Scleroderma and Raynaud Phenomenon
Heather A. Van Mater, C. Egla Rabinovich
Juvenile scleroderma encompasses a range of conditions unified by the presence of fibrosis of the skin. Juvenile scleroderma is divided into 2 major categories, juvenile localized scleroderma (JLS , also known as morphea ), which is largely limited to the skin, and juvenile systemic sclerosis (JSSc ), with multisystem organ involvement. Localized disease is the predominant type seen in pediatric populations (>95%), but systemic sclerosis is associated with mortality and severe multiorgan morbidity.
Etiology and Pathogenesis
The etiology of scleroderma is unknown, but the mechanism of disease appears to be a combination of a vasculopathy, autoimmunity, immune activation, and fibrosis. Triggers, including trauma, infection, and, possibly, subclinical graft-versus-host reaction from persistent maternal cells (microchimerism ), injure vascular endothelial cells, resulting in increased expression of adhesion molecules. These molecules entrap platelets and inflammatory cells, resulting in vascular changes with manifestations such as Raynaud phenomenon and pulmonary hypertension. Inflammatory cells infiltrate the area of initial vascular damage, causing further vascular damage and resulting in thickened artery walls and reduction in capillary numbers. Macrophages and other inflammatory cells then migrate into affected tissues and secrete cytokines that induce fibroblasts to reproduce and synthesize excessive amounts of collagen, resulting in fibrosis and subsequent lipoatrophy, dermal fibrosis, with loss of sweat glands and hair follicles. In late stages the entire dermis may be replaced by compact collagen fibers.
Autoimmunity is believed to be a key process in the pathogenesis of both localized and systemic scleroderma, given the high percentage of affected children with autoantibodies. Children with localized disease often have a positive antinuclear antibody (ANA) test result (42%), and 47% of this subgroup have antihistone antibodies. Children with JSSc have higher rates of ANA positivity (80.7%) and may have anti–Scl-70 antibody (34%, antitopoisomerase I). The relationship between specific autoantibodies and the various forms of scleroderma is not well understood, and all antibody test results may be negative, especially in JLS.
Classification
Localized scleroderma is distinct from systemic scleroderma and rarely progresses to systemic disease. The category of JLS includes several subtypes differentiated by both the distribution of the lesions and the depth of involvement (Tables 185.1 and 185.2 ). Up to 15% of children have a combination of 2 or more subtypes.
Epidemiology
Juvenile scleroderma is rare, with an estimated prevalence of 1 in 100,000 children. LS is much more common than SSc in children, by a 10 : 1 ratio, with linear scleroderma being the most common subtype. LS is predominantly a pediatric condition, with 65% of patients diagnosed before age 18 yr. After age 8 yr the female/male ratio for both LS and SSc is approximately 3 : 1, whereas in patients younger than 8 yr, there is no sex predilection.
Clinical Manifestations
Localized Scleroderma
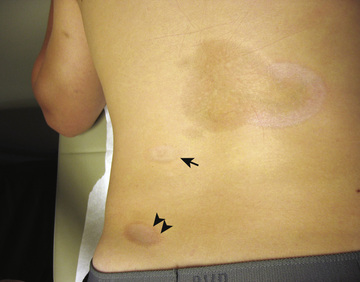
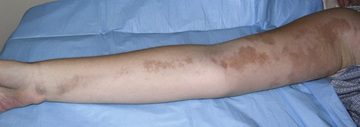
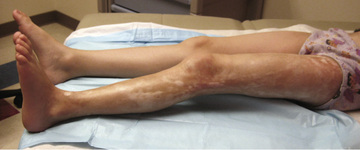
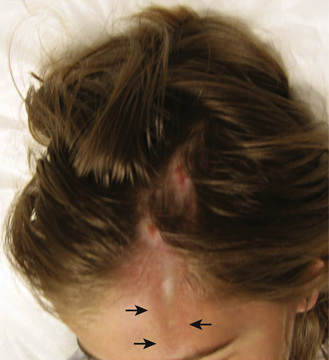
The onset of scleroderma is generally insidious, and manifestations vary according to disease subtype. The initial skin manifestations of localized disease usually include erythema or a bluish hue seen around an area of waxy induration; subtle erythema may be the only presenting sign (Fig. 185.1 ). Edema and erythema are followed by indurated, hypopigmented or hyperpigmented atrophic lesions (Fig. 185.2 ). LS varies in size from a few centimeters to the entire length of the extremity, with varying depth. Patients may present with arthralgias, synovitis, or flexion contractures (Fig. 185.3 ). Children also experience limb length discrepancies as a result of growth impairment caused by involvement of muscle and bone. Children with en coup de sabre may have symptoms unique to central nervous system (CNS) involvement, such as seizures, hemifacial atrophy, ipsilateral uveitis, and learning/behavioral changes (Fig. 185.4 ). Up to 25% of children with LS have extracutaneous manifestations, most frequently arthritis (47%) and neurologic symptoms (17%) associated with en coup de sabre.
Systemic Scleroderma
SSc also has an insidious onset with a prolonged course characterized by periods of remission and exacerbation, ending in either remission or, more often, chronic disability and death.
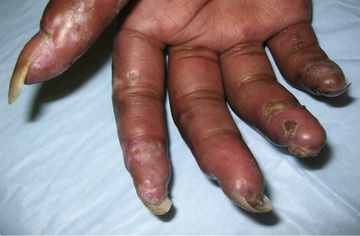
The skin manifestations of SSc include an early phase of edema that spreads proximally from the dorsum of the hands and fingers and includes the face. An eventual decrease in edema is followed by induration and fibrosis of skin, ultimately resulting in loss of subcutaneous fat, sweat glands, and hair follicles. Later, atrophic skin becomes shiny and waxy in appearance. As lesions spread proximally, flexion contractures develop at the elbows, hips, and knees associated with secondary muscle weakness and atrophy. In the face, this process results in a small oral stoma with decreased mouth aperture. Skin ulceration over pressure points, such as the elbows, may be associated with subcutaneous calcifications. Severe Raynaud phenomenon causes ulceration of the fingertips with subsequent loss of tissue pulp and tapered fingers (sclerodactyly ) (Fig. 185.5 ). Resorption of the distal tufts of the distal phalanges may occur (acroosteolysis ). Hyperpigmented postinflammatory changes surrounded by atrophic depigmentation gives a salt-and-pepper appearance to skin. Over years, remodeling of lesions sometimes results in focal improvement in skin thickening.
Pulmonary disease is the most common visceral manifestation of SSc and includes both arterial and interstitial involvement (alveolitis). Symptoms range from asymptomatic disease to exercise intolerance, dyspnea at rest, and right-sided heart failure. Pulmonary arterial hypertension is a poor prognostic sign, developing because of lung disease or independently as part of the vasculopathy. Clinical manifestations of pulmonary arterial hypertension in children appear late in the course, are subtle, and include cough and dyspnea on exertion. Pulmonary evaluation should include pulmonary function tests (PFTs) such as diffusion capacity of carbon monoxide (DLCO ), bronchoalveolar lavage (BAL), and high-resolution chest computed tomography (HRCT). PFTs reveal decreased vital capacity and decreased DLCO , whereas neutrophilia or eosinophilia on BAL suggest active alveolitis. Chest CT is much more sensitive than chest radiographs, which are often normal, showing typical basilar ground-glass abnormalities, reticular linear opacities, nodules, honeycombing, and mediastinal adenopathy.
Gastrointestinal tract disease is seen in 25% of children with SSc. Common manifestations include esophageal and intestinal dysmotility resulting in dysphagia, reflux, dyspepsia, gastroparesis, bacterial overgrowth, dilated bowel loops and pseudoobstruction, and dental caries, as well as malabsorption and failure to thrive. Renal arterial disease can cause chronic or severe episodic hypertension; unlike adult disease, renal crisis is rare. Cardiac fibrosis is associated with arrhythmias, ventricular hypertrophy, and decreased cardiac function. Mortality from JSSc is usually a result of cardiopulmonary disease. A scoring system helps identify the severity of the multiorgan involvement (Table 185.3 ).
Table 185.3
Medsger Systemic Sclerosis Severity Scale*
| ORGAN SYSTEM | 0 (NORMAL) | 1 (MILD) | 2 (MODERATE) | 3 (SEVERE) | 4 (END STAGE) |
|---|---|---|---|---|---|
| General | Wt loss <5% | Wt loss 5–10% | Wt loss 10–15% | Wt loss 15–20% | Wt loss 20%+ |
| Hct 37%+ | Hct 33–37% | Hct 29–33% | Hct 25–29% | Hct 25% | |
| Hb 12.3+ g/dL | Hb 11.0-12.2 g/dL | Hb 9.7-10.9 g/dL | Hb 8.3-9.6 g/dL | Hb <8.3 g/dL | |
| Peripheral vascular | No RP; RP not requiring vasodilators | RP requiring vasodilators | Digital pitting scars | Digital tip ulcerations | Digital gangrene |
| Skin | TSS 0 | TSS 1-14 | TSS 15-29 | TSS 30-39 | TSS 40+ |
| Joint/tendon | FTP 0-0.9 cm | FTP 1.0-1.9 cm | FTP 2.0-3.9 cm | FTP 4.0-4.9 cm | FTP 5.0+ cm |
| Muscle | Normal proximal muscle strength | Proximal weakness, mild | Proximal weakness, moderate | Proximal weakness, severe | Ambulation aids required |
| Gastrointestinal tract | Normal esophagogram; normal small bowel series | Distal esophageal hypoperistalsis; small bowel series abnormal | Antibiotics required for bacterial overgrowth | Malabsorption syndrome; episodes of pseudoobstruction | Hyperalimentation required |
| Lung |
DLCO 80%+ FVC 80%+ No fibrosis on radiograph sPAP <35 mm Hg |
DLCO 70–79% FVC 70–79% Basilar rales; fibrosis on radiograph sPAP 35-49 mm Hg |
DLCO 50–69% FVC 50–69% sPAP 50-64 mm Hg |
DLCO <50% FVC <50% sPAP 65+ mm Hg |
Oxygen required |
| Heart | ECG normal | ECG conduction defect | ECG arrhythmia | ECG arrhythmia requiring therapy | CHF |
| LVEF 50%+ | LVEF 45–49% | LVEF 40–44% | LVEF 30–40% | LVEF <30% | |
| Kidney | No history of SRC with serum creatinine <1.3 mg/dL | History of SRC with serum creatinine <1.5 mg/dL | History of SRC with serum creatinine 1.5-2.4 mg/dL | History of SRC with serum creatinine 2.5-5.0 mg/dL | History of SRC with serum creatinine >5.0 mg/dL or dialysis required |
* If 2 items are included for a severity grade, only 1 is required for the patient to be scored as having disease of that severity level.
CHF, Congestive heart failure; DLCO , diffusing capacity for carbon monoxide, % predicted; ECG, electrocardiogram; FTP, fingertip-to-palm distance in flexion; FVC, forced vital capacity, % predicted; Hb, hemoglobin; Hct, hematocrit; LVEF, left ventricular ejection fraction; RP, Raynaud phenomenon; sPAP, estimated pulmonary artery pressure by Doppler echo; SRC, scleroderma renal crisis; TSS, total skin score; Wt, weight.
Modified from Medsger TA Jr, Bombardieri S, Czirjak L, et al: Assessment of disease severity and prognosis, Clin Exp Rheumatol 21(3 Suppl 29):S51, 2003 (Table 1, p S-43).
Raynaud Phenomenon
Raynaud phenomenon (RP) is the most frequent initial symptom in pediatric systemic sclerosis, present in 70% of affected children months to years before other manifestations. RP refers to the classic triphasic sequence of blanching, cyanosis, and erythema of the digits induced by cold exposure and/or emotional stress. RP is typically independent of an underlying rheumatic disease (Raynaud disease) but can result from rheumatic diseases such as scleroderma, systemic lupus erythematosus (SLE), and mixed connective tissue disease (Fig. 185.6 ). The color changes are brought about by (1) initial arterial vasoconstriction, resulting in hypoperfusion and pallor (blanching), (2) venous stasis (cyanosis), and (3) reflex vasodilation caused by the factors released from the ischemic phase (erythema). The color change is classically reproduced by immersing the hands in iced water and reversed by warming. During the blanching phase, there is inadequate tissue perfusion in the affected area, associated with pain and paresthesias and resulting in ischemic damage only when associated with a rheumatic disease. The blanching usually affects the distal fingers but may also involve thumbs, toes, ears, and tip of the nose. The affected area is usually well demarcated and uniformly white. Digital ulcers associated with RP are indicative of underlying rheumatic disease.

Raynaud disease often begins in adolescence and is characterized by symmetric occurrence, the absence of tissue necrosis and gangrene, and the lack of manifestations of an underlying rheumatic disease. Children have normal nail fold capillaries (absence of periungual telangiectasias). RP should be distinguished from acrocyanosis and chilblains. Acrocyanosis is a vasospastic disorder resulting in cool, painless, bluish discoloration in the hands and sometimes feet despite normal tissue perfusion. It may be exacerbated by stimulant medications used to treat attention-deficit disorder. Chilblains is a condition with episodic color changes and the development of nodules related to severe cold exposure and spasm-induced vessel and tissue damage; it has been associated with SLE.
Diagnosis
The diagnosis of JLS is based on the distribution and depth of characteristic lesions. Biopsy is helpful to confirm the diagnosis. The diagnosis of JSSc requires proximal sclerosis/induration of the skin as well as the presence of 2 of 20 minor criteria (see Table 185.2 ).
Differential Diagnosis
The most important condition to differentiate from JLS is JSSc. Contractures and synovitis from juvenile arthritis can be differentiated from those caused by linear scleroderma by the absence of skin changes. Other conditions to consider include chemically induced scleroderma-like disease, diabetic cheiroarthropathy, pseudoscleroderma, and scleredema. Pseudoscleroderma comprises a group of unrelated diseases characterized by patchy or diffuse cutaneous fibrosis without the other manifestations of scleroderma. These include phenylketonuria, syndromes of premature aging, and localized idiopathic fibrosis. Scleredema is a transient, self-limited disease of both children and adults that has sudden onset after a febrile illness (especially streptococcal infections) and is characterized by patchy sclerodermatous lesions on the neck and shoulders and extending to the face, trunk, and arms.
Laboratory Findings
No laboratory studies are diagnostic of either localized or systemic scleroderma. Although the results of complete blood counts, serum chemistry analyses, and urinalysis are normal, children may have elevated erythrocyte sedimentation rate, eosinophilia, or hypergammaglobulinemia, all of which normalize with treatment. Elevations of muscle enzymes, particularly aldolase, can be seen with muscle involvement. Patients with JSSc may have anemia, leukocytosis, and eosinophilia and autoantibodies (ANA, anti–Scl-70). Imaging studies delineate the affected area and can be used to follow disease progression. MRI is useful in en coup de sabre and Parry-Romberg syndrome (facial hemiatrophy) for determination of CNS or orbital involvement. Infrared thermography uses the temperature variation between areas of active and inactive cutaneous disease to help differentiate active disease from damage. The role of ultrasound to examine lesion activity is evolving. HRCT, PFTs, echocardiography, and manometry are useful tools for diagnosing and monitoring visceral involvement in JSSc.
Treatment
Treatment for scleroderma varies according to the subtype and severity. Superficial morphea may benefit from topical corticosteroids or ultraviolet therapy. For lesions involving deeper structures, systemic therapy is recommended. A combination of methotrexate and corticosteroids is effective in treating JLS by preventing lesion extension and resulting in significant skin softening and improved range of motion of affected joints. The treatment plan for JLS includes (1) weekly subcutaneous (SC) methotrexate at 1 mg/kg (maximum dose 25 mg); (2) weekly SC methotrexate (1 mg/kg; max 25 mg) plus either 3 mo of high-dose intravenous (IV) corticosteroids (30 mg/kg; max 1,000 mg) for 3 consecutive days a month or weekly corticosteroids at the same dose for 3 mo; or (3) high-dose daily oral corticosteroids (2 mg/kg/day, max 60 mg) with a slow taper over 48 wk. Mycophenolate mofetil (MMF) is a second-line agent for recalcitrant disease. Physical and occupational therapy are important adjuncts to pharmacologic treatment. Eosinophilic fasciitis often responds well to corticosteroids and methotrexate.
Treatments for JSSc target specific disease manifestations. RP is treated with cold avoidance, and pharmacologic interventions are reserved for severe disease. Calcium channel blockers (nifedipine, 30-60 mg sustained-release form daily; amlodipine, 2.5-10 mg daily) are the most common pharmacologic interventions. Additional potential therapies for RP include losartan, prazosin, bosentan, and sildenafil. Angiotensin-converting enzyme (ACE) inhibitors (captopril, enalapril) are recommended for hypertension associated with renal disease. Methotrexate or MMF may be beneficial for skin manifestations. Cyclophosphamide and MMF are used to treat pulmonary alveolitis and prevent fibrosis. Corticosteroids should be used cautiously in SSc because of an association with renal crisis. Adults with SSc have been successfully treated with high-dose cyclophosphamide, antithymocyte globulin, and autologous stem cell transplantation.
The treatment of RP begins with avoiding cold stimuli, using hand and foot warmers, and avoiding carrying bags by their handles (impairs circulation). Nifedipine (10-20 mg 3 times daily adult dose) reduces but does not eliminate the number and severity of episodes. Side effects include headache, flushing, and hypotension. Topical nitrates may result in digital vasodilation and may reduce the severity of an episode.
Prognosis
JLS is generally self-limited, with initial inflammatory stage followed by a period of stabilization and then softening, for an average disease duration of 3-5 yr, although there are reports of active disease lasting up to 20 yr. Prolonged disease activity is associated primarily with linear and deep disease subtypes. JLS, especially linear and deep subtypes, can result in significant morbidity, disfigurement, and disability as a result of joint contractures, muscle atrophy, limb shortening, facial asymmetry, and hyper- and hypopigmentation. Death from a en coup de sabre lesion with progressive neurologic decline has been reported.
JSSc has a more variable prognosis. Although many children have a slow, insidious course, others demonstrate a rapidly progressive form with early organ failure and death. Skin manifestations reportedly soften years after disease onset. Overall, the prognosis of JSSc is better than that of the adult form, with 5-, 10-, and 15-year survival rates, respectively, in children of 89%, 80–87%, and 74–87%. The most common cause of death is heart failure caused by myocardial and pulmonary fibrosis.