Systemic Lupus Erythematosus
Rebecca E. Sadun, Stacy P. Ardoin, Laura E. Schanberg
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multisystem inflammation and the presence of circulating autoantibodies directed against self-antigens. SLE occurs in both children and adults, disproportionately affecting females of reproductive age. Although nearly every organ may be affected, most frequently involved are the skin, joints, kidneys, blood-forming cells, blood vessels, and central nervous system. Systemic signs of inflammation such as fever and lymphadenopathy can also be seen. Compared with adults, children and adolescents with SLE have more severe disease and more widespread organ involvement.
Etiology
The pathogenesis of SLE remains largely unknown, but several factors likely influence risk and severity of disease, including genetics, hormonal milieu, and environmental exposures. A genetic predisposition to SLE is suggested by the association with specific genetic abnormalities, including congenital deficiencies of C1q, C2, and C4, as well as several polymorphisms (e.g., interferon regulatory factor 5, protein tyrosine phosphatase N22), and familial clustering of SLE or other autoimmune disease. In addition, certain human leukocyte antigen (HLA) types (including HLA-B8, HLA-DR2, and HLA-DR3) occur with increased frequency in patients with SLE. Although SLE clearly has a genetic component, its occurrence is sporadic in families and its concordance incomplete (estimated at 2–5% among dizygotic twins and 25–60% among monozygotic twins), suggesting nonmendelian genetics as well as involvement of epigenetic and environmental factors. Patients with SLE often have family members, especially mothers and sisters, with SLE or other autoimmune diseases.
Because SLE preferentially affects females, especially during their reproductive years, it is suspected that hormonal factors are important in pathogenesis. Of individuals with SLE, 90% are female, making female sex the strongest risk factor for SLE. Estrogens are likely to play a role in SLE, and both in vitro and animal model studies suggest that estrogen exposure promotes B-cell autoreactivity. Estrogen-containing oral contraceptives do not appear to induce flares in quiescent SLE, although the risk of flares may be increased in postmenopausal women receiving hormone replacement therapy.
Environmental exposures that may trigger the development of SLE remain largely unknown; certain viral infections, including Epstein-Barr virus (EBV), may play a role in susceptible individuals, and ultraviolet light exposure is known to trigger SLE disease activity. Environmental influences also may induce epigenetic modifications to DNA, increasing the risk of SLE and drug-induced lupus. In mouse models, drugs such as procainamide and hydralazine can promote lymphocyte hypomethylation, causing a lupus-like syndrome.
Epidemiology
The reported prevalence of SLE in children and adolescents (1-6/100,000) is lower than that in adults (20-70/100,000). Prevalence of SLE is highest among blacks, Asians, Hispanics, Native Americans, and Pacific Islanders for both adult and pediatric populations. SLE predominantly affects females, with reported 2-5:1 ratio before puberty, 9:1 ratio during reproductive years, and return to near-prepubertal ratios in the postmenopausal period. Childhood SLE is rare before 5 yr of age and is usually diagnosed in adolescence, with a median age at diagnosis of 11-12 yr. Up to 20% of all individuals with SLE are diagnosed before age 16 yr. Some define pediatric-onset lupus as onset of symptoms before age 16, and others as onset before age 18.
Pathology
Histologic features most suggestive of SLE include findings in the kidney and skin. Renal manifestations of SLE are classified histologically according to the criteria of the International Society of Nephrology (see Chapter 538.2 ). The finding of diffuse proliferative glomerulonephritis (class IV) significantly increases risk for renal morbidity. Renal biopsies are helpful to establish the diagnosis of SLE and to stage disease. Immune complexes are typically found with “full house” deposition of immunoglobulin and complement.
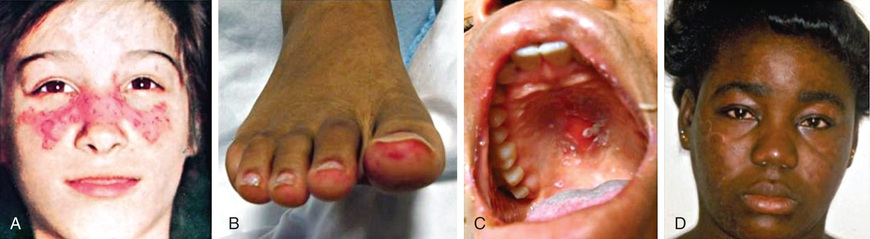
The characteristic discoid rash depicted in Figure 183.1D is characterized on biopsy by hyperkeratosis, follicular plugging, and infiltration of mononuclear cells into the dermal-epidermal junction (DEJ). The histopathology of photosensitive rashes can be nonspecific, but immunofluorescence examination of both affected and nonaffected skin may reveal deposition of immune complexes within the DEJ. This finding is called the lupus band test , which is specific for SLE.
Pathogenesis
A hallmark of SLE is the generation of autoantibodies directed against self-antigens, particularly nucleic acids. These intracellular antigens are ubiquitously expressed but are usually inaccessible and cloistered within the cell. During cell necrosis or apoptosis, the antigens are released. SLE skin cells are highly susceptible to damage from ultraviolet (UV) light, and the resulting cell death leads to release of cell contents, including nucleic antigens. Individuals with SLE may have greatly increased levels of apoptosis or significantly impaired ability to clear cell debris, causing prolonged exposure to nucleic antigens in the bloodstream and increased opportunity for recognition by immune cells, leading to B-cell stimulation and autoantibody production. Circulating autoantibodies form immune complexes and deposit in tissues, leading to local complement activation, initiation of a proinflammatory cascade, and, ultimately, tissue damage. Antibodies to double-stranded (ds) DNA can form immune complexes, deposit in glomeruli, and initiate inflammation leading to glomerulonephritis. However, many individuals with SLE have circulating antibodies to dsDNA but do not have nephritis, suggesting that autoantibodies are not the only pathway leading to end-organ damage in SLE.
Both the innate and the adaptive arms of the immune system have been implicated in the dysregulation of the immune system seen in SLE. High levels of interferon (IFN)-α production by plasmacytoid dendritic cells (DCs) promote expression of other proinflammatory cytokines and chemokines, maturation of monocytes into myeloid DCs, promotion of autoreactive B and T cells, and loss of self-tolerance. Almost 85% of patients with SLE exhibit this cytokine profile, known as the type I interferon signature . Other cytokines with increased expression in SLE include interleukin (IL)-1, IL-2, IL-6, IL-10, IL-12, IL-17, IL-21, anti–tumor necrosis factor-α, and IFN-γ, and B-lymphocyte stimulator (BLyS ), also known as B-cell activating factor (BAFF ). Both B and T cells demonstrate functional impairments in SLE. In active SLE, B-cell populations have impaired tolerance and increased autoreactivity, enhancing their ability to produce autoantibodies after exposure to self-antigen. In addition, cytokines such as BLyS/BAFF may promote abnormal B-cell number and function. T-cell abnormalities in SLE include increased numbers of memory T cells and decreased number and function of T-regulatory cells. SLE T cells display aberrant signaling and increased autoreactivity. As a result, they are resistant to attrition by normal apoptosis pathways. In addition, a neutrophil signature can be identified in 65% of adult SLE patients and has recently been recognized as a potential biomarker for active lupus nephritis.
Clinical Manifestations
Any organ system can be involved in SLE, so the potential clinical manifestations are myriad (Tables 183.1 and 183.2 ). The presentation of SLE in childhood or adolescence differs somewhat from that seen in adults. The most common presenting complaints of children with SLE include fever, fatigue, hematologic abnormalities, arthralgia, and arthritis. Arthritis is usually present in the 1st yr of diagnosis; arthritis may be painful or painless swelling, often with stiffness in the morning, and is usually a symmetric polyarthritis affecting large and small joints. Tenosynovitis is often present, but joint erosions or other radiographic changes are rare.
Table 183.1
Table 183.2
| CLINICAL FEATURE* | WITHIN 1 YR OF DIAGNOSIS (%) | ANY TIME (%) |
|---|---|---|
| Fever | 35-90 | 37-100 |
| Lymphadenopathy | 11-45 | 13-45 |
| Hepatosplenomegaly | 16-42 | 19-43 |
| Weight loss | 20-30 | 21-32 |
| Arthritis | 60-88 | 60-90 |
| Myositis | <5 | <5 |
| Any skin involvement | 60-80 | 60-90 |
| Malar rash | 22-68 | 30-80 |
| Discoid rash | <5 | <5 |
| Photosensitivity | 12-45 | 17-58 |
| Mucosal ulceration | 25-32 | 30-40 |
| Alopecia | 10-30 | 15-35 |
| Other rashes | 40-52 | 42-55 |
| Nephritis | 20-80 | 48-100 |
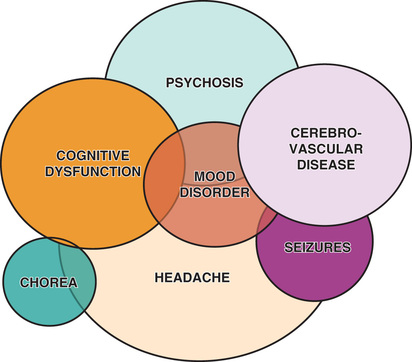
| Neuropsychiatric disease | 5-30 † | 15-95 ‡ |
| Psychosis | 5-12 | 8-18 |
| Seizures | 5-15 | 5-47 |
| Headache | 5-22 | 10-95 |
| Cognitive dysfunction | 6-15 | 12-55 |
| Acute confusional state | 5-15 | 8-35 |
| Peripheral nerve involvement | <5 | <5 |
| Cardiovascular disease | 5-30 | 25-60 |
| Pericarditis | 12-20 | 20-30 |
* Not all reports commented on all features or incidence in 1st yr.
† Had highest prevalence of central nervous system disease but did not describe incidence in 1st yr.
‡ Headache reported in 95% of patients.
From Petty RE, Laxer RM, Lindsley CB, Wedderburn LR, editors: Textbook of pediatric rheumatology, ed 7, Philadelphia, 2016, Elsevier (Table 23.5, p 291).
Renal disease in SLE is often asymptomatic, underscoring the need for careful monitoring of blood pressure and urinalyses; in adolescents, SLE can present with nephrotic syndrome and/or renal failure with the predominant symptoms being edema, fatigue, changes in urine color, and nausea/vomiting. Because SLE symptoms and findings may develop serially over several years, and not all may be present simultaneously, the diagnosis may require longitudinal follow up. SLE is often characterized by periods of flare and disease quiescence but may follow a more smoldering disease course. The neuropsychiatric complications of SLE may occur with or without apparently active SLE, posing a particularly difficult diagnostic challenge in adolescents, who are already at high risk for mood disorders (Fig. 183.2 ). Long-term complications of SLE and its therapy, including accelerated atherosclerosis and osteoporosis, become clinically evident in young to middle adulthood. SLE is a disease that evolves over time in each affected individual, and new manifestations arise even many years after diagnosis.
Diagnosis
The diagnosis of SLE requires a comprehensive clinical and laboratory assessment revealing characteristic multisystem disease and excluding other etiologies, including infection and malignancy. Presence of 4 of the 11 American College of Rheumatology (ACR) 1997 revised classification criteria for SLE simultaneously or cumulatively over time establishes the diagnosis (Table 183.3 ). Of note, although a positive antinuclear antibody (ANA) test result is not required for the diagnosis of SLE, ANA-negative lupus is extremely rare. ANA is very sensitive for SLE (95–99%), but it is not very specific (50%). The ANA may be positive many years before a diagnosis of SLE is established. However, most asymptomatic, ANA-positive patients do not have SLE or other autoimmune disease.
Antibodies against dsDNA and anti-Smith are specific for SLE (98%) but not as sensitive (40–65%). Hypocomplementemia , although common in SLE, is not one of the ACR classification criteria; however, hypocomplementemia has been added to updated criteria validated by the Systemic Lupus International Collaborating Clinics (SLICC) in 2012 (Table 183.4 ). Other differences in the SLICC criteria include the addition of nonscarring alopecia, additional cutaneous and neurologic manifestations of lupus, and a positive direct Coombs test in the absence of hemolytic anemia. The SLICC criteria have been validated in pediatric SLE and have been shown to have higher sensitivity (93% vs 77%) but lower specificity (85% vs 99%) than the ACR criteria.
Differential Diagnosis
Multiorgan disease is the hallmark of SLE. Given its wide array of potential clinical manifestations, SLE is in the differential diagnosis of many clinical scenarios, including unexplained fevers, joint pain or arthritis, rash, cytopenias, nephritis, nephrotic syndrome, pleural or pericardial effusions or other cardiopulmonary abnormalities, and new-onset psychosis, movement disorders, or seizures. For patients ultimately diagnosed with pediatric SLE, the initial differential diagnosis often includes infections (sepsis, EBV, parvovirus B19, endocarditis), malignancies (leukemia, lymphoma), poststreptococcal glomerulonephritis, other rheumatologic conditions (juvenile idiopathic arthritis, vasculitides), and drug-induced lupus.
Drug-induced lupus refers to the presence of SLE manifestations triggered by exposure to specific medications, including hydralazine, minocycline, many anticonvulsants, sulfonamides, and antiarrhythmic agents (Table 183.5 ). In individuals prone to SLE, these agents may act as a trigger for true SLE, but more often these agents provoke a reversible lupus-like syndrome. Unlike SLE, drug-induced lupus affects males and females equally. A genetic predisposition toward slow drug acetylation may increase the risk of drug-induced lupus. Circulating antihistone antibodies are often present in drug-induced SLE; these antibodies are only detected in up to 20% of individuals with SLE. Hepatitis, which is rare in SLE, is more common in drug-induced lupus. Individuals with drug-induced lupus are less likely to demonstrate antibodies to dsDNA, hypocomplementemia, and significant renal or neurologic disease. In contrast to SLE, manifestations of drug-induced lupus typically resolve after withdrawal of the offending medication; however, complete recovery may take several months to years, requiring treatment with hydroxychloroquine, NSAIDs, and/or corticosteroids.
Laboratory Findings
A positive ANA test is present in 95–99% of SLE patients. ANA has poor specificity for SLE, however, because up to 20% of healthy individuals also have a positive ANA test result, making the ANA a poor screen for SLE when used in isolation. High titers are more suggestive of underlying autoimmune disease, but ANA titers do not correlate with disease activity, so repeating ANA titers after diagnosis is not helpful. Antibodies to dsDNA are specific for SLE, and in many individuals, anti-dsDNA levels correlate with disease activity, particularly in those with significant nephritis. Anti-Smith antibody, although found specifically in patients with SLE, does not correlate with disease activity. Serum levels of total hemolytic complement (CH50 ), C3, and C4 are typically decreased in active disease and often improve with treatment. Table 183.6 lists autoantibodies found in SLE along with their clinical associations. Hypergammaglobulinemia is a common but nonspecific finding. Inflammatory markers, particularly erythrocyte sedimentation rate, are often elevated in active disease. C-reactive protein (CRP) correlates less well with disease activity; significantly elevated CRP values often reflect infection, whereas chronic mild elevation may indicate increased cardiovascular risk.
Table 183.6
Antiphospholipid antibodies , which increase clotting risk, can be found in up to 66% of children and adolescents with SLE. Antiphospholipid laboratory findings include the presence of anticardiolipin or anti–β2 -glycoprotein antibodies, prolonged phospholipid-dependent coagulation test results (partial thromboplastin time, dilute Russell viper-venom time), and circulating lupus anticoagulant (which confirms that a prolonged PPT is not corrected with mixing studies). When an arterial or venous clotting event occurs in the presence of an antiphospholipid antibody, antiphospholipid antibody syndrome is diagnosed, which can occur in the context of SLE (secondary) or independent of SLE (primary) (see Chapter 479 ).
Treatment
Treatment of SLE is tailored to the individual and is based on specific disease manifestations and medication tolerability. For all patients, sunscreen and avoidance of prolonged direct sun exposure and other UV light may help control disease and should be reinforced at every visit with the patient. Hydroxychloroquine is recommended for all individuals with SLE when tolerated. In addition to treating mild SLE manifestations such as rash and mild arthritis, hydroxychloroquine prevents SLE flares, improves lipid profiles, and may improve mortality and renal outcomes. Potential toxicities include retinal deposition and subsequent vision impairment; therefore, annual ophthalmology exams are recommended for patients taking hydroxychloroquine, including automated visual field testing as well as spectral-domain optical coherence tomography (SD-OCT). Given that risk factors for ocular toxicity include duration of use and dose, hydroxychloroquine in SLE should never be prescribed at doses >6.5 mg/kg (maximum 400 mg daily), and newer ophthalmology guidelines recommend limiting maintenance dosing to 4-5 mg/kg.
Corticosteroids are a treatment mainstay for significant manifestations of SLE and work quickly to improve acute deterioration; side effects often limit patient adherence, especially in adolescence, and potential toxicities are worrisome. It is important to limit dose and length of exposure to corticosteroids whenever possible. Potential consequences of corticosteroid therapy include growth disturbance, weight gain, striae, acne, hyperglycemia, hypertension, cataracts, avascular necrosis, and osteoporosis. The optimal dosing of corticosteroids in children and adolescents with SLE remains unknown; severe disease is often treated with high doses of intravenous (IV) methylprednisolone (e.g., 30 mg/kg/day to a maximum of 1,000 mg/day for 3 days, sometimes followed by a period of weekly pulses) and/or high doses of oral prednisone (1-2 mg/kg/day). As disease manifestations improve, corticosteroid dosages are gradually tapered over months. For most patients it is necessary to introduce a steroid-sparing immunosuppressive medication to limit cumulative steroid exposure.
Steroid-sparing immunosuppressive agents for the treatment of pediatric SLE include methotrexate, leflunomide, azathioprine, mycophenolate mofetil (MMF), tacrolimus, cyclophosphamide, rituximab, and belimumab. Methotrexate, leflunomide, and azathioprine are often used to treat persistent moderate disease, including arthritis, significant cutaneous or hematologic involvement, and pleural disease. Cyclophosphamide, MMF, and azathioprine are appropriate for the treatment of lupus nephritis, whereas MMF and rituximab are often used for significant hematologic manifestations, including severe leukopenia, hemolytic anemia, or thrombocytopenia.
Cyclophosphamide , usually administered intravenously, is reserved for the most severe, potentially life-threatening SLE manifestations, such as renal, neurologic, and cardiopulmonary disease. Although cyclophosphamide is highly effective in controlling disease, the potential toxicities are significant, including cytopenias, infection, hemorrhagic cystitis, premature gonadal failure, and increased risk of future malignancy. Attention to adequate hydration can attenuate the risk of hemorrhagic cystitis. Fortunately, young girls are at much lower risk of gonadal failure than older women, and the use of gonadotropin-releasing hormone agonists, such as leuprolide acetate, may help prevent gonadal failure.
The Childhood Arthritis Rheumatology Research Alliance (CARRA) consensus treatment plan for induction therapy of newly diagnosed proliferative lupus nephritis (class IV) is specific to the pediatric SLE population. The treatment is considered necessary for class IV lupus nephritis but also appropriate for certain patients with class III, V, or VI lupus nephritis. The CARRA treatment plans advise 6 mo of induction therapy with either cyclophosphamide (given per NIH Protocol as 500-1000 mg/m2 IV monthly) or MMF (600 mg/m2 , up to 1500 mg, twice daily), used in combination with 1 of 3 standardized glucocorticoid regimens. For patients who fail to achieve a partial response in 6 mo, it is appropriate to switch agents. For adult-weight adolescents, the cyclophosphamide dosing regimen used in the Euro-Lupus Nephritis Trial can be considered instead of the above 6 mo therapy to reduce toxicity from cyclophosphamide exposure. Per this protocol, a fixed dose of 500 mg is given every 2 wk for 3 mo; this regimen is thought to reduce adverse effects while maintaining comparable efficacy for lupus nephritis in adults, but has not been studied specifically in pediatric lupus. Oral medication adherence is very poor in pediatric SLE, which must be considered when weighing the benefits of an IV infusion vs a twice-daily oral medication such as MMF. Maintenance therapy of lupus nephritis consists of cyclophosphamide every 3 mo, or MMF, or azathioprine, typically for 36 mo after completion of induction therapy.
Clinical trial data on the use of rituximab in SLE with treatment-resistant glomerulonephritis has been largely disappointing, but results from the LUNAR study suggest a possible benefit for subpopulations of SLE patients. The U.S. Food and Drug Administration (FDA) has approved the use of belimumab , a monoclonal antibody against BLyS/BAFF, for the treatment of lupus in adults; when added to standard SLE therapy, belimumab improves multiple markers of disease severity. Other therapies being studied for treatment of lupus include rigerimod (a polypeptide corresponding to a sequence of the snRNP protein) and anifrolumab (monoclonal antibody to IFN-α receptor), all with encouraging phase II results.
Given the lifelong nature of SLE, optimal care of children and adolescents with this disease also involves preventive practices. Because of the enhanced risk of atherosclerosis in SLE, attention to cholesterol levels, smoking status, body mass index, blood pressure, and other traditional cardiovascular risk factors is warranted. Even though the Atherosclerosis Prevention in Pediatric Lupus Erythematosus (APPLE) study failed to support placing all children with SLE on a statin, post hoc analyses suggest that statins should be considered for primary prevention of atherosclerotic disease in certain clinical circumstances, particularly pubertal patients with an elevated CRP.
SLE patients with antiphospholipid antibody syndrome (antiphospholipid antibodies and a history of clot) are treated with long-term anticoagulation to prevent thrombotic events. For SLE patients who are antiphospholipid antibody positive without a history of clot, many pediatric rheumatologists prescribe aspirin (81 mg daily).
For all SLE patients, adequate intake of calcium and vitamin D is necessary to prevent future osteoporosis, particularly as vitamin D levels are lower in pediatric SLE patients compared to age-matched healthy controls. Studies suggest a link between hypovitaminosis D and SLE susceptibility, with a possible emerging role for vitamin D in immunomodulation.
Infections, particularly pneumococcal disease, frequently complicate SLE, so routine immunization is recommended, including the annual influenza vaccination. In addition, pediatric SLE patients age 6 or older should receive an additional pneumococcal 13 vaccination, followed by the pneumococcal 23 vaccination at least 2 mo later. It is important to note that many of the immunosuppressants used in SLE contraindicate live vaccines. Prompt attention to febrile episodes should include an evaluation for serious infections. Because pediatric SLE patients are at high risk for developing anxiety and depression, screening for depression is also essential. Peer support and cognitive-behavioral therapy interventions reduce pain and enhance resilience in pediatric SLE.
It should be remembered that pregnancy can worsen SLE, and obstetric complications are common. In addition, many medications used to treat SLE are teratogenic, so it is important to counsel adolescent girls about these risks and facilitate access to appropriate contraceptive options. Hydroxychloroquine is recommended throughout the pregnancy of all SLE patients, and other medications may need to be adjusted.
Complications
Within the first several years of diagnosis, the most common causes of death in SLE patients are infection and complications of glomerulonephritis and neuropsychiatric disease (Table 183.7 ). Over the long-term, the most common causes of mortality are complications of atherosclerosis and malignancy. The increased risk of premature atherosclerosis in SLE is not explained by traditional risk factors and is partly a result of the chronic immune dysregulation and inflammation associated with SLE. Increased malignancy rates may be caused by immune dysregulation as well as exposure to medications with carcinogenic potential.
Table 183.7
From Cassidy JT, Petty RE: Textbook of pediatric rheumatology, ed 6, Philadelphia, 2011, Elsevier Saunders.
Prognosis
The severity of pediatric SLE is notably worse than the typical course for adult-onset SLE. However, because of advances in the diagnosis and treatment of SLE, survival has improved dramatically over the past 50 yr. Currently, the 5 yr survival rate for pediatric SLE is approximately 95%, although the 10 yr survival rate remains 80–90%. Given their long burden of disease, children and adolescents with SLE face high risks of future morbidity and mortality from the disease and its complications, as well as medication side effects (see Table 183.7 ). Given the complex and chronic nature of SLE, it is optimal for children and adolescents with SLE to be treated by pediatric rheumatologists in a multidisciplinary clinic with access to a full complement of pediatric subspecialists.
Neonatal Lupus
Deborah M. Friedman, Jill P. Buyon, Rebecca E. Sadun, Stacy P. Ardoin, Laura E. Schanberg
Neonatal lupus erythematosus (NLE), an entity distinct from SLE, is one of the few rheumatic disorders manifesting in the neonate. NLE is not an autoimmune disease of the fetus but instead results from passively acquired autoimmunity, when maternal immunoglobulin G autoantibodies cross the placenta and enter the fetal circulation. In contrast to SLE, neonatal lupus is not characterized by ongoing immune dysregulation, although infants with neonatal lupus may be at some increased risk for development of future autoimmune disease. The vast majority of NLE cases are associated with maternal anti-Ro (also known as anti-SSA), anti-La antibodies (also known as anti-SSB), or anti-RNP (antiribonucleoprotein) autoantibodies. Despite the clear association with maternal autoantibodies, their presence alone is not sufficient to cause disease, because only 2% of offspring born to mothers with anti-Ro and anti-La antibodies develop neonatal lupus. Siblings of infants with NLE have a 15–20% chance of developing NLE. Neonatal lupus seems to be independent of maternal health since many mothers are asymptomatic and only identified to have anti-Ro/anti-La antibodies subsequent to the diagnosis of NLE. Half the infants with NLE are born to the mothers with a defined rheumatic disease, such as Sjögren syndrome or SLE.
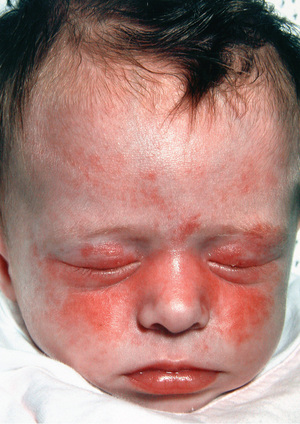
Clinical manifestations of neonatal lupus include a characteristic annular or macular rash typically affecting the face (especially the periorbital area), trunk, and scalp (Fig. 183.3 ). The rash can be present at birth but more often appears within the first 6-8 wk of life, after exposure to UV light, and typically lasts 3-4 mo. Infants may also have cytopenias and hepatitis, each occurring in approximately 25% of cases, but the most feared complication is congenital heart block .
Conduction system abnormalities range from prolongation of the PR interval to complete heart block, with development of progressive cardiomyopathy in the most severe cases. The noncardiac manifestations of NLE are usually reversible, whereas third-degree congenital heart block is permanent. Conduction system abnormalities can be detected in utero by fetal echocardiogram beginning at 16 wk gestational age. Neonatal lupus cardiac disease has a mortality rate of approximately 20%. Cardiac NLE can manifest as heart block, cardiomyopathy, valvular dysfunction, and endocardial fibroelastosis. Fetal bradycardia from heart block can lead to hydrops fetalis .
In vitro studies suggest that during cardiac development via apoptosis, Ro and La antigens may be exposed on the surface of cardiac cells in the proximity of the atrioventricular node, making the antigens accessible to maternal autoantibodies. Binding incites a local immune response, resulting in fibrosis within the conduction system as well as more extensive disease in fatal cases. In the skin, exposure to ultraviolet light results in cell damage and the subsequent exposure of Ro and La antigens, inducing a similar local inflammatory response that produces the characteristic rash.
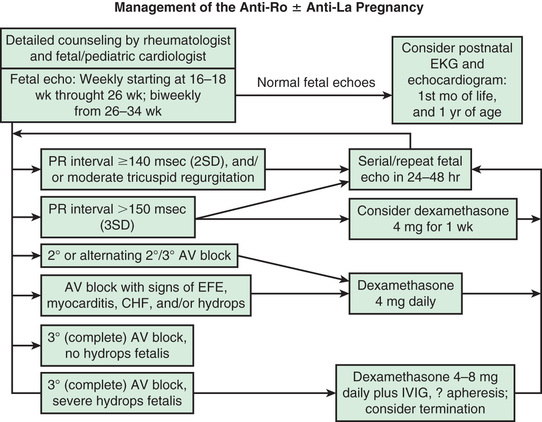
Although the scant clinical trial data have been mixed, fluorinated corticosteroids (dexamethasone or betamethasone), intravenous immune globulin (IVIG) at 1–2 g/kg maternal weight, plasmapheresis, hydroxychloroquine, and terbutaline (combined with steroids) have been used in pregnant women with anti-Ro or anti-La antibodies to prevent occurrence or progression of fetal cardiac abnormalities.
Most encouraging are retrospective cohort studies suggesting maternal treatment with hydroxychloroquine may reduce the frequency and recurrence of congenital heart block. In a case control study of women with lupus and known anti-Ro autoantibodies, maternal use of hydroxychloroquine decreased the rate of cardiac disease (odds ratio 0.28). This was confirmed in an expanded international study in which the recurrence rate of cardiac disease was 64% lower in pregnant women given hydroxychloroquine than controls (7.5% vs 21.2%). All clinical data on the use of hydroxychloroquine in pregnancy point to safety; prospective clinical studies are examining efficacy in the prevention of recurrent congenital heart block in pregnant women known to be anti-Ro and/or anti-La positive.
In utero, fluorinated corticosteroids seem to improve cases of hydrops fetalis. Furthermore, the addition of β-agonist therapy to increase fetal heart rate, used in combination with corticosteroids or IVIG, may help prevent hydrops in cases of severe fetal heart block. However, recent data from the Research Registry for Neonatal Lupus suggest that dexamethasone is not efficacious in preventing progression of isolated third-degree block, influencing the need for pacing after birth, or overall survival.
Significant conduction system abnormalities after birth are treated with cardiac pacing and occasionally IVIG and corticosteroids, whereas severe cardiomyopathy may require cardiac transplantation. If the conduction defect is not addressed, affected children are at risk for exercise intolerance, arrhythmias, and death. With cardiac pacing, children with conduction system disease in the absence of cardiomyopathy have an excellent prognosis.
Noncardiac manifestations are typically transient and are conservatively managed, often with supportive care alone. Topical corticosteroids can be used to treat moderate to severe NLE rash. Cytopenias may improve over time, but severe cases occasionally require IVIG. Supportive care is usually appropriate for hepatic and neurologic manifestation. As the neonate clears maternal autoantibodies over the 1st 6 mo of life, these inflammatory manifestations gradually resolve.
Because maternal autoantibodies gain access to the fetus through the placenta via FcRn at about 12 wk of gestation, all pregnant women with circulating anti-Ro and/or anti-La antibodies, or those with a history of offspring with NLE or congenital heart block, are monitored by a pediatric cardiologist, with screening fetal echocardiography performed weekly from 16-26 wk of gestation and then biweekly through 34 wk. The period of greatest vulnerability is usually 18-24 wk. If fetal bradycardia is found during in utero monitoring, and if fetal echocardiography confirms a conduction defect, screening for maternal anti-Ro and anti-La antibodies is warranted. Figure 183.4 presents a proposed management algorithm.
Bibliography
Brucato A. Prevention of congenital heart block in children of SSA-positive mothers. Rheumatology . 2008;47:iii35–iii37.
Clowse ME, Madger L, Witter F, et al. Hydroxychloroquine in lupus pregnancy. Arthritis Rheum . 2006;54(11):3640–3647.
Crucato A, Cimaz R, Caporali R, et al. Pregnancy outcomes in patients with autoimmune diseases and anti-Ro/SSA antibodies. Clin Rev Allergy Immunol . 2011;40(1):27–41.
Friedman DM, Kim MY, Copel JA, et al. Utility of cardiac monitoring in fetuses at risk for congenital heart block: the PR Interval and Dexamethasone Evaluation (PRIDE) prospective study. Circulation . 2008;117(4):485–493.
Friedman D, Kim MY, Copel JA, et al. Prospective evaluation of fetuses with autoimmune-associated congenital heart block followed in the PR interval and dexamethasone evaluation (PRIDE) study. Am J Cardiol . 2009;103:1102–1106.
Friedman DM, Llanos C, Izmirly PM, et al. Evaluation of fetuses in a study of intravenous immunoglobulin as preventive therapy for congenital heart block: results of a multicenter, prospective, open-label clinical trial. Arthritis Rheum . 2010;63(4):1138–1146.
Izmirly PM, Buyon JP, Saxena A. Neonatal lupus: advances in understanding pathogenesis and identifying treatments of cardiac disease. Curr Opin Rheumatol . 2012;24(5):466–472.
Izmirly PM, Costedoat-Chalumeau N, Pisoni CN, et al. Maternal use of hydroxychloroquine is associated with a reduced risk of recurrent anti-SSA/Ro–antibody-associated cardiac manifestations of neonatal lupus. Circulation . 2012;126:76–82.
Izmirly PM, Kim MY, Llanos C, et al. Evaluation of the risk of anti-SSA/Ro-SSB/La antibody-associated cardiac manifestations of neonatal lupus in fetuses of mothers with systemic lupus erythematosus exposed to hydroxychloroquine. Ann Rheum Dis . 2010;69:1827–1830.
Izmirly PM, Saxena A, Sahl SK, et al. Assessment of fluorinated steroids to avert progression and mortality in anti-SSA/Ro–associated cardiac injury limited to the fetal conduction system. Ann Rheum Dis . 2016;75(6):1161–1165.
Klein-Gitelman MS. Neonatal lupus: what we have learned and current approaches to care. Curr Rheumatol Rep . 2016;18(9):60.
Li YQ, Wang Q, Luo Y, et al. Neonatal lupus erythematosus: a review of 123 cases in China. Int J Rheum Dis . 2015;18(7):761–767.
Llanos C, Friedman DM, Saxena A, et al. Anatomical and pathological findings in hearts from fetuses and infants with cardiac manifestations of neonatal lupus. Rheumatology . 2012;51:1086–1092.
Silverman E, Jaeggi E. Non-cardiac manifestations of neonatal lupus erythematosus. Scand J Immunol . 2010;72(3):223–225.