Evaluation of Suspected Immunodeficiency
Kathleen E. Sullivan, Rebecca H. Buckley
Primary care physicians must have a high index of suspicion to diagnose immune system defects early enough to institute appropriate treatment before irreversible damage develops. Diagnosis can be difficult because most affected patients do not have abnormal physical features. The most typical manifestation of immunodeficiency in children is recurrent sinopulmonary infections. Although infections are common in children in general, an infection exceeding the expected frequency and usually involving multiple sites can suggest immunodeficiency. A single, severe, opportunistic, or unusual infection can also be the presentation of an immunodeficiency (Table 148.1 ). Increasingly recognized is the co-occurrence of autoimmune disease or inflammatory conditions and recurrent infections. Newborn screening for T-cell lymphopenia has been instituted in most states; this has led to the identification of some infants with immunodeficiency before any clear manifestations but is limited to T-cell deficiencies. Additional clues to immunodeficiency include failure to thrive with or without chronic diarrhea, persistent infections after receiving live vaccines, and chronic oral or cutaneous candidiasis (Tables 148.2 and 148.3 ).
Table 148.1
Predisposition to Specific Infections in Humans
| PATHOGEN | PRESENTATION | AFFECTED GENE/CHROMOSOMAL REGION | COMMENTS |
|---|---|---|---|
| BACTERIA | |||
| Streptococcus pneumoniae | Invasive disease | IRAK4, MyD88, C1QA, C1QB, C1QC, C4A+ C4B, C2, C3 | Also susceptible to other encapsulated bacteria |
| Neisseria | Invasive disease | C5, C6, C7, C8A, C8B, C8G, C9, properdin | Recurrent disease common |
| Burkholderia cepacia | Invasive disease not pulmonary colonization | CYBB, CYBA, NCF1, NCF2 | Also susceptible to staphylococcal and fungal infections |
| Nocardia | Invasive disease | CYBB, CYBA, NCF1, NCF2 | Also susceptible to staphylococcal and fungal infections |
| Mycobacteria | Usually nontuberculous mycobacteria | IL12B, IL12RB1, IKBKG, IFNGR1, IFNGR2, STAT1 (loss of function) | Also susceptible to Salmonella typhi infections |
| VIRUSES | |||
| Herpes simplex virus | Herpes simplex encephalitis | TRAF3, TRIF, TBK, UNC93B1, TLR3, STAT1 | Age of onset is typically outside the neonatal period. |
| Epstein-Barr virus | Severe infectious mononucleosis, hemophagocytic syndrome | SH2DIA, XIAP, ITK, CD27, PRF1, STXBP2, UNC13D, LYST, RAB27A, STX11, AP3B1 | Fulminant infectious mononucleosis, malignant and nonmalignant lymphoproliferative disorders, dysgammaglobulinemia, autoimmunity |
| Papillomavirus | Warts | RHOH, EVER1, EVER2, CXCR4, DOCK8, GATA2, STK4, SPINK5 | Warts are often progressive despite therapy. |
| Global susceptibility to viral infection | Severe, progressive viral infections | All types of severe combined immune deficiency, IFNAR2 | Presentation depends on virus and infected organ |
| FUNGI | |||
| Candida | Mucocutaneous candida | AIRE, STAT1 (gain of function), CARD9, STAT3, IL17F, IL17RC, IL17RA, ACT1 | AIRE deficiency is associated with endocrinopathies, STAT1 (GOF) is associated with autoimmunity |
| Dermatophytes | Tissue invasion | CARD9 | Autosomal recessive |
| Aspergillus | Deep infections | CYBB, CYBA, NCF1, NCF2 | |
| Environmental fungi | Deep infections | CYBB, CYBA, NCF1, NCF2, GATA2, STAT1 (gain of function), CD40L | |
Table 148.2
Characteristic Clinical Patterns in Some Primary Immunodeficiencies
| FEATURES | DIAGNOSIS |
|---|---|
| IN NEWBORNS AND YOUNG INFANTS (0-6 mo) | |
| Hypocalcemia, unusual facies and ears, heart disease | 22q11.2 deletion syndrome, DiGeorge anomaly |
| Delayed umbilical cord detachment, leukocytosis, recurrent infections | Leukocyte adhesion defect |
| Persistent thrush, failure to thrive, pneumonia, diarrhea | Severe combined immunodeficiency |
| Bloody stools, draining ears, atopic eczema | Wiskott-Aldrich syndrome |
| IN INFANTS AND YOUNG CHILDREN (6 mo to 5 yr) | |
| Recurrent staphylococcal abscesses, staphylococcal pneumonia with pneumatocele formation, coarse facial features, pruritic dermatitis | Hyper-IgE syndrome, PGM3 deficiency |
| Persistent thrush, nail dystrophy, endocrinopathies | Autoimmune polyendocrinopathy, candidiasis, ectodermal dysplasia |
| Short stature, fine hair, severe varicella | Cartilage hair hypoplasia with short-limbed dwarfism |
| Oculocutaneous albinism, recurrent infection, hemophagocytic syndrome | Chédiak-Higashi syndrome, Griscelli syndrome, Hermansky-Pudlak syndrome |
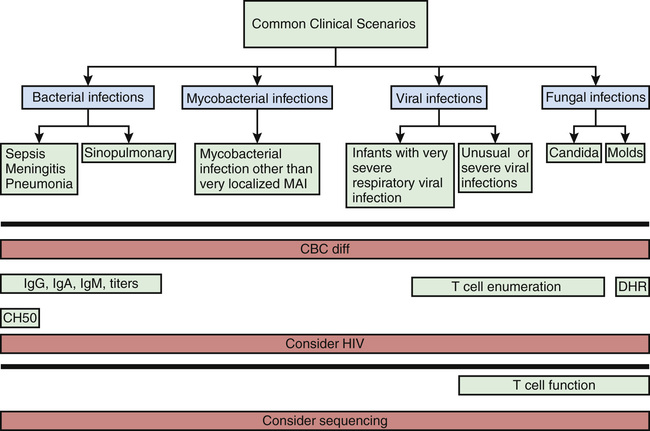
With >300 distinct primary immunodeficiencies, in order to focus the diagnostic approach and appropriate testing. it is often useful to consider 5 categories: T-cell disorders, B-cell and antibody disorders, complement disorders, phagocytic disorders, and natural killer cell disorders (Table 148.4 and Fig. 148.1 ).
Table 148.4
Characteristic Features of Primary Immunodeficiency
| CHARACTERISTIC | PREDOMINANT T-CELL DEFECT | PREDOMINANT B-CELL DEFECT | GRANULOCYTE DEFECT | CYTOLYTIC DEFECT | COMPLEMENT DEFECT |
|---|---|---|---|---|---|
| Age at onset of infection | Early onset, usually 2-6 mo | Onset after maternal antibodies diminish, usually after 5-7 mo, later childhood to adulthood | Early onset most frequently | Childhood onset generally | Onset at any age |
| Specific pathogens involved | Bacteria: common gram-positive and gram-negative bacteria and mycobacteria | Bacteria: pneumococci, streptococci, staphylococci, Haemophilus, Campylobacter, Mycoplasma | Bacteria: staphylococci, Serratia, Salmonella, mycobacteria | None usually | Bacteria: encapsulated organisms (C1, C4, C2, C3), Neisseria (FP, FD, FH, FI, C3, C5, C6, C7, C8, C9) |
| Viruses: CMV, EBV, adenovirus, parainfluenza 3, varicella, enterovirus | Viruses: enterovirus* | None generally | CMV, EBV | None generally | |
| Fungi: Candida and Pneumocystis jiroveci | Fungi and parasites: Giardia, Cryptosporidia | Fungi and parasites: Candida, Nocardia, Aspergillus | None generally | None generally | |
| Affected organs | Extensive mucocutaneous candidiasis, lungs, failure to thrive, protracted diarrhea | Recurrent sinopulmonary infections, chronic gastrointestinal symptoms, malabsorption, arthritis, enteroviral meningoencephalitis* |
Skin: abscesses, impetigo, cellulitis Lymph nodes: suppurative adenitis Oral cavity: gingivitis, mouth ulcers Internal organs: abscesses, osteomyelitis |
Hemophagocytic syndrome can affect any organ. | Deep or systemic infections |
| Special features |
Graft-vs-host disease caused by maternal engraftment or nonirradiated blood transfusion Postvaccination disseminated BCG or varicella Autoimmunity common in mild-moderate T-cell defects |
Autoimmunity Lymphoreticular malignancy: lymphoma, thymoma |
Prolonged attachment of umbilical cord, poor wound healing | SLE (C1, C4, C2), Glomerulonephritis (C3), atypical hemolytic-uremic syndrome (FH, FI, MCP, C3, FB) |
* X-linked (Bruton) agammaglobulinemia.
BCG, Bacille Calmette-Guérin; CMV, cytomegalovirus; EBV, Epstein-Barr virus; SLE, systemic lupus erythematosus.
The initial evaluation of immunologic function includes a thorough history, physical examination, and family history (Table 148.5 ). Over 10 immunodeficiencies are X-linked, and a growing number are autosomal dominant with variable expressivity and/or incomplete penetrance. Close attention to physical signs of autoimmune disease or end-organ effects from recurrent infections should be noted. The history of infections should include the age of onset, severity, involved locations, and assessment of the underlying microbial cause. Viral, bacterial, fungal, and mycobacterial infections all require distinct arms of the immune system for eradication; therefore identification of microbiologic causes of infection can be extremely helpful in defining the deficiency states in people with primary immunodeficiencies.
Table 148.5
Special Physical Features Associated With Immunodeficiency Disorders
| CLINICAL FEATURES | DISORDERS |
|---|---|
| DERMATOLOGIC | |
| Eczema | Wiskott-Aldrich syndrome, IPEX, hyper-IgE syndromes, hypereosinophilia syndromes, IgA deficiency |
| Sparse and/or hypopigmented hair | Cartilage-hair hypoplasia, Chédiak-Higashi syndrome, Griscelli syndrome |
| Ocular telangiectasia | Ataxia-telangiectasia |
| Oculocutaneous albinism | Chédiak-Higashi syndrome |
| Severe dermatitis | Omenn syndrome |
| Erythroderma | Omenn syndrome, SCID, graft-vs-host disease, Comel-Netherton syndrome |
| Recurrent abscesses with pulmonary pneumatoceles | Hyper-IgE syndromes |
| Recurrent organ granulomas or abscesses, lung, liver, and rectum especially | CGD |
| Recurrent abscesses or cellulitis | CGD, hyper-IgE syndrome, leukocyte adhesion defect |
| Cutaneous granulomas | Ataxia telangiectasia, SCID, CVID, RAG deficiency |
| Oral ulcers | CGD, SCID, congenital neutropenia |
| Periodontitis, gingivitis, stomatitis | Neutrophil defects |
| Oral or nail candidiasis | T-cell immune defects, combined defects (SCIDs); mucocutaneous candidiasis; hyper-IgE syndromes; IL-12, -17, -23 deficiencies; CARD9 deficiency; STAT1 deficiency |
| Vitiligo | B-cell defects, mucocutaneous candidiasis |
| Alopecia | B-cell defects, mucocutaneous candidiasis |
| Chronic conjunctivitis | B-cell defects |
| EXTREMITIES | |
| Clubbing of nails | Chronic lung disease caused by antibody defects |
| Arthritis | Antibody defects, Wiskott-Aldrich syndrome, hyper-IgM syndrome |
| ENDOCRINOLOGIC | |
| Hypoparathyroidism | DiGeorge syndrome, mucocutaneous candidiasis |
| Endocrinopathies (autoimmune) | Mucocutaneous candidiasis |
| Diabetes, hypothyroid | IPEX and IPEX-like syndromes |
| Growth hormone deficiency | X-linked agammaglobulinemia |
| Gonadal dysgenesis | Mucocutaneous candidiasis |
| HEMATOLOGIC | |
| Hemolytic anemia | B- and T-cell immune defects, ALPS |
| Thrombocytopenia, small platelets | Wiskott-Aldrich syndrome |
| Neutropenia | Hyper-IgM syndrome, Wiskott-Aldrich variant, CGD |
| Immune thrombocytopenia | B-cell immune defects, ALPS |
| SKELETAL | |
| Short-limb dwarfism | Short-limb dwarfism with T- and/or B-cell immune defects |
| Bony dysplasia | ADA deficiency, cartilage-hair hypoplasia |
ADA, Adenosine deaminase; ALPS, autoimmune lymphoproliferative syndrome; CGD, chronic granulomatous disease; CVID, common variable immunodeficiency; IPEX, X-linked immune dysfunction enteropathy polyendocrinopathy; SCID, severe combined immunodeficiency.
From Goldman L, Ausiello D: Cecil textbook of medicine, ed 22, Philadelphia, 2004, Saunders, p 1599.
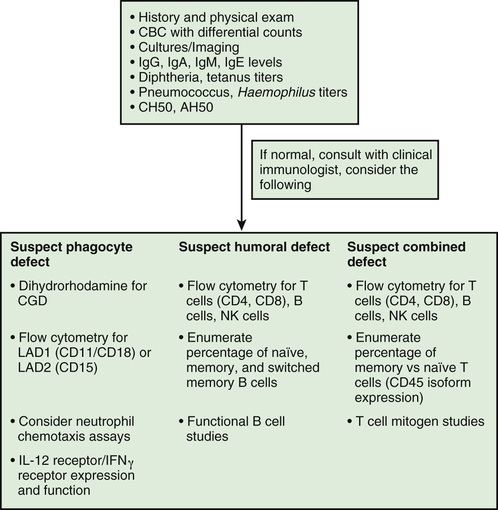
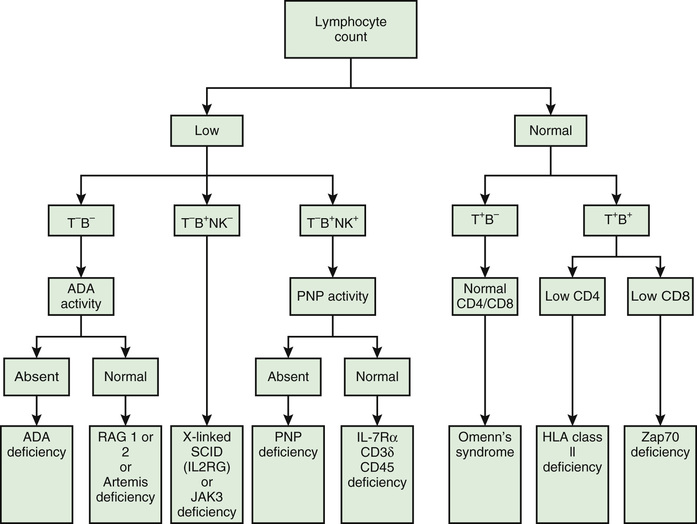
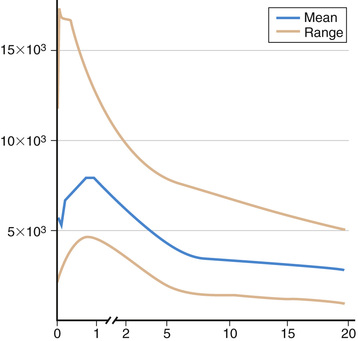
Most immunologic defects can be excluded at minimal cost with the proper choice of screening tests, which should be broadly informative, reliable, and cost-effective (Table 148.6 and Figs. 148.2 and 148.3 ). A complete blood count (CBC) with differential is the initial study if neutropenia is a consideration but is less recognized as a screening test for T-cell defects. Lymphopenia is seen the majority of T-cell defects. If an infant's neutrophil count is persistently elevated in the absence of any signs of infection, a leukocyte adhesion deficiency should be suspected. Normal lymphocyte counts are higher in infancy and early childhood than later in life (Fig. 148.4 ). Knowledge of normal values for absolute lymphocyte counts at various ages in infancy and childhood is crucial in the detection of T-cell defects. Additional clues from the CBC include absence of Howell-Jolly bodies, which argues against congenital asplenia. Normal platelet size or count excludes Wiskott-Aldrich syndrome. When immunodeficiency is suspected, obtaining IgG, IgA, IgM, and IgE levels can be a useful strategy, since antibody defects are the most common type of immunodeficiency. Immunoglobulin levels must be interpreted within the context of age-specific normative data.
Advanced Testing
Additional testing should be focused based on the phenotype and suspected category of immune deficiency (Table 148.7 ; see Figs. 148.2 and 148.3 ). For patients with recurrent sinopulmonary infections in whom an antibody defect is suspected, further attention to antibody testing may be revealing. In addition to Ig levels, responses to vaccines should also be pursued in this setting. A small but significant subset of patients with antibody deficiencies will have normal Ig levels but abnormal function, as detected by poor responses to vaccines. When hypogammaglobulinemia is identified, it is important to determine whether it is primary or secondary. Patients receiving corticosteroids or who have protein-losing states (nephrosis, protein-losing enteropathy) often have low serum IgG concentrations but produce normal responses to vaccines. Thus, if Ig levels are low, it is crucial before starting immune globulin replacement therapy that antibody titers to vaccines are measured. Antibody titers are not interpretable after the patient has received a blood transfusion, fresh-frozen plasma, or immune globulin therapy.
Table 148.7
Laboratory Tests in Immunodeficiency
| SCREENING TESTS | ADVANCED TESTS | RESEARCH/SPECIAL TESTS |
|---|---|---|
| B-CELL DEFICIENCY | ||
| IgG, IgM, IgA, and IgE levels | B-cell enumeration (CD19 or CD20) | Advanced B-cell phenotyping |
| Isohemagglutinin titers | Biopsies (e.g., lymph nodes) | |
| Ab response to vaccine antigens (e.g., tetanus, diphtheria, pneumococci, Haemophilus influenzae ) | Ab responses to boosters or to new vaccines | Ab responses to special antigens (e.g., bacteriophage φX174), mutation analysis |
| T-CELL DEFICIENCY | ||
| Lymphocyte count | T-cell subset enumeration (CD3, CD4, CD8) | Advanced flow cytometry |
| Chest x-ray examination for thymic size* | Proliferative responses to mitogens, antigens, allogeneic cells | Enzyme assays (e.g., ADA, PNP) |
| TRECs | 22q11.2 deletion analysis |
Mutation analysis T-cell activation studies |
| PHAGOCYTIC DEFICIENCY | ||
| WBC count, morphology | Adhesion molecule assays (e.g., CD11b/CD18, selectin ligand) |
Mutation analysis Macrophage functional testing |
| Respiratory burst assay | Mutation analysis | |
| COMPLEMENT DEFICIENCY | ||
| CH50 activity | AH50 , activity | Specific component assays |
* In infants only.
Ab, Antibody; ADA, adenosine deaminase; C, complement; CH, hemolytic complement; G6PD, glucose-6-phosphate dehydrogenase; HLA, human leukocyte antigen; Ig, immunoglobulin; MPO, myeloperoxidase; NADPH, nicotinamide adenine dinucleotide phosphate; PNP, purine nucleoside phosphorylase; TRECs, T-cell receptor rearrangement excision circle; WBC, white blood cell; φX, phage antigen.
One useful test for B-cell function is to determine the presence and titer of isohemagglutinins , or natural antibodies to type A and B red blood cell polysaccharide antigens. This test measures predominantly IgM antibodies. Isohemagglutinins may be absent normally in the first 2 yr of life and are always absent if the patient is blood type AB.
Because most infants and children are immunized with diphtheria-tetanus-pertussis (dTP), conjugated Haemophilus influenzae type b, and pneumococcal conjugate vaccine, it is often informative to test for specific antibodies to diphtheria, tetanus, H. influenzae polyribose phosphate, and pneumococcal antigens. If the titers are low, measurement of antibodies to diphtheria or tetanus toxoids before and 2-8 wk after a pediatric dTP or dT booster is helpful in assessing the capacity to form IgG antibodies to protein antigens. To evaluate a patient's ability to respond to polysaccharide antigens, antipneumococcal antibodies can be measured before and 4-8 wk after immunization with 23-valent unconjugated pneumococcal polysaccharide vaccine in patients ≥2 yr old. Antibodies detected in these tests are of the IgG isotype. These antibody studies can be performed in several different laboratories, but it is important to choose a reliable laboratory and to use the same laboratory for pre- and postimmunization titers.
Patients with defective B-cell maturation due to a class-switching defects produce neither IgA nor IgG antibodies normally. IgM is normal or elevated. Vaccine responses are uniformly low. If antibody responses to vaccines are normal and the IgG level is low, studies should be performed to evaluate the possible loss of immunoglobulins through the urinary or gastrointestinal tract (nephrotic syndrome, protein-losing enteropathies, intestinal lymphangiectasia). Another common confounder is the use of rituximab or corticosteroids, leading to hypogammaglobulinemia. Very high serum concentrations of 1 or more Ig classes suggest HIV infection, chronic granulomatous disease, chronic inflammation, or autoimmune lymphoproliferative syndrome.
IgG subclass measurements are seldom helpful in assessing immune function in young children with recurrent infections. They are strongly developmentally regulated with highly variable production in early childhood. It is difficult to know the biologic significance of the various mild to moderate deficiencies of IgG subclasses, particularly when completely asymptomatic individuals have been described as totally lacking IgG1, IgG2, IgG4, and/or IgA1 because of Ign heavy-chain gene deletions. Many healthy children have been described as having low levels of IgG2 but normal responses to polysaccharide antigens when immunized. In older children and adults, a low IgG2 maybe an antecedent finding before evolving into common variable immunodeficiency (CVID). Specific vaccine responses are usually much more useful than IgG subclass determinations.
Patients found to be agammaglobulinemic should have their blood B cells enumerated by flow cytometry using dye-conjugated monoclonal antibodies to B-cell–specific CD antigens (usually CD19 or CD20). Normally, approximately 5–10% of circulating lymphocytes are B cells. B cells are absent in X-linked agammaglobulinemia (XLA) and in several very rare autosomal recessive conditions, but they are usually present in CVID, IgA deficiency, and hyper-IgM syndromes. This distinction is important, because children with hypogammaglobulinemia from XLA and CVID can have different clinical problems, and the 2 conditions clearly have different inheritance patterns. Patients with CVID have more problems with autoimmune diseases and lymphoid hyperplasia. Molecular testing for XLA and other B-cell defects (see Chapter 150.1 ) is indicated in cases without a family history to aid genetic counseling.
T cells and T cell subpopulations can be enumerated by flow cytometry using dye-conjugated monoclonal antibodies recognizing CD antigens present on T cells (i.e., CD2, CD3, CD4, and CD8). This is a particularly important test to perform on any infant who is lymphopenic, because CD3+ T cells usually constitute 70% of peripheral lymphocytes. Regardless of molecular type, infants with SCID are unable to produce T cells, so are lymphopenic at birth. The flow cytometry for infants suspected of having SCID should also include monoclonal antibodies to naïve (CD45RA) and memory (CD45RO) T cells. In normal infants, >95% of the T cells are CD45RA+ (naïve) T cells. If the infant has SCID, there could be transplacentally transferred maternal T cells detected by flow cytometry, but they would be predominantly CD45RO+ T cells. SCID is a pediatric emergency that can be successfully treated by hematopoietic stem cell transplantation in more than 90% of cases if diagnosed before serious, untreatable infections develop. Normally, there are about twice as many CD4+ (helper) T cells as there are CD8+ (cytotoxic) T cells. Because some severe immunodeficiencies have phenotypically normal T cells, tests of T-cell function can be helpful. T cells can be stimulated directly with mitogens such as phytohemagglutinin, concanavalin A, or pokeweed mitogen. After 3-5 days of incubation with the mitogen, the proliferation of T cells is measured. Other stimulants that can be used to assess T-cell function in the same type of assay include antigens (Candida, tetanus toxoid) and allogeneic cells.
Natural killer (NK) cells can be enumerated by flow cytometry using monoclonal antibodies to NK-specific CD antigens, CD16 and CD56. NK function is assessed by killing of target cells and flow cytometry for CD107a, a marker of degranulation.
Chronic granulomatous disease should be suspected if a patient has recurrent staphylococcal abscesses or fungal infections. It can be evaluated by screening tests measuring the neutrophil respiratory burst after phorbol ester stimulation. Leukocyte adhesion defects (LAD) can be easily diagnosed by flow cytometric assays of blood lymphocytes or neutrophils, using monoclonal antibodies to CD18 or CD11 (LAD1) or to CD15 (LAD2). Neutrophil defects are most often associated with neutropenia or morphologic abnormalities visible by microscopy. Therefore, a combination of a CBC with differential, microscopic evaluation, and the flow cytometric approaches previously described often yields a diagnosis. The same is not true for macrophage defects , which are usually associated with susceptibility to mycobacteria, and testing requires advanced functional analyses or sequencing.
When invasive infection with encapsulated organisms or Neisseria leads to a suspicion of a complement defect, a CH50 test should be obtained. This bioassay measures the intactness of the entire complement pathway and yields abnormal results if classical pathway or terminal components are missing. Genetic deficiencies in the complement system often have a CH50 that is almost absent, although the most frequent cause of a slightly low CH50 is improper transport of the specimen. Complement proteins are highly labile and must be transported on ice. Specific factor assays are available at reference laboratories. Rare causes of neisserial susceptibility include alternative pathway defects, and the test for these deficiencies is the AH50 test. Identifying the specific component deficiency in the mode of inheritance is important for genetic counseling. Properdin deficiency is X-linked, and other deficiencies are autosomal recessive or autosomal dominant.