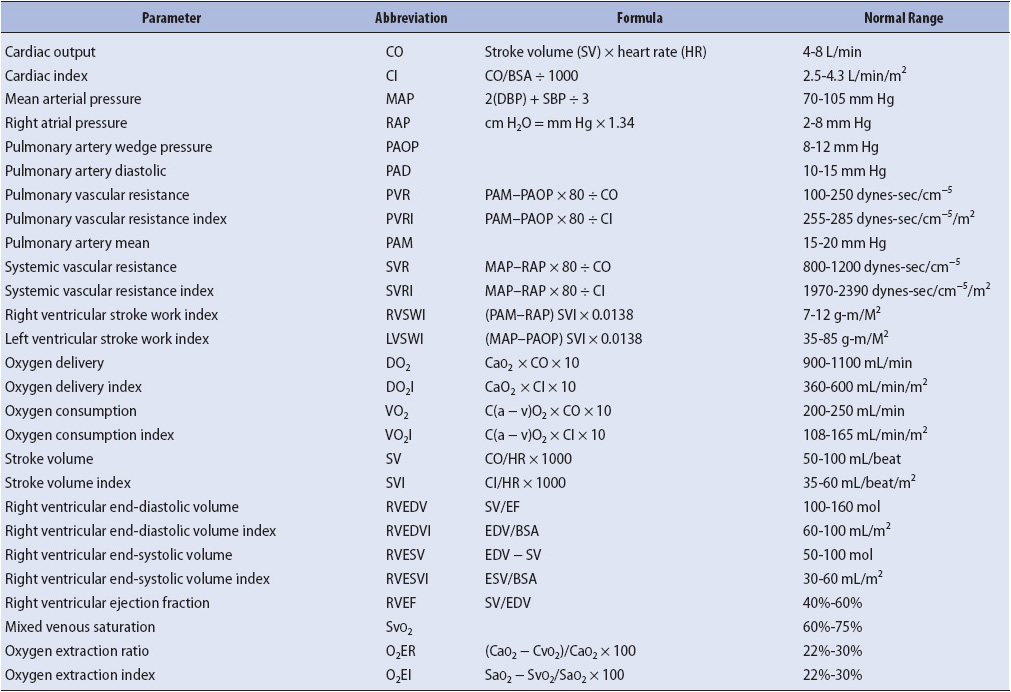
TABLE 4-1. NORMAL HEMODYNAMIC AND BLOOD FLOW PARAMETERS
KNOWLEDGE COMPETENCIES
1. Identify the characteristics of normal and abnormal waveform pressures for the following hemodynamic monitoring parameters:
• Central venous pressure
• Arterial blood pressure
2. Describe the basic elements of arterial and venous pressure-monitoring equipment and methods used to ensure accurate pressure measurements.
3. Discuss the indications, contraindications, and general management principles for the following common hemodynamic monitoring parameters:
• Central venous pressure
• Arterial blood pressure
4. Describe the clinical application of SVO2/SCVO2 monitoring.
5. Discuss the potential use of bioimpedance monitoring in a progressive care population.
The term hemodynamics refers to the interrelationship of blood pressure (BP), blood flow, vascular volumes, heart rate, ventricular function, and the physical properties of the blood. While a wide variety of hemodynamic monitoring systems are available in critical care units that are used to monitor these components, hemodynamic monitoring in the progressive care setting is generally limited to central venous pressure (CVP) monitoring and arterial monitoring. These two technologies yield a tremendous amount of useful data and thus it is essential that progressive care nurses have a working knowledge of how to obtain accurate data, analyze associated waveforms, and interpret and integrate the data. The end of this chapter focuses on these specific technologies in addition to providing a brief introductory description of other hemodynamic parameters obtained from a pulmonary artery catheter. The concepts are helpful to understand pathologic conditions discussed later in this book such as heart failure and sepsis. More extensive discussion of other hemodynamic monitoring systems such as the pulmonary artery catheter is found in the AACN Essentials of Critical Care Nursing.
Clinical examination can be a poor predictor of hemodynamics. Although noninvasive assessment techniques such as physical examination, history taking, and laboratory analysis are helpful and necessary, they do not provide the specific physiologic data that may be obtained with measurements such as the CVP. These technologies can directly measure the pressures and provide real-time data to clinicians so that appropriate interventions are ensured.
A description of the various parameters derived from the pulmonary artery catheter follows and reference values may be found in Table 4-1.
Cardiac output (CO) is the amount of blood pumped by the ventricles each minute. It is the product of the heart rate (HR) and the stroke volume (SV) (the amount of blood ejected by the ventricle with each contraction; Figure 4-1). This is evaluated with a pulmonary artery catheter and some noninvasive monitoring technologies such as bioimpedance systems.
CO = HR × SV
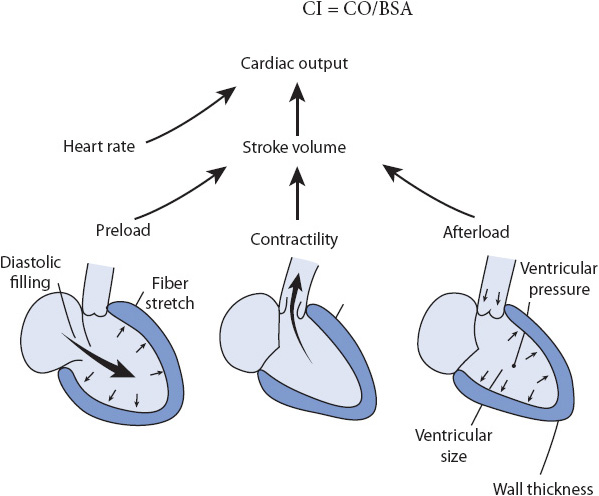
Figure 4-1. Factors affecting CO. (Reprinted from Price S, Wilson L. Pathophysiology: Clinical Concepts of Disease Processes. Philadelphia, PA: Mosby; 1992: 390, with permission from Elsevier.)
The normal value is 4.0 to 8.0 L/min. It is important to note that these values are relative to size. Values within the normal range for a person 5 ft tall weighing 100 lb, may be totally inadequate for a 6-ft, 200-lb individual. Cardiac index (CI) is the CO that has been adjusted to individual body size. It is determined by dividing the CO by the individual’s body surface area (BSA), which may be obtained from the DuBois body surface area chart or by pressing the CI button on the cardiac monitor. The normal value is 2.5 to 4.3 L/min/m2.
Cardiac Output measurements are used to assess the patient’s perfusion status, response to therapy, and as a rapid means to evaluate the patient’s hemodynamic status. As mentioned, CO is composed of HR and SV, or the amount of blood ejected with each contraction of the ventricle. Normal SV range is 60-100 mL/beat. SV depends on preload, afterload, and contractility. Therefore, CO is determined by:
1. HR (and rhythm)
2. Preload
3. Afterload
4. Contractility
As the SV of the left ventricle is a component used in the determination of CO, any condition or disease process which impairs the pumping (ejection) or filling of the ventricle may contribute to a decreased CO. Alterations that lead to diminished CO can be divided into two general categories: inadequate ventricular filling and inadequate ventricular emptying.
Factors that lead to inadequate ventricular filling include arrhythmias, hypovolemia, cardiac tamponade, mitral or tricuspid stenosis, constrictive pericarditis, and restrictive cardiomyopathy. Each of these abnormalities leads to a decrease in preload (the amount of volume in the ventricle at end diastole), which results in a decrease in CO.
Factors that lead to inadequate ventricular emptying include mitral/tricuspid insufficiency, myocardial infarction, increased afterload (hypertension, aortic/pulmonic stenosis), myocardial diseases (myocarditis, cardiomyopathy), metabolic disorders (hypoglycemia, hypoxia, severe acidosis), and use of negative inotropic drugs (beta-blockers, calcium channel blockers).
In theory, in the normal, healthy individual, any factor that increases HR and contractility and decreases afterload can contribute to an increase in CO. Hyperdynamic states, such as seen in sepsis, anemia, pregnancy, and hyperthyroid crisis, may cause CO values to be increased. Increased HR is a major component in hyperdynamic states; however, in sepsis a profound decrease in afterload also contributes to an increased CO.
Rate
Normal HR is 60 to 100 beats/min. In a normal, healthy individual, an increase in HR can lead to an increase in CO. In a person with cardiac dysfunction, increases in HR can lead to a decreased CO and often myocardial ischemia. The increase in HR decreases the ventricular filling time by reducing preload, which decreases SV and leads to decreased CO.
A lower HR does not necessarily result in a decrease in CO. Decreased heart rates with normal COs are often found in athletes. Their training and conditioning strengthens the myocardium such that each cardiac contraction produces an increased SV. In individuals with left ventricular (LV) dysfunction, a slow HR can produce a decrease in CO. This is caused by decreased contractility, as well as fewer cardiac contractions each minute.
Because CO is a product of SV times heart rate, any change in SV normally produces a change in the heart rate. If the SV is elevated, the HR may decrease (eg, as seen in adaptation to exercise). If the SV falls, the HR normally increases. Subsequently, evaluating the cause of the tachycardia becomes an essential component of hemodynamic assessment. Bradycardias and tachycardias are potentially dangerous because they may result in a decrease in CO if adequate SV is not maintained. Bradycardias that develop suddenly are almost always reflective of a falling CO. The cause of tachycardia, on the other hand, must be determined because it may not reflect a low output state but rather a normal physiologic response (eg, tachycardia secondary to fever). Heart rate varies between individuals and is related to many factors. Some are described below.
• Parasympathetic stimulation (vagus nerve stimulation) is a common occurrence in the acute care setting. It can occur with Valsalva maneuvers such as excessive bearing down during a bowel movement, vomiting, coughing, and suctioning.
• Conduction abnormalities, especially second- and third-degree blocks, are often seen in patients with cardiovascular diseases. Many drugs used in the progressive care setting may lead to a decreased HR, including digitalis, beta-blockers, calcium channel blockers, and phenylephrine (Neosynephrine).
• Athletes often have resting heart rates below 60 beats/min without compromising CO.
• The actual HR is not as important as the systemic effect of the heart rate. If the patient’s HR leads to diminished perfusion (decreased level of consciousness, decreased urinary output, hypotension, prolonged capillary refill, new-onset chest pain, and the like), treatment is initiated to increase the heart rate.
• Stress, anxiety, pain, and conditions resulting in compensatory release of endogenous catecholamines such as hypovolemia, fever, anemia, and hypotension may all produce tachycardia.
• Drugs with a direct positive chronotropic effect include epinephrine and dopamine.
Tachycardia is very common in acutely ill patients. When evaluating a rapid heart rate, each of the main sources for the tachycardia is evaluated; for example, if a patient has a HR of 120 beats/min, the clinician rules out such factors as fever, pain, and anxiety before assuming that the tachycardia is due to a reduced SV. Once these are ruled out, an investigation of the cause of a low SV is accomplished. The two most common reasons for a low SV are hypovolemia and LV dysfunction. Both causes of low SV can produce an increased HR if no abnormality exists in regulation of the HR (such as autonomic nervous system dysfunction or use of drugs that interfere with the sympathetic or parasympathetic nervous system such as beta-blockers).
An increased HR can compensate for a decrease in SV, although this compensation is limited. The faster the heart rate, the less time exists for ventricular filling. As an increased HR reduces diastolic filling time, the potential exists to eventually reduce the SV. There is no specific HR where diastolic filling is reduced so severely that SV decreases. However, as the HR increases, it is important to remember that SV may be negatively affected.
Increased HR also has the potential to increase myocardial oxygen consumption (MVO2). The higher the heart rate, the more likely it is that the heart consumes more oxygen. Some patients are more sensitive to elevated MVO2 than others; for example, a young person may tolerate a sinus tachycardia as high as 160 for several days, whereas a patient with coronary artery disease may decompensate and develop pulmonary edema with a HR in the 130s. Keeping heart rates as low as possible—particularly in patients with altered myocardial blood flow—is one way of protecting myocardial function.
Many of us have observed the deleterious effects produced by a supraventricular tachycardia, or a change from normal sinus rhythm to atrial fibrillation or flutter. Loss of “atrial kick” may contribute to decreased CO. Normally, atrial contraction contributes 20% to 40% of the ventricular filling volume. With tachycardia, that atrial contribution to SV may diminish significantly. Although those with normal cardiac function are unlikely to experience compromise, it is more likely in those with impaired cardiac function.
Stroke volume is the amount of blood ejected from each ventricle with each heartbeat. The right and left ventricle eject nearly the same amount, which normally is from 50 to 100 mL per heartbeat.
SV = CO/HR × 1000
Stroke Volume indexed to the patient’s BSA is SVI. Indexing helps compare values regardless of the patient’s size. Normal SVI is 33-47 mL/beat/m2. Common causes of decreased stroke volume/stroke volume index (SV/SVI) is inadequate blood volume (preload), impaired ventricular contractility (strength), increased systemic vascular resistance (SVR; afterload), and cardiac valve dysfunction. High SV/SVI occurs when the vascular resistance is low (sepsis, use of vasodilators, neurogenic shock, and anaphylaxis).
The ejection fraction (EF) is defined as how much blood is pumped with each contraction in relation to the volume of available blood; for example, assume the left ventricular end-diastolic volume (LVEDV is the amount of blood left in the heart just before contraction) is 100 mL. If the SV is 80 mL the EF is 80%; 80 mL of the possible 100 mL in the ventricle were ejected. Right ventricular volumes are roughly equal to those of the left ventricle (RVEF) (Table 4-1). A normal EF is usually over 60%. This is evaluated with a pulmonary artery catheter or with noninvasive methods such as the echocardiogram.
The EF may change before the SV in certain conditions, such as LV failure and sepsis; for example, the left ventricle may dilate in response to LV dysfunction from coronary artery disease, and LVEDV increases. Although the increase in LVEDV may prevent a drop in SV, EF may not be preserved. SV and SVI, then, are the best available measures to assess left and right ventricular dysfunction.
Preload is the volume of blood that exerts a force or pressure (stretch) on the ventricles during diastole. It may also be described as the filling pressure of the ventricles at the end of diastole or the amount of blood that fills the ventricles during diastole.
According to the Frank-Starling law of the heart, the force of contraction is related to myocardial fiber stretch prior to contraction. As the fibers are stretched, the contractile force increases up to a certain point. Beyond this point, the contractile force decreases and is referred to as ventricular failure (Figure 4-2). With increased preload there is an increase in the volume of blood delivered to the ventricle, the myocardium is stretched, and a more forceful ventricular contraction is produced. This forceful ventricular contraction yields an increase in SV, and therefore, CO. Too much preload causes the ventricular contraction to be less effective. A commonly referred to analogy uses the properties of a rubber band. The more a rubber band is stretched, the greater “snap” is produced when released. The rubber band may be stretched further and further, until it reaches a point where it loses its tautness and fails to recoil. Preload is measured with a CVP catheter and/or a pulmonary artery catheter.
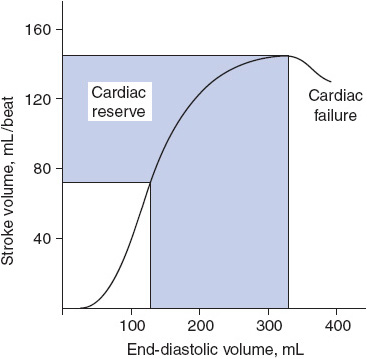
Figure 4-2. Ventricular function curve. As the end-diastolic volume increases, so does the force of ventricular contraction. The SV becomes greater up to a critical point after which SV decreases (cardiac failure). (From Langley LF. Review of Physiology. 3rd ed. New York, NY: McGraw-Hill; 1971.)
Preload is determined primarily by the amount of venous return to the heart. Venous constriction, venous dilation, and alterations in the total blood volume all affect preload. Preload decreases with volume changes. This can occur in hemorrhage (traumatic, surgical, gastrointestinal [GI], postpartum), diuresis (excessive use of diuretics, diabetic ketoacidosis, diabetes insipidus), vomiting and diarrhea, third spacing (ascites, severe sepsis, heart failure [HF]), redistribution of blood flow (use of vasodilators, neurogenic shock, severe sepsis), and profound diaphoresis. Venous dilatation also results in diminished preload. Etiologies that increase venous pooling and result in decreased venous return to the heart include hyperthermia, septic shock, anaphylactic shock, and drug administration (nitroglycerin, nitroprusside) (Table 4-2).
TABLE 4-2. HEMODYNAMIC EFFECTS OF CARDIOVASCULAR AGENTS
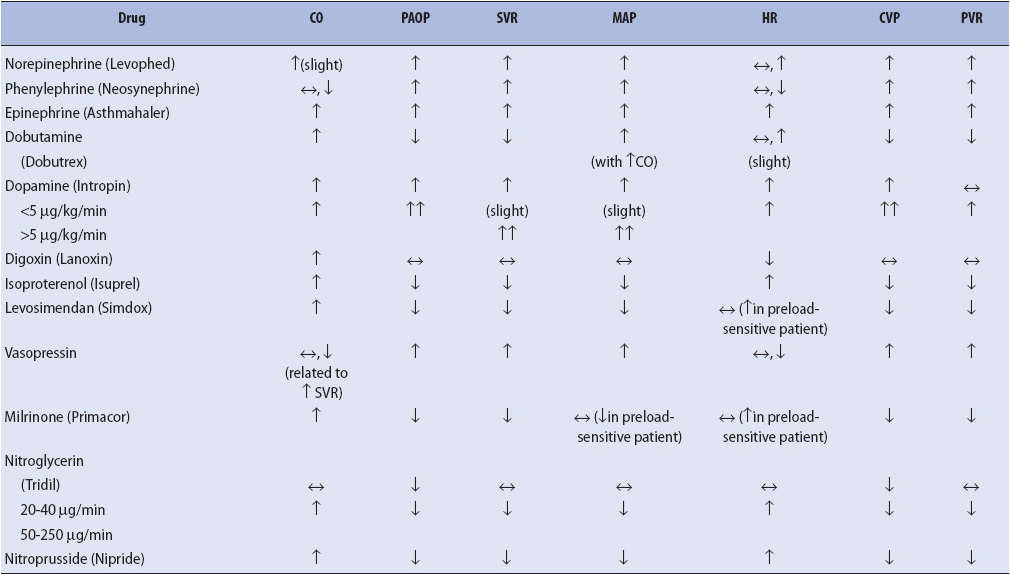
Factors leading to increased preload include excessive administration of crystalloids or blood products and the presence of renal failure (oliguric phase and/or anuria). Venous constriction results in the shunting of peripheral blood to the central organs (heart and brain). The increased venous return results in an increased preload. This may occur in hypothermia, some forms of shock (hypovolemic, cardiogenic, and obstructive) and with administration of drugs that stimulate the alpha receptors (epinephrine, dopamine at doses greater than 10 mcg/kg/min, norepinephrine) (see Table 4-2).
The right ventricle pumps blood into the pulmonary circulation and the left ventricle ejects blood into the systemic circulation. Both circulatory systems are affected by preload, afterload, and contractility. These are discussed below and, when appropriate, the clinical indicators are differentiated by right or left heart.
Normal right ventricular (RV) preload is 2 to 8 mm Hg or 2 to 10 cm H2O (CVP = central venous pressure; RAP = right atrial pressure). Right atrial pressures are measured to assess right ventricular function, intravascular volume status, and the response to fluid and drug administration. CVP/RAP pressures increase because of intravascular volume overload, cardiac tamponade (effusion, blood, and the like), restrictive cardiomyopathies, and RV failure. There are three etiologies of RV failure: (1) intrinsic disease such as RV infarct or cardiomyopathies; (2) secondary factors that increase pulmonary vascular resistance (PVR) such as pulmonary arterial hypertension, pulmonary embolism, hypoxemia, chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), sepsis; and (3) severe LV dysfunction as seen in mitral stenosis/insufficiency or LV failure. In contrast, the only clinically significant reason for a decreased CVP/RAP is hypovolemia. CVP/RAP is a late indicator of alterations in LV function therefore limiting its value in clinical decision making.
A 67-year-old woman is admitted to the progressive care unit with the diagnosis of hypotension secondary to presumed gastrointestinal bleeding. A history of melena for 3 days was provided by her daughter on admission. She is presently unresponsive but is breathing spontaneously. Breath sounds are clear, urine output is 15 mL in 8 hours, and her skin is cool. Laboratory work is pending and 3 U of packed red blood cells have been ordered stat. A CVP catheter is inserted to aid in the provision of fluids, blood, and medications, and to help interpret the situation. The following data are available:

Case Question 1. What findings reinforce that the patient is hypovolemic?
Case Question 2. Which hemodynamic findings are abnormal?
Case Question 3. What treatments are warranted in this patient?
1. The history of melena for the past 3 days, urinary output 15 mLs in 8 hours, and BP 86/54 mm Hg. Reviewing the patient’s BP history would be important–does she usually have a systolic BP less than 90? Lab tests that can be used to trend the volume status would be BUN and Hct. H/H would be especially important in this patient.
2. BP 86/54, PR 118/min, RR 30 breaths/min, CVP 3 mm Hg
3. The exact cause of the hypovolemia cannot be discerned from the CVP by itself but the history and the CVP value are consistent with the need for blood and aggressive fluid resuscitation. The CVP value can be used to target a reasonable preload level, then vasopressors may be added if necessary to further enhance blood pressure should the fluid and blood not do so.
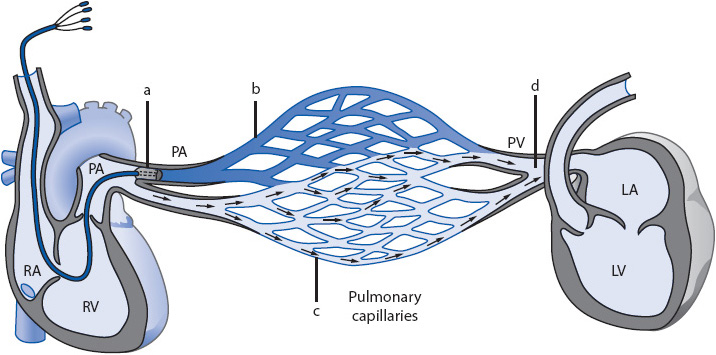
Normal LV preload is 8 to 12 mm Hg and is measured with a pulmonary artery catheter. There are many names that are synonymous with LAP and they include: PCWP = pulmonary capillary wedge pressure; PAOP = pulmonary artery occlusion pressure; and PAWP = pulmonary artery wedge pressure. The most commonly used term is PAOP. The normal PAOP is 8 to 12 mm Hg. The pressure is measured by inserting a small amount of air into the balloon port of the pulmonary artery (PA) catheter: the balloon becomes lodged in a portion of the PA that is smaller than the balloon. This occludes blood flow distal to the catheter tip. The pressure in the left atrium is sensed by the catheter tip. When the mitral valve is open during ventricular diastole, the pressure that is sensed is that of the left ventricle, the left ventricular end-diastolic pressure (LVEDP) or LV preload (Figure 4-3).
Figure 4-3. Schematic representation of the PA in the wedge position. From its position in small, occluded segment of the pulmonary circulation, the PA catheter in the wedged position allows the electronic monitoring equipment to “look through” a nonactive segment of the pulmonary circulation to the hemodynamically active pulmonary veins and left atrium. (Reprinted from Darovic GO. Hemodynamic Monitoring: Invasive and Noninvasive Clinical Application. Philadelphia, PA: WB Saunders; 2002:207, with permission from Elsevier.)
Pulmonary artery occlusion pressure increases because of conditions such as intravascular volume overload, cardiac tamponade (blood, effusion, etc), impaired ventricular relaxation (diastolic dysfunction, restrictive cardiomyopathy, and constrictive pericarditis), and LV dysfunction. Common etiologies of LV dysfunction include mitral stenosis/insufficiency, aortic stenosis/insufficiency, and diminished LV compliance (ischemia, fibrosis, hypertrophy). The only clinically significant reason for a decreased PAOP is hypovolemia.
Afterload is the resistance to ventricular emptying during systole. It is the pressure or resistance that the ventricles must overcome to open the aortic and pulmonary valves and to pump blood into the systemic and pulmonary vasculature. Vascular resistance is determined by the length of a vessel, its diameter or radius, and the viscosity of the blood. The length of the vessel is considered to be constant. The viscosity of the blood is relatively constant except when gross volume changes occur (eg, hemorrhage) or in polycythemia. Therefore, conditions that alter the diameter of the vessels, or the outflow tract, have a primary effect on the afterload of the ventricles.
As afterload increases (vasoconstriction or obstruction of the outflow tract), the heart must work harder to eject the volume. Afterload affects the isovolumetric contraction phase of the cardiac cycle. During this phase, the ventricular pressure rises so the ventricles are able to overcome the existing vascular resistance, open the semilunar valves, and eject the contents. Once the pressure within the ventricle is higher than the pressure in the aorta/pulmonary system, the valves open and the blood is ejected from the heart. With increased afterload, the heart works harder to eject the contents, leading to increased MVO2. This is a crucial period of myocardial susceptibility to ischemic injury and is a major reason to consider afterload reduction therapies.
Common causes of increased afterload include aortic/pulmonic stenosis, hypothermia, hypertension, compensatory response to hypotension and decreased CO, classic shock states (hypovolemic, cardiogenic, and obstructive), and response to drugs that stimulate the alpha receptors (epinephrine, norepinephrine, dopamine, phenylephrine) (see Table 4-2). Decreased afterload is seen in hyperthermia, the distributive shocks (septic, anaphylactic, and neurogenic), and after administration of vasodilating drugs (nitroprusside, nitroglycerin at higher doses, calcium channel blockers, beta-blockers, and the like) (see Table 4-2).
Afterload cannot be directly measured. It is evaluated with a pulmonary artery catheter and is a derived value (ie, is calculated based on other measured variables).
Systemic vascular resistance (SVR) is normally between 800 and 1200 dynes-sec/cm–5. If the SVR is elevated, the left ventricle faces an increased resistance to the ejection of blood. The SVR commonly elevates as a compensatory response to hypertension or a low CO, such as would occur in shock states. It is important for the clinician to know why the SVR is elevated; for example, if the SVR is elevated because of systemic hypertension, afterload-reducing agents are a critical part of the therapy. However, if the SVR is elevated secondary to a compensation for low CO, therapy should be directed toward the primary goal of improving CO by reducing SVR.
If the SVR is low, the left ventricle faces a lower resistance to the ejection of blood. Generally, the SVR only decreases as a pathologic response to inflammatory conditions (eg, sepsis, fever). The SVR can also be reduced in hepatic disease due to increased collateral circulation or from neurogenic induced central vasodilation. Generally, if the SVR is reduced, administration of fluids and or vasopressor drugs is considered. More important, treating the underlying condition is essential. If the underlying condition is not treated, the use of vasopressors provides only short-term success.
Pulmonary Vascular Resistance is lower in comparison to SVR. Normal PVR is about 100 to 250 dynes/s/cm–5 (see Table 4-1). Generally, only an elevated PVR is considered a problem, because it produces a strain on the right ventricle. If this strain is unrelieved, the right ventricle eventually fails. Failure of the right ventricle results in less blood entering the lungs and the left ventricle. Systemic hypotension follows due to RV dysfunction. The most common causes of an increase in PVR include pulmonary hypertension, hypoxia, end-stage COPD (cor pulmonale), and pulmonary emboli.
Contractility is the strength of the myocardial contraction, or the degree of myocardial fiber shortening with contraction. Contractility contributes significantly to CO. If the other determinants of CO were constant, then a heart with a greater contractile force would produce a greater CO. However, contractility depends on many variables including preload (Frank-Starling law of the heart) and afterload.
Electrolyte levels also have a major impact on the contractility of the heart. Monitoring and treating abnormal calcium, sodium, magnesium, potassium, and phosphorus levels is essential to ensure optimal contractility. Other factors that contribute to contractility include myocardial oxygenation (ischemia), amount of functional myocardium (infarction, cardiomyopathy), and administration of positive and negative inotropic drugs.
Myocardial contractility is reflected indirectly in the SVI, which is the SV adjusted according to body size, and the right and left stroke work index (RVSWI and LVSWI). The normal value for SVI is 33-47 mL/beat/m2; RVSWI is 5-10 gm/m/m2, and LVSWI is 50-62 gm/m/m2 (see Table 4-1). These are not direct indicators of contractility, but can be used to identify patients at risk for poor contractility and to monitor the effects of therapeutic management.
The basic components of hemodynamic monitoring systems include an indwelling catheter connected to a pressure transducer and flush system and a bedside monitor. All components that come in contact with the vascular system must be sterile, with meticulous attention paid to maintaining a closed sterile system during use.
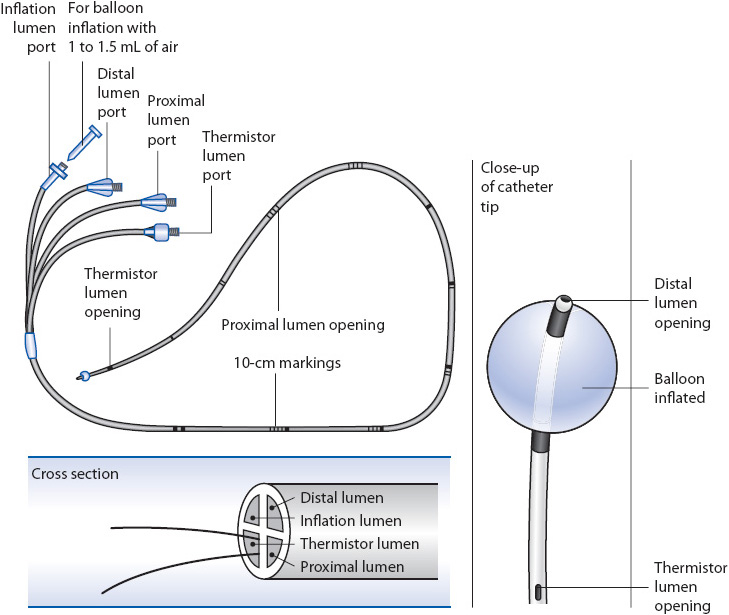
The PA catheter is a multilumen catheter inserted into the PA (Figures 4-3, 4-4). Each lumen or “port” has specific functions. The PA catheter typically is inserted through an introducer sheath (large-diameter, short catheter with a diaphragm) placed in a major vein. Veins used for PA catheter insertion include the internal jugular, subclavian, femoral, and less commonly, the brachial vein. More information on the PA catheter is found in AACN Essentials of Critical Care Nursing.
Figure 4-4. Flow-directed PA catheter (Swan-Ganz). (From Visalli F, Evans P. The Swan-Ganz catheter: a program for teaching safe, effective use. Nursing 1981. 1981;11:1.)
The arterial catheter, or “A-line,” has only one lumen, which is used for measuring arterial pressures, hemodynamic parameters, and for drawing arterial blood samples (Figure 4-5). Arterial catheters are inserted in any major artery, with the most common sites being the radial and femoral arteries.
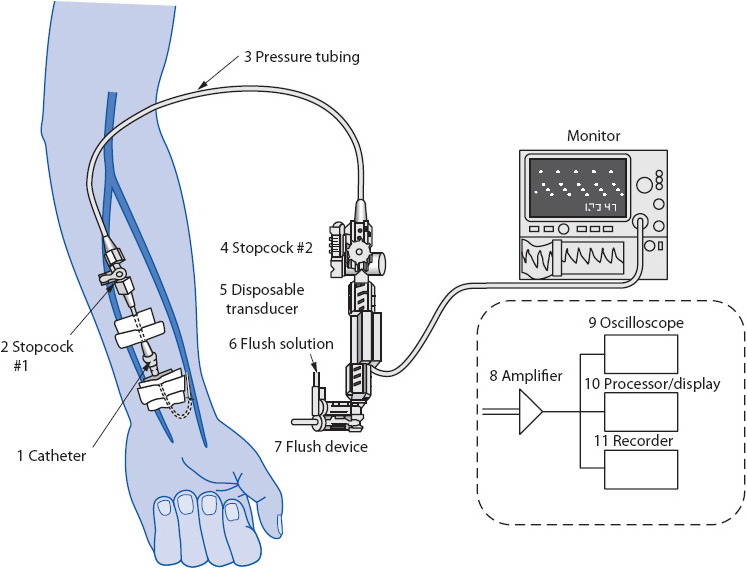
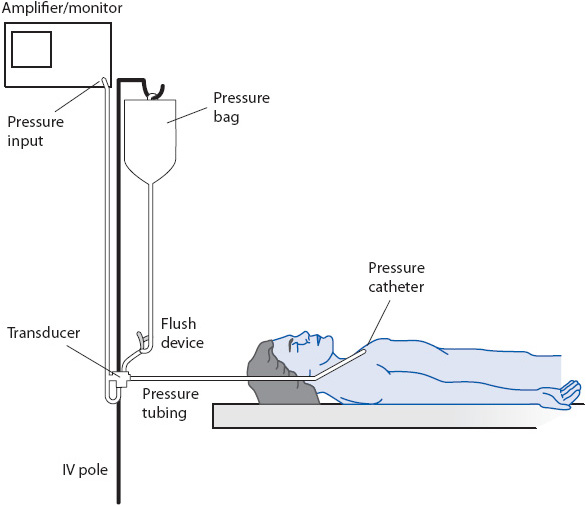
Figure 4-5. Components of a hemodynamic monitoring system. (Reprinted from Gardner R, Hollingsworth K. Electrocardiography and pressure monitoring: how to obtain optimal results. In: Shoemaker WC, Ayers S, Grenvik A, Holbrook P, eds. Textbook of Critical Care. 3rd ed. Philadelphia, PA: WB Saunders; 1995:272, with permission from Elsevier.)
The pressure tubing is a key component of any hemodynamic monitoring system (see Figure 4-5). It is designed to be a stiff (noncompliant) tubing to ensure accurate transfer of intravascular pressures to the transducer. The pressure tubing connects the intravascular catheter to the transducer. Many pressure tubings have stopcocks in line to facilitate blood sampling and zeroing the transducer (see below). Normally, the pressure tubing is kept as short as possible (no more than 3-4 ft), with a minimal number of stopcocks, to increase the accuracy of pressure measurements.
The pressure transducer is a small electronic sensor that has the ability to convert a mechanical pressure (vascular pressure) into an electrical signal (see Figure 4-5). This electrical signal can then be displayed on the pressure amplifier.
The pressure amplifier, or “bedside monitor,” augments the signal from the transducer and displays the converted vascular pressure as an electrical signal (see Figure 4-5). This signal is used to display a continuous waveform on the oscilloscope of the monitor and to provide a numerical display of the pressure measurement. Most bedside monitors also have a graphic recorder to print out the pressure waveform.
In addition to being attached to the pressure amplifier, the transducer is connected to an intravenous (IV) solution, which is placed in a pressure bag (Figure 4-6). The IV solution is normally 500 to 1000 mL of normal saline (NS), although 5% dextrose in water (D5W) can be used. The IV solution is placed under 300 mm Hg of pressure to provide a slow, continuous infusion of fluid through the vascular catheter.
Figure 4-6. Pressure bag and flush device connected to a pressure transducer and monitoring system. (Reprinted from Ahrens TS, Taylor L. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992: 210, with permission from Elsevier.)
The IV solution is placed under pressure for another reason. Included in most pressure systems is a flush device (see Figure 4-6). The flush device regulates fluid flow through the pressure tubing at a slow, continuous rate to prevent occlusion of the vascular catheter. Normally, the flush device restricts fluid flow to approximately 2 to 4 mL/h. If the flush device is activated, normally by squeezing or pulling the flush device, a rapid flow of fluid enters the pressure tubing. Flush devices are activated for two reasons: to rapidly clear the tubing of air or blood and to check the accuracy of the tubing/catheter system (square wave test). Measuring the fluid in the IV solution should be done on every shift to determine the amount of fluid infused from the pressure bag. Depending on hospital procedures, unfractionated heparin may be added to the IV solution to aid in keeping the system patent. If this is done, generally about 1 U of 1:1000 unfractionated heparin is added for every cubic centimeter (cc) of the IV solution.
Bedside monitors have alarms for each of the hemodynamic pressures being monitored. Normally, every parameter that is being monitored has high and low alarms, which can be set to detect variations from the current value. Alarm limits are generally set to detect significant decreases or increases in pressures or rates, typically ± 10% of the current values.
The information obtained from hemodynamic monitoring technology must be verified for accuracy by the bedside clinician.
A fundamental step in obtaining accurate hemodynamic values is to zero the transducer amplifier system. Zeroing is the act of electronically compensating for any offset (distortion) in the transducer. This is normally done by exposing the transducer to air and pushing an automatic zero button on the bedside monitor. This step is performed at least once before obtaining the first hemodynamic reading after catheter insertion. Because it is an electronic function, it normally has to be performed only once when the transducer and amplifier are first attached to the in situ catheter.
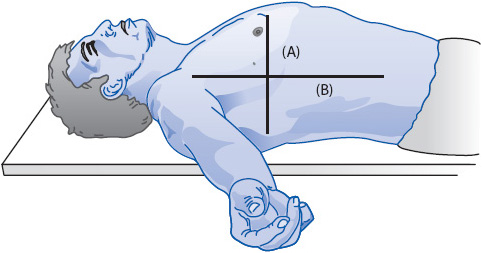
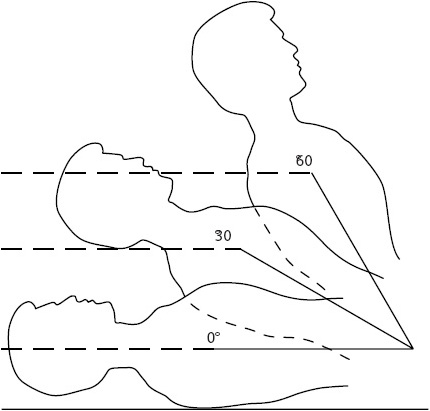
Leveling is the process of aligning the tip of the vascular catheter horizontal to a zero reference position, usually a stopcock in the pressure tubing close to the transducer. The reference point is the phlebostatic axis and is found at the intersection between the fourth intercostal space (ICS) and half the AP diameter of the chest (Figure 4-7).
Figure 4-7. Referencing and zeroing the hemodynamic monitoring system in a supine patient. The phlebostatic axis is determined by drawing an imaginary vertical line from the fourth ICS on the sternal border to the right side of the chest. (A) A second imaginary line is drawn horizontally at the level of the midpoint between the anterior and posterior surface of the chest. (B) The phlebostatic axis is located at the intersection of points A and B. (Reprinted from Keckeisen M, Chulay M, Gawlinski A, eds. Pulmonary artery pressure monitoring. In: Hemodynamic Monitoring Series. Aliso Viejo, CA: AACN; 1998:11, with permission from Elsevier.)
There are two basic methods for leveling. When the transducer and stopcocks are mounted on a pole close to the bed, the pole height is adjusted to have the stopcock opening horizontal to the external reference location of the catheter tip (Figure 4-8). To ensure horizontal positioning, a carpenter’s level is usually necessary. Each time the bed height or patient position is altered, this leveling procedure must be repeated (Figure 4-9).
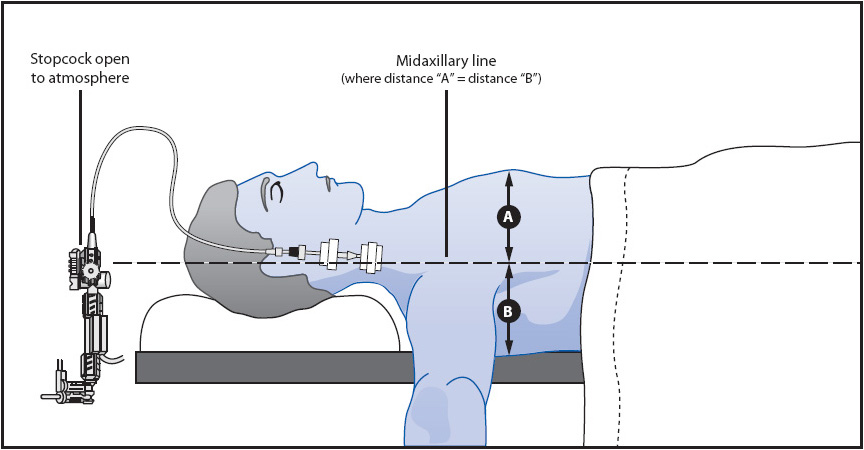
Figure 4-8. Typical leveling of CVP catheter with stopcock attached to the transducer for mounting on a pole. The stopcock close to the transducer is opened to atmospheric pressure (air) horizontal to the fourth ICS at the midaxillary line.
Figure 4-9. The level of the phlebostatic axis as the patient moves flat to a higher level of backrest. The level of the axis for referencing and zeroing the air-fluid interface rotates on the axis and remains horizontal as the patient moves from flat to increasingly higher backrest positions. For accurate hemodynamic pressure readings at different backrest elevations, the air-fluid interface must be at the level of the phlebostatic axis. (Reprinted from Bridges EJ, Woods SL. Pulmonary artery pressure measurement: state of the art. Heart Lung. 1993;22(2):101, with permission from Elsevier.)
The other method for leveling places the transducer and stopcock at the correct location on the chest wall or arm (Figure 4-10). Taping or strapping the transducer to the appropriate location on the body eliminates the need for repeating the leveling procedure when bed heights are changed. As long as the transducer/stopcock position remains horizontal to the external reference location, no releveling is required.
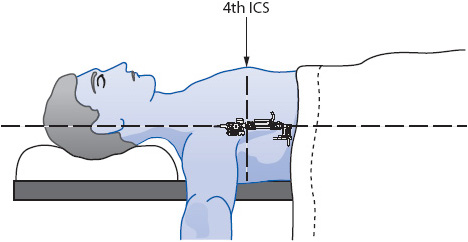
Figure 4-10. Leveling a transducer for mounting on the chest wall at the fourth ICS at the midaxillary line.
Leveling must be performed when obtaining the first set of values and any time the transducer is no longer horizontal to the external reference location. When obtaining the first set of readings, zeroing and leveling are frequently performed simultaneously. After this initial combined effort, zeroing does not need to be performed when leveling is done.
If the transducer/amplifier system is suspected of being inaccurate, calibration can be performed. Calibration is less important today because all disposable transducers are precalibrated by the manufacturer. If calibration needs to be checked prior to use, or if a reading is in doubt, a simple static pressure check can be done before the transducer is attached to the patient. Detailed descriptions of how to perform static pressure checks are found in most hemodynamic monitoring texts.
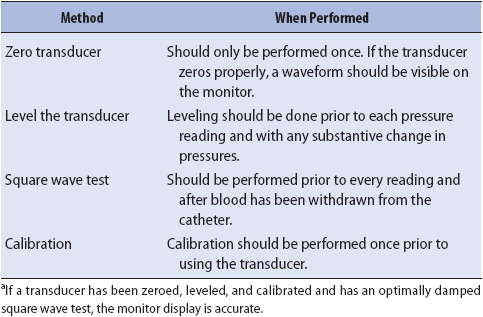
For hemodynamic monitoring to provide accurate information, the vascular pressure must be transmitted back to the transducer unaltered and then converted accurately into an electrical signal. For this waveform to be transmitted unaltered, no obstructions or distortions to the signal should be present along the transmission route. Distortion of the waveform leads to inaccurate pressure interpretations. A variety of factors can cause distortions to the waveform, including catheter obstructions (eg, clots, catheter bending, blood or air in tubing), excessive tubing or connectors, and transducer damage. Verification of an accurate transmission of the waveform to the transducer is checked by the bedside nurse by performing a square wave test. This occurs at the beginning of each shift.
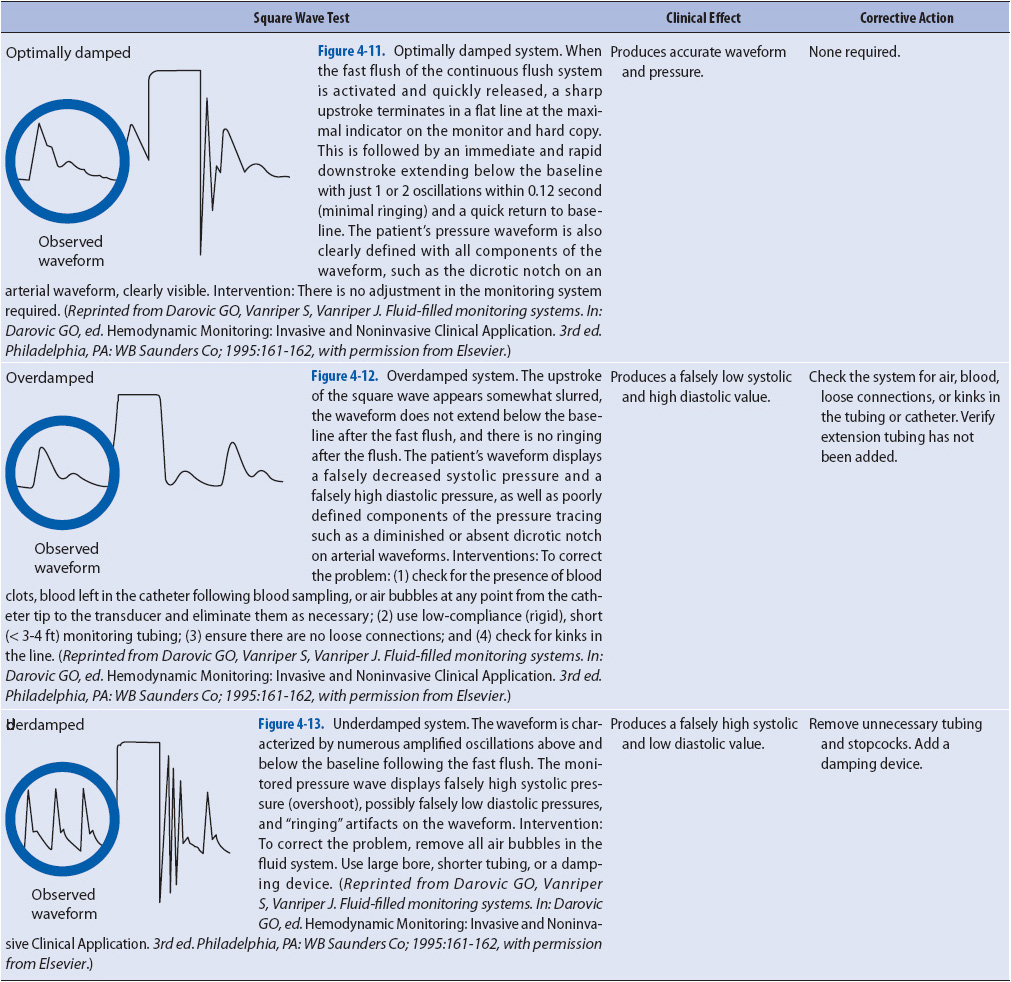
The square wave test is performed on all hemodynamic pressure systems before assuming that the waveforms and pressures obtained are accurate. The square wave test is performed by recording the pressure waveform while fast flushing the catheter (Table 4-3, Figure 4-11). The fast-flush valve is pulled or squeezed, depending on the model, for at least a second and then rapidly released. The tracing should show a rapid rise in the waveform to the top of the graph paper, with a square pattern. Release of the flush device should show a rapid decrease in pressure below the baseline of the pressure waveform (undershoot), followed immediately by a small increase above the baseline (overshoot) prior to resumption of the normal pressure waveform. Square wave tests with these characteristics are called optimally damped tests and represent an accurate waveform transmission. The square wave test is the best method available to the clinician to check the accuracy of hemodynamic monitoring equipment; for example, if an arterial line is to be examined for accuracy, a square wave test should be done. Do not compare the arterial line pressure with an indirect blood pressure reading with a sphygmomanometer, because the indirect method is usually less accurate than the direct method (arterial line pressure). If the square wave test indicates optimal damping, then the arterial line pressure is accurate.
Two problems may exist with waveform transmissions, and are referred to as overdamping and underdamping (see Table 4-3).
If something absorbs the pressure wave (like air or blood in the tubing, stopcocks, or connections), it is said to be overdamped. Overdamping decreases systolic pressures and increases diastolic pressures. An overdamped square wave test reflects the obstruction in waveform transmission. Characteristics of overdamping include a loss of the undershoot and overshoot waves after release of the flush valve and a slurring of the downstroke (Figure 4-12).
If something accentuates the pressure wave (like excessive tubing), it is said to be underdamped. Underdamping increases systolic pressures and decreases diastolic pressures (Figure 4-13). An underdamped square wave test reflects the amplification of pressure waves and includes large undershoot and overshoot waves after the release of the flush valve. Table 4-4 summarizes the methods of assessing and ensuring the accuracy of hemodynamic monitoring systems.
TABLE 4-4. SUMMARY OF METHODS FOR ASSESSING AND ENSURING ACCURACY OF HEMODYNAMIC MONITORING SYSTEMSa
Nosocomial infections related to the tubing/catheter system are usually caused by the entry of organisms through stopcocks. Stopcocks are opened for blood sampling and zeroing the transducer only when necessary. Closed, needleless systems are used whenever feasible to decrease the risks to the patient and clinician.
Tubing changes, including flush device, transducer, and flush solution, should occur every 72 hours. The frequency of catheter device changes is controversial, but must occur whenever the catheter is suspected as a source of an IV infection or by institutional policy.
Length/duration of indwelling catheter use varies depending upon the need for use, site accessed, patient clinical status, catheter type, and antibiotic coating (if any). There are widespread variations in site care techniques and materials used across hospitals. Current CDC recommendations state central lines should be removed as soon as their exclusive use is no longer required. A multidisciplinary approach to tracking “line days” with removal as soon as possible is supported by many professional organizations. Nurses should adhere to unit policy and work with the care team to remove invasive lines as expediently as reasonable.
Central venous catheters are frequently inserted to assess preload in acutely ill patients. However, because these catheters often have more than one port through which to infuse fluids, they may also be used to infuse vasoactive substances, other intravenous medications, and total parenteral nutrition.
Central venous catheters can be inserted into most large-diameter veins, with the internal jugular and subclavian veins being the most common insertion sites. Typically, the CVP catheter is advanced into the superior vena cava to a level above the right atrium. Following placement, location is verified with a chest radiograph to rule out the presence of a pneumothorax, kinking of the catheter, or other complications.
The waveform configuration associated with the CVP is readily identifiable (described later) and in combination with the value allows the clinician to monitor the patient’s status over time and with interventions.
Removing the CVP catheter, and/or discontinuing monitoring the CVP value is a clinical decision based on the assessment that the data from the catheter is no longer necessary for care management. This decision may be made anywhere from a few hours to several days after insertion. The removal of the CVP catheter is normally performed by a physician, nurse practitioner, or physician assistant—although in some institutions nurses perform this task. Nurses must be aware of their specific hospital and unit policies regarding removal of the CVP catheter.
Following the discontinuance of IV fluids, all stopcocks to the patient are turned off to avoid air entry into the vascular bed during catheter removal. The patient is placed in a supine position with the head of the bed flat. While the catheter is being gently withdrawn, the patient is instructed to exhale or hold his or her breath to further decrease the chance of air embolus. Resistance during catheter withdrawal may indicate catheter knotting (very rare). A chest x-ray is necessary to confirm the problem and special removal procedures are performed to avoid structural damage to the vessels.
Complications associated with CVP catheters include those associated with insertion, maintenance, and removal of the device. These include bleeding, pneumothorax or air emboli, and introduction of microorganisms and subsequent infection.
Blood pressure measurement with the indirect method (sphygmomanometer) is not as accurate as direct blood pressure measurement, particularly during conditions of abnormal blood flow (high or low CO states), SVR, or body temperature. The prevalence of these conditions in acutely ill patients may necessitate insertion of an arterial catheter to directly measure blood pressure.
Arterial catheters are short (< 4 in) catheters that can be inserted into radial, brachial, axillary, femoral, or pedal arteries. The most common site is the radial artery. Arterial catheters can be placed by cut down or with percutaneous insertion techniques, the latter being the most common insertion method.
General insertion steps for percutaneous insertion are similar to IV catheter insertion. Prior to insertion of a radial artery catheter, however, an Allen test is performed to verify the adequacy of circulation to the hand in the event of radial artery thrombosis. The Allen test is performed by completely obstructing blood flow to the hand by compressing the radial and ulnar arteries for a minute or two. If adequate collateral blood flow exists, there will be rapid return of color to the hand upon release of the ulnar artery (Figure 4-14).
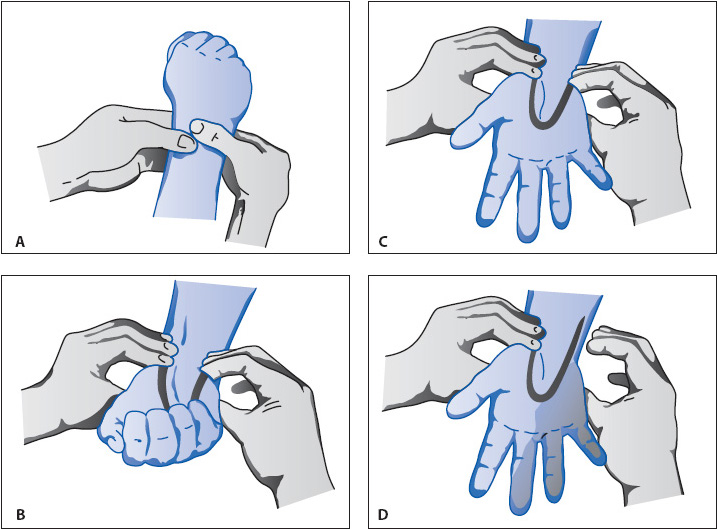
Figure 4-14. The Allen test. (From DeGroot KD, Damato MB. Monitoring intra-arterial pressure. Crit Care Nurs. 1986;6[1]:74-78.)
During insertion, care is exercised not to damage the arterial vessel by excessive probing or movement of the needle. Bleeding into the tissues occurs quite easily if the vessel is damaged, causing obstruction to distal blood flow and nerve pressure. Following artery cannulation, the catheter is connected to the pressure transducer and a high-pressure infusion system to prevent blood from backing up into the tubing and fluid container (see Figure 4-5).
The removal of the arterial catheter is warranted when an accurate blood pressure can be obtained via noninvasive methods, the blood pressure is no longer labile, or when frequent arterial blood samples are no longer indicated. Removal of arterial catheters is commonly performed by the nurse using procedures similar to IV catheter removal, but because they are in an artery, greater attention to achieving hemostasis is required. Following catheter removal, firm pressure is maintained over the site for at least 5 minutes or until hemostasis occurs. This prevents bleeding and hematoma formation. For patients with coagulation abnormalities, manual pressure may need to be applied for 10 minutes or longer. Pressure dressings, rather than manual pressure, at the site are not recommended as a means to achieve hemostasis. Once hemostasis is achieved, a pressure dressing may be used but is generally not needed.
Frequent assessment of the site after catheter removal is recommended to identify rebleeding and thrombosis of the artery. Checking the extremity for the presence of pulses, circulation, and bleeding is recommended for a few hours after catheter removal.
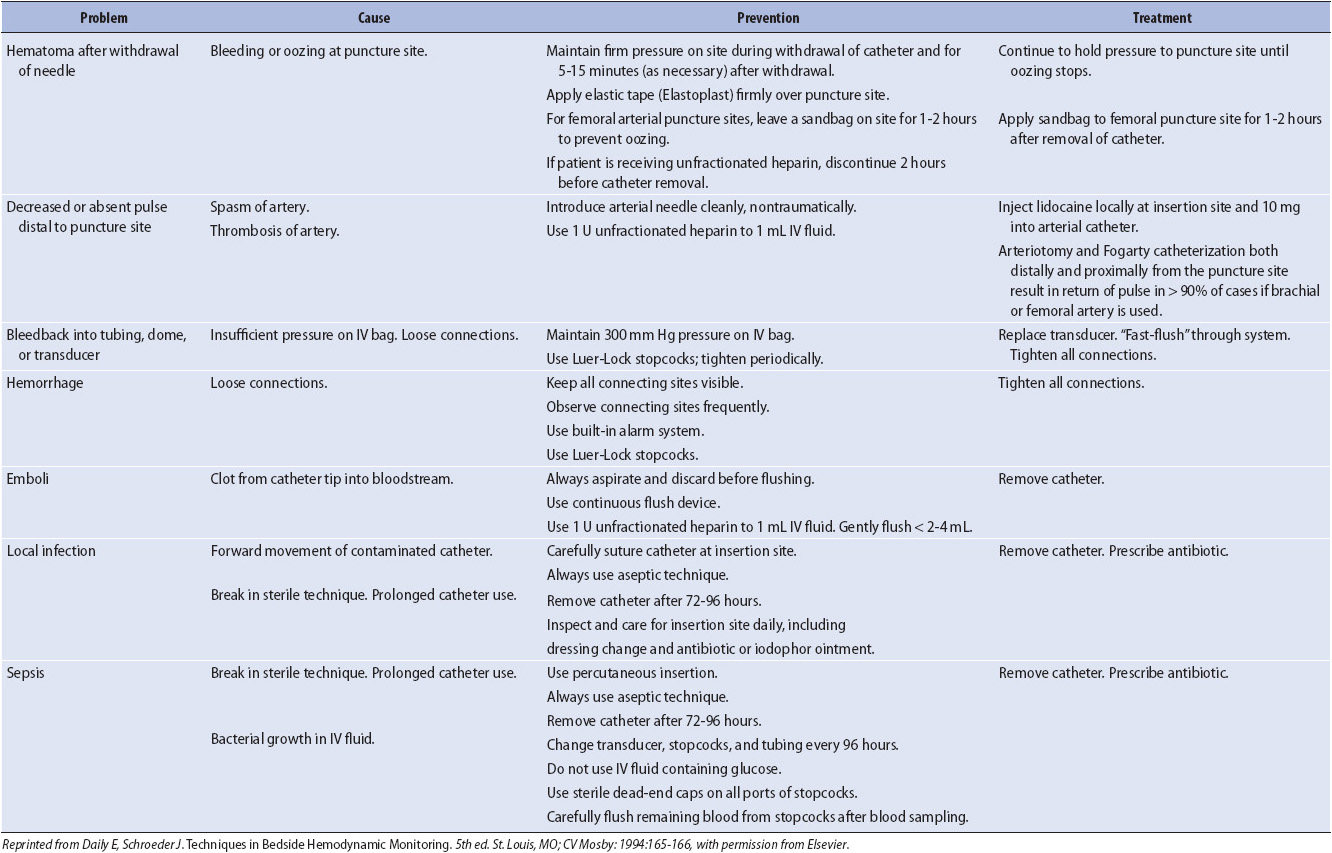
A variety of complications are associated with arterial catheters (Table 4-5). The most serious are related to bleeding from the arterial catheter and loss of arterial flow to the extremity from thrombus formation. Loose connections in the arterial system can lead to rapid and massive blood loss. The morbidity and mortality associated with these complications require stringent safeguards (Luer-Lock connections, minimum number of stopcocks, pressure alarm system activated at all times) to prevent bleeding and to rapidly identify disruptions in the arterial system. The catheters are removed as early as possible to prevent the potential for thrombus formation.
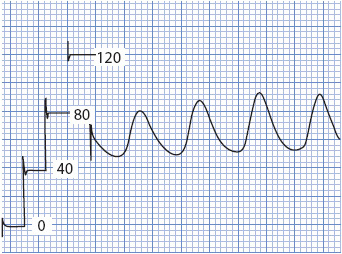
To obtain hemodynamic values, interpretation of waveforms is necessary. A multichannel strip recorder, which provides both an electrocardiographic (ECG) and pressure tracing, is the required element (Figure 4-15). Many institutions also use respiratory pressure waveforms, graphed simultaneously with the ECG, CVP, and arterial waveforms. The larger the scale, the easier is the interpretation of the wave. All waveforms are easily obtained simply by activating the record function of the bedside monitor. When obtaining waveforms for interpretation, make sure the calibration scales on the left side of the paper are properly aligned with the paper grid. Improperly aligned calibration marks increase the difficulty in reading the waveform and increase potential errors in interpretation.
Figure 4-15. Graphic tracing of an arterial waveform preceded by calibration scale markings (0/40/80/120 mm Hg). Note how the scale markers line up with the heavy line of the tracing paper. Each 1-mm line represents 4 mm Hg in this scale.
The patient is placed in the supine position, with the backrest elevated anywhere from 0° to 60° (see Figure 4-9). Generally, data should not be obtained if the patient is on his or her side, because it is difficult to identify the location of the catheter tip for purposes of leveling (Figure 4-16). Improper leveling distorts atrial and venous pressure readings.
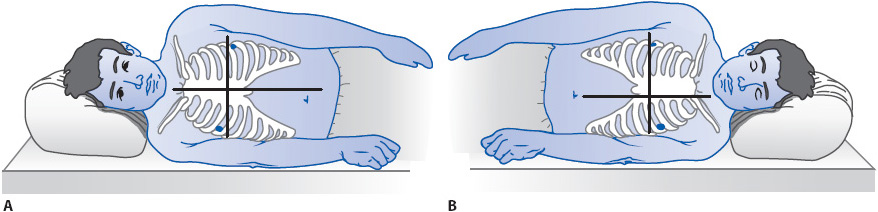
Figure 4-16. Referencing and zeroing the hemodynamic monitoring system in a patient in the lateral position. (A) For the right lateral position, the reference point is at the intersection of the fourth ICS and the midsternum. (B) For the left lateral position, the reference point is the intersection of the fourth ICS and the left parasternal border. (Reprinted from Keckeisen M, Chulay M, Gawlinski A, eds. Pulmonary artery pressure monitoring. In: Hemodynamic Monitoring Series. Aliso Viejo, CA: AACN; 1998:12, with permission from Elsevier.)
It is important to remember that patient comfort is a key issue when obtaining waveform readings. Do not position a spontaneously breathing patient with dyspnea flat for the sole reason of obtaining the readings. It is best to obtain values in the position in which the patient is most comfortable.
Correct interpretation of waveforms involves careful assessment of venous and arterial pressure waveforms. Normal values for each of the hemodynamic pressures are listed in Table 4-1.
The CVP (also known as right atrial pressure) is important because it is used to approximate the right ventricular end-diastolic pressure (RVEDP) and preload. A normal CVP is between 2 and 8 mm Hg. Low CVP values typically reflect hypovolemia or decreased venous return. High CVP values reflect overhydration, increased venous return, or right-sided cardiac failure. The CVP is obtained from the tip of central venous catheter. Measurement of CVP is done simultaneously with the ECG. Using the ECG allows the identification of the point where the CVP best correlates with the RVEDP.
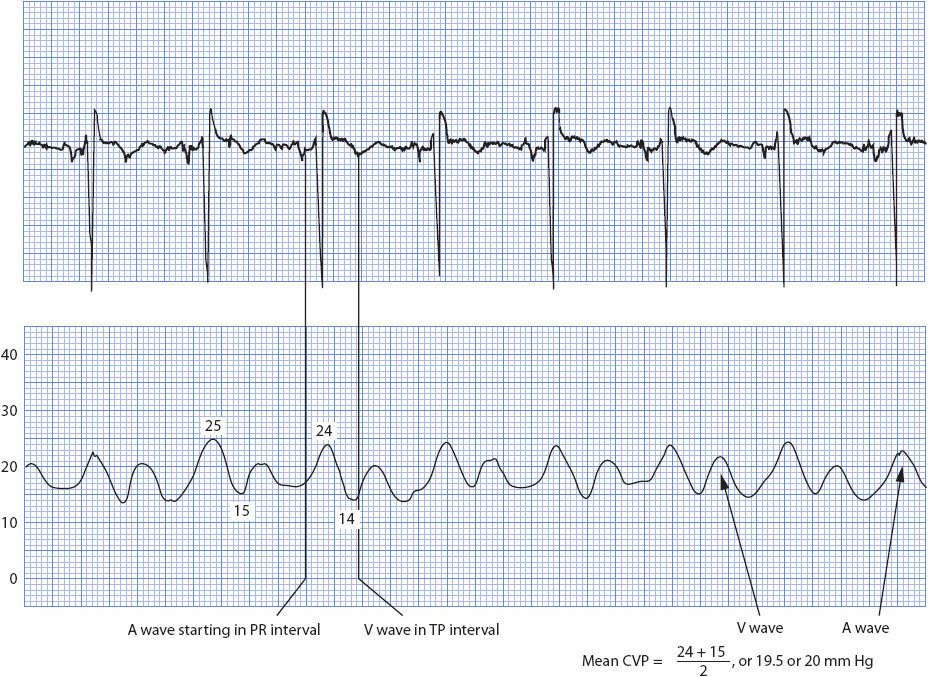
The CVP is read by one of two techniques. The first technique is to take the mean (average) of the A wave of the CVP waveform (Figure 4-17). Although three waves normally exist on atrial waveforms (A, C, and V waves), the mean of the A wave most closely approximates ventricular end-diastolic pressure. The A wave of the CVP waveform starts just after the P wave on the ECG is observed and represents atrial contraction. By taking the reading at the highest point of the A wave, adding it to the reading at the lowest point of that A wave, and dividing by 2, the average or mean CVP reading is obtained (generally a line is drawn though the middle of the A waves to derive a number).
Figure 4-17. Reading a CVP waveform by averaging the A wave. (Reprinted from Ahrens TS, Taylor L. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992:31, with permission from Elsevier.)
A second method, the Z-point technique, also can be used to estimate ventricular end-diastolic pressures (Figure 4-18). The Z-point is taken just before the closure of the tricuspid valve. This point is located on a CVP tracing in the mid to late QRS complex area. The Z-point technique is especially useful when an A wave does not exist; for example, in atrial fibrillation when atrial contraction is absent.
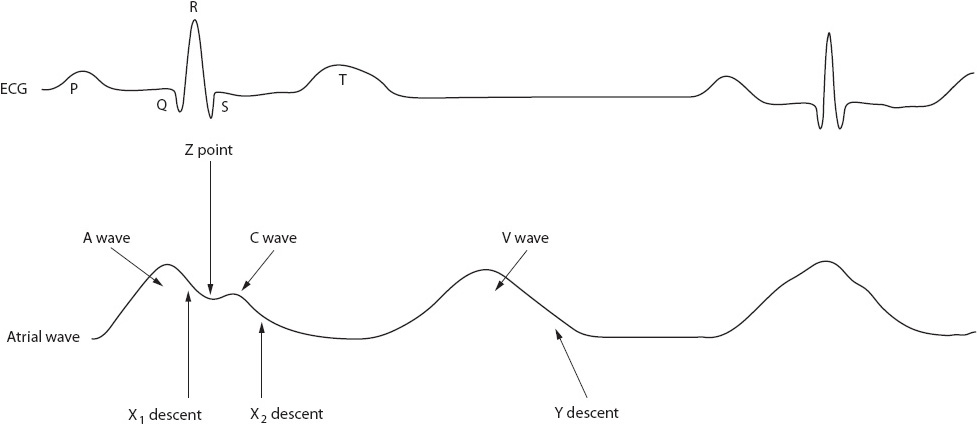
Figure 4-18. Use of the Z-point to read a CVP waveform. (Reprinted from Ahrens TS. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992:24, with permission from Elsevier.)
By isolating the A wave or using the Z-point technique, atrial pressures can reasonably estimate ventricular end-diastolic pressure. It is helpful to read these values off a multichannel strip recorder and not the digital display on the bedside monitor.
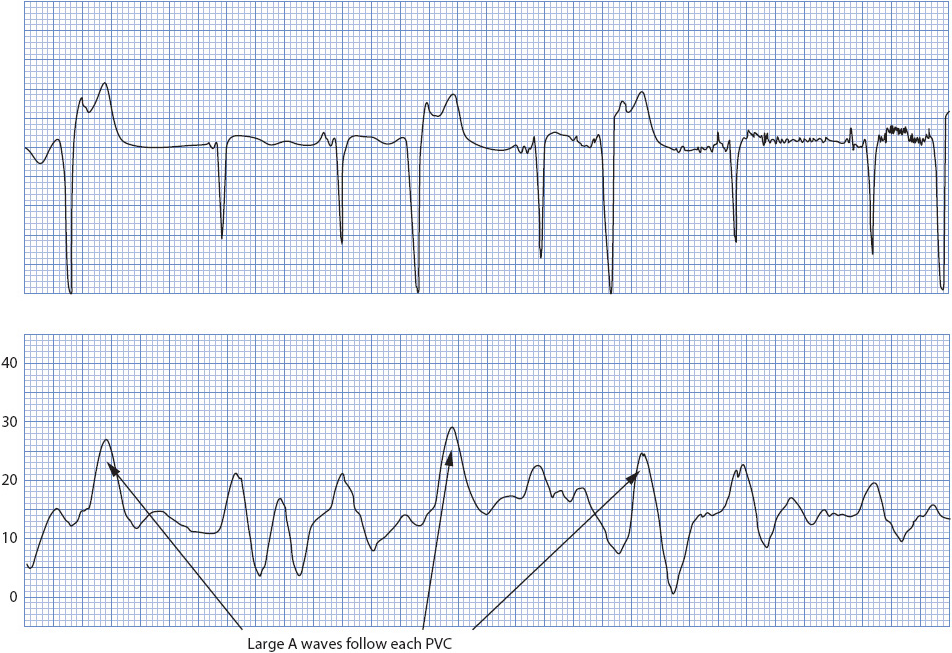
Two types of abnormal CVP waveforms are common. Large A waves (also called cannon A waves) occur when the atrium contracts against a closed tricuspid value (Figure 4-19). This occurs most commonly with arrhythmias like PVCs or third-degree heart block. Giant V waves are common in conditions such as tricuspid insufficiency or ventricular failure. Using the Z-point for CVP readings prevents incorrect interpretations associated with the use of large A or V waves.
Figure 4-19. Giant A waves with loss of atrioventricular synchrony. (Reprinted from Ahrens TS. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992:54, with permission from Elsevier.)
An arterial waveform, such as seen in systemic tracings, has three common characteristics: rapid upstroke, dicrotic notch, and progressive diastolic runoff (Figure 4-20). Diastole is read near the end of the QRS complex with systole read before the peak of the T wave. The mean arterial pressure can be calculated (see Table 4-1) or obtained from the digital display on the bedside monitor.
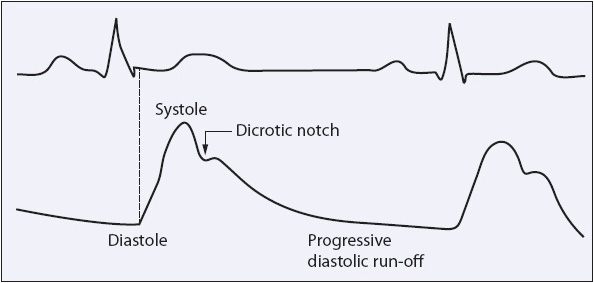
Figure 4-20. Characteristics of an arterial waveform. (From Ahrens TS, Prentice D. Critical Care: Certification Preparation and Review, 3rd ed. Stamford, CT: Appleton & Lange; 1993:82.)
Direct measurement of systemic arterial pressures is obtained from the tip of an arterial catheter and leveled to the phlebostatic axis (see Figure 4-10), with pressure waveforms interpreted as described. Normal pressures are generally in the region of 100 to 120 mm Hg systolic, 60 to 80 mm Hg diastolic, and 70 to 105 mm Hg mean (see Table 4-1).
Systemic arterial pressures are not interpreted without other clinical information. In general, however, hypotension is assumed if the mean arterial pressure drops below 60 mm Hg. Hypertension is assumed if the systolic blood pressure (SBP) is greater than 140 to 160 mm Hg or the diastolic pressure exceeds 90 mm Hg.
The arterial pressure is one of the most commonly used parameters for assessing the adequacy of blood flow to the tissues. Blood pressure is determined by two factors, CO and SVR. Blood pressure does not reflect early clinical changes in hemodynamics because of the interaction with CO and SVR.
In addition, the CO consists of HR and SV. These two interact to maintain a normal CO. Subsequently, if the SV begins to fall due to loss of volume (hypovolemia) or dysfunction (LV failure), the HR increases to offset the decrease in SV. The net effect is to maintain the CO at near-normal levels. If the CO does not change, then there is no change in the blood pressure.
A key point for the nurse to consider is that because of these compensatory mechanisms, blood pressure may not signal early clinical changes in hemodynamic status. If a patient begins to bleed postoperatively, the blood pressure generally does not reflect this change until compensation is no longer possible. In addition, hypotension is sometimes difficult to evaluate. It is possible that true hypotension exists only when tissue hypoxia is present and end organs are affected. Although tradition dictates that we identify hypotension using predefined levels of blood pressure, other measures such as mixed venous saturation of hemoglobin (SVO2) and lactate levels may be better indicators. SVO2 monitoring is described later in the Section Continuous Mixed and Central Venous Oxygen Monitoring (SVO2/SCVO2).
Although studies identify the role of hypertension in circulatory damage, the specific level of hypertension that results in the damage is unclear. Therefore, any SBP over 140 is considered potentially injurious to the vasculature.
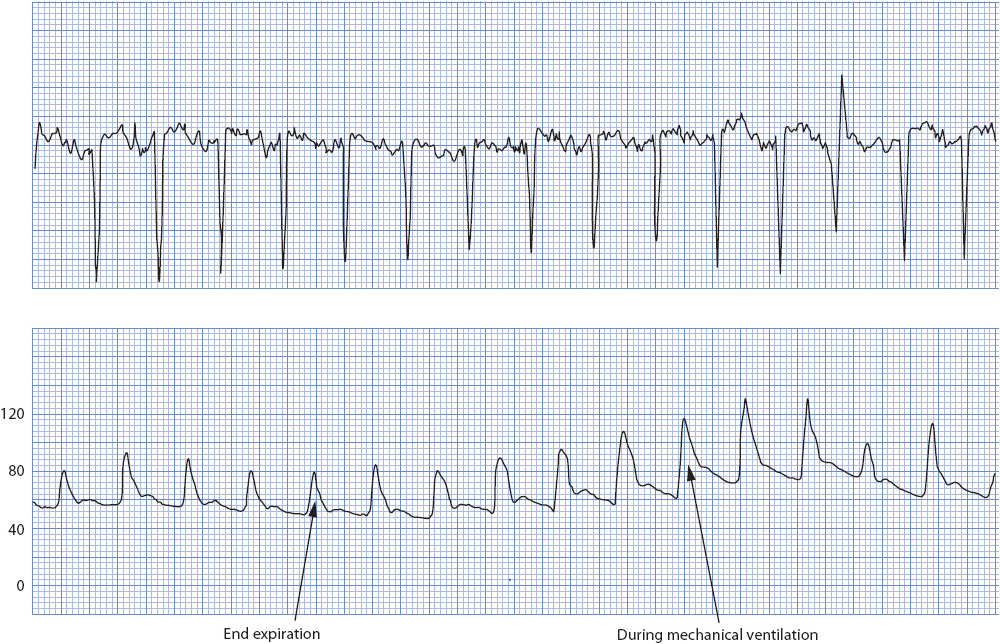
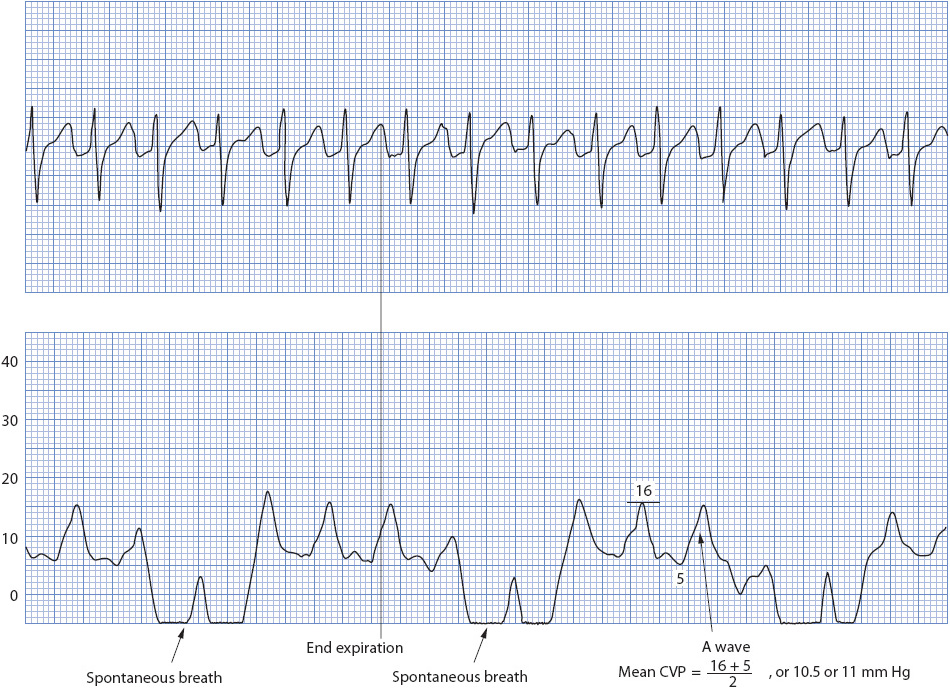
Respiration can physiologically change hemodynamic pressures. Spontaneous breathing augments venous return and slightly increases resistance to left ventricle filling. Mechanical ventilation does the opposite, potentially reducing venous return and reducing the resistance on the heart. The effect of mechanical ventilation on an arterial pressure is seen in Figure 4-21. The effect of spontaneous breathing on a CVP is noted in Figure 4-22.
Figure 4-21. Effect of respiration on arterial pressures. (Reprinted from Ahrens TS. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992:161, with permission from Elsevier.)
Figure 4-22. Effect of a spontaneous breath on a CVP waveform. (Reprinted from Ahrens TS. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992:165, with permission from Elsevier.)
A spontaneous breath or a patient-initiated ventilator breath produces a drop in the waveform because of the decrease in pleural pressure. A ventilator breath produces an upward distortion of the baseline due to an increase in pleural and intrathoracic pressure. The key to reading the waveform correctly is to isolate the point where pleural pressure is closest to atmospheric pressure. This point is usually at end expiration, just prior to inspiration (Figure 4-22).
Mixed venous oxygen saturation (SVO2) monitoring is generally done in a critical care unit and uses a specialized PA catheter. While a comprehensive discussion of the technology is not within the scope of this book, the associated concepts are important and are briefly discussed below. SVO2 catheters are different from other PA catheters in that they have two special fiber-optic bundles within the catheter that determine the oxygen saturation of hemoglobin by measuring the wavelength (color) of reflected light. Light is transmitted from the tip of the PA catheter down one bundle and is reflected off the oxygen-saturated hemoglobin, returning up the other bundle. This information is quantified by the bedside computer and numerically displayed as the percentage of saturation of the mixed venous blood.
A newer strategy is measurement of central venous oxygen saturation (SCVO2). This requires the placement of a central venous catheter, which is easily placed and has fewer complications than a PA catheter. Theoretically, it measures the degree of oxygen extraction from the brain and upper body and trends well with SVO2. The goal for SCVO2 is greater than 70%. The SCVO2 is usually less than the SVO2 except in shock states. This occurs because of redistribution of blood flow in classic shock states.
Continuous SVO2 monitoring is used as a diagnostic and therapeutic management tool. It provides early warning of alterations in hemodynamic status and a continuous monitor of the relationship between oxygen delivery and consumption. Many therapeutic strategies are added and adjusted in response to the changes in the SVO2. If a blood pressure is considered low but the SVO2 is above 60%, then the blood pressure is not contributing to a decrease in tissue perfusion. However, if the blood pressure and SVO2 are low, interventions to improve perfusion are essential.
SVO2 monitoring is used to continuously monitor how well the body’s demand for oxygen is being met under different clinical conditions. To understand this concept, an understanding of how the tissues are supplied with oxygen is necessary.
Blood leaves the left heart 100% saturated with oxygen and is transported to the tissues for cellular use based on the amount of perfusion (CO). Under normal conditions, only about 25% of the oxygen available on the hemoglobin is extracted by the tissues, with blood returning to the right heart with approximately 75% of the hemoglobin saturated with oxygen. Normal values for oxygen saturation are 60% to 80%.
In situations where tissue demands for oxygen increase, oxygen saturation of blood returning to the right heart will be lower than 70%. Clinical situations of increased tissue demand for oxygen include fever, pain, anxiety, infection, seizures, and some “routine” nursing activities like turning and suctioning. In contrast, hypothermia dramatically decreases oxygen consumption by the tissues. Interventions, then, are directed at decreasing or increasing the oxygen requirements as needed.
The concept of oxygen utilization is often referred to as supply and demand (or more accurately consumption) and is the essential concept inherent in SVO2 monitoring. Because tissue oxygenation depends on hemoglobin level, saturation of hemoglobin, oxygen consumption, and CO, the saturation of blood returning to the PA tells us much about the interaction of these four variables and can be used to assess the adequacy of interventions.
In some acute care units, new noninvasive technology may be used periodically to assess CO and other hemodynamic parameters. While research must still be accomplished to determine the efficacy of the devices, the noninvasive nature of the technologies makes them attractive for use in the progressive care setting. Two, thoracic bioimpedance and pulse contour measurement, are described below.
The resistance of current flow (impedance) across the chest is inversely related to the thoracic fluid. Using a current that flows from the electrodes on the chest and neck, the SV can be determined. Changes in impedance occur with changes in blood flow and velocity through the ascending aorta. The impedance changes reflect aortic flow, which is directly related to ventricular function (contractility).
Variables that change the bioimpedance and alter the relationship between impedance and SV are changes in hematocrit, lung water, lead contact, shivering, mechanical ventilation, and rhythm changes. Thoracic bioimpedance is a useful method for trend analysis but to date has not found to be accurate enough for diagnostic interpretation. Its major application has been outside the critical care setting (HF clinics, emergency department, pacemaker clinics).
Pulse contour measurement of hemodynamic parameters can be achieved invasively with an arterial line (PiCCO, LiDCO, Flotrac) and noninvasively with a finger pneumatic cuff (Nexfin). Various formulas are used to compute CCO values from the BP waveform.
The device provides a continuous beat-by-beat finger BP measure through the volume-clamp method (Figure 4-23). It then transforms the finger BP curve into a brachial arterial BP waveform and calculates CCO from the brachial pressure pulse contour.
Figure 4-23. Nexfin System for noninvasive continuous hemodynamic monitoring (Courtesy of: Edwards Lifesciences).
Hemodynamic parameters that can be measured with the noninvasive system are cardiac output/index, systolic/diastolic blood pressure, mean arterial pressure, HR, SV, stroke volume variation, pulse pressure variation, and systemic vascular resistance. It also includes pulse oximetry and non-invasive hemoglobin. The hemodynamic parameters will allow for the calculation of oxygen delivery.
The technology has been validated for measuring arterial pressure and has been found especially useful in cardiology clinics during tilt-test to detect orthostatic hypotension. It has also been used successfully in the perioperative management of patients. However, studies are needed to test the reliability of the measurement of cardiac index in critically ill patients. The sensor should only be continuously used on a finger for an 8 hour period then moved to another finger.
Ahrens TS. Hemodynamic Waveform Recognition. Philadelphia, PA: WB Saunders; 1993.
Ahrens TS, Taylor L. Hemodynamic Waveform Analysis. Philadelphia, PA: WB Saunders; 1992.
Alhashemi JA, Cecconi M, Hofer CK. Cardiac output monitoring: an integrative perspective. Crit Care. 2011;15(2):214.
Bigatello LM, George E. Hemodynamic monitoring. Minerva Anesthesiol. 2000;68(4):219-225.
Casserly B, Read R, Levy MM. Hemodynamic monitoring in sepsis. Crit Care Nurs Clin North Am. 2011;23(1):149-169.
Cocconi M, Johnston E, Rhodes A. What role does the right side of the heart play in circulation? Crit Care. 2006;10(suppl 3):1-7.
Cruz K, Franklin C. The pulmonary artery catheter: uses and controversies. Crit Care Clin. 2001;17(2):271-291.
Daily EK. Hemodynamic waveform analysis. J Cardiovasc Nurs. 2001;15(2):6-22.
Daily EK, Schroeder JS. Techniques in Bedside Hemodynamic Monitoring. St. Louis, MO: Mosby; 1994.
Darivuc DO. Hemodynamic Monitoring: Invasive and Noninvasive Clinical Application. Philadelphia, PA: Saunders; 2002.
Della Rocca G, Costa MG. Hemodynamic–volumetric monitoring. Minerva Anesthesiol. 2004;70(4):229-232.
deWaal EE, Wappler F, Buhre WF. Cardiac output monitoring. Curr Opin Anaesthesiol. 2009;22(1):71-77.
Frazier SK, Skinner GJ. Pulmonary artery catheter: state of the controversy. JCVN. 2008;32(2):113-121.
Gawlinski A. Cardiac output monitoring. In: Chulay M, Gawlinski A, eds. Hemodynamic Monitoring Series. Aliso Viejo, CA: AACN; 1998.
Glickman SW, Cairns CB, Otero RM, et al. Disease progression in hemodynamically stable patients presenting to the emergency department with sepsis. Acad Emerg Med. 2010;17(4):383-390.
Hadian M, Pinsky MR. Evidence-based review of the use of the pulmonary artery catheter: impact data and complications. Crit Care. 2006;10(suppl 3):1-11.
Imperial-Perez F, McRae M. Arterial pressure monitoring. In: Chulay M, Gawlinski A, eds. Hemodynamic Monitoring Series. Aliso Viejo, CA: AACN; 1998.
Keckeisen M. Pulmonary artery pressure monitoring. In: Chulay M, Gawlinski A, eds. Hemodynamic Monitoring Series. Aliso Viejo, CA: AACN; 1998.
Kim HK, Pinsky MR. Effect of tidal volume, sampling duration, and cardiac contractility on pulse pressure and stroke volume variation during positive pressure ventilation. Crit Care Med. 2008;36(10):2858-2862.
Kohli-Seth R, Oropello JM. The future of bedside monitoring. Crit Care Clin. 2000;16(4):557-578.
Latham HE, Rawson ST, Dwyer TT, et al. Peripherally inserted central catheters are equivalent to centrally inserted catheters in intensive care unit patients for central venous pressure monitoring. J Clin Monit Comput. 2012;26(2):85-90.
Leeper B. Monitoring right ventricular volumes. AACN Clin Issues. 2003;14(2):208-219.
Maar SP. Searching for the holy grail: a review of markers to tissue perfusion in pediatric critical care. Pediatr Emerg Care. 2008;24(12):883-887.
Monnet X, Richard C, Teboul JL. The pulmonary artery catheter in critically ill patients. Does it change outcomes? Minerva Anesthesiol. 2004;70(4):219-224.
Muller JC, Kennard JW, Browne JS, et al. Hemodynamic monitoring in the intensive care unit. Nutr Clin Pract. 2012;18(3):280-286.
Ott K, Johnson K, Ahrens T. New technologies in the assessment of hemodynamic parameters. J Cardiovasc Nurs. 2001;15(2):41-55.
Payen D, Gayat E. Which general intensive care unit patients can benefit from placement of the pulmonary artery catheter? Crit Care. 2006;10(suppl 3):1-6.
Pinsky MR. Hemodynamic monitoring in the intensive care unit. Clin Chest Med. 2003;24(4):549-560.
Pittman JA, Ping JS, Mark JB. Arterial and central venous pressure monitoring. Intl Anesthes Clin. 2004;42(1):13-30.
Plante A, Ro E., Rowbottom JR. Hemodynamic and related challenges: monitoring and regulation in the postoperative period. Anesthesiol Clin. 2012;30(3):527-554.
Polanco M, Pinsky M. Practical issues of hemodynamic monitoring at the bedside. Surg Clin North Am. 2006;86:1431-1456.
Prentice D, Ahrens T. Controversies in the use of the pulmonary artery catheter. J Cardiovasc Nurs. 2001;15(2):1-5.
Quaal SJ. Improving the accuracy of pulmonary artery catheter measurement. J Cardiovasc Nurs. 2001;15(2):71-82.
Rajaram SS, Desai NK, Kalra A, et al. Pulmonary artery catheters for adult patients in intensive care. Cochrane Database Syst Rev. 2013;28;2:CD003408.
Ranucci M. Which cardiac surgical patients can benefit from placement of a pulmonary artery catheter? Crit Care. 2006; 10(suppl 3):1-8.
Reinhart K, Bloos F. The value of venous oximetry. Curr Opin Crit Care. 2005;11:259-263.
Richard C, Monnet X, Teboul JL. Pulmonary artery catheter monitoring in 2011. Curr Opin Crit Care. 2011;17(3):296-302.
Robin E, Costecalde M, Lebuffe G, Vallet B. Clinical relevance of data from the pulmonary artery catheter. Crit Care. 2006; 10(suppl 3):1-10.
Saggar R, Sitbon O. Hemodynamics in pulmonary arterial hypertension: current and future perspectives. Am J Cardiol. 2012; 110(6 suppl):9S-15S.
Sandham JD. Pulmonary artery catheter use—refining the question. Crit Care Med. 2004;32(4):1070-1071.
Sivarajan VB, Bohn D. Monitoring of standard hemodynamic parameters: heart rate, systemic blood pressure, atrial pressure, pulse oximetry, and end–tidal CO2. Pediatr Crit Care Med. 2011;12(4 suppl):S2-S11.
Tucker D, Hazinski MF. The nursing perspective on monitoring hemodynamics and oxygen transport. Pediatr Crit Care Med. 2011;12(4 suppl):S72-S75.
Vender JS, Franklin M. Hemodynamic assessment of the critically ill patient. Intl Anesthesiol Clin. 2004;42(1):31-58.
Aherns T, Sona C. Capnography application in acute and critical care. AACN Clin Issues. 2003;14(2):123-132.
Akamatsu S, Oda A, Terazawa E. Automated cardiac output measurement by transesophageal color Doppler echocardiography. Anesth Analg. 2004;98(5):1232-1238.
Avolio AP, Butlin M, Walsh A. Arterial blood pressure measurement and pulse wave analysis—their role in enhancing cardiovascular assessment. Physiol Meas. 2010;31(1):R1-47.
Ayuela Azcarate JM, Clau Terré F, Ochagavia A, Vicho Pereira R. Role of echocardiography in the hemodynamic monitorization of critical patients. Med Intensiva. 2012;36(3):220-232.
Bartels SA, Stok WJ, Bezemer R, Boksem RJ, et al. Noninvasive cardiac output monitoring during exercise testing: Nexfin pulse contour analysis compared to an inert gas rebreathing method and respired gas analysis. J Clin Monit Comput. 2011 Oct;25(5): 315-321.
Bayram M, Yancy CW. Transthoracic impedance cardiography: a noninvasive method of hemodynamic monitoring. Heart Fail Clin. 2009;5(2):161-168.
Bogert LW, van Lieshout JJ. Non-invasive pulsatile arterial pressure and stroke volume changes from the human finger. Exp Physiol. 2005;90(4):437-446.
Boswell SA, Scalea TM. Sublingual capnometry. AACN Clin Issues. 2003;14(2):176-184.
Boyd JH, Walley KR. The role of echocardiography in hemodynamic monitoring. Curr Opin Crit Care. 2009;15(3):239-243.
Camporota L, Beale R. Pitfalls in hemodynamic monitoring based on the arterial pressure waveform. Crit Care. 2010;14:124.
Compton F, Schäfer JH. Noninvasive cardiac output determination: broadening the applicability of hemodynamic monitoring. Semin Cardiothorac Vasc Anesth. 2009;13(1):44-55.
Compton FD, Zukunft B, Hoffmann C, Zidek W, Schaefer JH. Performance of a minimally invasive uncalibrated cardiac output monitoring system (Flotrac/Vigileo) in haemodynamically unstable patients. Br J Anaesth. 2008;100:451-456.
Cottis R, Magee N, Higgins DJ. Haemodynamic monitoring with pulse-induced contour cardiac output (PiCCO) in critical care. Intensive Crit Care Nurs. 2003;19:301-307.
Creteur J. Gastric and sublingual capnometry. Curr Opin Crit Care. 2006;12:272-277.
de Jong RM, Westerhof BE, Voors AA, van Veldhuisen DJ. Noninvasive haemodynamic monitoring using finger arterial pressure waveforms. Neth J Med. 2009;67(11):372-375.
Dueck R, Goedje O, Clopton P. Noninvasive continuous beat-to-beat radial artery pressure via TL-200 applanation tonometry. J Clin Monit Comput. 2012;26(2):75-83.
Fellahi JL, Caille V, Charron C, Deschamps-Berger PH, Vieillard-Baron A. Noninvasive assessment of cardiac index in healthy volunteers: a comparison between thoracic impedance cardiography and Doppler echocardiography. Anesth Analg. 2009;108(5): 1553-1559.
Garwood S. Measuring renal blood flow with the intraoperative transesophageal echocardiography probe. Anesth Analg. 2009;108(5):1371-1376.
Ghanayem NS, Wernovsky G, Hoffman GM. Near infrared spectroscopy as a hemodynamic monitor in critical illness. Pediatr Crit Care Med. 2011:12(4 suppl):S27-S32.
Headley JM. Indirect calorimetry. AACN Clin Issues. 2003;14(2):155-167.
Heard SO. Gastric tonometry: the hemodynamic monitor of choice. Chest. 2003;123(5 suppl):469S-474S.
Hett DA, Jonas MM. Non-invasive cardiac output monitoring. Intensive Crit Care Nurs. 2004;20(2):103-108.
Horster S, Stemmler HJ, Sparrer J, et al. Mechanical ventilation with positive end-expiratory pressure in critically ill patients: comparison of CW-Doppler ultrasound cardiac output monitoring (USCOM) and thermodilution (PiCCO). Acta Cardiol. 2012;67(2):177-185.
Kim SH, Song JG, Park JH, et al. Beat-to-beat tracking of systolic blood pressure using noninvasive pulse transit time during anesthesia induction in hypertensive patients. Anesth Analg. 2013;116(1):94-100.
L’E Orme RM, Pigott DW, Mihm FG. Measurement of cardiac output by transpulmonary arterial thermodilution using a long radial artery catheter. A comparison with intermittent pulmonary artery thermodilution. Anaesthesia. 2004;59(6):590-594.
Lima A, Bakker J. Noninvasive monitoring of peripheral perfusion. Intensive Care Med. 2005;31:1316-1326.
Lima MV, Ochiai ME, Vieira KN, et al. Continuous noninvasive hemodynamic monitoring in decompensated heart failure. Arq Bras Cardiol. 2012;99(3):843-847.
Magder S. Central venous pressure: a useful but not so simple measurement. Crit Care Med. 2006;34(8):2224-2227.
Magder S. How to use central venous pressure measurements. Curr Opin Crit Care. 2005;11:264-270.
Marik PE. Regional carbon dioxide monitoring to assess the adequacy of tissue perfusion. Curr Opin Crit Care. 2005; 11: 245-251.
Marino R, Magrini L, Ferri E, Gagliano G, Di Somma, S. B-Type Natriuretic peptide and non-invasive haemodynamics and hydration status assessments in the management of patients with acute heart failure in the emergency department. High Blood Press Cardiovasc Prev. 2010;17 (4):1-7.
Marquél S, Cariou A, Chichel JD, Squara P. Comparison between Flotrac-Vigileo and bioreactance, a totally noninvasive method for cardiac output monitoring. Crit Care. 2009;13(3):1-6.
Martins S, Soares RM, Branco L. Non-invasive monitoring of pulmonary capillary wedge pressure in heart failure. Eur J Heart Fail. 2001;3(1):41-46.
Mathews L, Singh K. Cardiac output monitoring. Ann Card Anaesth. 2008;11(1):56-68.
Middleton PM, Davies SR. Noninvasive hemodynamic monitoring in the emergency department. Curr Opin Crit Care. 2011;17(4):342-350.
Monnet X, Picard F, Lidzborski E, et al. The estimation of cardiac output by the Nexfin device is of poor reliability for tracking the effects of a fluid challenge. Crit Care. 2012;16:R212.
Mutoh T, Kazumata K, Ishikawa T, Terasaka S. Performance of bedside transpulmonary thermodilution monitoring for goal-directed hemodynamic management after subarachnoid hemorrhage. Stroke. 2009;40:2368.
Napoli AM. Physiologic and clinical principles behind noninvasive resuscitation techniques and cardiac output monitoring. Cardiol Res Pract. Volume 2012 (2012), Article ID 531908, 12 pages http://dx.doi.org/10.1155/2012/531908
Napoli AM, Machan JT, Corl K, Forcada A. The use of impedance cardiography in predicting mortality in emergency department patients with severe sepsis and septic shock. Acad Emer Med. 2010;17(4):452-455.
Nelson MR, Stepanek J, Cevette M, et al. Noninvasive measurement of central vascular pressures with arterial tonometry: clinical revival of the pulse pressure waveform? Mayo Clin Proc. 2010;85(5):460-472.
Nguyen HB, Banta DP, Stewart G, et al. Cardiac index measurements by transcutaneous Doppler ultrasound and transthoracic echocardiography in adult and pediatric emergency patients. J Clin Monit Comput. 2010;24(3):237-247.
Noritomi DT, Vieira ML, Mohovic T, Bastos JF, et al. Echocardiography for hemodynamic evaluation in the intensive care unit. Shock. 2010;34 (suppl 1):59-62.
Nowak RM, Sen A, Garcia AJ, et al. The inability of emergency physicians to adequately clinically estimate the underlying hemodynamic profiles of acutely ill patients. Am J Emerg Med. 2011;30(6):954-960.
Odenstedt H, Stenqvist O, Lundin S. Clinical evaluation of a partial CO2 rebreathing technique for cardiac output monitoring in critically ill patients. Acta Anaesthesiol Scand. 2002;46(2):152-159.
Ospina-Tascon GA, Cordioli RL, Vincent JL. What type of monitoring has been shown to improve outcomes in acutely ill patients? Intensive Care Med. 2008;34:800-820.
Pearse RM, Ikram K, Barry J. Equipment review: an appraisal of the LiDCO trade mark plus method of measuring cardiac output. Crit Care. 2004;8(3):190-195.
Peyton PJ, Robinson JB, McCall PR. Noninvasive measurement of intrapulmonary shunting. J Cardiothorac Vasc Anesth. 2004;18(1):47-52.
Reinhart K, Kuhn HJ, Hartog C, Bredle D. Continuous central venous and pulmonary artery oxygen saturation monitoring in the critically ill. Intensive Care Med. 2004;30:1572-1578.
Silver MA, Cianci P, Brennan S. Evaluation of impedance cardiography as an alternative to pulmonary artery catheterization in critically ill patients. Congest Heart Fail. 2004;10 (suppl 2): 17-21.
Summers RL, Parrott CW, Quale C. Use of noninvasive hemodynamics to aid decision making in the initiation and titration of neurohormonal agents. Congest Heart Fail. 2004; 10 (suppl 2): 28-31.
Temporelli PL, Scapellato F, Eleuteri E, Imparato A, Giannuzzi P. Doppler echocardiography in advanced systolic heart failure: a noninvasive alternative to Swan-Ganz catheter. Circ Heart Fail. (2010);3(3):387-394
Truijen, J, VanLieshout JJ, Wesselink WA, Westerhof BE. Noninvasive continuous hemodynamic monitoring. J Clin Monit Comput. 2012;26(4):267-278.
Turner MA. Doppler-based hemodynamic monitoring. AACN Clin Issues. 2003;14(2):220-231.
van der Spoel AG, Voogel AJ, Folkers A, Boer C, Bouwman RA. Comparison of noninvasive continuous arterial waveform analysis (Nexfin) with transthoracic Doppler echocardiography for monitoring of cardiac output. J Clin Anesth. 2012;24(4): 304-309.
van Genderen ME, van Bommel J, Lima A. Monitoring peripheral perfusion in critically ill patients at the bedside. Curr Opin Crit Care. 2012;18(3):273-279.
Wooley JA. Indirect calorimetry: applications in practice. Respir Care Clin. 2006;12:619-633.
Young BP, Low LL. Noninvasive monitoring cardiac output using partial CO2 rebreathing. Crit Care Clin. 2010;26(2):383-392.
Yung GL, Fedullo PF, Kinninger K. Comparison of impedance cardiography to direct Fick and thermodilution cardiac output determination in pulmonary arterial hypertension. Congest Heart Fail. 2004;10(suppl 2):7-10.
Zhang Z, Xu X, Yao M, et al. Use of PiCCO system in critically ill patients with septic shock and acute respiratory distress syndrome. Trials. 2013;1:14-32.
Zimlichman E, Szyper-Kravitz M, Shinar Z, et al. Early recognition of acutely deteriorating patients in non-intensive care units: assessment of an innovative monitoring technology. J Hosp Med. 2012 Oct;7(8):628-633.
Adams KF. Guiding heart failure care by invasive hemodynamic measurements: possible or useful? J Cardiac Fail. 2002;8(2): 71-73.
Alvarez J, Bouzada M, Fernandez AL, et al. Hemodynamic effects of levosimendan compared with dobutamine in patients with low cardiac output after cardiac surgery. Rev Esp Cardiol. 2006;59(4):338-345.
Bagshaw SM, Brophy PD, Cruz D, Ronco C. Fluid balance as a biomarker: impact of fluid overload on outcome in critically ill patients with acute kidney injury. Crit Care. 2008;12(4):1-7.
Bauer SR, Lam SW. Arginine vasopressin for the treatment of septic shock in adults. Pharmacotherapy. 2010;30(10):1057-1071.
Bayer O, Reinhart K, Kohl M, Kabisch B, et al. Effects of fluid resuscitation with synthetic colloids or crystalloids alone on shock reversal, fluid balance, and patient outcomes in patients with severe sepsis. Crit Care Med. 2012;40(9):2543-2551.
Brazdzionyte J, Macas A, Sirvinskas E. Application of methods for hemodynamic monitoring in critical cardiac pathology (an experimental model for assessment of hemodynamics). Medicina. 2002;38(8):835-842.
Buerke M, Lemm H, Dietz S, Werdan K. Pathophysiology, diagnosis, and treatment of infarction–related cardiogenic shock. Herz. 2011;36(2):73-83.
Debacker D, Cretekr J, Dubois M, et al. The effects of dobutamine on microcirculatory alterations in patients with septic shock are independent of its systemic effects. Crit Care Med. 2006;34(2):403-408.
Deedwania PC, Carbajal E. Evidence-based therapy for heart failure. Med Clin North Am. 2012;96(5):915-931.
Di Giantomasso D, Morimatsu H, May CN. Increasing renal blood flow: low-dose dopamine or medium-dose norepinephrine. Chest. 2004;125(6):2260-2267.
Dünser MW, Mayr AJ, Ulmer H, et al. The effects of vasopressin on systemic hemodynamics in catecholamine-resistant septic and postcardiotomy shock: a retrospective analysis. Anesth Analg. 2001;93(1):7-13.
Faybik P, Hetz H, Baker A. Iced versus room temperature injectate for assessment of cardiac output, intrathoracic blood volume, and extravascular lung water by single transpulmonary thermodilution. J Crit Care. 2004;19(2):103-107.
Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797-805.
Ferguson-Myrthil N. Vasopressor use in adult patients. Cardiol Rev. 2012;20(3):153-158.
Havel C, Arrich J, Losert H, Gamper G, et al. Vasopressors for hypotensive shock. Cochrane Database Syst Rev. 2011;11(5): CD003709.
Heart Failure Society of America, Lindenfield J, Albert NM, et al. HFSA 2010 Comprehensive heart failure practice guideline. J Card Fail. 2010;16:e1.
Hollenberg SM. Inotrope and vasopressor therapy of septic shock. Crit Care Clin. 2009;25(4):781-802.
Jain M, Canham M, Upadhyay D. Variability in interventions with pulmonary artery catheter data. Intensive Care Med. 2003;29(11):2059-2062.
Kampmeier TG, Rehberg S, Westphal M, Lange M. Vasopressin in sepsis and septic shock. Minerva Anestesiol. 2010;76(10):844-850.
Kapoor PM, Kakani M, Chowdhury U, et al. Early goal-directed therapy in moderate to high-risk cardiac surgery patients. Ann Card Anaesth. 2008;11(1): 27-34.
Khot UN, Novaro GM, Popovic ZB. Nitroprusside in critically ill patients with left ventricular dysfunction and aortic stenosis. N Engl J Med. 2003;348(18):1756-1763.
Krejci V, Hiltebrand LB, Higurdsson GH. Effects of epinephrine, norepinephrine and phenylephrine on microcirculatory blood flow in the gastrointestinal tract in sepsis. Crit Care Med. 2006;34(1):1456-1463.
Kumar A, Anel R, Bunnell E. Pulmonary artery occlusion pressure and central venous pressure fail to predict ventricular filling volume, cardiac performance, or the response to volume infusion in normal subjects. Crit Care Med. 2004;32(3):691-699.
Landoni G, Biondi-Zoccai G, Greco M, Greco T, et al. Effects of levosimendan on mortality and hospitalization. A meta-analysis of randomized controlled studies. Crit Care Med. 2012;40(2):634-646.
Leuchte HH, Schwaiblmair M, Baumgartner RA. Hemodynamic response to sildenafil, nitric oxide, and iloprose in primary pulmonary hypertension. Chest. 2004;125(2):580-586.
Liu SS, Monti J, Kargbo HM, Athar MW, et al. Frontiers of therapy for patients with heart failure. Am J Med. 2013;126(1):6-12.
Malliotakis P, Xenikakis T, Linardakis M, Hassoulas J. Haemodynamic effects of levosimendan for low cardiac output after cardiac surgery: a case series. Hellenic J Cardiol. 2007;48(2):80-88.
Myburgh J. Norepinephrine: more of a neurohormone than a vasopressor. Crit Care. 2010;14(5):196.
O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32-43.
Papp A, Uusaro A, Parviainen I. Myocardial function and haemodynamics in extensive burn trauma: evaluation by clinical signs, invasive monitoring, echocardiography and cytokine concentrations. A prospective clinical study. Acta Anaesthesiol Scand. 2003;47(10):1257-1263.
Parissis JT, Rafouli-Stergiou P, Stasinos V, et al. Intropes in cardiac patients: update 2011. Curr Opin Crit Care. 2010;16(5):432-441.
Pestel GJ, Fukui K, Kimberger O, et al. Hemodynamic parameters change earlier than tissue oxygen tension in hemorrhage. J Surg Res. 2010 May 15;160(2):288-293.
Pinto BB, Rehberg S, Etmer C, Westphal M. Role of levosimendan in sepsis and septic shock. Curr Opin Anaesthesiol. 2008;21(2):168-177.
Puskarich MA. Emergency management of severe sepsis and septic shock. Curr Opin Crit Care. 2012;18(4):295-300.
Richards AM, Troughton RW. Use of natriuretic peptides to guide and monitor heart failure therapy. Clin Chem. 2012;58(1):62-71.
Rivers E, Nguyen B, Havestad S, et al. Early goal-directed therapy on the treatment of severe sepsis and septic shock. NEJM. 2001;345(19):1368-1377.
Ruggiero M. Effects of vasopressin in septic shock. AACN Adv Crit Care. 2008;19(3):281-287.
Russell JA. Bench-to-bedside review: vasopressin in the management of septic shock. Crit Care. 2011;15(4):226.
Sandifer JP, Jones AE. Dopamine versus norepinephrine for the treatment of septic shock EBEM commentators. Ann Emerg Med. 2012;60(3):372-373.
Shoemaker WC, Wo CC, Yu S. Invasive and noninvasive hemodynamic monitoring of acutely ill sepsis and septic shock patients in the emergency department. Eur J Emerg Med. 2000;7(3):169-175.
Szokol JW, Murphy GS. Transesophageal echocardiographic monitoring of hemodynamics. Intl Anesthesiol Clin. 2004;42(1):59-81.
Teerlink JR, Metra M, Zacà V, Sabbah HN, Cotter G, et al. Agents with inotropic properties for the management of acute heart failure syndromes. Traditional agents and beyond. Heart Fail Rev. 2009;14(4):243-253.
Vollman KM. Understanding critically ill patients hemodynamic response to mobilization. Crit Care Nurs Q. 2013;56(1):17-27.
Zafir B, Amir O. Beta blocker therapy, decompensated heart failure, and inotropic interactions: current perspectives. Isr Med Assoc J. 2012;14(3):184-189.
Zanotti Cavazzoni SL, Dellinger RP. Hemodynamic optimization of sepsis-induced tissue hypoperfusion. Crit Care. 2006; 10(suppl 3): 1-8.
AACN Hemodynamic Monitoring Practice Alert. Aliso Viejo, CA: AACN; 2004. http://www.aacn.org. Accessed January 1, 2010.
American Association of Critical-Care Nurses (AACN). Practice Alert: Pulmonary Artery Pressure Measurement. Aliso Viejo, CA: AACN; 2009. http://classic.aacn.org/AACN/practiceAlert.nsf/Files/PAP/$file/PAP%20Measurement%2005-2004.pdf. Accessed January 1, 2010.
Gawlinski A. AACN Protocol for Practice: Cardiac Output Monitoring. Aliso Viejo, CA: AACN; 1998.
Imperial-Perez F, McRae M. AACN Protocol for Practice: Arterial Pressure Monitoring. Aliso Viejo, CA: AACN; 1998.
Jesurum JT. AACN Protocol for Practice: SVO2 Monitoring. Aliso Viejo, CA: AACN; 1998.
Keckeisen M. AACN Protocol for Practice: Pulmonary Artery Pressure Monitoring. Aliso Viejo, CA: AACN; 1998.