Disorders of the Complement System
Evaluation of the Complement System
Richard B. Johnston Jr.
Testing for total hemolytic complement activity (CH50 ) effectively screens for most of the common diseases of the complement system. A normal result in this assay depends on the ability of all 11 components of the classical pathway and membrane attack complex to interact and lyse antibody-coated sheep erythrocytes. The dilution of serum that lyses 50% of the cells determines the end-point. In congenital deficiencies of C1 through C8, the CH50 value is 0 or close to 0; in C9 deficiency the value is approximately half-normal. Values in the acquired deficiencies vary with the type and severity of the underlying disorder. This assay does not detect deficiency of mannose-binding lectin (MBL), factor D or B of the alternative pathway, or properdin (Fig. 160.1 ). Deficiency of factor I or H permits persistence of the classical and alternative pathway convertase and thus consumption of C3, with reduction in the CH50 value. When clotted blood or serum sits at room temperature or warms, CH50 activity begins to decline, leading to values that are falsely low but not zero. It is important to separate the serum and freeze it at −70°C (−94°F) by no more than 1 hr after blood draw.
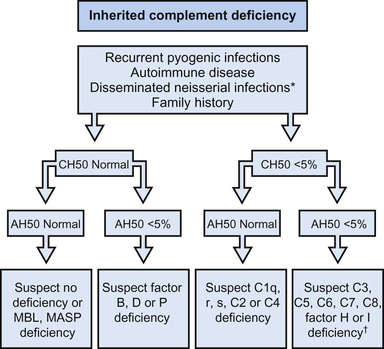
In hereditary angioedema , depression of C4 and C2 during an attack significantly reduces the CH50 . Typically, C4 is low and C3 normal or slightly decreased. Concentrations of C1 inhibitor protein will be normal in 15% of cases; but C1 acts as an esterase, and the diagnosis can be established by showing increased capacity of the patient's sera to hydrolyze synthetic esters.
A decrease in serum concentration of both C4 and C3 suggests activation of the classical pathway by immune complexes. Decreased C3 and normal C4 levels suggest activation of the alternative pathway. This difference is particularly useful in distinguishing nephritis secondary to immune complex deposition from that caused by NeF (nephritic factor). In the latter condition and in deficiency of factor I or H, factor B is consumed and C3 serum concentration is low. Alternative pathway activity can be measured with a relatively simple and reproducible hemolytic assay that depends on the capacity of rabbit erythrocytes to serve as both an activating (permissive) surface and a target of alternative pathway activity. This assay, AP50 , detects deficiency of properdin, factor D, and factor B. Immunochemical methods can be used to quantify individual components and split products of all 3 pathways, guided by results of the screening hemolytic assays.
A defect of complement function should be considered in any patient with recurrent angioedema, autoimmune disease (especially SLE), chronic nephritis, hemolytic-uremic syndrome, or partial lipodystrophy, or with recurrent pyogenic infections, disseminated meningococcal or gonococcal infection, or a 2nd episode of bacteremia at any age. A previously well adolescent or young adult with meningococcal meningitis caused by an uncommon serotype (not A, B, or C) should undergo screening for a late-component or alternative pathway deficiency with CH50 and AP50 assays.
Bibliography
Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J . 2015;34:2735–2757.
Giclas PC. Evaluation of complement activation and its clinical relevance. Detrick B, Hamilton RG, Folds JD. Manual of molecular and clinical laboratory immunology . ed 7. ASM Press: Washington, DC; 2006:115–117.
Genetic Deficiencies of Complement Components
Richard B. Johnston Jr.
Congenital deficiencies of all 11 components of the classical–membrane attack pathway and of factors D and B and properdin of the alternative pathway are described in Table 160.1 . All components of the classical and alternative pathways except properdin and factor B are inherited as autosomal recessive co-dominant traits. Each parent transmits a gene that codes for synthesis of half the serum level of the component. Deficiency results from inheritance of 1 null gene from each parent; the hemizygous parents typically have low normal CH50 levels and no consequences of the partial deficiency. Properdin deficiency is transmitted as an X-linked trait. Factor B is an autosomal recessive non–co-dominant trait.
Table 160.1
| DISEASE | GENETIC DEFECT/PRESUMED PATHOGENESIS | INHERITANCE | FUNCTIONAL DEFECT | ASSOCIATED FEATURES |
|---|---|---|---|---|
| C1q deficiency | Mutation in C1QA , C1QB , C1QC: classical complement pathway components | AR | Absent CH50 hemolytic activity; defective activation of the classical pathway, diminished clearance of apoptotic cells | SLE, infections with encapsulated organisms |
| C1r deficiency | Mutation in C1R: classical complement pathway component | AR | Absent CH50 hemolytic activity; defective activation of the classical pathway | SLE, infections with encapsulated organisms |
| C1s deficiency | Mutation in C1S: classical complement pathway component | AR | Absent CH50 hemolytic activity; defective activation of the classical pathway | SLE, infections with encapsulated organisms |
| C4 deficiency | Mutation in C4A , C4B: classical complement pathway components | AR | Absent CH50 hemolytic activity; defective activation of the classical pathway, defective humoral immune response to carbohydrate antigens in some patients | SLE, infections with encapsulated organisms |
| C2 deficiency | Mutation in C2: classical complement partway component | AR | Absent CH50 hemolytic activity; defective activation of the classical pathway | SLE, infections with encapsulated organisms, atherosclerosis |
| C3 deficiency | Mutation in C3: central complement component | AR, gain-of-function AD | Absent CH50 and AH50 hemolytic activity; defective opsonization, defective humoral immune response | Infections; glomerulonephritis, aHUS with gain-of-function mutations |
| C5 deficiency | Mutation in C5: terminal complement component | AR | Absent CH50 and AH50 hemolytic activity; defective bactericidal activity | Neisserial infections |
| C6 deficiency | Mutation in C6: terminal complement component | AR | Absent CH50 and AH50 hemolytic activity; defective bactericidal activity | Neisserial infections |
| C7 deficiency | Mutation in C7: terminal complement component | AR | Absent CH50 and AH50 hemolytic activity; defective bactericidal activity | Neisserial infections |
| C8 α-γ deficiency | Mutation in C8A, C8G: terminal complement components | AR | Absent CH50 and AH50 hemolytic activity; defective bactericidal activity | Neisserial infections |
| C8b deficiency | Mutation in C8B: terminal complement component | AR | Absent CH50 and AH50 hemolytic activity. defective bactericidal activity | Neisserial infections |
| C9 deficiency | Mutation in C9: terminal complement component | AR | Reduced CH50 and AH50 hemolytic activity; deficient bactericidal activity | Mild susceptibility to neisserial infections |
| C1 inhibitor deficiency | Mutation in C1NH: regulation of kinins and complement activation | AD | Spontaneous activation of the complement pathway with consumption of C4/C2; spontaneous activation of the contact system with generation of bradykinin from high-molecular-weight kininogen | Hereditary angioedema |
| Factor B | Mutation in CFB: activation of the alternative pathway | AD | Gain-of-function mutation with increased spontaneous AH50 | aHUS |
| Factor D deficiency | Mutation in CFD: regulation of the alternative complement pathway | AR | Absent AH50 hemolytic activity | Neisserial infections |
| Properdin deficiency | Mutation in CFP: regulation of the alternative complement pathway | XL | Absent AH50 hemolytic activity | Neisserial infections |
| Factor I deficiency | Mutation in CFI: regulation of the alternative complement pathway | AR | Spontaneous activation of the alternative complement pathway with consumption of C3 | Infections, neisserial infections, aHUS, preeclampsia, membranoproliferative glomerulonephritis |
| Factor H deficiency | Mutation in CFH: regulation of the alternative complement pathway | AR | Spontaneous activation of the alternative complement pathway with consumption of C3 | Infections, neisserial infections, aHUS, preeclampsia, membranoproliferative glomerulonephritis |
| MASP-1 deficiency | Mutation in MASP1: cleaves C2 and activates MASP-2 | AR | Deficient activation of the lectin activation pathway, cell migration | Infections, 3MC syndrome |
AD, Autosomal dominant; aHUS, atypical hemolytic-uremic syndrome; AR, autosomal recessive; SLE, systemic lupus erythematosus; XL, X-linked; 3MC, previously Carnevale, Mingarelli, Malpuech, and Michels syndromes.
From Kliegman RM, Lye PS, Bordini BJ, et al, editors: Nelson pediatric symptom-based diagnosis, Philadelphia, 2018, Elsevier, Table 41.11, p 765.
Most patients with primary C1q deficiency have systemic lupus erythematosus (SLE); some have an SLE-like syndrome without typical SLE serology, a chronic rash with underlying vasculitis, or membranoproliferative glomerulonephritis (MPGN). Some C1q-deficient children have serious infections, including septicemia and meningitis. Individuals with C1r, C1s, combined C1r/C1s, C4, C2, or C3 deficiency also have a high incidence of autoimmune syndromes (see Table 160.1 ), especially SLE or an SLE-like syndrome, without an elevated antinuclear antibody level.
C4 is encoded by 2 genes, C4A and C4B . C4 deficiency represents absence of both gene products. Complete deficiency of only C4A, present in approximately 1% of the population, also predisposes to SLE, although C4 levels are only partially reduced. Patients with only C4B deficiency may be predisposed to infection. A few patients with C5, C6, C7, or C8 deficiency have SLE, but recurrent meningococcal infections are much more likely to be the major problem.
There are at least 2 possible reasons for the concurrence of complement component deficiencies, especially C1, C4, C2, or C3 deficiency, and autoimmune–immune complex diseases. First, deposition of C3 on autoimmune complexes facilitates their removal from the circulation through binding to complement receptor 1 (CR1) on erythrocytes and transport to the spleen and liver. Second, the early components, particularly C1q and C3, expedite the clearance of necrotic and apoptotic cells, which are sources of autoantigens.
Individuals with C2 deficiency carry the risk of life-threatening septicemic illnesses, usually caused by pneumococci. However, most have not had problems with other increased susceptibility to infection, presumably because of the protective function of the alternative pathway, particularly if enhanced by pneumococcal and Haemophilus influenzae immunization. The genes for C2, factor B, and C4 are situated close to each other on chromosome 6, and a partial depression of factor B levels can occur in conjunction with C2 deficiency. Persons with a deficiency of both proteins may be at particular risk. One percent of European Caucasians carry 1 null gene for C2.
Because C3 can be activated by C142 or by the alternative pathway, a defect in the function of either pathway can be compensated for, at least to some extent. Without C3, however, opsonization of bacteria is inefficient, and the chemotactic fragment from C5 (C5a) is not generated. Some organisms must be well opsonized in order to be cleared, and genetic C3 deficiency has been associated with recurrent, severe pyogenic infections caused by pneumococci, H. influenzae , and meningococci.
More than half the individuals reported to have congenital C5, C6, C7, or C8 deficiency have had meningococcal meningitis or extragenital gonococcal infection. C9 deficiency is most often reported in individuals of Japanese or Korean descent. C9-deficient individuals retain about one-third normal CH50 titers; some have had Neisseria disease. In studies of patients ≥10 yr old with systemic meningococcal disease, 3–15% have had a genetic deficiency of C5, C6, C7, C8, C9, or properdin. Among patients with infections caused by the uncommon Neisseria meningitidis serogroups (X, Y, Z, W135, 29E, or nongroupable; but not A, B, or C), 33–45% have an underlying complement deficiency. It is not clear why patients with a deficiency of one of the late-acting components have a particular predisposition to Neisseria infections. It may be that serum bacteriolysis is uniquely important in defense against this organism. Many persons with such a deficiency have no significant illness.
A few individuals have been identified with deficiency of factor D or Factor B of the alternative pathway, all with recurrent infections, most often neisserial or pneumococcal. Hemolytic complement activity and C3 levels in their serum were normal, but alternative pathway activity was markedly deficient or absent.
Mutations in the structural gene encoding MBL or polymorphisms in the promoter region of the gene result in pronounced interindividual variation in the level of circulating MBL. More than 90% of individuals with MBL deficiency do not express a predisposition to infection. Those with a very low level of MBL have a predisposition to recurrent respiratory infections in infancy and to serious pyogenic and fungal infections if there is another underlying defect of host defense. MBL-associated serine protease (MASP)-2 deficiency has been reported with SLE-like symptoms and recurrent pneumococcal pneumonia. Homozygous ficolin-3 deficiency has been associated with repeated pneumonia since early childhood, cerebral abscesses, and bronchiectasis.
Bibliography
Botto M, Kirschfink M, Macor P, et al. Complement in human diseases: lessons from complement deficiencies. Mol Immunol . 2009;46:2774–2783.
Manian FA, Alame D. Case 11-2015: A 28-year-old woman with headache, fever, and a rash. N Engl J Med . 2015;372:1454–1462.
Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol . 2013;190:3831–3838.
Sfyroera G, Ricklin D, Reis ES, et al. Rare loss-of-function mutation in complement component C3 provides insight into molecular and pathophysiological determinants of complement activity. J Immunol . 2015;194:3305–3316.
Slade C, Bosco J, Unglik G, et al. Deficiency in complement factor B. N Engl J Med . 2013;369:1667–1669.
Tebruegge M, Curtis N. Epidemiology, etiology, pathogenesis, and diagnosis of recurrent bacterial meningitis. Clin Microbiol Rev . 2008;21:519–537.
Deficiencies of Plasma, Membrane, or Serosal Complement Control Proteins
Richard B. Johnston Jr.
Congenital deficiencies of 5 plasma complement control proteins have been described (see Table 160.1 ). Factor I deficiency was reported originally as a deficiency of C3 resulting from hypercatabolism. The 1st patient described had suffered a series of severe pyogenic infections similar to those associated with agammaglobulinemia or congenital deficiency of C3. Factor I is an essential regulator of both pathways. Its deficiency permits prolonged existence of C3b as a part of the C3 convertase of the alternative pathway, C3bBb. This results in constant activation of the alternative pathway and cleavage of more C3 to C3b, in circular fashion. Intravenous infusion of plasma or purified factor I induced a prompt rise in serum C3 concentration in the patient and a return to normal of C3-dependent functions in vitro, such as opsonization.
The effects of factor H deficiency are similar to those of factor I deficiency because factor H also assists in dismantling the alternative pathway C3 convertase. A trigger event such as infection initiates uninhibited continuous activation of the alternative pathway, which consumes C3, factor B, total hemolytic activity, and alternative pathway activity. Patients have sustained systemic infections due to pyogenic bacteria, particularly Neisseria meningitidis. Many have had glomerulonephritis or atypical hemolytic-uremic syndrome (aHUS) (see Chapter 538.5 ). Mutations in genes encoding membrane cofactor protein (MCP, CD46), factors I or B, C3, or the endothelial antiinflammatory protein thrombomodulin, or autoantibodies to factors H or B, are also associated with aHUS. The majority of patients with factor H deficiency and aHUS, typically <2 yr old, develop end-stage renal disease, and many die.
The few patients thus far reported as having C4-binding protein deficiency have approximately 25% of the normal levels of the protein and no typical disease presentation, although one patient had angioedema and Behçet disease.
Persons with properdin deficiency have a striking predisposition to N. meningitidis meningitis. All reported patients have been male. The predisposition to infection in these patients demonstrates clearly the need for the alternative pathway in defense against bacterial infection. Serum hemolytic complement activity is normal in these patients, and if the patient has specific antibacterial antibody from immunization or prior exposure, the need for the alternative pathway and properdin is greatly reduced. Several patients have had dermal vasculitis or discoid lupus.
Hereditary angioedema occurs in persons unable to synthesize normal levels of functional C1 inhibitor (C1 INH). In 85% of affected families, the patient has markedly reduced concentrations of inhibitor, averaging 30% of normal; the other 15% have normal or elevated concentrations of an immunologically cross-reacting but nonfunctional protein. Both forms of the disease are transmitted as autosomal dominant traits. C1 INH suppresses the complement proteases C1rs and MASP-2 and the activated proteases of the contact and fibrinolysis systems. In the absence of full C1 INH function, activation of any of these proteases tips the balance toward the protease. This activation leads to uncontrolled C1 and kallikrein activity with breakdown of C4 and C2 and release of bradykinin, which interacts with vascular endothelial cells to cause vasodilation, producing localized, nonpitting edema. The biochemical triggers that induce attacks of angioedema in these patients are not well understood.
Swelling of the affected part progresses rapidly, without urticaria, itching, discoloration, or redness and often without severe pain. Swelling of the intestinal wall, however, can lead to intense abdominal cramping, sometimes with vomiting or diarrhea. Concomitant subcutaneous edema is often absent, and patients have undergone abdominal surgery or psychiatric examination before the true diagnosis was established. Laryngeal edema can be fatal. Attacks last 2-3 days and then gradually abate. They may occur at sites of trauma, especially dental, after vigorous exercise, or with menses, fever, or emotional stress. Attacks begin in the 1st 5 yr of life in almost half of patients, but are usually not severe until late childhood or adolescence. Acquired C1 INH deficiency can occur in association with B-cell cancer or autoantibody to C1 INH. SLE and glomerulonephritis have been reported in patients with the congenital disease (for treatment see Chapter 160.5 ).
Three of the membrane complement control proteins—CR1, MCP (CD46), and decay-accelerating factor (DAF)—prevent the formation of the full C3-cleaving enzyme, C3bBb, which is triggered by C3b deposition. CD59 (membrane inhibitor of reactive lysis) prevents the full development of the membrane attack complex that creates the “hole.” Paroxysmal nocturnal hemoglobinuria (PNH) is a hemolytic anemia that occurs when DAF and CD59 are not expressed on the erythrocyte surface. The condition is acquired as a somatic mutation in a hematopoietic stem cell of the PIGA gene on the X chromosome. The product of this gene is required for normal synthesis of a glycosyl-phosphatidylinositol molecule that anchors about 20 proteins to cell membranes, including DAF and CD59. One patient with genetic isolated CD59 deficiency had a mild PNH-like disease despite normal expression of membrane DAF. In contrast, genetic isolated DAF deficiency has not resulted in hemolytic anemia (for treatment see Chapter 160.5 ).
Bibliography
Banerji A, Riedl MA, Bernstein JA, et al. Effect of lanadelumab compared with placebo on prevention of hereditary angioedema attacks. JAMA . 2018;320(20):2108–2121.
Berger TD, Garty BZ. Hereditary angioedema presenting as recurrent acute pancreatitis. Pediatrics . 2017;137(2):e20150620.
Geerdink LM, Westra D, van Wijk JA, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol . 2012;27:1283–1291.
Jokiranta TS. HUS and atypical HUS. Blood . 2017;129:2847–2856.
Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol . 2013;33:508–530.
Martinez-Saguer I, Farkas H. Erythema marginatum as an early symptom of hereditary angioedema: case report of 2 newborns. Pediatrics . 2017;137(2):e20152411.
Sekar A, Bialas AE, De Rivera H, et al. Schizophrenia risk from complex variation of complement component 4. Nature . 2016;530:177–183.
Zuraw BL, Christiansen SC. HAE pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol . 2016;51:216–229.
Secondary Disorders of Complement
Richard B. Johnston Jr.
Partial deficiency of C1q has occurred in patients with severe combined immunodeficiency disease or hypogammaglobulinemia, apparently secondary to the deficiency of IgG, which normally binds reversibly to C1q and prevents its rapid catabolism.
Chronic membranoproliferative glomerulonephritis can be caused by nephritic factor (NeF), an IgG autoantibody to the C3-cleaving enzyme of the classical pathway (C4b2a) or alternative pathway (C3bBb). NeF protects the enzyme from inactivation and promotes consumption of C3 and decreased concentration of serum C3. Pyogenic infections, including meningitis, may occur if the serum C3 level drops to <10% of normal. This disorder has been found in children and adults with dense-deposit disease or partial lipodystrophy. Adipocytes are the main source of factor D and synthesize C3 and factor B; exposure to NeF induces their lysis. The IgG NeF that inhibits the classical pathway C3 convertase has been described in acute postinfectious nephritis and in SLE. The consumption of C3 that characterizes poststreptococcal nephritis and SLE could be caused by this factor, by immune complex activation, or by both.
Newborn infants have mild to moderate reductions in all plasma components of the complement system. Opsonization and generation of chemotactic activity in serum from full-term newborns can be markedly deficient through either the classical or the alternative pathway. Complement activity is even lower in preterm infants. Patients with severe chronic cirrhosis of the liver, hepatic failure, malnutrition, or anorexia nervosa can have significant deficiency of complement components and functional activity. Synthesis of components is depressed in these conditions, and serum from some patients with malnutrition also contains immune complexes that could accelerate depletion.
Patients with sickle cell disease have normal activity of the classical pathway, but some have defective function of the alternative pathway in opsonization of pneumococci, in bacteriolysis and opsonization of Salmonella, and in lysis of rabbit erythrocytes. Deoxygenation of erythrocytes from patients with sickle cell disease alters their membranes to increase exposure of phospholipids that can activate the alternative pathway and consume its components. This activation is accentuated during painful crisis. Children with nephrotic syndrome may have decreased serum levels of factors B and D and subnormal serum opsonizing activity.
Immune complexes initiated by microorganisms or their by-products can induce complement consumption. Activation occurs primarily through fixation of C1 and initiation of the classical pathway. Formation of immune complexes and consumption of complement have been demonstrated in lepromatous leprosy, bacterial endocarditis, infected ventriculojugular shunts, malaria, infectious mononucleosis, dengue hemorrhagic fever, and acute hepatitis B. Nephritis or arthritis can develop as a result of deposition of immune complexes and activation of complement in these infections. In SLE, immune complexes activate C142, and C3 is deposited at sites of tissue damage, including kidneys and skin; depressed synthesis of C3 is also noted. The syndrome of recurrent urticaria, angioedema, eosinophilia, and hypocomplementemia secondary to activation of the classical pathway may be caused by autoantibody to C1q and circulating immune complexes. Circulating immune complexes and decreased C3 have been reported in some patients with dermatitis herpetiformis, celiac disease, primary biliary cirrhosis, and Reye syndrome.
Circulating bacterial products in sepsis or tissue factors released after severe trauma can initiate activation of the classical and alternative pathways, leading to increased serum levels of C3a, C5a, and C5b-9 and systemic inflammatory response syndrome (SIRS) and multi-organ failure. C5a and its receptors, particularly on neutrophils, appear to be central to the pathogenesis of SIRS. Intravenous injection of iodinated roentgenographic contrast medium can trigger a rapid and significant activation of the alternative pathway, which may explain the occasional reactions that occur in patients undergoing this procedure.
Burns can induce massive activation of the complement system, especially the alternative pathway, within a few hours after injury. Resulting generation of C3a and C5a stimulates neutrophils and induces their sequestration in the lungs, leading to shock lung. Cardiopulmonary bypass, extracorporeal membrane oxygenation, plasma exchange, or hemodialysis using cellophane membranes may be associated with a similar syndrome as a result of activation of plasma complement, with release of C3a and C5a. In patients with erythropoietic protoporphyria or porphyria cutanea tarda , exposure of the skin to light of certain wavelengths activates complement, generating chemotactic activity. This chemotactic activity leads to lysis of capillary endothelial cells, mast cell degranulation, and the appearance of neutrophils in the dermis.
Some tumor cells can avoid complement-mediated lysis by overexpressing DAF, MCP, CD59, CR1, or factor H, or by secreting proteases that cleave tumor-bound C3b. Microorganisms have evolved similar evasive mechanisms; for example, HIV-1 particles budding from infected cells acquire the membrane proteins DAF and CD59, and staphylococci can produce multiple complement inhibitors.
Bibliography
Banerji A, Busse P, Shennak M, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med . 2017;376:717–728.
Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol . 2012;34:129–136.
Sethi S, Sullivan A, Smith RJ. C4 dense-deposit disease. N Engl J Med . 2014;370:784–786.
Wang RH, Phillips G Jr, Medof ME, et al. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. J Clin Invest . 1993;92:1326–1355.
Wood JH, Partrick DA, Johnston RB Jr. The inflammatory response to injury in children. Curr Opin Pediatr . 2010;22:315–320.
Treatment of Complement Disorders
Richard B. Johnston Jr.
No specific therapy is available at present for genetic deficiencies of the components of the classical, alternative, and lectin complement pathways. Much can be done, however, to protect patients with any of these disorders from serious complications; and specific treatment is available for 3 disorders caused by control-protein deficiencies: hereditary angioedema, aHUS, and PNH.
Management of hereditary angioedema starts with avoidance of precipitating factors, usually trauma. Infusion of C1 INH concentrate (nanofiltered C1-esterase inhibitor ) was approved by the U.S. Food and Drug Administration (FDA) for use in children in 2016. An inhibitor of kallikrein (ecallantide ) that blocks bradykinin production and an antagonist of the bradykinin receptor (icatibant ) are approved in the United States for use in adolescents and adults for long-term prophylaxis, preparation for surgery or dental procedures, or treatment of acute attacks. The synthetic androgen oxandrolone increases the level of functional C1 INH several-fold and is approved for cautious use in children. Antihistamines, epinephrine, and corticosteroids have no effect.
Lanadelumab, a selective inhibitor of kallikein, has potential as a prophylactic agent. Eculizumab , a humanized monoclonal antibody to C5, prevents generation of the membrane-attack complex C5b9 and is an effective treatment for PNH and aHUS .
Effective supportive management is available for other primary diseases of the complement system, and identification of a specific defect in the complement system can have an important impact on management. Concern for the associated complications, such as autoimmune disease and infection, should encourage vigorous diagnostic efforts and earlier institution of therapy. Individuals with SLE and a complement defect generally respond as well to therapy as do those without complement deficiency. With the onset of unexplained fever, cultures should be obtained and antibiotic therapy instituted more quickly and with less stringent indications than in a normal child.
The parent or patient should be given letters describing any predisposition to systemic bacterial infection or autoimmune disease associated with the patient's deficiency, along with the recommended initial approach to management, for possible use by school, camp, or emergency department physicians. The patient and close household contacts should be immunized against H. influenzae, Streptococcus pneumoniae, and N. meningitidis. High titers of specific antibody might opsonize effectively without the full complement system, and immunization of household members could reduce the risk of exposing patients to these particularly threatening pathogens. Repeat immunization of patients is advisable since complement deficiency can be associated with a blunted or shorter-lived antibody response than normal.
Heparin , which inhibits both classical and alternative pathways, has been used to prevent “postpump syndrome.”
Bibliography
Banerji A, Busse P, Shennak M, et al. Inhibiting plasma kallikrein for hereditary angioedema prophylaxis. N Engl J Med . 2017;376:717–728.
Frank MM, Zuraw B, Banerji A, et al. Management of children with hereditary angioedema due to C1 inhibitor deficiency. Pediatrics . 2017;138:e20160575.
Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med . 2013;368:2169–2181.
Longhurst H, Cicardi M, Craig T, et al. Prevention of hereditary angioedema attacks with a subcutaneous C1 inhibitor. N Engl J Med . 2017;376:1131–1140.
Lumry W, Manning ME, Hurewitz DS, et al. Nanofiltered C1-esterase inhibitor for the acute management and prevention of hereditary angioedema attacks due to C1-inhibitor deficiency in children. J Pediatr . 2013;162:1017–1022.
MacGinnitie AJ, Davis-Lorton M, Stolz LE, et al. Use of ecallantide in pediatric hereditary angioedema. Pediatrics . 2013;132:381–382 [e490–e497].
Zuraw BL, Bernstein JA, Lang DM, et al. A focused parameter update: hereditary angioedema, acquired C1 inhibitor deficiency, and angiotensin-converting enzyme inhibitor–associated angioedema. J Allergy Clin Immunol . 2013;131:1491–1518.