for 5 days per cycle. A recent study in elderly patients (age 65 and older) with GBM compared a short course of RT (40 Gy in 15 fractions) with this radiation schedule and concurrent temozolomide; demonstrating an improvement in survival with the combined treatment.
Although concurrent radiation and temozolomide chemotherapy followed by maintenance temozolomide improves survival, tumor recurrence is inevitable. A variety of second-line therapies are used, although none have clearly demonstrated a survival benefit. They include:
Implantation of carmustine-containing wafers.
Cytotoxic chemotherapy agents such as lomustine, procarbazine, irinotecan, carboplatin.
Bevacizumab, a monoclonal antibody against circulating VEGF, has reported response rates in the 30% to 50% range and prolongation of progression-free survival (PFS) in recurrent disease but a recent phase III study did not show a survival benefit.
Targeted molecular therapies against regulatory signaling pathways, such as EGFR, PDGF, and mammalian target of rapamycin (mTOR) have been tested; however, none have demonstrated clinical efficacy.
Immunotherapies are under evaluation for glioblastoma. These treatments include checkpoint inhibitors, peptide and dendritic cell vaccines, and tumor injections with oncolytic virus.
Established prognostic factors for GBM include patient age, performance status, extent of tumor resection, and tumor MGMT methylation status.
Median survival with standard treatment in GBMs is 15 to 18 months with a 2-year survival of 26.5% to 35%.
Prognosis is improved for patients with MGMT-methylated GBM that have median survival of 21 to 23 months and 2-year survival rate of 40% to 50%.
Up to 10% of patients with GMBs may live 5 years or longer.
Children with high-grade tumors (grade 3 to 4) tend to do better than adults, with 5-year survival of 25%.
Diffuse midline gliomas, H3-K27M-mutant is a new recognized entity in 2016 WHO classification that distinguish pediatric GBM from adult GBM that look alike histologically but have a different molecular profile and unfavorable outcome.
Anaplastic astrocytoma (AA) affect younger people than GBM with median age of 45 years old. The definition of the median survival is evolving. Patients with AA harboring an IDH mutation have a median survival of 8 to 10 years, whereas those without an IDH mutation have a median survival of 2 to 3 years with current treatments.
The optimal treatment of AA remains controversial. There is increasing consensus that grade 3 tumors (anaplastic gliomas, AG) that do not have an IDH mutation are pre-GBM and should be treated with chemoradiation following the standard therapy for GBM. AG that are IDH mutated have a much better prognosis and it is unclear whether concurrent radiation and chemotherapy is optimal as this treatment is associated with greater risk of brain injury. Early results from the CATNON study demonstrated that radiation plus postradiation temozolomide was superior to radiation alone in anaplastic glioma without 1p/19q codeletion. The benefit of combined radiation and chemotherapy has not yet been determined. This issue is even more complicated in AG with IDH mutation and 1p/19q codeletion, now defined as anaplastic oligodendroglioma and discussed below.
Anaplastic Oligodendroglioma (AO)
Oligodendroglial tumors represent 5% to 20% of all glial tumors, with typical age peak at 40 to 60 years and anaplastic tumors preferring older age of onset. Survival time is prolonged and striking differences in oligodendroglial tumors subgroups point to completely different biologic entities and stress the importance of genetic profiling to individualize treatment.
AOs are more sensitive to chemotherapy than astrocytic tumors. Molecular subtype and patient characteristics provide the basis for patient management ramification following surgery and RT. Indeed, chemosensitivity relates to 1p/19q status, the response rate report to be almost 100% response rate in 1p 19q codeleted tumors.
The management of patients with recurrent disease despite treatment is unclear, but patients failing an agent do still present some response rate for another chemotherapy regime or second-line treatment.
Low grade gliomas (LGG) encompass a heterogeneous group of tumors with astrocytic or oligodendroglial features that usually affects younger patient population and have longer survival. Because the prognosis is better, the long-term consequences of treatment are a critical aspect of determining optimal therapy.
Clinical presentation:
Seizures are the presenting sign in over 50% cases, and more than 80% of patients have a seizure during the disease trajectory. Seizures often originate from the brain tissue adjacent to the brain tumor and some locations as the motor strip correlate with resistance to antiepileptic drugs (AEDs). Radiotherapy, chemotherapy, and tumor resection may improve seizure control.
Other presenting symptoms include gradual loss of motor, sensory, language function or visual field loss, depending upon tumor location.
Grade 1 (Pilocytic Astrocytoma)
Although the designation of low grade astrocytomas encompass both grade 1 (pilocytic astrocytomas) and grade 2 diffuse astrocytomas, they are biologically very different and must be considered separately. Most pilocytic astrocytomas develop before the age of 20 with peak of age around the end of the first decade, and often occur as midline posterior fossa lesion involving the cerebellum, although could manifest in the optic-hypothalamic region and in the brainstem as dorsally exophytic lesions. Surgery is the primary treatment and can be curative. Characteristically, these tumors
Are cystic and well demarcated.
Almost always enhance on MRI with brightly enhancing mural nodule appearance.
Complete surgical excision is typically curative accounting for the excellent prognosis with a 10-year survival rate of 95%.
Malignant transformation is uncommon, but may be associated with radiation treatment.
Grade 2 (Diffuse Low-Grade Astrocytomas)
Diffuse low-grade astrocytomas are classified as WHO grade 2. These tumors are typically slow growing but local infiltration of surrounding brain parenchyma prevents cure with surgical resection alone. These tumors commonly occur in young-middle aged adults with median age at diagnosis at 35 to 45 years.
Nearly all grade 2 gliomas have an IDH mutation; 1p19q co-deletion is diagnostic of a grade 2 oligodendroglioma whereas p53 and/or ATRX mutations define an astrocytic lineage.
IDH wild-type has been reported, but increasingly this is associated with a misdiagnosis such as a pilocytic astrocytoma or other rare variants.
T2 hyperintense signal that follows the white matter distribution on MRI
Typically, the tumor is nonenhancing MRI. When enhancement is seen, it may indicate that there has been malignant transformation to a higher grade.
There are a wide variety of treatment options, ranging from observation to aggressive combined treatment.
Maximum safe tumor resection is often pursued as this has important diagnostic and prognostic impact. These tumors may harbor regions demonstrating more malignant cells, thereby altering the diagnosis, prognosis, and treatment. Additionally, there is increasing evidence that extent of resection impacts survival.
Increasingly, grade 2 astrocytomas are being treated with radiation therapy followed by chemotherapy with either temozolomide or the PCV combination regimen. The RTOG 9802 study compared radiation with radiation followed by PCV and demonstrated an almost doubling of survival with the combination regimen.
Median survival of 8 to 10 years
Potential to transform into higher grade, more aggressive tumors
Size greater than 6 cm, crossing midline, presurgery neurological deficits, age >40 years at disease onset are poor prognostic factors
Low-grade oligodendrogliomas are three times more frequent than anaplastic tumors, accounting for 2% to 5% of primary brain tumors and up to 15% of all gliomas. They do occur more frequently in young adult males with a peak of incidence between 30 and 40 years, and although not common, may present with intracerebral hemorrhage due to thin-walled capillary network. They have better prognosis than astrocytomas as they are more chemosensitive.
Oligodendrogliomas are defined by 1p/19q codeletion and IDH mutation
Neither ATRX nor p53 are mutated
Occur more frequently commonly along the convexity in subcortical areas particularly in frontotemporal lobes
Appear as partially calcified mass lesions, easily detected as hyperintense on CT imaging particularly along the cortical ribbon as a gyriform pattern
On MRI demonstrate a high signal on T2 and T2/FLAIR sequences
Contrast enhancement is not typical for grade 2 oligodendroglioma and suggests a higher-grade tumor
Despite the grade 2 designation, leptomeningeal spread has been reported in 1% to 2% of cases
Optimal treatment remains controversial, centered around when to perform a surgical procedure and when to initiate therapy after surgical resection. Gross total resection may provide prolongation of PFS but the impact on overall survival (OS) has not been proven. Patients with gross total resection who are under age 40 are often carefully monitored without additional treatment until progression. Patients over age 40 or with residual tumor after surgery are typically treated. Results from RTOG 9802 suggest that radiation followed by chemotherapy may be better than radiation alone. Grade 2 oligodendroglioma are chemotherapy sensitive, therefore early use of chemotherapy to delay radiation treatment has been used, but this has not been proven to be a comparable approach.
Median survival of 15 years
Presence of contrast enhancement on MRI reduces the median survival, likely as this finding indicates a higher-grade tumor.
Ependymomas range from grade I tumors (subependymoma and myxopapillary ependymoma) to grade 3. They are frequent in children especially below the age of 3, representing 10% of all intracranial tumors in pediatric population, and although rare in adults they represent the most common adult tumor of the spinal cord.
The commonest location is the fourth ventricle, especially in children
There is positive association with neurofibromatosis type II
On neuroimaging they have typically heterogeneous appearances in all modalities due to areas of necrosis, calcification, cystic change and hemorrhage
Symptoms at presentation depends on tumor localization, with supratentorial ependymomas causing more frequently increased intracranial pressure symptoms, infratentorial location giving raise to cranioneuropathies, ataxia and hydrocephalus, and spinal ependymomas often manifesting with back/radicular pain.
Incidence of spinal seeding, that is, subarachnoid dissemination along CSF pathways with metastases along the spine, ranges from 10% to 22% with higher rate from infratentorial tumors origin site and higher tumor grade.
The molecular classification stratifies patient risks better than histopathological grading and 2016 WHO classification describes nine molecular subgroups associated with specific age groups and risk stratification for both OS and PFS. This classification also incorporates a genetically defined variant characterized RELA fusion positive profile and accounting for the majority of supratentorial ependymomas in children with a worse prognosis than tumors not harboring the RELA fusion.
Surgery is the standard treatment as a complete resection can be curative, particularly for grade 1 ependymomas.
Most of data relate to pediatric population. Current therapeutic strategy includes maximal safe surgical resection, followed by adjuvant radiotherapy with exception of selected cases of supratentorial tumors with no ventricular communication and undergoing gross total resection. Adjuvant chemotherapy has been pursued especially in young children in attempt to avoid or delay radiation therapy, but multiple clinical trials have failed to show a survival benefit.
In adults, surgery is the initial approach for low-grade ependymomas. This may be followed by radiation beam therapy depending on diagnostic findings, resection success, and determination of risk of recurrence. Clinical and neuroimaging monitoring including spine imaging are advised as follow up. In case of recurrence, radiotherapy has been utilized using photon or proton radiation, with focal or craniospinal approach depending on individual risks, previous treatment and response, and dissemination findings. There are few established chemotherapy regimens, although carboplatin, cisplatin, and temozolomide have reported responses. Recent combinations temozolomide with lapatinib and carboplatin with bevacizumab have shown activity in recent clinical trials.
The 10-year OS is about 64% in pediatric patients, with older patients doing better than younger ones, and ranges from 70% to 89% in adult patients.
Two molecular subgroups of ependymoma (posterior fossa EPN-A subgroup that is characterized by DNA hypermethylation and supratentorial RELA-fusion) have been associated with poor outcome with 10-year OS of 50% and PFS of 20%.
Recurrence rate is variable, usually occurring between 18 and 45 months, and traditionally with local relapses.
Nonglial tumors of the brain are more commonly meningiomas and acoustic schwannomas, followed by embryonal tumors, pituitary tumors, primary CNS lymphoma as well as tumors of the pineal gland and choroid plexus tumors. Rare tumors are slightly more common in men than in women, occurring across age ranges depending on tumor types and being characterized by longer survival rates than glial tumors. Detailed discussion of rare tumors is beyond the scope of this chapter. The following section will focus on the most common and those of relevance for specific age group or population subgroup.
Meningiomas are extra-axial tumors, that is, they belong to tumors that arise from structures and tissue adjacent to the brain, like meninges. They account for 33.8 % of all brain and CNS tumors and are the most common brain tumors diagnosed above the age of 34 years old. Although they are usually benign, they can be associated with significant morbidity. Female sex, age, inherited susceptibility in DNA repair genes, genetic condition as neurofibromatosis type II, breast cancer and ionizing radiation exposure are recognized factor risks. Other possible predisposing factors are hormones, increased body index, and immunological factors. Meningiomas are classified as benign (grade 1), atypical (grade 2), and anaplastic (grade 3). The 2016 WHO classification has defined brain invasion as criteria for the diagnosis of grade 2 atypical meningioma.
Common feature in sporadic meningiomas is deletion and inactivation of NF2 on chromosome 22.
Malignant meningiomas have more genomic instability with multiple chromosomal copy number alterations, including loss of 1p, 10q, and 14q, and less frequently 6q and 18q.
Familial meningiomas usually have germline defect in NF2 and other predisposing mutations.
Specific mutations have been recently defined in subsets of meningioma. These include SMO and AKT, representing potential therapeutic targets.
Epigenetic aberration as DNA methylation events may be predominant in meningioma biology.
They are usually benign and slow-growing tumors, with clinical insidious onset. Most often present with focal neurological signs, headache, visual field defects, and seizures (up to 50% of patients). In addition to symptoms caused by compression and increased intracranial pressure, parasagittal meningiomas can cause symptoms due to dural venous sinus obstruction. The most common locations in descending order are convexity, parasagittal, sphenoid, and middle cranial fossa.
Presence of broad dural base, dural tails, diffuse contrast enhancement, preservation of arachnoid plane
Calcification of T2 signal iso or hypointensity may predict decreased growth potential
X ray and CT can display hyperostosis or lytic lesions by direct invasion or primary intraosseous meningiomas
Alanine peak, decreased N-acetylaspartate (NAA) and distinct peak of the chemical substance resonating at 3.8 ppm detected by MR spectroscopy are unique to meningiomas
Cerebral angiography and MR venogram to assess patency of dural-based blood sinuses are useful in planning treatment timing and evaluation
Incidental finding of meningioma may only require observation until become symptomatic. In particular, observational management with no treatment intervention may be pertinent in asymptomatic tumors in elderly as they are exposed to increased morbidity risks from treatment. Careful evaluation and consideration should be given to women of child-bearing potential with meningiomas because it may lead to tumor growth as a consequence of excessive hormone production during pregnancy. In addition, conflicting data have been published regarding the link between meningioma and hormonal replacement therapy (HRT) use but larger studies seem to confirm this positive association raising questions on HRT use in women.
Treatment goal is complete surgical resection, as it has a main impact on preventing recurrence.
Radiotherapy is typically performed for grade 3 meningiomas and incompletely resected grade 2 tumors. Radiation treatment for incompletely resected grade 1 and completely resected grade 2 meningiomas remains controversial.
Chemotherapy is considered in recurrent meningiomas refractory to other treatment or when there are no other treatment options. Despite the presence of hormonal receptors, meningiomas are nonresponsive to hormonal therapy. Treatment regimens include α-interferon, somatostatin receptor agonists, and vascular endothelial growth factor (VEGF) signaling pathway inhibitors
95% of meningiomas are benign
Deletion of 1p is associated with higher recurrence rate
Recurrence rate varies from 5% to 20% within 10 years and increases with the length of follow-up
Loss of 14q and loss of 9p with a specific CDKN2A impairment is associated with worse prognosis
Medulloblastomas are the most common malignant brain tumor of childhood, accounting for up to 25% of all pediatric CNS tumors and 40% of pediatric posterior fossa tumors. Around 80% of patients present between the age of 1 and 10 years old. Most commonly they present as midline masses in the roof of the fourth ventricle. In the adult population, where they account for 0.4% to 1% of brain tumors, they present in the third or fourth decade in atypical location. Medulloblastomas are associated with a variety of genetic syndromes as Coffin-Siris, Cowden, Gardner, Gorlin, Li-Fraumeni, Turcot, and Rubinstein-Tyabi syndromes.
Medulloblastoma classification has undergone major reconstruction in the 2016 WHO revision. The combination of four histological groups and four genetic variants stratifies the prognostic risks and outcome of patients from low-risk tumors (WNT-activated) to high-risk tumors (SHH-activated/TP53 mutant, and non WNT/non-SHH).
Up to 40% of medulloblastomas present abnormalities of chromosome 17
P53 mutation has no prognostic implication on WNT subgroup while it does affect prognosis on SHH subgroup almost doubling 5-year OS in the p53 wild-type
Half of the patients have a short symptomatic interval of 6 weeks prior to diagnosis and the main symptoms are linked to intracranial hypertension and hydrocephalus, with a combination of the most frequent symptoms and signs being papilledema, headache, recurrent vomiting, ataxia, nystagmus, and appendicular dysmetria. Risk stratification is based on localized disease at diagnosis and total or near-total resection for average risk group versus disseminated disease at diagnosis and/or partial resection for high risk group.
Typically, CT and MRI reveal a contrast enhancing posterior fossa mass on the midline
94% of pediatric medulloblastomas localize in the cerebellum and three quarter in the vermis
Adult medulloblastomas localize in the cerebellar hemispheres
Iso to hyperintense to grey matter in T2/FLAIR sequences with heterogeneous appearance due to cystic formation and presence of calcification and necrosis
Standard therapy consists of surgical resection followed by craniospinal irradiation (36 Gy or reduced 24 Gy in localized disease) with boost to the primary tumor site (32.4 Gy) and metastatic sites. Treatment in pediatric population with medulloblastoma is stratified on risk-adapted strategies and studies are being carried out to evaluate the risk/benefit ratio of reducing further radiation dose to the neuraxis in children to 18 Gy. Radiotherapy in children below 3 years of age is controversial because of the more severe neurodevelopmental effect of the treatment, and chemotherapy is often used to fill the interval gap before radiation therapy could be given with less long-term side effects burn.
In average-risk patients with nondisseminated disease, reduced dose RT with adjuvant chemotherapy consisting in 8 cycles of lomustine (CCNU), vincristine, and cisplatin regimen, has been beneficial showing 3-year PFS rate of around 80%.
In high risk patients, chemotherapeutic agents typically used are cisplatin, carboplatin, cyclophosphamide, and vincristine.
In recurrent disease, attempts with high dose chemotherapy (cyclophosphamide) and with stem cell harvest for possible transplant has been tested, and treatments targeting molecular pathways such as SHH are under investigation.
There are few evidence-based guidelines for treatment in the adult population, and treatment considerations are modeled by data extrapolation from the pediatric experience. Surgery followed by craniospinal radiotherapy at 36 Gy with boost of 18.8 Gy to the origin tumor site in partial resection is the mainstream treatment approach. Proton beam RT of the neuraxis should always been considered as preferred option in adults as it significantly decreases the long-term side effects, spares the bone marrow reserve and ovary toxicity in women, and it is better tolerated. New therapies are evaluated in clinical trials based on subgroup molecular profiling of medulloblastomas.
Disease-wide 5-year survival stands at 60% to 70%.
17p loss has been associated with poor outcome.
MYC gene amplification and TP53 mutations are prognostic factors of poor outcome.
Metastatic disease at diagnosis (seeding in one third of patients at diagnosis), age <3 years and disease relapse, are very poor prognostic factors.
Staging evaluation is important and has been historically based on tumor size and extent of metastatic disease (spinal dissemination, bone marrow invasion) by Harisiadis and Chang in 1977.
Despite 5-year survivorship with current therapies up to 80%, current treatment toxicity and long-term sequelae significantly impact the neurological and neurocognitive development of pediatric patients.
Primary CNS Lymphomas (PCNLS) are extranodal high grade non-Hodgkin B-cell lymphomas (NHL) arising in the CNS (brain, eyes, leptomeninges, spinal cord) that are diagnosed in the absence of systemic lymphomas and that typically remains in the brain. They account for 3% to 5% of all brain tumors and 1% of NHL. After a steady increase in incidence since the end of the 20th century, over the past decade, there has been a plateau or even a decrease in incidence of PCNLS among immunodeficient patients, likely linked to improved treatment and outcome of HIV/AIDS patients. Nonetheless, the incidence among immunocompetent elderly population remains high, with median age at diagnosis of 60 years old.
A prominent risk factor for the development of PCNSL is immunodeficiency, due to congenital disorders, iatrogenic immunosuppression, and most notably, HIV that historically increased the risk of developing PCNLS by 3600-fold. It is also strongly associated with Epstein-Barr virus (EBV) infection in immunosuppressed patients and immunocompetent elderly patients treated with mychopenolate mofetil, methotrexate, or azathioprine.
Presentation is usually with focal neurologic symptoms and symptoms of increased intracranial pressure. Elderly patients more commonly present with change in behavior and personality. A profound steroid-induced response is classic but may prevent a tissue diagnosis; therefore, steroids should be withhold until tissue confirmation of diagnosis. CSF analysis highlights lymphomatous cells in 10% to 30% of patients. High suspicion is raised in HIV/AIDS patients with classic lesion on brain imaging and positive EBV DNA in the CSF. As systemic involvement is rare, staging is often achieved through neuroimaging, HIV testing, CSF analysis, ocular slit-lamp examination, and clinical assessment. In selected cases body CT scan and bone marrow biopsy are pursued. For occult lymphoma, FDG body PET is required.
CNS lymphoma is typically manifest on MRI or CT as homogeneously enhancing solitary (two third of cases) or multiple lesions in the periventricular areas.
Ring enhancement is more commonly seen in immunodeficient patients.
Rapid leakage of contrast medium is reflected by distinct signal-time intensity curves.
Significant elevation of lipid resonance at spectroscopy studies.
Despite poor overall prognosis, the treatment of primary brain lymphoma has made advances and 20% to 30 % of patients can achieve long-term remission. The standard of care for PCNSL is systemic chemotherapy with or without whole brain radiotherapy (WBRT) or intrathecal chemotherapy. Traditionally surgery has been discouraged apart from diagnostic biopsy but this paradigm has been recently challenged by a German PCNSL study group that showed increased PFS and OS in patients undergoing subtotal or gross total resection. WBRT usually at dosage of 40 to 50 Gy has several limitations including delayed neurotoxicity especially on neurocognitive functions, while low dose radiation (23.4 Gy) in patients older than 60 years old, the group most prone to late radiation effects does not seem to have comparable efficacy to the higher dose regimens. High dose methotrexate has been used as induction chemotherapeutic regimen, and it is usually coupled with preventive measures to limit its toxicity side effects as hydration, urine alkalization, and avoidance of interacting agents (penicillin derivatives). High-dose chemotherapeutic consolidation has been investigated in a further attempt to decrease the need for radiation. Although several regimens are in use, most centers incorporate high-dose cytarabine and etoposide. For newly diagnosed PCNSL patients, a novel program has evaluated immunochemotherapy combination regimen (induction consisting of methotrexate, temozolomide, and rituximab followed by consolidative infusional etoposide plus high-dose cytarabine), with promising results. In recurrent disease, a key consideration is whether the lymphoma is methotrexate-sensitive, while other salvage treatments including autologous stem-cell transplantation are under study.
The significant advances in PCNSL treatment lead to anticipate that between 40% to 50% of PCNSL patients will exhibit long-term survival and a significant proportion may be cured. Nonetheless, research stresses the importance of future developments as at least 40% to 50% of PCNSL patients will develop disease refractory to the current treatment agents
Most pineal region masses are malignant cell tumors that occur in young male patients, the most frequent being germinoma. Given the location, these tumors can compress the aqueduct resulting in hydrocephalus. Symptoms are therefore related to increased intracranial pressure with headache, nausea and vomiting, and cranial nerve palsies. When tumor compress the superior colliculi, it leads to Parinaud syndrome characterized by impaired upgaze, convergence and retraction nystagmus, eyelid retraction also called Collier sign and pupillary light-near dissociation. Tumor markers may be increased as α -fetoprotein, α-human chorionic gonadotropin, and placental alkaline phosphatase.
Pineal region masses give rise of a wide differential due to the variety of cell types in the region. Several characteristics may help differentiating them, although biopsy is indicated for diagnosis confirmation:
Germ cell tumors, half of which being germinoma, usually appear as homogeneous mass with signal intensity and attenuation similar to those of gray matter; engulfing a densely calcified pineal gland.
Pineal parenchyma tumors are usually either pineocytomas (grade 1) or pineoblastomas (grade 4). Recently, a third category has been described, pineal parenchymal tumor of intermediate differentiation, graded as a WHO II-III depending upon cellular characteristics.
Pineal parenchymal tumors demonstrate calcifications dispersed peripherally to the mass on neuroimaging.
Pineal cysts often have a rim thin enhancement at contrast imaging.
Tentorial meningioma tends to depress cerebral veins while intrinsic pineal tumors tend to cause upward displacement of the internal cerebral veins.
Tectal astrocytoma are slightly hyperintense on T2 weighted images, can present cystic spaces and calcifications, and usually do not or minimally enhance.
Diagnostic evaluation should include craniospinal MRI and CSF analysis. Biopsy should be pursued depending on the suspected lesion with stereotactic or endoscopic approach in germinoma or tectal glioma and microsurgical techniques for open biopsy in other cases. Radiation therapy is first-line treatment for germinomas. Craniospinal radiotherapy associated with adjuvant chemotherapy is pursued when there is evidence of CSF seeding or malignant tumor.
METASTATIC CNS TUMORS
Tumour cells detaching from systemic tumours spread hematogenously into the CNS by producing and secreting angiogenic substances that enable them to open the BBB locally. Different cancers show different intracranial compartment tropism.
Brain Metastases
Brain metastases occur in 15% to 40% of patients with systemic cancer. Although the true incidence of metastatic brain tumors remains unknown, it is following an increasing trend likely due to better control of systemic cancer and prolonged survival. Still, it remains undetected in 15% of patients. Aging is a factor risk and the most frequent primary tumor origin is lung, breast, melanoma, and colorectal. Hematological tumors constitute 10% of brain metastases and primarily affect the leptomeninges. Presentation is usually with focal neurologic deficits related to mass compression, edema, and increased intracranial pressure. From a neuroimaging point of view lesions are characteristically localized at the grey/white matter junction and are surrounded by significant edema with higher edema/tumor size ratio. Most metastatic brain lesions are hypointense on T1 weighted images on MRI that could indicate haemorrhage and necrosis, and hyperintense on T2 weighted images.
Medical management usually comprise the use of oral steroids to decrease the edema at common dose range of dexamethasone 4 to 8 mg/day and up to 16mg/day for severe symptoms. One quarter of patients do present with seizure and antiepileptic treatment is indicated. There is no evidence based role for seizure prophylactic treatment, although short prophylactic treatment with a one week course and rapid tapering off scheme has been adopted for the perioperative period. Often there is an increased risk of venous thromboembolism partly due to chemotherapeutic agents use, and in high-risk patients, perioperative prophylaxis with heparin has reduced the related mortality without increasing the risk of intracranial haemorrhage.
The treatment of brain metastases is individualised and tailored in the context of the primary tumor and the patient’s overall systemic options. Depending on the primary tumor staging and grading, brain metastases treatment may encompass surgery, radiotherapy and in certain cases chemotherapy for chemosensitive tumors as small cell lung carcinoma, germ cell tumors, and lymphoid neoplasms. The other predictive factor of chemotherapy response to be considered is the BBB permeability of the agent, although its importance is much less critical than chemotherapy agent choice based on primary tumor efficacy. The overall prognosis is often below a year of survival but there is a wide heterogeneity that can be stratified depending on primary disease control, age of the patient (>65 years old), functional status of the patients, Karnofsky performance score (KPS< 70), single or multiple brain metastases. Interestingly, there is now evidence that brain metastases can be prevented by targeted therapy of the primary brain tumor. In patients with good prognosis, emphasis is placed on balancing treatment effectiveness against neurotoxicity and the goal of therapy has shifted from short-term palliation to long-term survival and quality of life (QoL). Hence, whole-brain radiation therapy (WBRT) is less preferable in situations where stereotactic radiosurgery and systemic agents are reasonable options. Surgical treatment with removal of brain parenchyma adjacent to the metastatic lesion confers better local control than gross total resection. The pathologic confirmation of tumor-free resection margins provides rate of local recurrence comparable to standard gross total resection and adjuvant radiotherapy.
Spinal Metastases
Metastases of the spine most frequently involve vertebral elements and epidural space and arise from lung, breast, liver, skeletal, and prostate primary tumors. Management includes tailored approach depending on individual localization, clinical symptomatology, and previous treatments, and may include combination of steroids, surgery, radiation, and chemotherapies. Spinal cord compression because of these metastases to the spine with extension into the spinal canal represents a true oncologic emergency as neurologic deficits may not improve particularly if the compression results in vascular compromise to the spinal cord. Metastases that involve the subarachnoid space are leptomeningeal metastases. Intradural metastases are far less common but are typically referred to as drop metastases as they are extramedullary and either compress the cord or invade into the spinal cord parenchyma.
Neoplastic Meningitis
Meningeal involvement can occur by local infiltration or by dissemination of tumor cells by the cerebrospinal flow. Seeding of the leptomeninges by malignant cells may occur in primary brain tumor patients as well as in cancer patients for both hematological (more frequent) and solid (breast, lung, and melanoma) tumors in a percentage that is variable from 1% to 15%. Extensive investigations with contrast MRI of brain and spine, as well as repeated lumbar punctures for CSF analysis are needed as CSF cytology may be negative in almost half of the patients. Neoplastic meningitis causes progressive neurological dysfunction. Treatment with focal radiation to areas of bulk disease or neurologic symptoms from involvement of cranial or spinal nerves may improve function. In individual cases, intrathecal chemotherapy may be of benefit and more recently selected chemotherapy agents have been administered at high doses systemically to generate therapeutic concentrations within the CSF. The optimal treatment of patients with leptomeningeal cancer is based on consideration of the primary cancer type, patient’s performance status, CSF disease burden, and extent of systemic disease.
SUMMARY
Treatment of cancer in the central nervous system is complicated. Primary brain tumors, while rarely spreading outside of the CNS, are typically invasive therefore not curable with surgery. Radiation and chemotherapy regimens have been developed for most primary brain tumors and are being increasing refined by tumor type and recently molecular subtypes, underscoring the importance of accurate histologic and molecular classification. Secondary CNS cancers (brain and leptomeningeal metastases) are often late complications of systemic cancer and optimal treatment is based on the cancer type and stage or extent of the systemic disease. In all patients, realistic appraisal of treatment outcomes in the context of both short- and long-term toxicities highlights the need for systematic evaluation of patient outcomes to provide patients with cancer information necessary for informed decision making.
Suggested Readings
1.Amirian ES, Scheurer ME, Zhou R, et al. History of chickenpox in glioma risk: a report from the glioma international case-control study (GICC). Cancer Med. 2016;5(6):1352–1358.
2.Blakeley JO, Grossman SA. Management of pienal region tumors. Curr Treat Options Oncol. 2006;7(6):505–516.
3.Blitshten, S, Crook JE, Jaeckle KA. Is there an association between meningioma and hormone replacement therapy? J Clin Oncol. 2008;26(2):279–282.
4.Bouffet E, Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv Syst. 1999;15(10):563–570.
5.CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors diagnosed in the United States in 2004-2008. Central Brain Tumor Registry of the United States; 2008.
6.Chamberlain MC. Treatment of meningioma, including in cases with no further surgical or radiotherapy options. Oncology. 2015;29(5):369–371.
7.Crocetti E, Trama A, Stiller C, et al. Epidemiology of glial and non-glial brain tumors in Europe. Eur J Cancer. 2012;48(10):1532–1542.
8.Demir MK, Iplikcioglu AC, Dincer A, et al. Single voxel proton MR spectroscopy findings of typical and atypical intracranial meningiomas. Eur J Radiol. 2006;60(1):48–55.
9.Fox BD, Cheung VJ, Patel AJ, et al. Epidemiology of metastatic brain tumors. Neurosurg Clin N Am. 2011;22(1):1–6.
10.Fraser E, Gruenberg K, Rubenstein JL, et al. New approaches in primary central nervous system lymphoma. Chin Clin Oncol. 2015;4(1):11.
11.Gottardo NG, Gajjar A. Chemotherapy for malignant brain tumors of childhood. J Child Neurol. 2008;23(10):1149–1159.
12.Jun P, Hong C, Wong JM, et al. Epigenetic silencing of the kinase tumor suppressor WNK2 is tumor-type and tumor-grade specific. Neuro Oncol. 2009;11(4):414–422.
13.Kleinschmidt-DeMasters BK, Damek DM, Lillehei K, et al. Epstein Barr virus-associated primary CNS lymphomas in elderly patients on immunosuppressive medications. J Neuropathol Exp Neurol. 2008;67(11):1103–1111.
14.Korshunov A, Ryzhova M, Hovestadt V, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015;129(5):669–678.
15.Kousi E, Tsougos I, Fountas K, et al. Distinct peak at 3.8 ppm observed by 3T MR spectroscopy in meningiomas, while nearly absent in high-grade gliomas and cerebral metastases. Mol Med Rep. 2012;5(4):1011–1018.
16.Kuratsu J, Kochi M, Ushio Y. Incidence and clinical features of asymptomatic meningiomas. J Neurosurg. 2000;92(5):766–770.
17.Lafay-Cousin L, Mabbott DJ, Halliday W, et al. Use of ifosfamide, carboplatin, and etoposide chemotherapy in choroid plexus carcinoma. J Neurosurg Pediatr. 2010;5(6):615–621.
18.Louis, DN, Perry A, Reinfeberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neurpathol. 2016;131(6):803–820.
19.Ma C, Cao L, Zhao J, et al. Inverse association between Prediagnostic IgE Levels and the risk of brain tumors: A systematic review and meta-analysis. BioMed Res Int. 2015;2015:94213.
20.McCabe MG, Backlund LM, Leong HS, et al. Chromosome 17 alterations identify good-risk and poor-risk tumors independently of clinical factors in medulloblastoma. Neuro Oncol. 2011;13(4):376–383.
21.Metha M, Vogelbaum MA, Chang S, et al. Neoplasms of the central nervous system. In L. T. DeVita Jr, Cancer: Principles and Practice of Oncology. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2011:1700–1749.
22.Mohile NA, DeAngelis LM, Abrey LE. The utility of body FDG PET in staging primary central nervous system lymphoma. Neuro Oncol. 2008;10(2):223–228.
23.Ostrom QT, Gittleman H, Liao P, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2014;16(4):iv1–63.
24.Pajtler KW, Witt H, Sill M, et al. Molecular classification of Ependymal Tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cel. 2015;27(5):728–743.
25.Palma L, Celli P, Mariottini A, et al. The importance of surgery in supratentorial ependymomas. Long-term survival in a series of 23 cases. Childs Nerv Syst. 2000;16(3):170–175.
26.Perreault S, Ramaswamy V, Achrol AS, et al. MRI surrogates for molecular subgroups of medulloblastoma. Am J Neuroradiol. 2014;35(7):1263–1269.
27.Perry JR, Lapierre N, O’Callaghan CJ, et al. Short-course radiation plus temozolamide in elderly patients with glioblastoma. N Engl J Med. 2017;376:1027–1037.
28.Pizer BL, Clifford SC. The potential impact of tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg. 2009;23(4):364–375.
29.Sjostrom S, Hjalmars U, Juto P, et al. Human immunoglobulin G levels of viruses and associated glioma risk. Cancer Causes Control. 2011;22(9):1259–1266.
30.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant Temozolomide verusu radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC study. Lancet Oncol. 2009;10(5):459–466.
31.Sundeep D, Lynch CF, Sidenaller ZA, et al. Trends in brain cancer incidence and survival in the United States: Surveillance, Epidemiology, and End Results Program, 1973 to 2001. Neurosurg Focus. 2006;20(4):E1.
32.Weller M, Martus P, Roth P, et al. Surgery for primary CNS lymphoma? Challenging a paradigm. Neuro Oncol. 2012;14(12):1481–1484.
33.Wiemesl J, Wrensch M, Claus EB. Epidemiology and etiology of meningioma. J Neuro Oncol. 2010;99(3):307–314.
34.Yew A, Trang A, Nagasawa DT, et al. Chromosomal alterations, prognostic factors, and targeted molecular therapies for malignant meningiomas. J Clin Neurosci. 2013;20(1):17–22.
35.Zhukova N, Ramaswamy V, Remke M, et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol. 2013;31(23):2927–2935.