Neurologic Disease
NIGHT TERRORS (PAVOR NOCTURNAS)
Acute Disseminated Encephalomyelitis (ADEM)
ANTI-NMDA RECEPTOR ENCEPHALITIS
INCREASED INTRACRANIAL PRESSURE (ICP)
Arteriovenous Malformations (AVMs)
CLINICALLY RELEVANT TYPES OF STROKE
AGENESIS OF THE CORPUS CALLOSUM
Seizures
DEFINITION
 WARD TIP
WARD TIP
The diagnosis of clinical epilepsy requires two or more unprovoked seizures.
 A paroxysmal electrical discharge of neurons in the brain resulting in an alteration
of function or behavior.
A paroxysmal electrical discharge of neurons in the brain resulting in an alteration
of function or behavior.
The most common neurologic disorder in children:
4–10% of children.
1% of all ED visits.
Highest incidence: <3 years.
ETIOLOGY
 EXAM TIP
EXAM TIP
Recurrence risk after a first unprovoked episode is 45% (27–52%).
The risk of epilepsy is >70% after two unprovoked episodes.
Multiple etiologies have been identified for seizures. Provoked causes include:
Fever.
Metabolic:
Hypoglycemia.
Hyponatremia.
Hypocalcemia.
Inborn errors of metabolism.
Medications and illegal drugs.
EXAM TIP
In most children with seizures, an underlying cause cannot be determined and a diagnosis of idiopathic epilepsy is given.
Trauma (intracranial hemorrhage).
Infections (encephalitis, meningitis, abscess).
Vascular events (strokes).
Hypoxic ischemia encephalopathy.
Idiopathic.
TYPES OF SEIZURES
EXAM TIP
Partial seizures: Onset in one brain region.
Generalized seizures: Onset simultaneously in both cerebral hemispheres.
See Table 17-1.

Partial (Focal) Seizures
Begin in one brain region.
1. Simple partial seizures:
Average duration is 10–20 seconds.
Restricted at onset to one focal cortical region.
Consciousness is not altered.
EXAM TIP
Aura: Abnormal perception or hallucination, which occurs before consciousness is lost and for which memory is returned afterwards. In seizure, as opposed to migraine, the aura is part of the seizure.
Tend to involve the face, neck, and extremities.
Patients may complain of aura, which is characteristic for the brain region involved
in the seizure (i.e., visual aura, auditory aura, etc.).
Seizures can also be somatosensory/visual or auditory.
2. Complex partial seizures:
EXAM TIP
Both simple and complex partial seizures may become generalized.
Average duration is 1–2 minutes.
Hallmark feature is alteration or loss of consciousness.
Automatisms are seen in 50–75% of cases (psychic, sensory, or motor phenomena).
3. Secondarily generalized seizures:
Starts as a partial seizure in a focal area of the brain and then spread to the
entire brain leading to a generalized seizure.
Sometimes the person does not recall the first part of the seizure.
Occurs in >30% of people with partial epilepsy.
Generalized Seizures
WARD TIP
The first step in evaluating any seizure disorder is determining the type of seizure.
Begins simultaneously in both cerebral hemispheres. Consciousness is impaired from seizure onset.
1. Typical absence seizures (formerly “petit mal”):
Generalized seizure.
EXAM TIP
Motor activity is the most common symptom of simple partial seizures.
Characterized by sudden cessation of motor activity or speech.
Brief stares (usually <10 seconds), rarely longer than 30 seconds.
More common in girls. Male-to-female ratio: 2:1.
Onset 4-10 years.
Can occur many times throughout the day.
There is no aura.
There is no postictal state.
Seizure can be elicited by hyperventilation.
Childhood absence epilepsy is associated with characteristic 3-Hz spike-and-wave
pattern (Figure 17-1) on EEG.

FIGURE 17-1. Absence seizure EEG. Characteristic 3-Hz spike and wave pattern.
EXAM TIP
The presence of an aura always indicates a focal onset of the seizure. Physiologically, an aura is simply the earliest conscious manifestation of a seizure and corresponds with area of brain involved.
2. Generalized tonic-clonic (GTC, formerly “grand mal”) seizures:
Extremely common and may follow a partial seizure with focal onset.
Patients suddenly lose consciousness, their eyes roll back, and their entire musculature
undergoes tonic contractions, rarely arresting breathing.
Gradually, the hyperextension gives way to a series of rapid clonic jerks.
Finally, a period of flaccid relaxation occurs, during which sphincter control
is often lost (incontinence).
Prodromal symptoms (not aura) often precede the attack by several hours and include
mood change, apprehension, insomnia, or loss of appetite. (Unclear if these are warning
signs or part of the cause.)
Absence Versus Complex Partial Seizures
WARD TIP
Automatisms are a common symptom of complex partial seizures.
 While examining an 8-year-old girl in your office, the child suddenly develops a
blank stare and flickering eyelids. Twenty seconds later she returns to normal and
acts as if nothing out of the ordinary has occurred. Think: Absence seizure.
While examining an 8-year-old girl in your office, the child suddenly develops a
blank stare and flickering eyelids. Twenty seconds later she returns to normal and
acts as if nothing out of the ordinary has occurred. Think: Absence seizure.
You are reviewing the history before seeing a patient. She is a 7-year-old bright
girl with no significant past medical history. The schoolteacher noted that she sometimes
does not respond when her name is called. Also, she stares in space with a blank look
momentarily. Think: Absence seizures.
PEDIATRIC SEIZURE DISORDERS
EXAM TIP
Absence Seizures
Shorter (seconds)
Automatism –
More frequent (dozens)
Quick recovery
Hyperventilation +
EEG: 3/sec spikes and waves
Complex Partial Seizures
Longer (minutes)
Automatism +
Less frequent
Gradual recovery
Hyperventilation –
EEG: Focal spikes
Simple Febrile Seizure
The most common seizure disorder during childhood.
Occurs in approximately 2–4% of children younger than 5 years with a peak incidence
between 12 and 18 months.
Present as a brief tonic-clonic seizure associated with a fever.
Risk of recurrence is 30% after first episode and 50% after second episode.
Highest recurrence if episode before 1 year of age (50%).
Antipyretics do not appear to prevent the onset of future febrile seizures.
There are no long-term sequelae, and most children will outgrow by age 6.
Risk of epilepsy (1–2% as opposed to 0.5–1% in the general population) not statistically
significant.
↑ risk of epilepsy (up to 13%) in the presence of:
Abnormal neurologic examination.
Complex febrile seizure (lasting >15 minutes, focal in nature, and/or recurrent
seizure within 24 hours).
Family history of epilepsy.
Among first-degree relatives 10–20% of parents and siblings also have had febrile
seizures. An autosomal-dominant inheritance pattern with incomplete penetrance is
demonstrated in some families (19p and 8q13–21).
Consider meningitis or toxin exposures for a febrile seizure >15 minutes. Have
a greater consideration for spinal tap in infants <1 year of age or in those with
clinical signs of meningitis.
Neonatal Seizure
The most common neurologic manifestation of impaired brain function.
Occurs in 1.8–3.5 of every 1000 newborns.
Higher incidence in low-birth-weight infants.
Metabolic, toxic, hypoxic, ischemic, and infectious diseases are commonly present
during the neonatal period, placing the child at an ↑ risk for seizures.
Myelination is not complete at birth; thus, GTC seizures are very uncommon in the
first month of life.
May manifest as tonic, myoclonic, clonic, or subtle (prolonged nonnutritive sucking,
nystagmus, color change, autonomic instability).
EEG may show burst suppression (alternating high and very low voltages), low-voltage
invariance, diffuse or focal background slowing, and focal or multifocal spikes.
WARD TIP
Benign neonatal familial convulsions (“fifth-day fits”) are a brief self-limited autosomal-dominant condition with generalized seizures beginning in the first week of life and subsiding within 6 weeks. There is a normal interictal EEG. There is a 10–15% chance of future epilepsy, but otherwise carries an excellent prognosis. Always elicit a family history in neonatal seizures usually revealed after interviewing grandparents.
Neonatal seizures are typically treated acutely with phenobarbital (drug of choice),
fosphenytoin, or benzodiazepines.
Phenytoin not a first-line agent due to depressive effect on the myocardium and
variable metabolism in newborns.
Other antiseizure drugs, such as levetiracetam or topiramate, are being increasingly
used for treatment of neonatal seizures but are not yet considered evidence-based
first-line agents.
Infantile Spasm
Onset: 4–7 months.
EXAM TIP
Immature neonatal brain is more excitable than older children.
Clusters of brief rapid symmetric flexor/extensor contractions of the neck, trunk,
and extremities up to 100 per day. Clusters can last <1 minute to 10–15 minutes.
Symptomatic type is most commonly seen with central nervous system (CNS) malformations,
brain injury, tuberous sclerosis, or inborn errors of metabolism, and typically has
a poor outcome.
Cryptogenic type has a better prognosis and children typically have an uneventful
birth history and reach developmental milestones before the onset of the seizures.
WARD TIP
If you are present during a tonic–clonic seizure:
Keep track of the duration.
Place the patient between prone and lateral decubitus to allow the tongue and secretions
to fall forward.
Loosen any tight clothing or jewelry around the neck.
Do not try to force open the mouth or teeth!
Treated with adrenocorticotropic hormone (ACTH) in the United States.
Vigabatrin (equally as effective as ACTH therapy).
EEG has characteristic hypsarrhythmia pattern: Large-amplitude chaotic multifocal
spikes and slowing (see Figure 17-2).
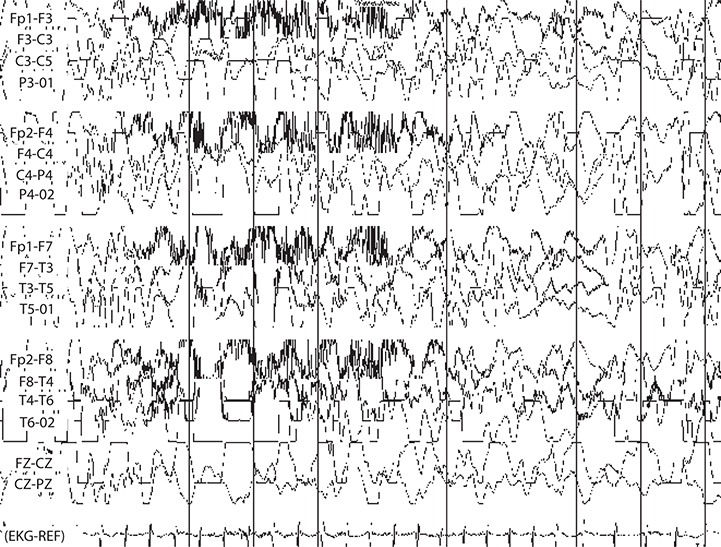
FIGURE 17-2. EEG demonstrating hypsarrhythmia pattern. Often seen in tuberous sclerosis, for example.
EPILEPSY
DEFINITION
A history of two or more unprovoked seizures.
After a nebulous period (on the order of 5–10 years) of seizure freedom without
the aid of antiepileptic medications or devices, the epilepsy can be considered to
have resolved, particularly if the patient fits an epilepsy syndrome that is known
typically to resolve.
EPIDEMIOLOGY
WARD TIP
If the seizure is brief with fever and immediate complete recovery consistent with febrile seizure, then only good examination and clinically indicated laboratory evaluation are needed to find the cause of fever. CT/ EEG/LP are not routinely indicated.
Epilepsy occurs in 0.5–1% of the population and begins in childhood in 60% of the cases.
SIGNS AND SYMPTOMS
Vary depending on the seizure pattern. See above discussion of types of seizures.
A seizure is defined electrographically as a hypersynchronous, hyperrhythmic, high-amplitude
signal that evolves in both frequency and space.
An aura is a stereotyped symptom set that immediately precedes the onset of a clinical
seizure and does not affect consciousness.
Physiologically, the aura is the true beginning of the seizure, and as such its
character can be quite useful for localizing seizure onset.
EXAM TIP
Etiologies of neonatal seizure:
Hypoxic-ischemic encephalopathy (35–42%)
Intracranial hemorrhage/infarction (15–20%)
CNS infection (12–17%)
Metabolic and inborn errors of metabolism (8–25%)
CNS malformation (5%)
A seizure prodrome is a set of symptoms, much less stereotyped than an aura, that
precedes a seizure by hours to days. Symptoms such as headache, mood changes, and
nausea are reported by over 50% of patients in some series.
TREATMENT
Therapy is directed at preventing the attacks.
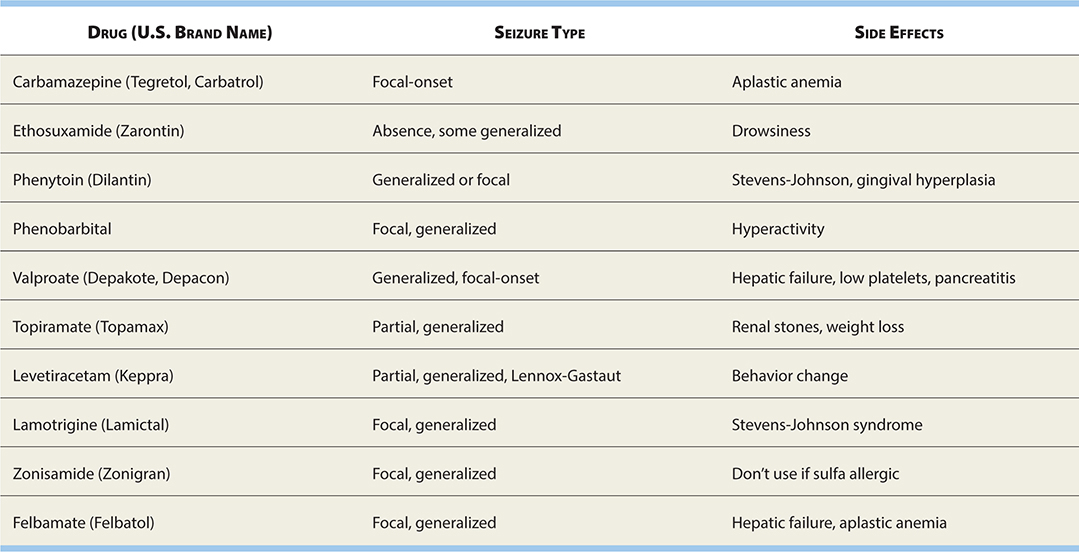
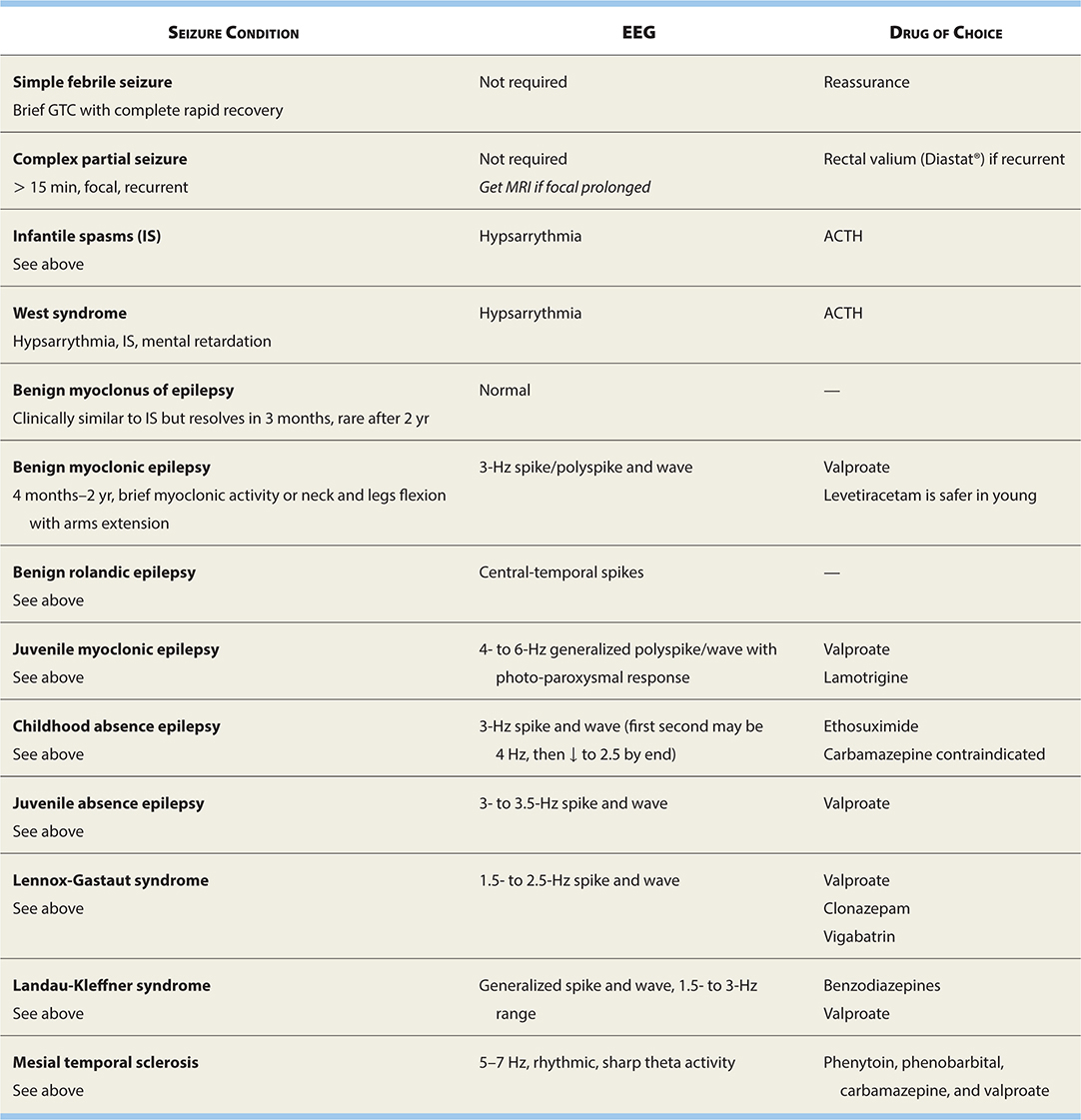
See Table 17-2 for current pharmacologic treatments for epilepsy.
TABLE 17-2. Epilepsy Drugs and Their Use in Different Seizure Types
COMMON EPILEPSY SYNDROMES
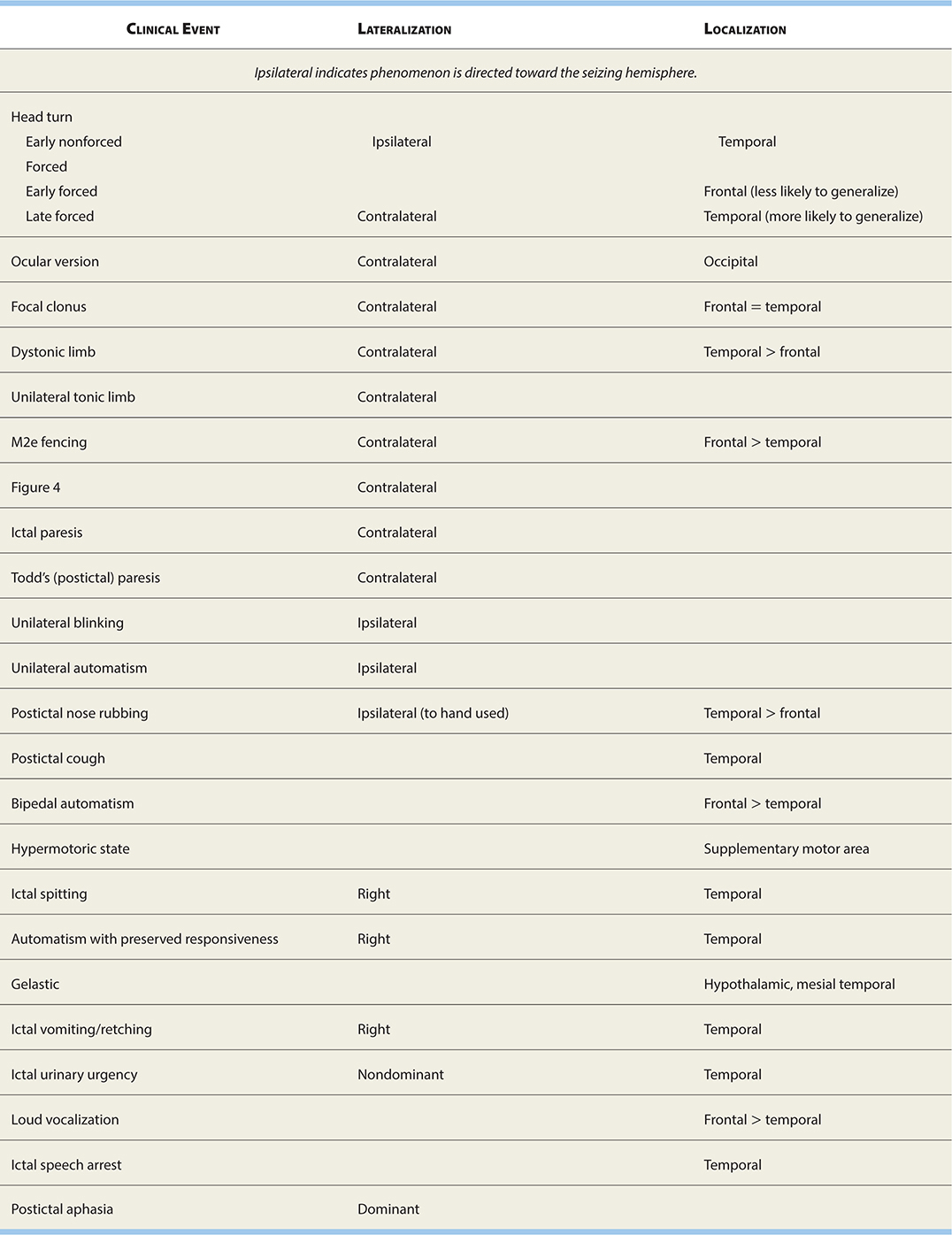
See Table 17-3 for localizing/lateralizing seizure semiologies.
TABLE 17-3. Localizing/Lateralizing Seizure Semiologies
Localization-Related Epilepsy
EXAM TIP
Unprovoked seizure: Unrelated to current acute CNS insult such as infection, ↑ intracranial pressure (ICP), trauma, toxin, etc.
Seizures secondary to a focal CNS lesion, not necessarily visible on imaging, best
candidates for epilepsy surgery.
Common examples include masses (particularly cortical tubers of
tuberous sclerosis [TS]), cortical dysplasia, postencephalitic gliosis, and arteriovenous
malformations (AVMs).
Benign Epilepsy with Centrotemporal spikes (BECTS)
A 5-year-old boy was noted to have facial twitching and facial drooling at a day
care center during a nap followed by generalized shaking of the body lasting 1–2 minutes.
The mother also reported noticing facial twitching during sleep. In the ED, he is
awake and his neurological examination is normal. You order an EEG, which shows centrotemporal
spikes. Think: BECTS formerly known as benign rolandic epilepsy.
BECTS is a partial epilepsy of childhood. The usual age of presentation is 3–13 years. Typical presentation: Seizure occurs during sleep (nighttime) with facial involvement. EEG shows central temporal spikes. Seizures typically resolve spontaneously by early adulthood.
Most common partial epilepsy.
Onset 3–13 years.
Particularly nocturnal (early morning hours before awakening).
EEG: Central temporal spikes (Figure 17-3).
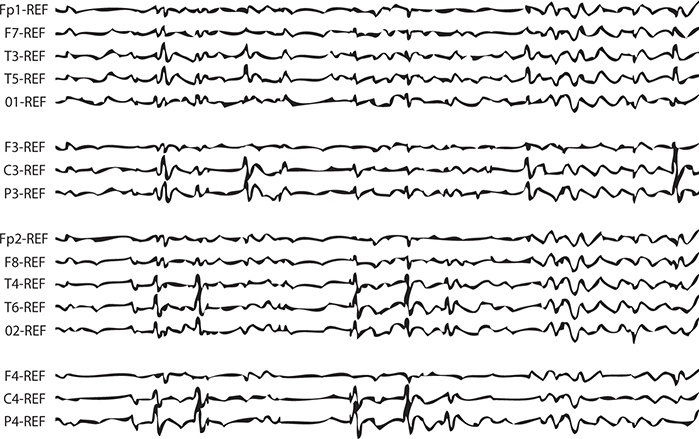
FIGURE 17-3. EEG demonstrating central temporal spikes characteristic of benign Rolandic epilepsy.
Excellent prognosis; most resolve by age 16 years.
Treatment: Carbamazepine, phenytoin, and valproic acid.
West Syndrome
Two percent of childhood epilepsies, but 25% of epilepsy with onset in the first
year of life.
Onset is at age 4–8 months.
Triad: Infantile spasms, mental retardation (MR), and hypsarrhythmia.
Boys are more commonly affected but not significantly; generally poor prognosis.
WARD TIP
Epilepsy History
Age, sex, handedness
Seizure semiology (what the seizures look like, details about right/left). If more
than one type, the pattern of progression (if any)
Seizure duration/history of status epilepticus
Postictal lethargy or focal neurologic deficits
Current frequency/tendency to cluster
Age at onset
Date of last seizure
Longest seizure-free interval
Known precipitants (don’t forget to ask if the seizures typically arise out of
sleep)
History of head trauma, difficult birth, intrauterine infection, hypoxic/ischemic
insults, meningoencephalitis, or other CNS disease
Developmental history (delay strongly correlated with poorer prognosis)
Family history of epilepsy, febrile seizures
Psychiatric history
Current AEDs
AED history (maximum doses, efficacy, reason for stopping)
Previous EEG, MRI findings
Differential includes TS (largest group), CNS malformation, intrauterine infection,
inborn metabolic disorders, and idiopathic. Idiopathic group fares the best.
Treatment in the United States is restricted to ACTH.
Juvenile Myoclonic Epilepsy (JME)
Onset: 12–16 years.
Characteristic history: Usually early morning on awakening, while hair combing
and tooth brushing.
Seizures: Myoclonus, absence, GTC.
EEG: 4- to 6-Hz irregular spike-and-wave pattern (Figure 17-4 and Table 17-4).
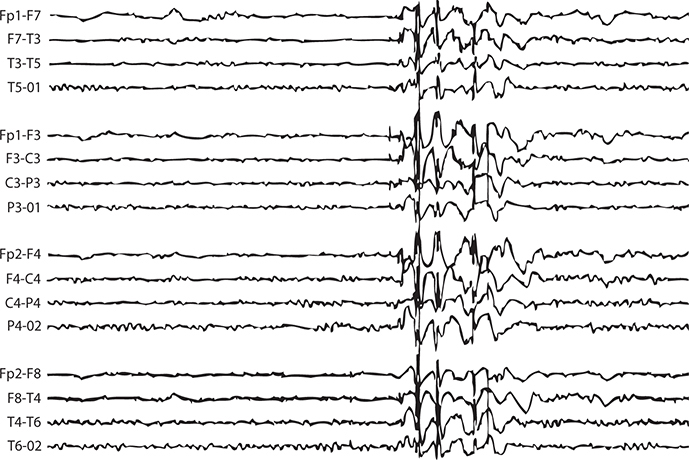
FIGURE 17-4. EEG demonstrating characteristic pattern of juvenile myoclonic epilepsy.
TABLE 17-4. Characteristic EEG Patterns in Various Seizure Conditions
Treatment: Valproate, lamotrigine.
Prognosis: Good Rx response but lifelong.
High rate of recurrence if antiepileptic drug (AED) discontinued.
Childhood Absence Epilepsy (CAE; Pyknolepsy)
See absence seizures above. GTC seizures often develop in adolescence; spontaneous
resolution is the rule, however.
Juvenile absence epilepsy (JAE): Similar to CAE except beginning in adolescence
and have more GTC seizures, sexes affected equally, EEG spike and wave often faster
than 3 Hz.
Lennox-Gastaut Syndrome (LGS)
A generalized epilepsy syndrome.
Multiple seizure types (tonic, atonic, absence, and myoclonic seizures).
EEG: 1.5- to 2.5-Hz spike-and-wave pattern.
Cognitive impairment.
Infantile spasms may evolve to LGS (30%).
Seizures are frequent and resistant to treatment with AEDs.
Landau-Kleffner Syndrome (LKS; Acquired Epileptic Aphasia)
WARD TIP
Loss of language skills in a previously normal child with seizure disorder. Think: LKS.
Language regression.
Aphasia (primarily receptive or expressive).
Seizures of several types (focal or GTC, atypical absence, partial complex).
EEG: High-amplitude spike-and-wave discharges. Obtain EEG during sleep (more apparent
during non-rapid eye movement sleep).
Differential diagnosis: Autism.
Treatment: Valproic acid.
Progressive Myoclonic Epilepsies
WARD TIP
Evaluate patients following their first seizure (for mass, lesion, etc.) prior to diagnosing and treating epilepsy.
This group of diseases includes Unverricht-Lundborg disease, myoclonic epilepsy
with ragged-red fibers (MERRF), Lafora disease, neuronal ceroid lipofuscinosis, and
sialidosis/mucolipidosis, and Ramsay Hunt syndrome.
Begin in late childhood to adolescence, and entail progressive neurologic deterioration
with myoclonic seizures, dementia, and ataxia. Death within 10 years of onset is common,
but survival to old age occurs.
Mesial Temporal Sclerosis/Temporal Lobe Epilepsy
Gliotic scarring and atrophy of the hippocampal formation, creating a seizure focus.
Abnormality is often apparent on high-resolution magnetic resonance imaging (MRI).
Rhythmic, 5–7 Hz, sharp theta activity.
Phenytoin, phenobarbital, carbamazepine, and valproate are equally effective. Curative
resection is often possible if refractory to treatment.
Rett Syndrome
DEFINITION
A neurodegenerative disorder of unknown cause.
EPIDEMIOLOGY
X-linked recessive with MECP2 gene mutation occurs almost exclusively in females. Rett syndrome does exist in males
with 47,XXY and MEP2 gene mutation. However, males with 46,XY and MECP2 gene mutation do not survive.
Prevalence: 1 in 15,000 to 1 in 22,000.
ETIOLOGY
EXAM TIP
The hallmark of Rett syndrome is repetitive hand-wringing and loss of purposeful and spontaneous hand movements.
Most cases result from defect in MECP2. Gene testing is available.
CDKL5 gene mutations can also cause Rett syndrome.
SIGNS AND SYMPTOMS
Normal development until 12–18 months (can appear as early as 5 months).
The first signs are deceleration of head growth, lack of interest in environment,
and hypotonia, followed by a regression of language and motor milestones.
Ataxia, hand-wringing, reduced brain weight, and episodes of hyperventilation are
typical.
Autistic behavior.
PROGNOSIS
After the initial period of regression, the disease appears to plateau.
Death occurs during adolescence or the third decade of life (cardiac arrhythmias).
Status Epilepticus (SE)
DEFINITION
Any seizure or recurrent seizures without return to baseline lasting >20 minutes.
ETIOLOGY
EXAM TIP
In children under age 3, febrile seizures are the most likely etiology of status epilepticus.
Febrile seizures, idiopathic status epilepticus, and symptomatic SE.
Febrile SE accounts for 5% of febrile seizures and one-third of all episodes of
SE.
PATHOPHYSIOLOGY
Prolonged neural firing may result in neuronal cell death, called excitotoxicity.
TREATMENT
Initial treatment includes assessment of the respiratory and cardiovascular systems
(ABCs).
Obtain rapid bedside glucose level.
MANAGEMENT
EXAM TIP
Neonatal status that is refractory to the usual measures may respond to pyridoxine. This is seen in pyridoxine dependency (due to diminished glutamate decarboxylase activity, a rare autosomal- recessive condition) or pyridoxine deficiency in children born to mothers on isoniazid.
Stabilization phase (0–5 minutes of seizure activity): Airway, breathing, circulation (ABCs); give O2; obtain IV acess, monitor vital signs, obtain rapid bedside glucose; obtain additional
labs and cultures as indicated.
Initial therapy phase (5–20 minutes of seizure activity): Administer a benzodiazepine (specifically IM
midazolam, lorazepam, or diazepam, OR Intranasal [IN] midazolam or buccal midazolam
if IV access not obtained). Benzodiazepines are recommended as the initial therapy
of choice, given its demonstrated efficacy, safety, and tolerability.
Second therapy phase (20–40 minutes of seizure activity): If seizure continues, other options include
fosphenytoin, valproic acid, and levetiracetam. IV phenobarbital is an alternative
if none of the three recommended therapies are available, but can worsen respiratory
depression.
Third therapy phase (40+ minutes of seizure activity): If seizure continues, treatment considerations
should include repeating second-line therapy or anesthetic doses of either thiopental,
midazolam, pentobarbital, or propofol (all with continuous EEG monitoring).
Neuro-Cutaneous Syndromes
STURGE-WEBER SYNDROME
Dermato-oculo-neural syndrome.
EPIDEMIOLOGY
Occurs sporadically in 1 in 50,000.
ETIOLOGY
WARD TIP
If you see “port-wine stain,” think Sturge-Weber syndrome.
Abnormal development of the meningeal vasculature, resulting in hemispheric vascular
steal phenomenon and resultant hemiatrophy.
Facial capillary hemangioma usually accompanies in V1 distribution.
SIGNS AND SYMPTOMS
Cutaneous facial nevus flammeus (distribution of the trigeminal nerve) → port-wine
stain.
Ipsilateral diffuse cavernous hemangioma of the choroid → glaucoma.
Ipsilateral meningeal hemangiomatosis (seizures and mental retardation).
The lesions in the eye, skin, and brain are always present at birth.
Contrast-enhanced MRI to look for meningeal angioma.
Seizures are usually refractory, and hemispherectomy improves the prognosis.
It is very unlikely to have meningeal involvement without port-wine stain, but
most children with a facial port-wine nevus do not have an intracranial angioma.
VON HIPPEL–LINDAU DISEASE
DEFINITION
A neurocutaneous syndrome (usually no cutaneous involment) affecting many organs, including the cerebellum, spinal cord, medulla, retina, kidneys, pancreas, and epididymis.
SIGNS AND SYMPTOMS
The major neurologic manifestations are:
Cerebellar/spinal hemagioblastomas: Present in early adult life with signs of ↑ ICP.
EXAM TIP
Renal carcinoma is the most common cause of death associated with von Hippel–Lindau disease.
Retinal angiomata: Small masses of thin-walled capillaries in the peripheral retina.
Multiple congenital cysts of the pancreas and polycythemia are also associated
with it.
Early detection and resection is the best management.
Photocoagulation for retinal detachment.
NEUROFIBROMATOSIS (NF)
EPIDEMIOLOGY
Both types display autosomal-recessive inheritance patterns.
Type 1: The most prevalent type (~90%) with an incidence of 1 in 4000 (chromosome 17).
Type 2: Accounts for 10% of all cases of NF, with an incidence of 1 in 40,000 (chromosome
22).
EXAM TIP
About 50% of NF-1 results from new mutations. Parents should be carefully screened before counseling on the risk to future children.
CLINICAL MANIFESTATIONS
Type 1
Diagnosis is made by the presence of two or more of the following:
Six or more café-au-lait macules (must be >5 mm prepuberty, >15 mm postpuberty).
Axillary or inguinal freckling (Crowe sign).
WARD TIP
NF-1: Café-au-lait spots, childhood onset.
NF-2: Bilateral acoustic neuromas, teenage onset, multiple CNS tumors.
Two or more iris Lisch nodules (melanocytic hamartomas).
Two or more cutaneous neurofibromas.
A characteristic osseous lesion (sphenoid dysplasia, thinning of long-bone cortex).
Optic glioma.
A first-degree relative with confirmed NF-1.
EXAM TIP
Café-au-lait is French for “coffee with milk,” which is the color of these lesions.
Learning disabilities, abnormal speech development, and seizures are common.
Patients are at a higher risk for other tumors of the CNS such as meningiomas and
astrocytomas (optic nerve gliomas in 20%) (but not as significantly as in NF-2).
Risk of malignant transformation to neurofibrosarcoma is <5%.
Type 2
Diagnosis is made when one of the following is present:
Bilateral CN VIII masses (most of the cases).
A parent or sibling with the disease and either a neurofibroma, meningioma, glioma,
or schwannoma.
Café-au-lait spots and skin neurofibromas are not common findings.
Patients are at significantly higher risk for CNS tumors than in NF-1 and typically
have multiple tumors.
TREATMENT
EXAM TIP
Prenatal diagnosis and genetic confirmation of diagnosis are available in familial cases of both NF-1 and NF-2, but not new mutations.
Treatment is mainly aimed at preventing future complications and early detection of malignancies. Resection of the schwannomas can be done to preserve hearing.
TUBEROUS SCLEROSIS
EPIDEMIOLOGY
Inherited as an autosomal-dominant trait, with a frequency of 1:6,000.
Two-thirds are new mutations.
PATHOLOGY
EXAM TIP
In general, the younger that a child presents with signs and symptoms, the greater the likelihood of mental retardation.
Characteristic brain lesions consist of tubers, which are located in the convolutions
of the cerebrum, where they undergo calcification and project into the ventricles.
There are two recognized genes: TSC1 on chromosome 9, encoding a protein called
hamartin; and TSC2 on chromosome 16, encoding a protein called tuberin.
Tubers may obstruct the foramen of Monro, → hydrocephalus.
CLINICAL MANIFESTATIONS
Hypopigmented macules (Ash leaf skin lesions) are seen in 90% and are best viewed
under a Wood’s lamp (violet/ultraviolet light source).
CT scan shows calcified hamartomas (tubers) in the periventricular region.
Seizures and infantile spasms (IS) are common. Seizures usually present as IS before
age 1 and are difficult to control. Children develop autistic features and have developmental
disabilities and learning difficulties.
Adenoma sebaceum—small, raised papules resembling acne that develop on the face
in butterfly pattern between 4 and 6 years of age, actually are small hamartomas.
A Shagreen patch (rough, raised, leathery lesion with an orange-peel consistency
in the lumbar region) is also a classic finding; typically does not develop until
adolescence.
Fifty percent of children also have rhabdomyomas of the heart, which may → CHF
or arrhythmias. They can be found on prenatal ultrasonography but usually regress
after birth.
Hamartomas of the kidneys and the lungs are also frequently present.
EXAM TIP
Tuberous sclerosis is the most common cause of infantile spasms, an ominous seizure pattern in infants.
DIAGNOSIS
A high index of suspicion is needed, but all children presenting with infantile
spasms should be carefully assessed for skin and retinal lesions.
CT or MRI will confirm the diagnosis.
Genetic testing is available for mutations in TSC1 and TSC2.
EXAM TIP
Hamartoma: A tumor-like overgrowth of tissue normally found in the area surrounding it.
SLEEP DISORDERS
Parasomnias
As a group, these disorders are:
Paroxysmal.
Predictable in their appearance in the sleep cycle.
Nonresponsive to environmental manipulation .
Characterized by retrograde amnesia.
A thorough history makes the diagnosis and an extensive workup is rarely needed.
NIGHT TERRORS (PAVOR NOCTURNAS)
DEFINITION
Transient, sudden-onset episodes of terror in which the child cannot be consoled
and is unaware of the surroundings, usually lasting for 5–15 minutes.
There is total amnesia following the episodes.
EPIDEMIOLOGY
Occur in 1–3% of the population, primarily in boys between ages 5 and 7.
PATHOPHYSIOLOGY
Fifty percent complete recovery by age 8.
Fifty percent are also sleepwalkers.
Often, incontinence and diaphoresis.
Occur in stage 4 (deep) sleep, which is first third of night sleep.
DIAGNOSIS
PSG (polysomnography).
TREATMENT
Reassurance; usually self-limited and resolve by age 6.
SOMNAMBULANCE (SLEEPWALKING)
Occurs during slow-wave sleep.
EXAM TIP
Night terrors, sleepwalking, and nightmares are associated with disturbed sleep, but have no known neurologic disorder.
Occurs during first third of the night.
Onset: 8–12 years.
Awakened only with difficulty and may be confused when awakened.
Fifty percent also have night terrors.

INSOMNIA
WARD TIP
Sleep deprivation causes attention deficit, hyperactivity, and behavior disturbances in children—often mistaken for attention deficit/hyperactivity disorder (ADHD).
WARD TIP
Obstructive sleep apnea due to adenotonsillar hyperplasia is an indication for tonsillectomy and adenoidectomy.
Affects 10–20% of adolescents.
Depression is a common cause and should be ruled out.
OBSTRUCTIVE SLEEP APNEA (OSA)
EXAM TIP
Since obstructive sleep apnea causes hypoxia, it may be associated with polycythemia vera, growth failure, and serious cardiorespiratory pathophysiology.
Occurs in 2–5 % of children, most often between ages 2 and 6.
Characterized by chronic partial airway obstruction with intermittent episodes
of complete obstruction during sleep, resulting in disturbed sleep.
Snoring is the most common symptom, occurring in most of them (12% of general pediatric
population has snoring without OSA).
Symptoms: Fatigue/hyperactivity, headache, daytime somnolence.
Signs: Narrow airway, tonsillar hypertrophy, often obese.
Diagnosis: History and physical examination, polysomnography (>1 apnea/hypopnea per hour).
Coma
Consciousness refers to the state of awareness of self and environment.
Pediatric evaluation of consciousness is dependent on both age and developmental
level.
DEFINITION
Pathologic cause of loss of normal consciousness.
PATHOPHYSIOLOGY
Consciousness is the result of communication between the cerebral cortex and the
ascending reticular-activating system.
Coma can be caused by:
Lesions of the medullary reticular-activating system or its ascending projections.
Ventral pontine lesions → locked-in syndrome, which is not coma.
ETIOLOGY
WARD TIP
Herniation is a result of increased intracranial pressure and often leads to coma or death.
Herniation syndromes that may result in coma:
Uncal herniation: Pressure on CN 6 with diploplia and inability to abduct eye
Central (trans-tentorial herniation): Blown (fixed and dilated) pupil, ptosis, CN 3 compression (down and out eye), ipsilateral
hemiplegia
Structural causes include trauma, vascular conditions, and mass lesions involving
directly or mass effects.
Metabolic and toxic causes include hypoxic-ischemic injury, toxins, infectious
causes, and seizures.
EVALUATION
Administer glucose via IV line so that the brain has an adequate energy supply.
Treat underlying cause (toxin antidote, reduce ICP, antibiotics, etc.).
PROGNOSIS
WARD TIP
Prognosis depends on the etiology of the insult and the rapid initiation of treatment!
Overall, children tend to do better than adults.
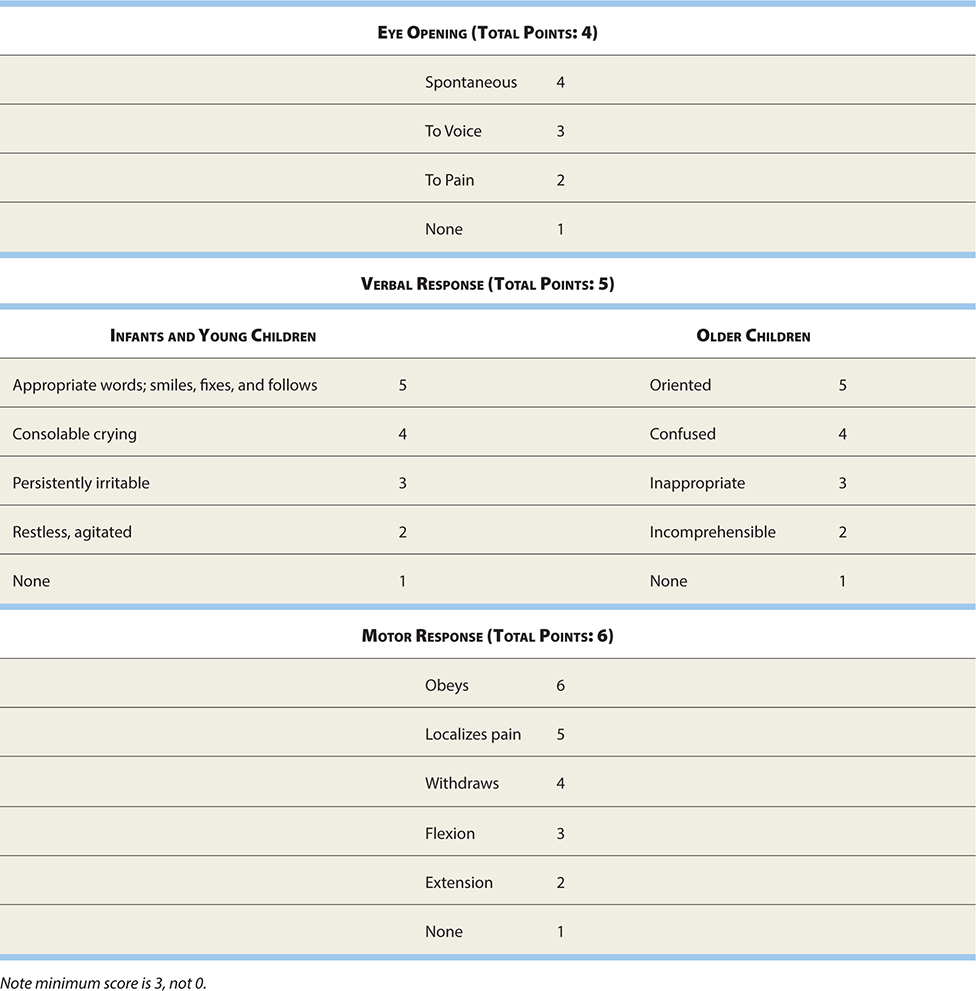
Several measurement scales have been published attempting to predict outcome. The
most widely accepted is the Glasgow Coma Scale (see Table 17-5).
TABLE 17-5. Glasgow Coma Scale (GCS)
Another scale that you should know exists is the Pediatric Cerebral Performance
Category Scale, which, unlike the Glasgow, was specifically designed for pediatric
patients.
ENCEPHALOPATHIES
CNS Infection
MENINGITIS
DEFINITION
Diffuse inflammation of the meninges, particularly arachnoids and pia mater.
Bacteria:
<3 months: Group B streptococci and gram-negative organisms, Escherichia coli, Listeria.
>3 months: Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis (two life-threatening clinical syndromes: meningococcemia and meningococcal meningitis).
Virus: The term aseptic meningitis is used to describe the syndrome of meningism and CSF leukocytosis usually caused
by viruses or bacteria.
SIGNS AND SYMPTOMS
If immunocompromised, these signs and symptoms will be not prominent.
Fever, headache, and nuchal rigidity (most important features).
Photophobia or myalgia may be present.
Meningism (Brudzinski and Kernig signs) (see Figures 17-5 and 17-6).
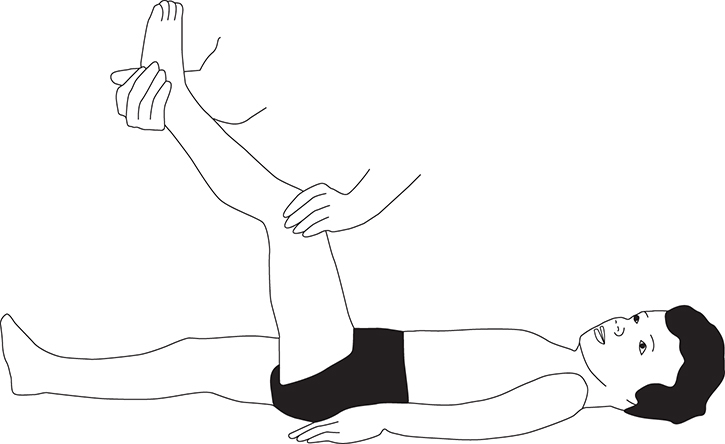
FIGURE 17-5. Kernig sign. Flex patient’s leg at both hip and knee, and then straighten knee. Pain on extension is a positive sign.
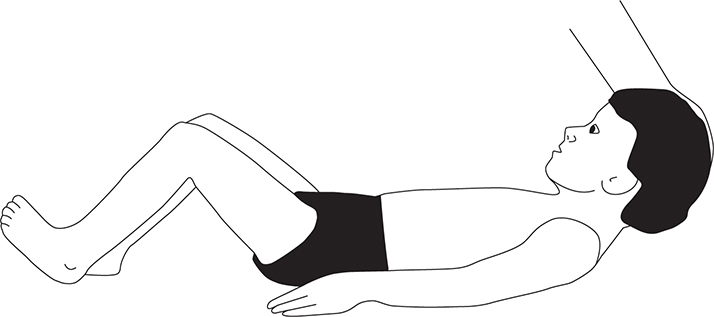
FIGURE 17-6. Brudzinski sign. Involuntary flexion of the hips and knees with passive flexion of the neck while supine.
Altered consciousness, petechial rash, seizures, cranial nerve, or other abnormal
neurological findings.
DIAGNOSIS
EXAM TIP
Take some time to familiarize yourself with Tables 17-6 and 17-7: You will be asked this!
TABLE 17-6. Cerebrospinal Fluid (CSF) Findings in Meningitis

TABLE 17-7. Common Causes of Pediatric Bacterial Meningitis
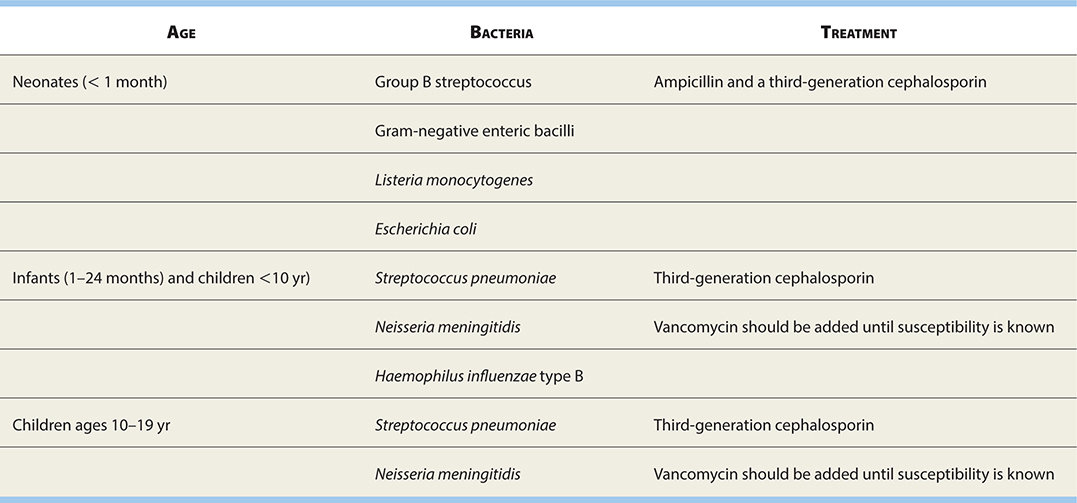
Analysis of the CSF is not always predictive of viral or bacterial infection since there is considerable overlap in the respective CSF findings, especially at the onset of the disease (Table 17-6).
Bacterial Meningitis
See Table 17-7 for common meningitis-causing bacteria.
Associated with high rate of complications and chronic morbidity and death.
Pathogenesis: 95% blood-borne. Organism enters the CSF, multiplies, and stimulates
an inflammatory response. Direct toxin from organism, hypotension, or vasculitis →
thrombotic event; vasogenic/cytotoxic edema causes ↑ ICP and ↓ blood flow, which all
may contribute to further damage.
Viral Meningitis
EXAM TIP
Chronic meningitis: Subacute symptoms of meningitis for >4 weeks: Infectious, autoimmune, or neoplastic.
Enterovirus (85%): Echovirus, coxsackievirus, and nonparalytic poliovirus.
Other classic causes are herpes simplex virus type 1 (HSV-1), Epstein-Barr virus
(EBV), mumps, influenza, arboviruses, and adenoviruses.
Clinical presentation is similar but symptoms usually are less severe than that
of bacterial meningitis.
Children may not be toxic appearing.
Children show typical viral-type infectious signs (fever, malaise, myalgia, nausea,
and rash) as well as meningeal signs.
Typically is a self-limited process with complete recovery, and treatment is supportive.
Fungal Meningitis
Although relatively uncommon, the classic organism is Cryptococcus.
Encountered primarily in the immunocompromised patient (with transplants, AIDS,
or on chemotherapy).
May be rapidly fatal (as quickly as 2 weeks) or evolve over months to years.
Tends to cause direct lymphatic obstruction, → hydrocephalus.
TREATMENT
WARD TIP
Nuchal rigidity. Think: Meningitis.
Third-generation cephalosporin (cefotaxime/ceftriaxone).
Add ampicillin for Listeria in neonates. Neonates can be treated with ampicillin + gentamicin or ampicillin +
cefotaxime.
Add vancomycin, considering the increasing resistance of pneumococci to cephalosporins
and carbapenems until the sensitivities are known.
If viral etiology is suspected or CSF is not clearly differentiating between bacterial
and viral etiologies, consider adding acyclovir until viral polymerase chain reaction
(PCR) comes back negative.
Steroid use is controversial.
Encephalitis
DEFINITION
WARD TIP
Congenital syphilis may manifest around age 2 with Hutchinson’s triad:
Interstitial keratitis
Peg-shaped incisors
Deafness (cranial nerve [CN] VIII)
A disease process in the brain primarily affecting the brain parenchyma.
Because patients often have symptoms of both meningitis and encephalitis, the term
meningoencephalitis is often applied.
ETIOLOGY
Chronic Bacterial Meningoencephalitis
1. Mycobacterium tuberculosis, M. bovis, and M. avium-intracellulare.
Nonspecific features develop over days to weeks. Patients have generalized complaints
of headache, malaise, and weight loss initially.
This is followed by confusion, focal neurological signs, cranial nerve palsies,
and seizures or, in advanced cases, hemiparesis, hemiplegia, or coma.
EXAM TIP
Argyll Robertson pupil is discrepancy in pupil size seen in neurosyphilis.
Serious complications include arachnoid fibrosis, → hydrocephalus, and arterial
occlusion, → infarcts.
M. avium-intracellulare is common in AIDS patients.
2. Neurosyphilis (tabes dorsalis).
Causative organism is Treponema pallidum.
May present with aseptic meningitis only.
Tertiary syphilis (late-stage syphilis) manifests with neurologic, cardiovascular,
and granulomatous lesions.
EXAM TIP
The transmission rate of syphilis from infected mother to infant is nearly 100%. Treat infant with IV penicillin G.
Congenital syphilis presents with a maculopapular rash, lymphadenopathy, and mucopurulent
rhinitis.
Routine prenatal screening for syphilis is now mandatory in most states to prevent
congenital syphilis.
Viral Meningoencephalitis
1. Herpes simplex virus:
HSV-1: Most cases after the neonatal period.
HSV-2: Usually blood-borne and results in diffuse meningoencephalitis and other
organ involvement. It is the congenitally acquired form, transmitted to 50% of babies
born to a mother with active vaginal lesions.
2. Herpes zoster virus:
Can occur after primary infection or as a result of reactivation later in life.
Usually with a rash, but outcome is poor in those without a rash.
In immunocompetent hosts, after 2–6 months of primary infection, the dormant virus
in the ganglia becomes activated in and causes large-vessel vasculitis → infarcts.
In immunocompromised hosts, the dormant virus causes small-vessel vasculitis and
results in hemorrhagic infarcts of gray and white matter.
EEG will show diffuse slowing and periodic lateralizing epileptiform discharges
(PLEDs).
WARD TIP
Acyclovir is the treatment of choice for herpetic meningitis.
3. Rabies:
Causes severe encephalitis, coma, and death due to respiratory failure.
Transmitted via bite or saliva from an infected animal, usually associated with
dogs, bats, skunks, raccoons, or squirrels.
The virus travels up the peripheral nerves from the bite site and enters the brain.
Nonspecific symptoms (fever, malaise) and paresthesia around the bite site are
pathognomonic. This is followed by more specific neurologic symptoms of hydrophobia,
aerophobia, agitation, hypersalivation, and seizures. This proceeds to coma and death.
Prophylaxis is indicated when there is potential exposure with rabies immunoglobulin
and rabies vaccine (multiple doses).
Transverse Myelitis
DEFINITION
An acute focal infectious or immune-mediated illness causing swelling and demyelination
of the spinal cord. This most commonly affects the thoracic spinal cord (80%) followed
by cervical cord.
It is a neurological emergency and requires prompt diagnosis and treatment to prevent
permanent damage.
SIGNS AND SYMPTOMS
Fever, lethargy, malaise, muscle pains.
Begins acutely and progresses within 1–2 days.
Back pain at the level of the involved cord and paresthesias of the legs are common.
Anterior horn involvement may cause lower motor neuron dysfunction.
Bladder and bowel dysfunction is present.
DIAGNOSIS
MRI: Enhanced T2 signals.
EXAM TIP
Numerous viruses as well as the rabies vaccination and smallpox vaccination have been linked to transverse myelitis.
CSF: Pleocytosis.
Electromyogram (EMG): Anterior horn-cell dysfunction in involved segments.
TREATMENT
IV steroids, intravenous immune globulin (IVIG), may require surgical intervention.
PROGNOSIS
Most make good recovery; however, it is slow.
Acute Disseminated Encephalomyelitis (ADEM)
DEFINITION
An immune-mediated demyelinating encephalopathy in which there is a sudden widespread
attack of the inflammation of the brain, spinal cord, and nerves, with destruction
of white matter.
Two-thirds with history of an antecedent viral infection.
Some features resemble multiple sclerosis.
SIGNS AND SYMPTOMS
Abrupt onset of change in consciousness or behavior changes unexplained by fever.
Often associated with at least one fever free day before onset of symptoms.
Irritability and lethargy are common first signs of acute disseminated encephalomyelitis.
Fever returns and headache are present in up to half of the cases. Meningismus
is also detected in approximately one-third of the cases. Over the course of minutes
to weeks, multifocal neurologic abnormalities develop which can include weakness,
ataxia, and cranial nerve abnormalities.
DIAGNOSIS
MRI: ADEM lesions are characteristically multiple, bilateral but asymmetric, and widespread within the CNS.
TREATMENT
WARD TIP
ADEM is associated with bilateral optic neuritis and MS is usually unilateral.
First-line high-dose IV steroids, also intravenous immune globulin (IVIG).
PROGNOSIS
2–10% mortality but most recover completely or with mild sequelae.
Tetanus
A 1-week-old child born to an immunocompromised mother presents with difficulty feeding,
trismus, and other rigid muscles. Think: Tetanus.
Tetanus is a toxin-mediated disease characterized by severe skeletal muscle spasms. It is a serious infection in neonatal life. Initial symptoms can be nonspecific. Inability to suck and difficulty in swallowing are important clinical features followed by stiffness and seizures. Neonatal tetanus can be prevented by immunizing mothers before or during pregnancy and providing sterile care throughout the delivery.
DEFINITION
An acute illness with painful muscle spasms and hypertonia caused by the neurotoxin
produced by Clostridium tetani.
These symptoms usually start in the jaw and facial muscles and progressively involve
other muscle groups.
SIGNS AND SYMPTOMS
Trismus (masseter muscle spasm) is the characteristic sign and is present in 75%
of cases.
Risus sardonicus, a grin caused by facial spasm, is also classic.
Dysphagia due to pharyngeal spasm develops over a few days; laryngo-spasm may result
in asphyxia.
Descending paralysis and when involves the trunk and thigh, the patient may exhibit
an arched posture in which only the head and heels touch the ground.
Late stages manifest with recurrent seizures consisting of sudden severe tonic
contractions of the muscles with fist clenching, flexion, and adduction of the upper
limb and extension of the lower limb and is associated with poor prognosis.
Autonomic dysfunction may be seen as ↑ sweating, heart rate, blood pressure, and
temperature.
Can also present with localized spasms at the site of infection or with abdominal
pain mimicking acute abdomen.
Incubation period varies from 2 to 14 days (average 7 days).
DIAGNOSIS
Diagnosis is clinical: Trismus, dysphagia, ↑ rigidity, and muscle spasms.
Laboratory studies are usually normal, but a moderate leukocytosis may be present.
CSF is normal.
Gram stain is positive in only one-third of the cases.
TREATMENT
Prophylactic intubation.
WARD TIP
Tetanic contractions can be triggered by minor stimuli, such as a flashing light. Patients should be sedated, intubated, and put in a dark room in severe cases.
Rapid administration of human tetanus immune globulin.
IV penicillin G, metronidazole, or doxycycline.
Surgical excision and debridement of the wound.
Muscle relaxants such as diazepam and phenobarbital should be used to promote relaxation
and seizure control. Neuromuscular blocking agents like vecuronium are also used.
PROGNOSIS
EXAM TIP
Tetanus is an entirely preventable disease via immunization.
Mortality rate: 5–35%.
Neonatal tetanus mortality ranges from 10% to 75%, depending on quality of care
received.
Other Encephalopathies
ANTI-NMDA RECEPTOR ENCEPHALITIS
BACKGROUND
An acute form of encephalitis that is potentially lethal but has a high probability
for recovery with treatment.
Autoimmune reaction, antibodies against NR1-NR2 NMDA receptors (N-methyl-D-aspartate receptor).
Often associated with tumors, classically ovarian teratomas, but many have no tumor
association.
SIGNS AND SYMPTOMS
Abrupt onset of change in consciousness or behavior changes unexplained by fever.
Initially, symptoms are nonspecific including fever, headache, and fatigue.
This is followed by a stage of psychosis with agitation, paranoia, psychosis, and
violent behaviors and can be associated with bizarre behavior, hallucinations.
Symptoms can progress to altered level of consciousness, hypoventilation, seizures,
autonomic instability, and dyskinesias.
DIAGNOSIS
Serum NMDA-receptor antibodies in the CSF.
Pelvic ultrasound to rule out tumor.
Exclude other causes of encephalopathy.
TREATMENT
EXAM TIP
The diagnosis of anti-NMDA receptor encephalitis is often delayed due to resemblance to other conditions, particularly psychiatric disorders.
Early removal of tumor if present.
IV corticosteroids, IVIG, and plasma exchange therapy in severe cases.
PROGNOSIS
The recovery process from anti-NMDA encephalitis can take many months.
~50% will fully recover. Some will recover with variable sequelae or deficits.
<10% mortality.
MITOCHONDRIAL ENCEPHALOPATHY
A group of disorders that can be caused by mutations in either nuclear or mitochondrial DNA, resulting in a variety of symptoms:
1. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes (MELAS):
The most common of mitochondrial encephalopathies.
Onset between ages 2 and 10 years; initial development normal, but short stature
is present.
The most initial feature is GTC seizure (often associated with hemiparesis and
cortical blindness), recurrent headache, and vomiting.
The neurologic abnormalities are transient initially, but later become progressive
and → coma and death.
MRI shows multiple strokes not in vascular distribution pattern. ↑ lactic acid
in blood and CSF. Muscle biopsy is diagnostic (ragged-red fibers).
2. Myoclonic epilepsy with ragged-red fibers (MERRF):
EXAM TIP
MELAS and MERRF are caused by point mutations in transfer RNA (tRNA) in mitochondrial DNA.
MELAS = leucine
MERRF = lycine
MERRF is often confused with Friedreich ataxia
Onset may be in childhood or adult life.
Four cardinal features are myoclonus, myoclonic epilepsy, ataxia, and ragged-red
fibers on muscle biopsy.
The initial feature is progressive insidious decline in school performance. GTC
seizures or myoclonus is usually the first symptom to seek medical attention. Later,
they develop progressive epilepsy, cerebellar ataxia, and dysarthria. Clinical myopathy
may not be present.
Diagnosis is by gene testing and muscle biopsy.
3. Reye syndrome:
A disorder of mitochondrial dysfunction associated with viral infection and aspirin
ingestion.
Sporadic syndrome can occur with varicella-zoster or influenza B infection.
Recurrent Reye-like syndrome is seen in children with inborn errors of metabolism,
medium-chain acyl Co-A dehydrogenase (MCAD) deficiency, urea cycle disorders, pyruvate
metabolism disorders.
WARD TIP
In general, salicylates should be avoided in children to prevent Reye syndrome.
Diagnosis: Liver biopsy is diagnostic. ↓ blood glucose, ↑ ammonia and liver enzymes without
jaundice.
HEPATIC ENCEPHALOPATHY
Acute hepatic failure caused by viral hepatitis, drugs, toxins, or Reye syndrome
results in altered consciousness (due to cerebral edema and accumulation of toxins,
ammonia).
In children, most commonly related to fulminant viral hepatitis (50–75%).
Early symptoms are malaise, lethargy, jaundice, dark urine, and abnormal liver
function tests (LFTs). The encephalopathy can be acute or chronic.
Other features include sleep disturbance, change in affect, drowsiness, asterixis
(flapping tremor). Decerebrate posturing may occur in the terminal stages.
Hepatic encephalopathy is reversible with treatment, and most therapies are aimed
at controlling the cerebral, renal, and cardiovascular functions until the liver regenerates
or liver transplantation can be done. These are achieved by lowering:
Ammonia level (↓ dietary protein, stop gastrointestinal [GI] bleed, treat constipation).
Cerebral edema with fluid restriction and the use of hyperosmolar agents (mannitol).
Patients who recover typically have no long-term sequelae.
HIV/AIDS ENCEPHALOPATHY
There is a 40–90% incidence of CNS involvement in perinatally infected children.
Ninety percent of infected infants are symptomatic by 18 months of age.
Develops 2–5 months after infection.
Commonly presents with progressive encephalopathy and hepatosplenomegaly, → failure
to meet developmental milestones, impaired brain growth, and symmetrical motor dysfunction.
Imaging techniques reveal cerebral atrophy in 85% of children and ventricular enlargement.
Basal ganglia calcifications may be present.
Opportunistic infections such as toxoplasmosis typically occur later in adolescence.
PCR analysis of HIV DNA or RNA is used to detect HIV infection in infants <18 months.
EXAM TIP
Old lead paint is the number one cause of lead toxicity.
Diagnosis: Via immunoglobulin G (IgG) antibody to HIV for patients >18 months and a confirmatory
test HIV DNA PCR.
Treatment: Highly active antiretroviral therapy (HAART).
All pregnant mothers are tested for HIV infection and are treated to ↓ the transmission.
ADRENOLEUKODYSTROPHY
A progressive disease, characterized by demyelination of the CNS and peripheral
nerves and adrenal insufficiency.
X-linked recessive, peroxisomal disorder, defect in the ability to catabolize long-chain
fatty acids (LCFAs).
It presents between 4 and 10 years with behavioral and cognitive decline with visual
loss, followed by motor symptoms.
Diagnosis: White matter abnormality on MRI, ↑ serum very-long-chain fatty acids (VLCFA), labs
for adrenal insufficiency.
Treatment: Bone marrow transplantation if only radiological changes are present and no appearance
of the neurological symptoms.
LEAD ENCEPHALOPATHY
There is no direct correlation to the level of lead and clinical manifestations.
Blood lead level >5 µg/dL is considered toxic. Lead interferes with porphyrin metabolism
in red blood cells (RBCs).
Acute: Vomiting, abdominal pain, seizures, impaired consciousness, and respiratory arrest
are common.
Chronic: Gradual confusion, behavior changes, sleep problems, seizures, ataxia. Peripheral
neuropathy, while common in adults, is rarely seen in children unless they also have
sickle cell anemia.
Pica is common in these children (e.g., eating paint chips).
Diagnosis is made primarily through history and also via blood lead testing. Microcytic
hypochromic anemia, basophilic stippling, and azotemia also present.
Treatment: Removing the source of lead, and chelation therapy when blood lead level
>45 µg/dL.
Movement disorders
Can be classified by a paucity of movement (hypokinetic) versus excessive or exaggerated movement (hyperkinetic). Hyperkinetic movement disorders predominate in children.
SYDENHAM’S CHOREA
Most frequent cause of new onset chorea in children.
Rapid, brief, unsustained, nonstereotypical movements of the body.
Autoimmune mediated.
Twice as common in females.
Onset: Age 3–17 years.
Postinfectious chorea appearing 4–8 weeks after a group A streptococcal pharyngitis.
Resolves after 8–9 months; 50% have persistent chorea.
Diagnosis: Recent throat infection (anti-streptolysin O, DNase B), ↑ T2 signals in basal ganglia.
EXAM TIP
Methylphenidate may unmask Tourette syndrome but does not cause it.
Treatment:
Valproate: First choice.
Dopamine-blocking agents: Second choice.
Also treat primary infection.
TOURETTE SYNDROME
A lifelong condition affecting 1 in 2000 that presents before age 15.
Diagnostic criteria: Multiple motor and vocal tics for >1 year with tic-free period
not more than 3 consecutive months.
Often associated with other conditions like obsessive-compulsive disorder (OCD),
attention deficit/hyperactivity disorder (ADHD).
Symptoms are enhanced by stress and anxiety.
Treatment with medications should be avoided.
Treat when tics interfere with child’s developmental learning or cause undue social
stress. Also treat comorbid conditions.
PANS AND PANDAS
PANS (Pediatric Acute-onset Neuropyschiatric Syndrome) and PANDAS (Pediatric Autoimmune
Neuropsychiatric Disorder Associated with Streptococci) have nearly identical presentations.
PANS is associated with a variety of infections and PANDAS is always associated
with streptococci.
Manifested by the development or exacerbation of tics and/or obsessive-compulsive
disorder (OCD).
Diagnosis is considered controversial by some authorities.
EXAM TIP
Titubations are a disturbance of body equilibrium in standing or walking, resulting in an uncertain gait and trembling, especially resulting from diseases of the cerebellum.
Ataxias
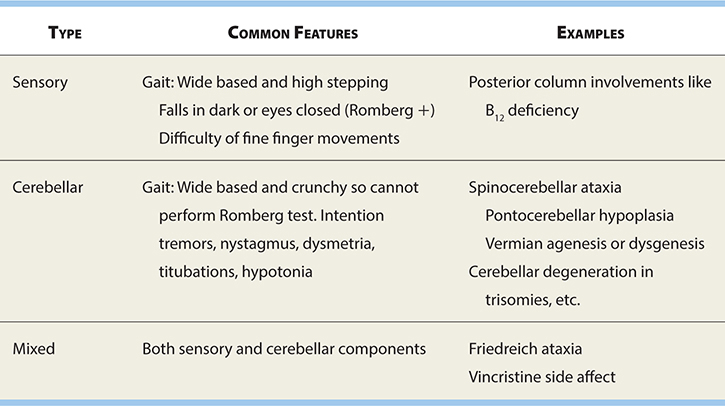
Inability to coordinate muscle activities to regulate posture and also strength and direction of extremity movements (see Table 17-8).
TYPES
Acute Cerebellar Ataxia
A diagnosis of exclusion occurring in children 2–7 years old.
Often follows viral infection by 2–3 weeks; thought to be autoimmune response and
has been seen with live inactivated vaccines like varicella vaccine.
Sudden onset of severe truncal ataxia; often, the child cannot stand or sit.
Severity is maximum at the onset with clear sensorium.
Horizontal nystagmus in 50%.
Diagnosis: Diagnosis of exclusion; exclude other serious causes first.
Treatment: Self-limited disease.
Prognosis: Complete recovery typically occurs within 2 months (1–5 months).
Freidreich’s Ataxia
Autosomal-recessive mutation (usually a triplet expansion) in Frataxin gene on
chromosome 9.
Degeneration of the dorsal columns and rootlets, spinocerebellar tracts, and, to
a lesser extent, the pyramidal tracts and cerebellar hemispheres.
Onset before age 10 (2–16 years).
Slow progression of ataxia involving the lower limbs > upper limbs associated with
dysarthria, ↓ tendon reflexes, positive Babinski sign, high-arch foot with loss of
dorsal column sensations.
Romberg test is positive.
Associated abnormalities include skeletal abnormalities (scoliosis), cardiomyopathy,
and optic atrophy.
Elevated α-fetoprotein (AFP).
Clinical features establish the diagnosis, which is confirmed with genetic testing.
There is no curative treatment available but symptomatic treatment to improve quality
of life.
WARD TIP
Myoclonic epilepsy with ragged-red fibers (MERRF) is often confused with Friedreich ataxia.
Ataxia-Telangiectasia
Autosomal-recessive disorder of nervous and immune system due to gene mutation
at chromosome 11.
The most common degenerative ataxia.
A slowly progressive ataxia beginning during first year of life resulting in inability
to walk by adolescence.
Oculomotor apraxia is present in 90% of the patients.
Telangiectasia becomes evident after 2 years or in the teenage years and is most
prominent on the bulbar conjunctiva (first), bridge of nose, and exposed surfaces
of the extremities. Sun exposure exacerbates the telangiectasia.
Sinopulmonary infection is another important feature. ↓ or absent IgA, IgE, and
especially IgG2 subclass. IgM may be increased.
↑ AFP and peripheral acanthocytes.
Have a 50- to 100-fold greater chance of brain tumors and lymphoid tumors, so avoid
radiation exposure by limiting imaging studies.
Peripheral Neuropathies
Injuries to the peripheral nerves may be either:
Demyelinating (injury to Schwann cells).
Degenerating (injury to the nerve or axon).
Peripheral neuropathy is the most common cause of progressive distal weakness.
Most common are hereditary causes and slow progression.
The most common acquired cause is Guillain-Barré syndrome (GBS) with rapid progression.
TYPES
Guillain-Barré Syndrome
A 6-year-old boy with no significant past medical history presents to the ED with
difficulty walking for past few days and is now unable to walk. He also has some weakness
in his upper extremities but he does not have any respiratory distress. There is no
clear history of any recent illness, vaccination, or sick contacts. He had upper respiratory
infection symptoms a few weeks ago. On examination, he is weaker more in the lower
extremities than upper, and deep tendon reflexes are absent at knee and ankle. Think: Guillain-Barré syndrome (GBS).
GBS is an ascending paralysis. History of prior upper respiratory tract or viral infection or recent vaccination may be present. Initial symptoms are pain, numbness, paresthesia, or weakness in the lower extremities, which rapidly progresses to bilateral and relatively symmetric weakness. ↓ or absent deep-tendon reflexes are often present. Lumbar puncture typically shows ↑ protein with normal CSF and white cell count (cytoalbuminologic dissociation).
A postinfection demyelinating neuropathy affecting predominantly the motor neurons.
It is due to immune cross-reactivity to a secondary illness within 4 weeks. Most
commonly seen after upper respiratory infection (URI), Campylobacter jejuni, Mycoplasma pneumoniae, cytomegalovirus (CMV), Epstein-Barr virus (EBV), varicella, influenza, hepatitis
A and B infection.
Weakness begins in the legs and progresses symmetrically upward to the trunk, arms,
then bulbar and ocular muscles.
Tendon reflexes are absent.
Respiratory muscles in 50%, autonomic dysfunction, pain, paresthesias can be present.
↑ proteins in CSF with no ↑ in lymphocytes.
Nerve conduction will be slow with conduction blocks, and enhancement of nerve
roots can be seen on MRI.
Treatment includes close monitoring for respiratory weakness and IVIG or plasmapheresis
in more severe cases.
Botulism
EXAM TIP
It is not possible to have botulism without having multiple cranial nerve palsies.
Botulinum toxin is disseminated through the blood and, due to the rich vascular
network in the bulbar region, symmetric flaccid paralysis of the cranial nerves is
the typical manifestation.
Infant botulism: The first sign is usually absence of defecation. The head control
is lost and the weakness descends.
Most dreaded complication is respiratory paralysis, and approximately 50% of patients
are intubated.
Prognosis is good in noncomplicated cases.
Antibiotics and blocking antibodies have not been shown to affect the course of
the disease.
Electromyogram (EMG) with high frequency (20–50 Hz) reverses the presynaptic blockade
and produces an incremental response.
Myasthenia Gravis
EXAM TIP
Infantile botulism traditionally associated with ingestion of honey (honey contains botulism spores) which is why honey is not given in the first year of life. Most cases are due to ingestion of environmental dust or soil from home canned foods or construction at or near the home.
↓ in postsynaptic acetylcholine receptors due to autoimmune degradation, resulting
in rapid fatigability of muscles.
Ptosis and extraocular eye weakness are the earliest and most diagnostic symptoms.
Onset usually after age 8, as early as 6 months. Prepubertal male bias, postpubertal
female bias.
WARD TIP
Children with myasthenic syndromes cannot tolerate neuromuscular blocking drugs, such as succinylcholine, and various other drugs. Most offenders are in the antibiotic, cardiovascular, and psychotropic categories.
Diagnosis is made by EMG with repetitive stimulation, edrophonium (Tensilon) test,
a quick test (acetylcholinesterase inhibitor). Acetylcholine receptor-binding or receptor-blocking
antibodies are detected in the seropositive forms and are an indication for thymectomy.
May be associated with autoimmune thyroid disease and seizures.
Cholinesterase drugs are the mainstay of treatment, with oral steroids used as
needed for immune suppression (initially may exacerbate the disease).
Prognosis varies, with some children undergoing spontaneous remission, while in
others the disease persists into adulthood.
Transitory Neonatal Myasthenia
WARD TIP
Remember, rapid correction of hyponatremia can result in cerebellar pontine myelinosis.
Passive transfer of antibodies from myasthenic mothers (10–15% incidence).
Self-limited disease consisting of generalized weakness and hypotonia for 1 week
to 2 months. Symptoms develop a few hours after birth. If develop after 3 days, then
are unlikely.
Poor suck and respiratory problems are addressed with supportive care. Neostigmine
or exchange transfusion can be used in more severe cases.
Electrolyte Imbalances
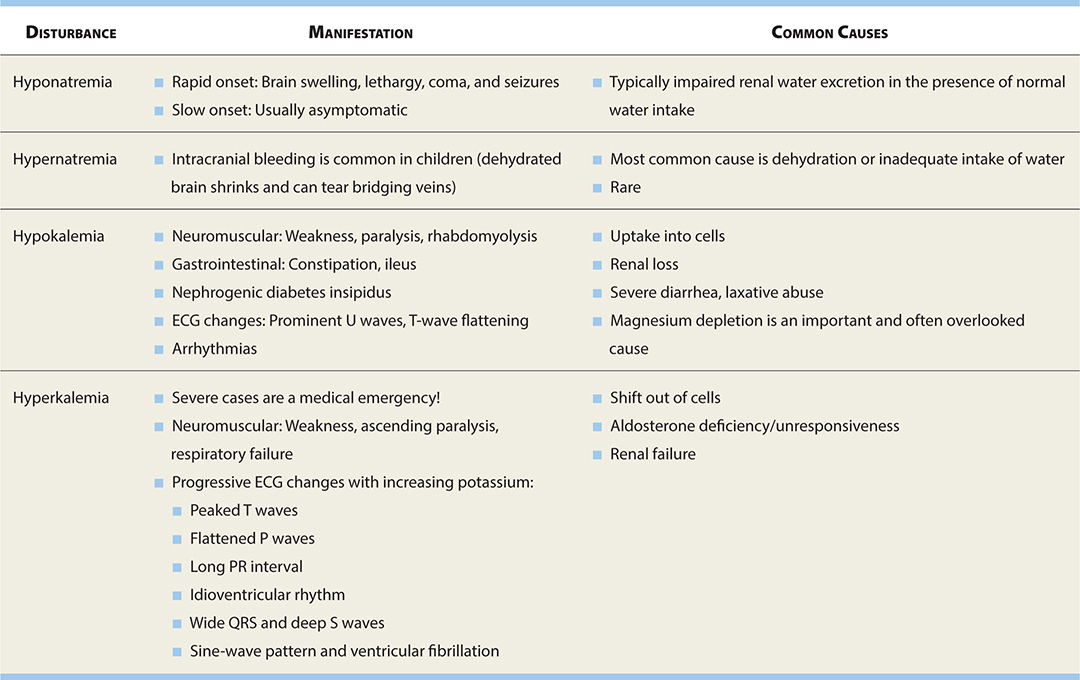
See Table 17-9 for common electrolyte imbalances affecting the nervous system.
TABLE 17-9. Electrolyte Disturbances and the Nervous System
Headaches
MIGRAINE
The most common type of headache in the pediatric population with female predominance.
DEFINITION
A recurrent headache with symptom-free intervals and can be associated with the following:
Abdominal pain.
Nausea and/or vomiting.
Throbbing headache.
Often bilateral (versus unilateral in adults).
Associated aura.
Relieved by sleep.
Family history of migraines.
Diagnosis of migraine is clinical and no neuroimaging is necessary unless it is persistently occipital or with abnormal neurologic examination.
CLASSIFICATION
Migraines may be classified into the following subgroups:
Migraine without Aura
Headache lasting 4–72 hours.
Two of the following: Unilateral, pulsating, moderate/severe pain, aggravation
of routine physical activity.
Headache may have associated nausea, vomiting, photophobia, phonophobia.
Migraine with Aura
Headache with fully reversible aura symptoms:
Visual, sensory, speech, motor, brainstem, retinal.
Aura is accompanied or followed by headache within 60 minutes, and may last 5–60
minutes.
Aura symptoms may spread gradually or two or more symptoms occur in succession.
Chronic Migraine
Defined as headache on >15 days per month for more than 3 months.
Daily headaches of less severity with less prominent migrainous features.
Complicated Migraine
WARD TIP
Episodic syndromes that may be associated with migraines include cyclic vomiting syndrome, abdominal migraine, benign paroxysmal vertigo, and benign paroxysmal torticollis.
Transient neurologic signs develop during a headache and persist after the resolution of the headache for a few hours to days.
TREATMENT
Avoid the possible triggers: Often, migraines occur in response to specific triggers,
such as psychological stress, strenuous exercise, sleep deprivation, cheese, chocolate,
processed meat, or moving vehicles, and minimizing these factors may have great therapeutic
effect.
Consider nonpharmacologic treatment with biofeedback techniques in chronic stress
headache.
For acute attacks:
Dark, quiet environment and sleep.
Adequate fluid intake.
Pharmacologic therapy: Acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs)
are first line.
Second-line drugs include triptans, caffeine, and ergot alkaloids (status migrinosus).
Antiemetics are helpful at the start of headache.
Treatment should be instituted as early as possible in an attack.
PROPHYLAXIS
WARD TIP
Prophylaxis should be offered to children with two or more migraines per month that interfere with activities such as school or recreation.
Antiepileptic drugs, such as topiramate, valproate, levetiracetam.
Tricyclic antidepressants such as amitriptyline.
β-blockers such as propranolol.
CLUSTER HEADACHE
Brief, severe, unilateral stabbing headaches that occur multiple times daily over
a period of several weeks and tend to be seasonal.
Onset after 10 years of age.
Male predominance.
Conjunctival injection, tearing, rhinorrhea.
Prophylaxis with lithium or calcium channel blocker.
Acute treatment with 100% oxygen or sumatriptan and dihydroergotamine (DHE).
TENSION HEADACHE
Tension or stress headaches are rare in children prior to puberty and are often difficult to differentiate from migraines.
PRESENTATION
Most often occur with a stressful situation, such as an exam.
Described as “hurting” but not “throbbing.”
It presents like a band around the head. It is present most of the times of the
day.
WARD TIP
Headaches can occur in children secondary to refractive errors. It is therefore imperative to perform a visual acuity determination.
Unlike migraines and ↑ intracranial pressure, tension headaches are not associated
with nausea and vomiting.
However, it is sometimes difficult to differentiate them from migraine.
DIAGNOSIS
Diagnosis of exclusion.
EEG or CT is not necessary.
TREATMENT
WARD TIP
Normal ICP
Newborns: 6 mm Hg
Children: 6–13 mm Hg
Adolescents/adults: 0–15 mm Hg
Steps should be taken to minimize anxiety and stress:
Mild analgesics often are ample.
Other options include counseling and biofeedback.
Sedatives or antidepressants are rarely necessary.
INCREASED INTRACRANIAL PRESSURE (ICP)
Headache due to tension of the blood vessels or dura may be the first symptom of an ↑ in intracranial pressure.
SYMPTOMS
It usually presents as headache, nausea, vomiting, diplopia, personality changes.
WARD TIP
Any time you see papilledema, think ↑ ICP.
It can present as bulging fontanelle, impaired upward gaze in infants.
The presentation depends also on rate at which the ICP increases. If it increases
slowly, then the intracranial structures have time to accommodate for the change.
Coughing or Valsalva maneuver tends to make the pain worse by increasing ICP further.
ETIOLOGY
WARD TIP
Cushing triad: A sign of increased intracranial pressure and impending herniation of the brain
1. Irregular respirations
2. ↓ heart rate
3. ↑ BP (actually seen in 20–30%)
Common causes include posterior fossa brain tumors (and other brain tumors), obstructive hydrocephalus, hemorrhage, meningitis, venous sinus thrombosis, pseudotumor cerebri, abscesses, and chronic lead poisoning.
DIAGNOSIS
Thorough history and physical exam are vital.
Papilledema (if ↑ pressure is present for some time) and nuchal rigidity are helpful
signs.
Obtain CBC, erythrocyte sedimentation rate (ESR), and CT/MRI to narrow the differential.
If CT/MRI is negative, consider lumbar puncture (LP).
TREATMENT
Varies with particular diagnosis, and should be directed at the underlying etiology.
Techniques to lower ICP acutely are as follows:
1. Intubation and subsequent hyperventilation results in cerebral vasoconstriction, effective for about 30 minutes.
2. Elevating the head 30 degrees facilitates venous return.
3. Hyperosmolar agents such as mannitol (osmotic diuretic) or hypertonic 3% saline, avoid hypovolemia.
4. Extraventricular drain provides temporary relief and can provide continuous monitoring of ICP.
5. Surgical decompression if persistently remains ↑.
Aneurysms
The pathogenesis of the aneurysms is multifactorial and controversial; however,
it is believed that focal congenital weakness of the internal elastic lamina and muscular
layers in the cerebral arteries → to aneurysmal formation.
Most common in internal carotid artery followed by middle cerebral artery, anterior
communicating artery, and basilar artery.
WARD TIP
Never perform an LP if papilledema is present. Must obtain CT before LP if suspicious of ↑ ICP.
Saccular aneurysms are the most common type and often at bifurcation of the internal
carotid artery.
Early warning signs are headaches or localized cranial nerve compression.
Most common presentation is subarachnoid hemorrhage (SAH).
More likely to rupture in patients <2 years of age or >10 years.
More common in males 2:1.
Familial occurrence is common.
ETIOLOGY
Most often are related to congenital diseases:
Ehlers-Danlos syndrome.
Marfan syndrome, tuberous sclerosis.
AVMs.
Coarctation of the aorta.
WARD TIP
CT does not always reveal a subarachnoid hemorrhage (SAH) so must consider LP to make definitive diagnosis. LP reveals ↓ RBCs in tube 4 and xanthochromia in SAH.
Polycystic kidney disease (likely develop secondary to hypertension in this condition);
called berry aneurysms.
Acquired aneurysms are most often related to bacterial endocarditis:
Embolization of bacteria results in mycotic aneurysms in the cerebral vasculature.
Twenty-five percent present with bleeding, such as a subarachnoid or intraparenchymal
hemorrhage.
DIAGNOSIS
EXAM TIP
Relatively more children have aneurysms in the vertebrobasilar circulation (23%) compared to adults (12%).
Angiography is the gold standard for aneurysms in both children and adults.
Magnetic resonance angiography (MRA) may also be used and is becoming more reliable.
TREATMENT
Surgical clipping or endovascular coiling is the treatment of choice.
Risk for rebleeding.
Arteriovenous Malformations (AVMs)
True AVMs consist of an abnormal communication of arteries and veins without intervening
capillaries that arises during development in prenatal period or just after birth.
It grows in size with time and varies in size from several millimeters to several
centimeters.
The larger ones create a significant atrioventricular (AV) shunt (steal phenomenon)
and considerable damage if they rupture.
Supratentorial (90%).
PRESENTATION
EXAM TIP
Gamma knife radiation typically takes up to 2 years to see resolution of the AVM, during which time the patient is at risk for hemorrhage; thus, surgery is the treatment of choice.
Small unruptured malformations present with headache or seizures.
Larger malformations may present with progressive neurologic deficit.
Hemorrhage is most often presentation (subarachnoid or intraparenchymal).
DIAGNOSIS
Angiography is the test of choice and is required to direct the future therapy.
MRA is also available.
MRI or CT with contrast can demonstrate an AVM but provide less information than
angiography.
Photon knife is the treatment.
COMMON AVM VARIANTS
Vein of Galen Malformations
Normal vein of Galen does not develop from its primitive vein, which persists and
communicates with superior saggital sinus.
Typically present during infancy with high-output congestive heart failure (CHF),
failure to thrive, or enlarging head size.
Mortality is 50%.
Treatment in difficult embolization is preferred over surgery.
A cranial bruit is often present with vein of Galen malformations.
Cavernous Hemangiomas
Low-flow AVM with tendency to leak (cause seizure) but usually do not result in
massive intracerebral hemorrhage.
Retinal cavernous hemangiomas may be also present.
Surgical resection is indicated if symptomatic.
Venous Angiomas
Rarely symptomatic (seizures are the most common presenting sign).
Surgery is not indicated unless complications arise.
TREATMENT
Treatment consists of surgical resection or embolization.
Focused gamma knife radiation has some benefits in smaller lesions.
Stroke
Transient ischemic attacks (TIAs): Neurologic deficits that resolve in <24 hours.
Stroke: Neurological deficits persist beyond 24 hours.
EPIDEMIOLOGY
2.6–13 cases per 100,000 per year.
Hemorrhagic stroke 1.5–5 per 100,000 children per year.
Ischemic stroke 0.6–8 per 100,000 children per year.
SIGNS AND SYMPTOMS
Sudden onset of neurologic deficit or seizures in neonates.
Headache, neck pain, and visual symptoms.
ETIOLOGY
Pediatric causes of stroke differ from those in the adult population.
Types of stroke include:
Ischemic: Thrombosis (both arterial and venous) or embolic (arterial).
Hemorrhage.
A variety of conditions or risk factors exist for stroke, including:
AVMs.
Antiphospholipid antibodies/lupus anticoagulant.
Congenital coagulopathies such as factor V Leiden and deficiencies of protein C,
S, and antithrombin III.
Hemoglobinopathies, sickle cell disease (SCD).
Sickle cell anemia at risk for ischemic stroke (sickling RBCs may → thrombosis
or endothelial injury).
EXAM TIP
Cardiac abnormalities are the most common cause of thromboembolic stroke in children.
Cardiac conditions: Arrhythmias, myxoma, paradoxical emboli through a patent foramen
ovale, and septic emboli from bacterial endocarditis.
Blunt trauma to the head and neck → arterial dissection.
Vasculitis, such as Kawasaki, hemolytic-uremic syndrome, systemic lupus erythematosus
(SLE), meningitis.
Mitochondrial diseases.
Extracorporeal membrane oxygenation (ECMO) is a risk for both intracranial hemorrhage
and embolic ischemic stroke.
CLINICALLY RELEVANT TYPES OF STROKE
Arterial Thrombosis/Embolism
Intracerebral arterial dissection after trivial trauma to head and neck due to
a tear in the intima.
The cerebral area supplied by the vessel distal to lesion undergoes infarction
and produces symptoms (loss of functions).
Cerebral symptoms such as a progressive hemiplegia, lethargy, or aphasia result
from the shedding of small emboli into the carotid circulation.
Seizures are the most common presenting symptom in neonates.
Cardiac source usually.
Venous Thrombosis
WARD TIP
A typical workup for a stroke syndrome will include head CT or MRI scan, followed by an angiogram (if the CT/MRI is nondiagnostic), and a cardiac echo to exclude cardiac causes.
May be subdivided into septic and nonseptic causes.
Septic causes include bacterial meningitis, otitis media, and mastoiditis.
Aseptic causes are numerous and include severe dehydration, hypercoagulable states,
congenital heart disease, and hemoglobinopathies (SCD).
Neonates present with diffuse neurologic signs and seizures.
In children, focal neurologic signs are more common.
Closed Head Trauma
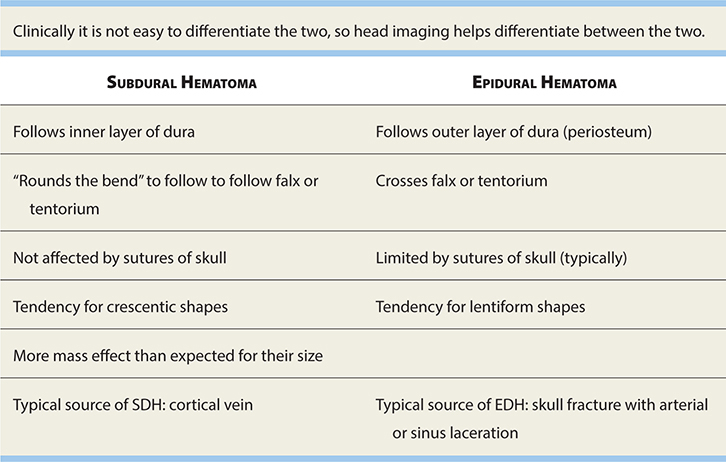
See Table 17-10 for a comparison of subdural and epidural hematomas.
TABLE 17-10. Features of Acute Epidural and Subdural Hematomas
SUBDURAL HEMATOMA (SDH)
EPIDEMIOLOGY
The most frequent focal brain injury in sports and the most common form of sports-related intracranial hemorrhage. Seen most often in infants, with a peak at 6 months.
EXAM TIP
Low-molecular-weight heparin has been shown to be safe, effective, and well tolerated in children with strokes.
ETIOLOGY
Occurs when a bridging vein is torn between the dura and the brain.
In neonates due to a tear in tentorium near its junction with the falx.
Trauma is usually the cause. Skull fracture is not seen commonly.
An SDH should be ruled out if changes in conscious level are present after head
injury.
Typically frontoparietal location. It can be acute, subacute, or chronic.
SIGNS AND SYMPTOMS
These depend on age of the child and also severity of the SDH.
Neonates: Seizures, a bulging fontanelle, and ↓ activity.
Retinal and preretinal hemorrhages common in children, especially in abused children.
↑ ICP (irritability, lethargy, vomiting, papilledema, headache).
WARD TIP
Subdural hematomas appear crescent shaped (concave) on CT and will not cross the midline, but will cross ipsilateral suture lines.
DIAGNOSIS
Gold standard is CT scan.
EPIDURAL HEMATOMA
EPIDEMIOLOGY
Seen most often in children >2 years of age.
ETIOLOGY
Most commonly results from a fracture in the temporal bone, lacerating the middle
meningeal artery.
Can be acute (arterial bleed) or chronic (venous bleed).
Skull fracture is seen commonly.
Nearly always unilateral; however, bilateral case has been described.
SIGNS AND SYMPTOMS
WARD TIP
Lucid interval. Think: Epidural hematoma.
Classic progression involves an initial loss of consciousness, followed by a lucid
interval, and then abrupt deterioration and death (not as helpful in younger children).
Hemorrhage and acute brain swelling cause ↑ ICP that can result in herniation with
ispilateral ptosis, dilated pupil, and ipsilateral hemiparesis due to contralateral
compression of crus cerebri.
Retinal and preretinal hemorrhages are not common.
WARD TIP
Epidural hematomas appear lens shaped (convex) on CT and will not cross the midline or other cranial sutures.
↑ ICP is seen (irritability, lethargy, vomiting, papilledema, headache).
DIAGNOSIS
Gold standard is CT scan.
TREATMENT
EXAM TIP
Old contusions develop an orange color secondary to hemosiderin deposition and are referred to as plaques jaunes by pathologists.
Epidural hematomas may progress rapidly, and immediate neurosurgical treatment is indicated.
COUP/CONTRECOUP INJURIES
Cerebral contusion injury mainly occurs when the head is subjected to a sudden acceleration or deceleration.
Coup Injuries
Located directly at the point of impact.
More common in acceleration injuries such as being hit with a baseball bat.
Multiple microhemorrhages as blood leaks into the brain tissue.
Contrecoup Injuries
EXAM TIP
Contrecoup injuries tend to be more severe than coup injuries.
Located opposite (180 degrees) from the point of impact.
More common in deceleration injuries, such as striking one’s head on the pavement
after a fall.
DIFFUSE AXONAL INJURY
EPIDEMIOLOGY
Tissues with differing elastic properties shear against each other, tearing axons.
Caused by rapid deceleration/rotation of head.
Locations:
Cerebral hemispheres near gray-white junction.
Basal ganglia.
Corpus callosum, especially splenium.
Dorsal brain stem.
WARD TIP
Diffuse axonal injury is best visualized on a T2-weighted MRI.
High morbidity and mortality—common cause of posttraumatic vegetative state.
Initial CT often normal despite poor GCS.
Lesions often nonhemorrhagic and seen only on MRI.
Survivors often have substantial long-term cognitive and behavioral morbidity.
Concussion
A teenage girl experienced a head-to-head collision with another player during soccer
4 hours ago. She reports headache, dizziness, nausea, and difficulty concentrating
and focusing since the injury. She has no focal neurologic findings on examination.
Think: Concussion.
Trauma-induced brain dysfunction without demonstrable structural injury on standard
neuroimaging.
The signs and symptoms are nonspecific and may include:
Headache.
Fatigue.
Dizziness.
Nausea/vomiting.
Unsteadiness.
Mental fogginess.
Anterograde or retrograde amnesia.
Difficulties with concentration.
Sleep disturbances.
Emotional lability.
Neuroimaging is normal in concussions and should only be used judiciously to rule
out other pathology such as intracranial bleeds.
The mainstay of management is physical and neurocognitive rest; however, low level
of activity is not harmful.
EXAM TIP
The PECARN head trauma prediction rules have been shown to be accurate in the identification of children at very low risk for clinically important traumatic brain injuries and has resulted in decreased head CT utilization in children.
In addition, efforts to prevent additional injury though return to play guidelines
that limit sports until full recovery is evident.
Most pediatric patients will recover fully from concussions but caution is advised
with repeat concussions.
Hydrocephalus
Head circumference >2 SD above the mean is macrocephaly, and if due to ↑ CSF in the CSF spaces, it is called hydrocephalus.
PHYSIOLOGY
CSF is made by the choroid plexus in the walls of the lateral, third, and fourth
ventricles.
CSF flows in the following direction: lateral ventricles → foramen of Monro → third
ventricle → cerebral aqueduct → fourth ventricle → foramina of Magendie and Luschka
→ subarachnoid space of spinal cord and brain → arachnoid villi.
CSF is absorbed primarily by the arachnoid villi through tight junctions.
ETIOLOGY
Obstructive (noncommunicating) hydrocephalus:
Most commonly due to stenosis or narrowing of the aqueduct of Sylvius.
An obstruction in the fourth ventricle is a common cause in children, including
posterior fossa brain tumors, Arnold-Chiari malformations (type II), and Dandy-Walker
syndrome.
Also seen in brain abscess, hematoma, infectious, vein of Galen malformation.
Nonobstructive (communicating) hydrocephalus:
EXAM TIP
Pneumococcal and tuberculous meningitis produce a thick exudate that can obstruct the basal cisterns, → communicating hydrocephalus.
Most commonly follows a subarachnoid hemorrhage or meningitis.
Blood in the subarachnoid spaces may obliterate the cisterns or arachnoid villi
and obstruct CSF flow.
Venous sinus thrombosis, meningeal malignancy, and intrauterine infections are
other causes.
Ex vacuo: Hydrocephalus resulting from ↓ brain parenchyma.
CLINICAL MANIFESTATIONS
Infants:
Accelerated rate of enlargement of the head is most prominent sign.
Bulging anterior fontanelle (fontanelles can provide some pressure relief in infants,
delaying symptoms of ↑ ICP). Widening of cranial sutures, sun-setting sign, and Parinaud
syndrome.
Upper motor neuron signs such as brisk reflexes are common findings due to stretching
of the descending cortical spinal tract.
↑ ICP signs (lethargy, vomiting, headache, etc.) may be present, especially acutely.
Children and adolescents:
Signs are more subtle because the cranial sutures are partially closed.
↑ ICP signs may be present. Visual fields particularly peripheral fields are involved
gradually. Papilledema can be present.
A gradual change in school performance may be the first clue to a slowly obstructing
lesion.
DIAGNOSIS
EXAM TIP
Preemies with intraventricular hemorrhage frequently develop hydrocephalus.
A detailed history and physical exam is key to discovering the underlying etiology.
Ultrasound and head CT/MRI are the most important studies to identify the cause
of hydrocephalus.
Familial cases of aqueductal stenosis have been reported and have an X-linked pattern
of inheritance.
Neurofibromatosis and meningitis have also been linked to aqueductal stenosis.
TREATMENT
Medical management with acetazolamide (may ↓ CSF production) and furosemide may
provide temporary relief.
Placement of an extraventricular drain (EVD) or ventriculoperitoneal shunt (VPS),
if the etiology is permanent, may be required.
Neoplasms
PEDIATRIC BRAIN TUMORS
EPIDEMIOLOGY
EXAM TIP
Glioblastoma multiforme (GBM) is a high-grade glioma common in adults but rare in children.
Most common solid tumors of the childhood.
Third most common pediatric tumors (#1 leukemia, #2 lymphoma).
Supratentorial tumors are as common as infratentorial tumors.
Glial cell tumors are the most common tumors in childhood and consist of astrocytomas,
ependymomas, oligodendrioglioma, and primitive neuroectodermal tumor (PNET).
Medulloblastoma is a common PNET only in childhood.
CLINICAL MANIFESTATIONS
Generally present with either signs and symptoms of ↑ ICP (infants) or with focal
neurologic signs (adolescents).
Alterations in personality are often the first symptoms of a brain tumor.
Nystagmus is the classic finding in posterior fossa tumors.
Clinical signs depending on location of the tumor (loss/alteration in the functions
of the brain area).
Tumors in the posterior fossa tend to result in hydrocephalus secondary to CSF
flow obstruction.
Cerebellar Astrocytoma
The most common posterior fossa tumor of childhood.
It is a slow-growing pilocytic astrocytoma and more benign than the adult-onset
astrocytomas.
Histologically shows fibrillary astrocytes with dense cytoplasmic inclusions called
Rosenthal fibers.
Associated with neurofibromatosis type 2 (NF2).
Good prognosis; 5-year survival >90% after gross total resection which is achieved
in 70% of the cases.
Treatment is surgical resection.
Medulloblastoma (PNET)
The second most common posterior fossa tumor and the most prevalent brain tumor
in children under the age of 7 years. More common in males.
Rapidly growing malignant tumor, arises from the undifferentiated neural cells
in the region of cerebellar vermis.
Tends to invade the fourth ventricle and spread along CSF pathways and involves
the spine, so consider imaging the spine.
Histologic analysis shows deeply staining nuclei with scant cytoplasm arranged
in pseudorosettes.
Presents with intracranial hypertension and ataxia, symptoms evolving in few weeks;
papilledema is absent.
WARD TIP
MRI is the best test for a posterior fossa tumor.
MRI: Brightly enhancing mass with cystic lesion.
Treatment: Surgical resection followed by irradiation.
Prognosis: Dependent on size and dissemination of the tumor, 5-year survival rate is >80%.
Craniopharyngioma
One of the most common supratentorial brain tumors of childhood which arises from
cells in the Rathke’s pouch.
It is locally aggressive and recurs.
Short stature or other endocrine-associated problems are common initial signs.
Typically slow growing and benign.
The tumor may be confined to the sella turcica or extend through the diaphragma
sellae and compress the optic nerve or, rarely, obstruct CSF flow.
Due to location, surgical resection is often subtotal.
DIAGNOSIS
Ninety percent of craniopharyngiomas show calcification on CT scan; MRI provides
better images of surrounding structures.
Baseline endocrine studies and visual fields should be done prior to surgery.
Neuroblastoma (NB)
EPIDEMIOLOGY
EXAM TIP
Infants tend to have localized NB in the cervical or thoracic region, whereas older children tend to have disseminated abdominal disease.
A common tumor of neural crest origin, representing the most common neoplasm in
infants and 8% of all childhood malignancies.
Malignant tumor that arises from the neural crest cells.
Ninety percent are diagnosed before age 5, with a peak at 2 years.
PATHOGENESIS
NB is a small, round blue cell tumor with varying degrees of neuronal differentiation.
CLINICAL PRESENTATION
The tumor may arise at any site of sympathetic nervous tissue.
The adrenals, retroperitoneal sympathetic ganglia, and abdomen are the most common
sites.
Thirty percent arise in the cervical or thoracic region and may present with Horner
syndrome.
Opsoclonus-myoclonus: “Dancing eyes, dancing feet”—the telltale symptom of this
disease (secondary to paraneoplastic antibodies).
DIAGNOSIS
Typically, a mass is seen on CT or MRI.
Ninety-five percent of cases have elevated tumor markers, most often homovanillic
acid (HVA) and vanillylmandelic acid (VMA) in the urine.
Metaiodobenzylguanidine (MIBG) radioisotope scan for detecting small primaries
and metastases.
Stage 4: Infantile form, self-limited with good prognosis.
Unfavorable prognosis is associated with ↑ neuron-specific enolase and amplification
of N-Myc gene.
Treatment is surgical resection followed by radio + chemotherapy.
Congenital Malformations
AGENESIS OF THE CORPUS CALLOSUM
Associated with numerous syndromes and several inborn errors of metabolism, including
patients with lissencephaly, Dandy-Walker syndrome, Arnold-Chiari type 2 malformations,
and Aicardi syndrome.
Imaging techniques reveal that the lateral ventricles are shifted laterally.
Normal intelligence is not unusual, and often only mild clinical signs are seen.
The severity of the disease varies greatly, from only mild deficits to marked retardation
and severe epilepsy.
SYRINGOMYELIA
A teenage girl has a headache and a cape-like distribution of pain and temperature
sensory loss that developed after a minor motor vehicle accident. Think: Cervical syringomyelia with undiagnosed Chiari I.
The Chiari type I malformation is characterized by herniation of the cerebellar tonsils through the foramen magnum and may → the development of syringomyelia. Common presentations include headache, neck pain, vertigo, sensory changes, and ataxia. Typical scenario is occipital pain precipitated by cough or Valsalva maneuver. MRI is the modality of choice.
A slowly progressive paracentral cavity formation within the brain or spinal cord,
most often in the cervical or lumbar regions.
Thought to arise from incomplete closure of the neural tube during the fourth week
of gestation.
MRI is the test of choice for diagnosis.
Often develops post-traumatically in the setting of an undiagnosed Chiari I malformation
or tethered cord.
Symptoms include bilateral impaired pain and temperature sensation due to decussation
of these fibers near the central canal. Also weakness of the hand muscles and progressive
symptoms as the cavity enlarges. It contains a yellow fluid.
Called syringobulbia when present in brain stem.
DANDY-WALKER MALFORMATION
Results from a developmental failure of the roof of the fourth ventricle to form,
resulting in a cystic expansion into the posterior fossa.
Ninety percent of patients have hydrocephalus.
Agenesis of the cerebellar vermis and corpus callosum is also common.
Infants present with a rapid ↑ in head size.
Management is via shunting of the cystic cavity to prevent hydrocephalus.
ARNOLD-CHIARI MALFORMATIONS
Four variations exist (see Figure 17-7), with type 2 being the most common, in which the cerebellum and medulla are shifted
caudally, resulting in crowding of the upper spinal column.
FIGURE 17-7. The Chiari malformations. Schematic representations of the Chiari malformations. Commonly associated hydrocephalus and syringomyelia not depicted.
Type 2 is also associated with meningomyelocele in >95% of cases.
Syringomyelia is associated in 70% of type 1, and 20–50% overall.
Management includes close observation with serial MRIs and surgery as required.
Cerebral Palsy (CP)
DEFINITION
A non-progressive disorder of movement and posture resulting from damage to the developing brain prior
to or surrounding birth. If progressive, consider another diagnosis.
Most cases occur in the absence of identifiable causes.
ETIOLOGY
EXAM TIP
CP is a static disorder, meaning that it does not result in the loss of previously acquired milestones.
Prematurity with intraventricular hemorrhage.
Birth or other asphyxia.
Intrauterine growth retardation (IUGR), placental insufficiency.
Infection: Prenatal/postnatal.
Twin pregnancy.
Chromosomal and genetic disorders.
Head trauma.
SIGNS AND SYMPTOMS
Prenatal and perinatal history.
Delayed motor, language, or social skills.
Not losing skills previously acquired.
Feeding difficulties.
Late-onset dystonia (ages 7–10 years).
EXAMINATION
Hypertonia.
Hyperreflexia.
Posture and movement: May be spastic, ataxic, choreoathetoid, and dystonic.
Abnormal primitive reflexes.
Abnormal gait.
Impaired growth of affected extremity.
ASSOCIATED PROBLEMS
Seizure disorder.
Mental retardation.
Developmental disorders.
CLASSIFICATION
WARD TIP
When there are no risk factors, family history of neurologic disease, presents late infancy or early childhood, ataxic CP, or atypical features, then consider other diagnosis.
Hemiplegic cerebral palsy: Upper limb involvement > lower limb; many walk before
2 years.
Diplegic cerebral palsy.
Quadriplegic cerebral palsy: Majority does not walk.
Dystonic/athetoid cerebral palsy.
Ataxic cerebral palsy.
Monoplegic cerebral palsy: Usually lower limb and appears late.
TREATMENT
WARD TIP
Extensor plantar response (presence of Babinski sign) can be present up to 1 year of age, but should be present symmetrically.
Multidisciplinary approach with goals of maximizing function and minimizing impairment.
Team includes general pediatrician, physiotherapist, occupational therapist, language
therapist, neurologist, and social and educational support services.
Orthopedic interventions are sometimes helpful.
Mental Retardation (MR)
EXAM TIP
DQ is often used as a rough estimator of IQ in infants and younger children. It is simply the mental age (estimated from historical milestones and exam) divided by the chronologic age, × 100.
DEFINITION
Below average intellectual functioning in association with deficits in adaptive
behavior prior to 18 years of age.
Intelligence quotient (IQ) or developmental quotient (DQ) <70 or <2 standard deviations
(SDs).
EPIDEMIOLOGY
Affects 1–3% of the population.
Approximately 75% are mild cases.
Males are affected more than females.
SIGNS AND SYMPTOMS
EXAM TIP
The IQ is scaled such that the mean is 100 and the standard deviation (SD) is 15. So MR is simply defined as an IQ two SDs below the mean.
Significant delay in reaching developmental milestones.
Delayed speech and language skills in toddlers with less severe MR.
The child will continue to learn new skills depending on severity of MR.
DIAGNOSIS
Classification is based on IQ:
Mild: IQ 55–70, 85% of cases.
Moderate: IQ 40–55, 10% of cases.
Severe: IQ 25–40, 3–5% of cases.
Profound: IQ < 25, 1–2% of cases.
Learning Disability (LD)
Significant discrepancy between a person’s intellectual ability and academic achievement.
Often learn best in unconventional ways.
Often restricted to a particular realm such as reading or mathematics with correspondingly
discrepant scores on standardized measures of intelligence or academic achievement.
Significant improvement with appropriate interventions.
