Principles and Clinical Indications of Hematopoietic Stem Cell Transplantation
Rachel A. Phelan, David Margolis
Allogeneic (from a donor) or autologous (from the same individual) hematopoietic stem cells have been used to cure both malignant and nonmalignant disorders. Autologous transplantation is employed as a rescue strategy after delivering otherwise lethal doses of chemotherapy with or without radiotherapy in children with hematologic malignancies such as relapsed lymphoma or selected solid tumors (e.g., neuroblastoma, brain tumors). Allogeneic transplantation is used to treat children with genetic diseases of blood cells, such as hemoglobinopathies, primary immunodeficiency diseases, various inherited metabolic diseases, and bone marrow failure. Allogeneic transplant is also used as treatment for hematologic malignancies, such as leukemia and myelodysplastic syndromes. Bone marrow had represented the only source of hematopoietic progenitors employed. Growth factor (granulocyte colony-stimulating factor)–mobilized peripheral blood hematopoietic stem cells and umbilical cord blood hematopoietic progenitors have now also been regularly used in clinical practice to perform hematopoietic stem cell transplantation (HSCT ).
An HLA-matched sibling was once the only type of donor employed. Currently, matched unrelated volunteers, full-haplotype mismatched family members, and unrelated cord blood donors have been largely employed to transplant patients lacking an HLA-identical relative.
Protocols for allogeneic HSCT consist of 2 parts: the preparative regimen and transplantation itself. During the preparative conditioning regimen , chemotherapy, at times associated with irradiation, is administered to destroy the patient's hematopoietic system and to suppress the immune system, especially T cells, so that graft rejection is prevented. In patients with malignancies, the preparative regimen also serves to significantly reduce the tumor burden. The patient then receives an intravenous infusion of hematopoietic cells from the donor. Less aggressive conditioning regimens, known as reduced-intensity conditioning regimens , are also used in pediatric patients. These regimens are mainly immunosuppressive and aim at inducing a state of reduced immune competence of the recipient to avoid the rejection of donor cells.
The immunology of HSCT is distinct from that of other types of transplant because, in addition to stem cells, the graft contains mature blood cells of donor origin, including T cells, B cells, natural killer cells, and dendritic cells. These cells repopulate the recipient's lymphohematopoietic system and give rise to a new immune system, which helps eliminate residual leukemia cells that survive the conditioning regimen. This effect is known as the graft-versus-leukemia (GVL) effect.
The donor immune system exerts its T-cell–mediated GVL effect through alloreactions directed against histocompatibility antigens displayed on recipient leukemia cells. Because some of these histocompatibility antigens are also displayed on tissues, however, unwanted T-cell–mediated alloreactions may ensue. Specifically, donor alloreactive cytotoxic CD8+ effector T cells may attack recipient tissues, particularly the skin, gastrointestinal (GI) tract, and liver, causing acute graft-versus-host disease (GVHD) , a condition of varying severity that in some cases can be life threatening or even fatal (see Chapter 163 ).
The success of allogeneic HSCT is undermined by diversity between donors and recipients in major and minor histocompatibility antigens. The human leukocyte antigens (HLA) , including HLA-A, HLA-B, and HLA-C major histocompatibility complex (MHC) class I molecules, present peptides to CD8+ T cells, whereas the HLA-DR, HLA-DQ, and HLA-DP MHC class II molecules present peptides to CD4+ T cells. There are 100s of variant forms of each class I and class II molecule, and even small differences can elicit alloreactive T-cell responses that mediate graft rejection and/or GVHD. Disparities for HLA-A, -B, -C, or -DRB1 alleles in the donor-recipient pair are independent risk factors for both acute and chronic GVHD. There is also increasing evidence that HLA-DQ and HLA-DP may play a role, prompting some transplant centers to also explore matching at these alleles.
Minor histocompatibility antigens derive from differences between the HLA-matched recipient and donor in peptides that are presented by the same HLA allotype. These antigens result from polymorphisms of non-HLA proteins, differences in the level of expression of proteins, or genetic differences between males and females. An example of the latter is represented by the H-Y antigens encoded by the Y chromosome, which can stimulate GVHD when a female donor is employed to transplant an HLA-identical male recipient. Thus, from this evidence, it is clear that GVHD may occur even when the donor and recipient are HLA identical.
The preferred donor for any patient undergoing HSCT is an HLA-identical sibling. Because polymorphic HLA genes are closely linked and usually constitute a single genetic locus, any pair of siblings has a 25% chance of being HLA identical . Thus, also in view of the limited family size in the developed countries, <25–30% of patients in need of an allograft can receive their transplant from an HLA-identical sibling. This percentage is even lower in patients with inherited disorders since affected siblings will not be considered donor candidates.
HSCT From an HLA-Identical Sibling Donor
Allogeneic HSCT from an HLA-compatible sibling is the treatment of choice for children with hematologic malignancies and various congenital or acquired diseases (Table 161.1 ). Best results are achieved in patients with congenital or acquired nonmalignant disorders because the risk of disease recurrence is low and the cumulative transplantation-related mortality is lower than in children receiving transplants for hematologic malignancies.
Acute Lymphoblastic Leukemia
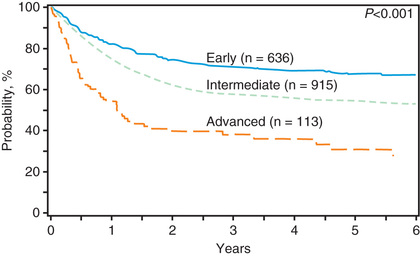
Allogeneic HSCT is used for pediatric patients with acute lymphoblastic leukemia (ALL ), either in the 1st complete remission when a child is considered to be at high risk of leukemia recurrence (e.g., those carrying poor-risk cytogenetic characteristics or with high levels of minimal residual disease), or in 2nd or further complete remission after previous marrow relapse. ALL is the most common indication for HSCT in childhood. Several patient-, donor-, disease-, and transplant-related variables may influence the outcome of patients with ALL given an allogeneic HSCT. The long-term probabilities of event-free survival (EFS) for patients with ALL transplanted in the 1st or 2nd complete remission is 60–70% and 40–60%, respectively. Inferior results are obtained in patients receiving transplants in more advanced disease phases. The use of total body irradiation (TBI) during the preparative regimen offers an advantage in terms of better EFS compared to a regimen consisting of cytotoxic drugs alone (Fig. 161.1 ), but it can induce more long-term side effects. This has prompted more investigation into TBI-sparing alternatives. Less intensive GVHD prophylaxis is also associated with a better outcome. Bone marrow is generally still the preferred source of stem cells to be employed for transplantation, although this differs among transplant centers.
Although the main benefit for allogeneic HSCT recipients with leukemia derives from the GVL effect displayed by immunocompetent cells, disease recurrence remains the main cause of treatment failure. The risk of failing to eradicate leukemia is influenced by many variables, including disease phase, molecular lesions of tumor cells, and disparity for major or minor histocompatibility antigens in the donor/recipient pairs. To overcome the hurdle of tumor elusion caused by HLA loss on malignant cells, the use of non-HLA–restricted chimeric antigen receptors (CARs) has been proposed. This therapeutic strategy is based on genetic reprogramming of T cells through artificial immune receptors that reproducibly and efficiently redirect the antigen specificity of polyclonal T lymphocytes toward target antigens expressed by leukemic cells. When expressed by T cells, CARs mediate antigen recognition and tumor cytolysis in an MHC-unrestricted fashion and can target any molecule (protein, carbohydrate, or glycolipid) expressed on the surface of tumor cells, thus bypassing one of the major tumor escape mechanisms based on the downregulation of MHC molecules. CARs are composed of an extracellular specific antigen-binding moiety, obtained from the variable regions of a monoclonal antibody, linked together to form a single-chain antibody (scFv), and of an intracellular signaling component derived from the ζ chain of the T-cell–receptor (TCR)–CD3 complex. The addition to the CAR gene construct of co-stimulation signals and cytokines promoting T-cell expansion and survival improves the antitumor efficiency of the engineered T cells and their survival in the tumor milieu. Gamma retrovirus and lentiviruses are usually used to transduce CARs into T lymphocytes to be employed in the clinical setting. These vectors have been shown to efficiently infect T lymphocytes, integrate into the host genome, and produce robust expression of the gene in human T cells and their progeny.
Acute Myeloid Leukemia
Allogeneic HSCT from an HLA-identical sibling is largely employed as postremission treatment of pediatric patients with acute myeloid leukemia (AML ). Children with AML in 1st complete remission who are given allogeneic HSCT as consolidation therapy have a better probability of EFS than those treated with either chemotherapy alone or autologous transplantation. Results obtained in patients given HSCT from an HLA-identical sibling after either a TBI-containing or a chemotherapy-based preparative regimen are similar, the probability of EFS being in the order of 70%. Therefore, for AML, conditioning regiments generally omit the use of TBI because of associated long-term side effects. Children with acute promyelocytic leukemia in molecular remission at the end of treatment with chemotherapy and all-trans -retinoic acid, or with AML and translocation t(8;21); inversion of chromosome 16 (inv16), translocation t(16;16), or normal cytogenetics and presence of NPM1 or CEPB α mutation are no longer considered eligible for allogeneic HSCT in 1st complete remission in view of their improved prognosis with alternative treatments. Studies suggest restricting the use of HSCT to those patients with poor molecular lesions, such as FLT3-internal tandem duplication or mixed-lineage leukemia abnormalities, or with high levels of minimal residual disease at the end of induction therapy. Approximately 40–60% of pediatric patients with AML in the 2nd complete remission can be rescued by HSCT.
Chronic Myelogenous Leukemia
For many years, allogeneic HSCT has been considered the only proven curative treatment for children with Philadelphia-positive (Ph+) chronic myelogenous leukemia. Leukemia-free survival of chronic myelogenous leukemia patients after an allograft is 45–80%. The phase of disease (chronic phase, accelerated phase, blast crisis), recipient age, type of donor employed (related or unrelated), and time between diagnosis and HSCT are the main factors influencing the outcome. The best results are obtained in children transplanted during the chronic phase from an HLA-identical sibling within 1 yr from diagnosis. Unlike other forms of pediatric leukemia, infusion of donor leukocytes can reinduce a state of complete remission in a large proportion of patients experiencing leukemia relapse.
Treatment with the specific BCR-ABL tyrosine protein kinase inhibitors (imatinib mesylate, dasatinib, nilotinib), targeting the enzymatic activity of the BCR-ABL fusion protein, has modified the natural history of the disease and thus the indications for transplantation. The indication for HSCT in this population is thus evolving and is generally reserved for patients with a poor response to tyrosine kinase inhibitors or those who do not tolerate their side effects.
Juvenile Myelomonocytic Leukemia
Juvenile myelomonocytic leukemia (JMML ) is a rare hematopoietic malignancy of early childhood, representing 2–3% of all pediatric leukemias. JMML is characterized by hepatosplenomegaly and organ infiltration, with excessive proliferation of cells of monocytic and granulocytic lineages. Hypersensitivity to granulocyte-macrophage colony-stimulating factor (GM-CSF) and pathologic activation of the RAS-RAF-MAP (mitogen-activated protein) kinase signaling pathway play an important role in the pathophysiology. JMML usually runs an aggressive clinical course, with a median duration of survival for untreated children of <12 mo from diagnosis. Rare patients with CBL1 or N-RAS mutations can survive for years without an allograft.
HSCT is able to cure approximately 50–60% of patients with JMML. Patients who receive a transplant from an unrelated donor have comparable outcome to those given HSCT from an HLA-compatible related donor. Cord blood transplantation represents a suitable alternative option. Leukemia recurrence is the main cause of treatment failure in children with JMML after HSCT, with the relapse rate as high as 40–50%. Because children with JMML frequently have massive spleen enlargement, splenectomy has been performed before transplantation. However, spleen size at the time of HSCT and splenectomy before HSCT do not appear to affect the posttransplantation outcome. Unlike in CML, donor leukocyte infusion is not useful to rescue patients experiencing disease recurrence; a 2nd allograft can induce sustained remission in approximately one third of children with JMML relapsing after a 1st HSCT.
Myelodysplastic Syndromes Other Than Juvenile Myelomonocytic Leukemia
Myelodysplastic syndromes are a heterogeneous group of clonal disorders characterized by ineffective hematopoiesis leading to peripheral blood cytopenia and a propensity to evolve toward AML. HSCT is the treatment of choice for children with refractory anemia with excess of blasts (RAEB) and for those with RAEB in transformation (RAEB-t). The probability of survival without evidence of disease for these children is 65–70%. It is still unclear whether patients with myelodysplastic syndromes and a blast percentage >20% benefit from pretransplantation chemotherapy. HSCT from an HLA-identical sibling is also the preferred treatment for all children with refractory cytopenia. Transplantation from an alternative donor is also employed in children with refractory cytopenia associated with monosomy 7, complex karyotype, life-threatening infections, profound neutropenia, or transfusion dependency. For children with refractory cytopenia, the probability of EFS after HSCT may be as high as 80%, disease recurrence being rarely observed. This observation has provided the rationale for testing reduced-intensity regimens in these patients.
Non-Hodgkin Lymphoma and Hodgkin Disease
Childhood non-Hodgkin lymphoma (NHL) and Hodgkin disease (HD) are very responsive to conventional chemoradiotherapy, but some patients have refractory disease or are at high risk for relapse. HSCT can cure a proportion of patients with relapsed NHL and HD and should be offered early after relapse, while the disease is still sensitive to therapy. If an HLA-matched donor is available, allogeneic transplantation can be offered to patients with NHL to take advantage of the GVL effect. Patients with sensitive disease and limited tumor burden have favorable outcomes, with EFS rates of 50–60%. Studies also suggest that patients with relapsed or refractory HD do well after autologous HSCT, with EFS of 50–60%. HD patients may also benefit from a GVL effect when given an allograft.
Acquired Aplastic Anemia
Because the probability of long-term survival for a matched-sibling bone marrow transplant (BMT) is reproducibly >80% for children and young adults, BMT is the treatment of choice for children and young adults with acquired severe aplastic anemia. Historically, the treatment of choice for children and young adults without an HLA-matched sibling has been intensive immunosuppression. Because the outcomes of matched unrelated donor transplant for children with acquired aplastic anemia have improved to probability of survival rates >75%, the use of unrelated donor HSCT upfront without prior immunosuppressive therapy is being considered more frequently; 2-year overall survival can be as high as 96% in upfront, matched unrelated donor recipients.
For patients who do not have a matched-sibling donor or well-matched unrelated donor, historically the transplant options were very disappointing. Fortunately, there is hope in current studies looking at haploidentical transplant for this disease. Although numbers are small, the use of posttransplant cyclophosphamide has shown significant improvement over prior experiences. There is hope that all children and young adults who need a transplant for severe aplastic anemia will have the opportunity to do well with a BMT.
Inherited Bone Marrow Failure Syndromes
Fanconi anemia and dyskeratosis congenita are genetic disorders associated with a high risk of developing pancytopenia. Fanconi anemia (FA) is an autosomal recessive disease characterized by spontaneous chromosomal fragility, which is increased after exposure of peripheral blood lymphocytes to DNA cross-linking agents, including clastogenic compounds such as diepoxybutane, mitomycin C, and melphalan. Patients with FA, besides being at risk for pancytopenia, show a high propensity to develop clonal disorders of hematopoiesis, such as myelodysplastic syndromes and AML. HSCT can rescue aplastic anemia and prevent the occurrence of clonal hematopoietic disorders. In view of their defects in DNA repair mechanisms, which are responsible for the chromosomal fragility, FA patients have an exquisite sensitivity to alkylating agents and radiation therapy. Thus, they must be prepared for the allograft with reduced doses of cyclophosphamide and only judicious use of radiation. Many FA patients were once successfully transplanted after receiving low-dose cyclophosphamide and thoracoabdominal irradiation. However, the use of this regimen is associated with an increased incidence of posttransplantation head and neck cancers. Low-dose cyclophosphamide combined with fludarabine has been very well tolerated in patients with FA who have a matched-related donor. The addition of low-dose TBI and antithymocyte globulin (ATG) for those with an unrelated donor has shown similar success. Currently, the 5-yr overall survival is >90% in patients with FA who receive HSCT before the transformation to hematologic malignancy. Because of their underlying disorder, however, patients with FA must be monitored closely in the years after transplant to assess for late effects, including secondary malignancies and endocrinopathies.
Allogeneic HSCT remains the only potentially curative approach for severe bone marrow failure associated with dyskeratosis congenita , a rare congenital syndrome characterized also by atrophy and reticular pigmentation of the skin, nail dystrophy, and leukoplakia of mucous membranes. Results of allograft in these patients have been relatively poor, with 10-yr survival of 20–30%, because of both early and late complications, reflecting increased sensitivity of endothelial cells to radiotherapy and alkylating agents.
Thalassemia
Conventional treatment (i.e., regular blood transfusion and iron-chelation therapy) has dramatically improved both the survival and the quality of life of patients with thalassemia, changing a previously fatal disease with early death to a chronic, slowly progressive disease compatible with prolonged survival. However, HSCT remains the only curative treatment for patients with thalassemia. In these patients the risk of dying from transplant-related complications depends primarily on patient age, iron overload, and concomitant hepatic viral infections. Adults, especially when affected by chronic active hepatitis, have a poorer outcome than children. Among children, 3 classes of risk have been identified on the basis of 3 parameters: regularity of previous iron chelation, liver enlargement, and presence of portal fibrosis. In pediatric patients without liver disease who have received regular iron chelation (class 1 patients), the probability of survival with transfusion independence is >90%, whereas for patients with low compliance with iron chelation and signs of severe liver damage (class 3 patients), the probability of survival has been 60%.
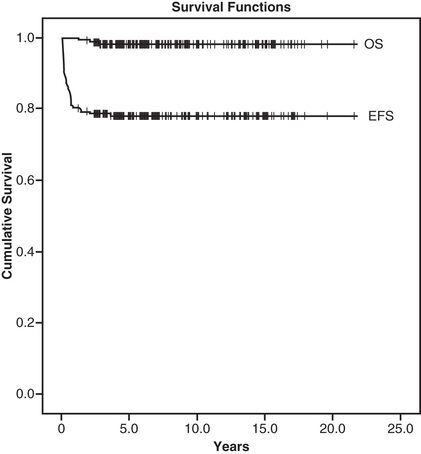
With improvements in supportive care and conditioning regimens, even patients with more advanced liver disease have had excellent outcomes (Fig. 161.2 ). The most effective pharmacologic combinations (e.g., including cyclosporine and methotrexate) should be employed to prevent GVHD. The outcome of patients transplanted from an unrelated donor has been reported similar to that of HLA-identical sibling recipients. The increased use of umbilical cord blood and haploidentical donors in this population is being explored to expand the number of patients eligible for HSCT. Also, advancements in gene therapy are being made in thalassemia in early trials, which may eventually change the approach to this disease.
Sickle Cell Disease
Disease severity varies greatly among patients with sickle cell disease, with 5–20% of the overall population suffering significant morbidity from vasoocclusive crises and pulmonary, renal, or neurologic damage. Hydroxyurea, an agent favoring the synthesis of fetal hemoglobin, reduces the frequency and severity of vasoocclusive crises and improves the quality of life for patients with sickle cell disease; however allogeneic HSCT is the only curative treatment for this disease at this time. Although HSCT can cure homozygous hemoglobin S, hemoglobin Sβ0, or hemoglobin SC disease, selecting appropriate candidates for transplantation is difficult. Patients with sickle cell disease may survive for decades, but some patients have a poor quality of life, with repeated hospitalizations for painful vasoocclusive crises and central nervous system (CNS) infarcts. The main indications for performing HSCT in patients with sickle cell disease are history of strokes, MRI of CNS lesions associated with impaired neuropsychologic function, failure to respond to hydroxyurea as shown by recurrent acute chest syndrome, and/or recurrent vasoocclusive crises, severe anemia, or osteonecrosis. The results of HSCT are best when performed in children with an HLA-identical sibling, with a probability of cure of 80–90%. However, the use of alternative donor transplants in this population, including matched unrelated donors and haploidentical donors, is being investigated through a number of clinical trials and may increase the number of patients eligible to undergo potentially curative HSCT. Reduced-intensity and reduced-toxicity regimens are also being explored to further decrease transplant-related morbidity and mortality, although graft failure remains an important issue in this patient population.
Immunodeficiency Disorders
HSCT is the treatment of choice for children affected by severe combined immunodeficiency (SCID), as well as for other inherited immunodeficiencies, including Wiskott-Aldrich syndrome, leukocyte adhesion deficiency (LAD), and chronic granulomatous disease (see Table 161.1 ). With an HLA-identical sibling, the probability of survival approaches 100%, with less favorable results for patients transplanted from an HLA–partially matched relative. Some children with SCID, mainly those without residual natural killer activity or maternal T-cell engraftment, may be transplanted without receiving any preparative regimen, the donor lymphoid cells usually being the only elements that engraft. Sustained donor engraftment is more difficult to achieve in children with Omenn syndrome, hemophagocytic lymphohistiocytosis , or LAD. Life-threatening opportunistic fungal and viral infections occurring before the allograft adversely affect the patient's outcome after HSCT. Because of this, patients with the most severe immunodeficiencies must be transplanted as early as possible to prevent infectious complications.
Inherited Metabolic Diseases
Inherited metabolic diseases are a broad group of diseases that result from the accumulation of substrate within tissues caused by dysfunction of the lysosome or peroxisome. The use of HSCT has been established for a variety of inherited metabolic diseases, including mucopolysaccharidosis type 1 (Hurler syndrome) and adrenoleukodystrophy (ALD). Although some of these diseases are treatable with exogenous enzyme replacement therapy, the clinical manifestations of disease tend to progress over time, especially disease in the CNS, where enzyme is unable to be reliably delivered. It is thought that undergoing HSCT results in the engraftment of microglial cells that are able to provide new enzyme to the areas where enzyme replacement therapy, if available, cannot have a substantial impact. Multiple studies have shown significantly improved outcomes for patients who are diagnosed with their underlying conditions relatively early and are able to undergo HSCT expeditiously, before significant damage from accumulated substrate that may be irreversible.