Lymphocyte Development and Function
Kathleen E. Sullivan, Rebecca H. Buckley
Defense against infectious agents is secured through a combination of anatomic physical barriers, including the skin, mucous membranes, mucous blanket, and ciliated epithelial cells, and the components of the immune system. The immune system of vertebrates integrates 2 fundamental response mechanisms. Innate (natural) immunity is rapid and utilizes receptors encoded in the germline. The innate defenses comprise cell-intrinsic responses to viral infections, leukocyte responses to pathogens, and soluble mediators such as complement proteins. Acquired (adaptive) immunity is specific to T and B cells. These cells undergo DNA recombination to generate receptors and require an education process to minimize autoreactive cells. In addition, there are lymphocyte subsets that are innate in nature and either do not require DNA recombination or utilize a single recombination event to generate a monospecific receptor.
Lymphopoiesis in the Fetus
Pluripotential hematopoietic stem cells first appear in the yolk sac at 2.5-3 wk of gestational age, migrate to the fetal liver at 5 wk gestation, and later reside in the bone marrow, where they remain throughout life (Fig. 149.1 ). Lymphoid stem cells develop and differentiate into T, B, or natural killer (NK) cells, depending on the organs or tissues to which the stem cells traffic. Development of the primary lymphoid organs —thymus and bone marrow—begins during the middle of the 1st trimester of gestation and proceeds rapidly. Development of the secondary lymphoid organs —spleen, lymph nodes, tonsils, Peyer patches, and lamina propria—soon follows. These organs serve as sites of differentiation of T, B, and NK lymphocytes from stem cells throughout life. Both the initial organogenesis and the continued cell differentiation result from the interaction of a vast array of lymphocytic and microenvironmental cell surface molecules and proteins secreted by the involved cells. Clusters of differentiation (CD) refer to cellular protein (Table 149.1 ), whereas cytokines and chemokines refer to soluble mediators of immune function (Table 149.2 ).
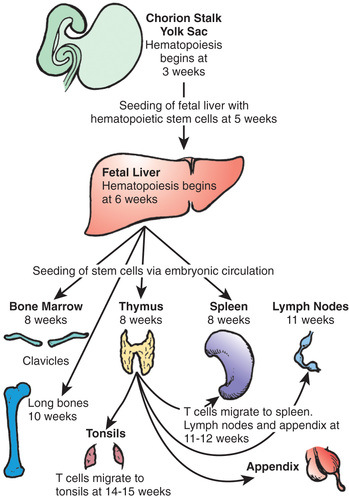
Table 149.1
AMP, Adenosine monophosphate; EBV, Epstein-Barr virus; ICAMs, intracellular adhesion molecules; IL, interleukin; LFA, leukocyte function–activating antigen; NK, natural killer; TCR, T-cell receptor.
Table 149.2
| CATEGORY | CYTOKINE | FUNCTION |
|---|---|---|
| Interferons | IFN-α | Antiviral defense |
| IFN-β | Antiviral defense | |
| IFN-γ | Antiviral defense | |
| Innate responses | TNF | Regulates endothelial adhesion molecules for recruitment of neutrophils; activates macrophages for killing |
| IL-1β | Drives the inflammatory response, fever | |
| IL-12 | Polarizes T cells toward Th1; activates NK cells | |
| Lymphocyte regulation | IL-2 | Key growth factor for T cells |
| IL-4 | Polarizes T cells toward Th2 | |
| IL-6 | Growth factor for B cells | |
| IL-7 | T-cell homeostatic factor | |
| IL-10 | Growth factor for B cells, immunosuppressive | |
| IL-12 | Polarizes T cells toward Th1, activates NK cells | |
| IL-17 | Polarizes T cells toward Th17, stimulates antimicrobial peptide expression | |
| IL-21 | Supports B-cell class switching |
IL, Interleukin; NK, natural killer; Th, T-helper cell; TNF, tumor necrosis factor.
T-Cell Development and Differentiation
The primitive thymic rudiment is formed from the ectoderm of the 3rd branchial cleft and endoderm of the 3rd branchial pouch at 4 wk gestation. Beginning at 7-8 wk, the right and left rudiments fuse in the midline. Bloodborne T-cell precursors from the fetal liver then begin to colonize the perithymic mesenchyme at 8 wk gestation and move into the thymus at 8.0-8.5 wk. The earliest cells to enter the thymus are found in the subcapsular region and do not express CD3, CD4, CD8, or either type of T-cell receptor (TCR). These lymphoid cell precursors are triggered to proliferate and become thymocytes through interactions with the thymic stroma. The cells are arrested at this stage until they productively rearrange the β-chain locus of the TCR. The β chain then pairs with the surrogate pre-T α chain. This tests the function of the β chain, and if signaling occurs, β-chain rearrangement ceases. CD4 and CD8 are then expressed simultaneously (i.e., they are double-positive thymocytes). Fetal cortical thymocytes are among the most rapidly dividing cells in the body and increase in number by 100,000-fold within 2 wk after stem cells enter the thymus. As these cells proliferate and mature, they migrate deeper into the thymic cortex. The double-positive thymocytes begin efficient rearrangement of the α-chain locus. TCR gene rearrangement occurs by a process in which large, noncontiguous blocks of DNA are spliced together. V (variable) , D (diversity) , and J (joining) blocks exist in families of minimally different segments. Random combinations of the segments account for much of the enormous diversity of TCRs that enables humans to recognize millions of different antigens. TCR gene rearrangement requires the presence of recombinase-activating genes , RAG1 and RAG2, as well as other recombinase components.
As immature cortical thymocytes begin to express TCRs, the processes of positive and negative selection take place. Positive selection occurs in immature thymocytes, recognizing major histocompatibility complex (MHC) antigens present on cortical thymic epithelial cells. Some cells are selected to mature into CD4 or CD8 single-positive cells. Negative selection occurs next in the thymic medulla on medullary thymic epithelial cells. Autoreactive T cells undergo apoptosis and die. T cells begin to emigrate from the thymus to the spleen, lymph nodes, and appendix at 11-12 wk of embryonic life and to the tonsils by 14-15 wk. They leave the thymus via the bloodstream and are distributed throughout the body, with the heaviest concentrations in the paracortical areas of lymph nodes, the periarteriolar areas of the spleen, and the thoracic lymph duct. Recent thymic emigrants co-express the CD45RA isoforms and CD62L (L -selectin).
Rearrangement of the TCR locus during intrathymic T-cell development results in the excision of DNA and the excised elements form circular episomes as a by-product. These TCR recombination excision circles can be detected in T cells that are recent thymic emigrants. TCR recombination excision circles detected in dried-blood spots collected from infants shortly after birth is the test used for newborn screening for severe combined immunodeficiency (SCID). By 12 wk gestation, T cells can proliferate in response to plant lectins, such as phytohemagglutinin and concanavalin A. Antigen-specific T cells have been found by 20 wk gestation. Hassall corpuscles (bodies), which are swirls of terminally differentiated medullary epithelial cells, are first seen in the thymic medulla at 16-18 wk of embryonic life.
B-Cell Development and Differentiation
B-cell development begins in the fetal liver by 7 wk gestation. Fetal liver CD34 stem cells are seeded to the bone marrow of the clavicles by 8 wk of embryonic life and to that of the long bones by 10 wk (see Fig. 149.1 ). As B cells differentiate from primitive stem cells, they proceed through stages that are marked by the sequential rearrangement of immunoglobulin gene segments to generate a diverse repertoire of antigen receptors. The early pro-B cell is the first descendent of the pluripotential stem cell committed to B-lineage development, and in this stage the heavy chain locus rearranges first. In the early pro-B cell, D-J rearrangements are made on both chromosomes. In the late pro-B cell, the V segment rearranges to a D-J gene segment. The next stage is the pre-B cell , during which immunoglobulin (Ig) light-chain genes are rearranged. The pre-B cell is distinguished by the expression of cytoplasmic µ heavy chains but no surface IgM (sIgM), because Ig light chains are not yet produced. Next is the immature B-cell stage, during which the light-chain genes have already been rearranged, and sIgM but not sIgD is expressed. Immature B cells leave the bone marrow for secondary lymphoid organs. The last stage of antigen-independent B-cell development is the mature naïve B cell, which co-expresses both sIgM and sIgD. Pre-B cells can be found in fetal liver at 7 wk gestation, sIgM+ and sIgG+ B cells at 7-11 wk, and sIgD+ and sIgA+ B cells by 12-13 wk. By 14 wk of embryonic life, the percentage of circulating lymphocytes bearing sIgM and sIgD is the same as in cord blood and slightly higher than in the blood of adults.
Antigen-dependent stages of B-cell development are those that develop after the mature B cell is stimulated by antigen in secondary lymphoid organs. Once antigen stimulation has occurred, the mature B cells can become memory B cells or plasmablasts. Both outcomes require the presence of T-cell help.
There are 5 immunoglobulin isotypes , which are defined by unique heavy chains: IgM, IgG, IgA, IgD, and IgE. IgG and IgM, the only complement-fixing isotypes, are the most important immunoglobulins in the blood and other internal body fluids for protection against infectious agents. IgM is confined primarily to the intravascular compartment because of its large size, whereas IgG is present in all internal body fluids. IgA is the major protective immunoglobulin of external secretions—in the gastrointestinal, respiratory, and urogenital tracts—but it is also present in the circulation. IgE, present in both internal and external body fluids, has a major role in host defense against parasites. Because of high-affinity IgE receptors on basophils and mast cells, however, IgE is the principal mediator of allergic reactions of the immediate type. The significance of IgD is still not clear. There are also immunoglobulin subclasses , including 4 subclasses of IgG (IgG1, IgG2, IgG3, and IgG4) and 2 subclasses of IgA (IgA1 and IgA2). These subclasses each have different biologic roles. Secreted IgM and IgE have been found in as young as 10 wk gestation, and IgG as early as 11-12 wk.
Even though these B-cell developmental stages have been described in the context of B-cell ontogeny in utero, it is important to recognize that the process of B-cell development from pluripotential stem cells goes on throughout postnatal life. Plasma cells are not usually found in lymphoid tissues of a fetus until about 20 wk gestation, and then only rarely, because of the sterile environment of the uterus. Intestinal lymphoid development occurs relatively late. Peyer patches have been found in significant numbers by the 5th intrauterine mo, and plasma cells have been seen in the lamina propria by 25 wk gestation. Before birth there may be primary follicles in lymph nodes, but secondary follicles are usually not present.
A human fetus begins to receive significant quantities of maternal IgG transplacentally at around 12 wk gestation, and the quantity steadily increases until, at birth, cord-blood serum contains a concentration of IgG comparable to or greater than that of maternal serum. IgG is the only class to cross the placenta to any significant degree. All 4 IgG subclasses cross the placenta, but IgG2 does so least well. A small amount of IgM (10% of adult levels) and a few nanograms of IgA, IgD, and IgE are normally found in cord blood serum. Because none of these proteins crosses the placenta, they are presumed to be of fetal origin. These observations suggest that certain antigenic stimuli normally cross the placenta to provoke responses, even in uninfected fetuses. Some atopic infants occasionally have IgE antibodies to antigens, such as egg white, to which they have had no known exposure during postnatal life, suggesting that synthesis of these antibodies could have been induced in the fetus by antigens ingested by the mother.
Natural Killer–Cell Development
NK cell activity is found in human fetal liver cells at 8-11 wk of gestation. NK lymphocytes are also derived from bone marrow precursors. Thymic processing is not necessary for NK-cell development, although NK cells have been found in the thymus. After release from bone marrow, NK cells enter the circulation or migrate to the spleen, with very few NK cells in lymph nodes. In normal individuals, NK cells represent 8–10% of lymphocytes. Certain tissues harbor large numbers of NK cells.
Unlike T and B cells, NK cells do not rearrange antigen receptor genes during their development but are defined by their functional capacity to mediate non–antigen-specific cytotoxicity. NK cells have killer inhibitory receptors that recognize certain MHC antigens and inhibit the killing of self tissues. NK-activating receptors recognize stress protein, and the balance of activating and inhibitory receptor engagement determines the action of the NK cells. If a viral infection drives down MHC class I expression, the loss of inhibitory function drives cytotoxicity. High levels of stress proteins, typically seen in viral infections, can also activate cytotoxicity.
Lymphocyte Choreography
The main functions of T cells are to signal B cells to make antibody, to kill virally infected cells or tumor cells, and to activate macrophages for intracellular killing. The subset of regulatory T cells (Tregs), is critical in the prevention of autoimmune responses. T cells are activated by antigen presented by antigen-presenting cells (APCs) . These are usually dendritic cells, macrophages, or B cells. For high-affinity binding of T cells to APCs, several molecules on T cells, in addition to TCRs, bind to molecules on APCs or target cells. The CD4 molecule binds directly to MHC class II molecules on APCs. CD8 on cytotoxic T cells binds the MHC class I molecule on the target cell. Lymphocyte function–associated antigen 1 (LFA-1 ) on the T cell binds a protein called ICAM-1 (intracellular adhesion molecule 1), designated CD54 , on APCs. CD2 on T cells binds LFA-3 (CD58) on the APCs. With the adhesion of T cells to APCs (the immunologic synapse), T-helper (Th) cells are stimulated to make interleukins and upregulate cell surface molecules, such as the CD40 ligand (CD154), that provide help for B cells, and cytotoxic T cells are stimulated to kill their targets. A key safety net to ensure appropriate activation of T cells in the setting of a true threat is the requirement for co-stimulation of the T cells. APCs that have encountered a pathogen express CD80 and CD86. Engagement of these molecules provides the 2nd, co-stimulatory, signal. Without co-stimulation, the T cell will be rendered anergic, or nonfunctional.
In the primary antibody response , native antigen is carried to a lymph node draining the site, captured by complement, taken up by specialized cells called follicular dendritic cells (FDCs) , and expressed on their surfaces. Mature B cells bearing sIgM specific for that antigen then bind to the antigen on the surfaces of the FDCs. If the affinity of the B-cell sIgM antibody for the antigen present on the FDCs is sufficient, and if other signals are provided by activated T cells, the B cell develops into a memory B cells or antibody-producing plasma cell. The signals from activated T cells include several cytokines (IL-4, IL-5, IL-6, IL-10, IL-13, and IL-21) that they secrete (see Table 149.2 ) and a surface T-cell molecule, the CD40 ligand or CD154, which, on contact of the activated CD4+ T cell with the B cell, binds to CD40 on the B-cell surface. Binding of CD40 on B cells by CD154 on T cells in the presence of certain cytokines causes the B cells to undergo proliferation and to initiate immunoglobulin synthesis. In the primary immune response, only IgM antibody is usually made, and most of it is of relatively low affinity. Some B cells become memory B cells during the primary immune response. The secondary antibody response occurs when these memory B cells again encounter that antigen. Developing memory B cells switch their Ig genes so that IgG, IgA, and/or IgE antibodies of higher affinity are formed on a secondary exposure to the same antigen. Plasma cells form, just as in the primary response; however, many more cells are rapidly generated, and IgG, IgA, and IgE antibodies are made. In addition, genetic changes in Ig genes (somatic hypermutation) lead to increased affinity of those antibodies.
The exact pattern of isotype response to antigen in normal individuals varies, depending on the type of antigen and the cytokines present in the microenvironment. Both class switching and somatic hypermutation are completely dependent on T-cell help. Thus, T cells represent a kind of gatekeeper for specific antibody production.
Postnatal Lymphocyte Behavior
Virtually all T cells in cord blood bear the CD45RA (naïve) isoform, and a dominance of CD45RA over CD45RO T cells persists during childhood. After mid-adulthood, the CD45RO (memory) T cells predominate. CD4 T cells can be further subdivided according to the cytokines they produce when activated. Th1 cells produce interleukin (IL)-2 and interferon (IFN)-γ, which promote cytotoxic T-cell or delayed hypersensitivity types of responses, whereas Th2 cells produce IL-4, IL-5, IL-6, IL-13, and IL-21 (see Table 149.2 ), which promote B-cell responses and allergic sensitization, Th17 cells produce IL-17, and Tregs produce IL-10 (Fig. 149.2 ). Differentiation into these memory subsets is dictated by the cytokine milieu regulating specific transcription factors and epigenetic changes. In vivo, these subsets are largely stable but in some circumstances can change to a different subset. The importance of these subsets is that memory cells respond to antigen more quickly and are primed to produce the cytokines most likely to drive pathogen clearance.

Newborn infants have increased susceptibility to infections with gram-negative organisms because IgM antibodies, powerful opsonins that enhance phagocytosis, do not cross the placenta. The other major opsonin, C3b, is also lower in newborn serum than in adults. These factors probably account for impaired phagocytosis of some organisms by newborn polymorphonuclear cells. Maternally transmitted IgG antibodies serve quite adequately for most gram-positive bacteria, and IgG antibodies to viruses offer protection against those agents. Because there is a relative deficiency of the IgG2 subclass in infancy, antibodies to capsular polysaccharide antigens may be deficient. Because premature infants have received less maternal IgG by the time of birth than full-term infants, their serum opsonic activity is low for all types of organisms.
Neonates begin to synthesize antibodies of the IgM class at an increased rate very soon after birth in response to the immense antigenic stimulation of their new environment. Premature infants appear to be as capable of doing this as are full-term infants. At about 6 days after birth, the serum concentration of IgM rises sharply. This rise continues until adult levels are achieved by approximately 1 yr of age. Cord serum from noninfected normal newborns does not contain detectable IgA. Serum IgA is normally first detected at around the 13th day of postnatal life but remains low throughout infancy. Cord serum contains an IgG concentration comparable to or greater than that of maternal serum. Maternal IgG gradually disappears during the 1st 6-8 mo of life, while the rate of infant IgG synthesis increases (IgG1 and IgG3 faster than IgG2 and IgG4 during the 1st yr) until adult concentrations of total IgG are reached and maintained by 7-8 yr. IgG1 and IgG4 reach adult levels first, followed by IgG3 at 10 yr and IgG2 at 12 yr. The serum IgG level in infants usually reaches a low point at about 3-4 mo of postnatal life. The rate of development of IgE generally follows that of IgA.
After adult concentrations of each of the 3 major immunoglobulins are reached, these levels remain remarkably constant for a normal individual. The capacity to produce specific antibodies to protein antigens is intact at birth, but infants cannot usually produce antibodies to polysaccharide antigens until after 2 yr of age unless the polysaccharide is conjugated to a protein carrier, as is the case for the conjugate Haemophilus influenzae type b and Streptococcus pneumoniae vaccines.
The percentage of NK cells in cord blood is usually lower than in the blood of children and adults, but the absolute number of NK cells is approximately the same because of the higher lymphocyte count. The capacity of cord blood NK cells to mediate target lysis in either NK-cell assays or antibody-dependent cellular cytotoxicity assays is about two-thirds that of adults.
Lymphoid Organ Development
Lymphoid tissue is proportionally small but rather well developed at birth and matures rapidly in the postnatal period. The thymus is largest relative to body size during fetal life and at birth is ordinarily two-thirds its mature weight, which it attains during the 1st yr of life. It reaches its peak mass, however, just before puberty, then gradually involutes thereafter. By 1 yr of age, all lymphoid structures are mature histologically. Absolute lymphocyte counts in the peripheral blood also reach a peak during the 1st yr of life (see Fig. 149.2 ). The spleen, however, gradually accrues its mass during maturation and does not reach full weight until adulthood. The mean number of Peyer patches at birth is one-half the adult number, and gradually increases until the adult mean number is exceeded during adolescent years.
Inheritance of Abnormalities in T-, B-, and NK-Cell Development
More than 300 immunodeficiency syndromes have been described. Specific molecular defects have been identified for most diseases. Most are recessive traits with X-linked, autosomal dominant loss of function, and autosomal dominant gain of function also is seen. Defects include those associated with absence of a cell type, either a lineage (e.g., absence of T cells in SCID), absence of a subset of cells (e.g., absence of Tregs in immune dysregulation, polyendocrinopathy, X-linked syndrome), or dysfunction of a cell (e.g., the hemophagocytic lymphohistiocytosis disorders). In some cases, multiple cell types are affected, and in some syndromes, excess of a certain cell type or function disrupts the critical balance needed for immune homeostasis.