Behçet Disease
Seza Özen
Behçet disease (BD) is classified as a primary variable vessel vasculitis , emphasizing the involvement of any size and type (arterial, venous) of blood vessel. BD is also recognized as an autoinflammatory disease. Originally described with recurrent oral ulcerations, uveitis, and skin abnormalities, the BD spectrum is much broader.
Epidemiology
Behçet disease has a high prevalence in countries along the Silk Road, extending from Japan to the eastern Mediterranean. It is increasingly recognized among people of European ancestry. BD has a prevalence of 5-7 per 100,000 adults, which makes it more frequent than the other vasculitides such as granulomatosis polyangiitis (Wegener disease). The increased disease recognition might have had a role in the rising prevalence of BD as well as the immigrations of the 20th century. Prevalence in children is probably not more than 10% of the adult counterparts in eastern Mediterranean countries; boys and girls are equally affected. Family history of BD is present in approximately 20% of the cases. Onset in children is 8-12 yr of age. Newborns of affected mothers have demonstrated symptoms of BD.
Etiology and Pathogenesis
Behçet disease is a polygenic autoinflammatory disorder. Genetic contribution to BD is evident through the well-known association with HLA-B5101, the familial cases, the sibling and twin recurrence rate, the specific frequency of the disease among people along the Silk Road, evidence for genetic anticipation, and genome-wide analysis. Genome wide analysis studies among Turkish and Japanese BD patients confirm the marked association with HLA-B5101. Other significant associations include interleukin (IL)-10 and IL-23R/IL-12Rβ2 genes. Other possible susceptibility loci in a Turkish cohort demonstrate associations with STAT4 (a transcription factor in a signaling pathway related to cytokines such as IL-12, type I interferons, and IL-23) and ERAP1 (an endoplasmic reticulum–expressed aminopeptidase that functions in processing of peptides onto major histocompatibility complex class I).
The autoinflammatory nature of BD is suggested by its episodic nature, the prominent innate immune system activation, the absence of identifiable autoantibodies, and the co-association with the MEFV (Mediterranean fever) gene. An infectious agent may be responsible for inducing the aberrant innate immune system attacks in the genetically predisposed host. A number of infectious agents have been implicated and include streptococci, herpes simplex virus type 1, and parvovirus B19.
Clinical Manifestations and Diagnosis
The course of BD is characterized by exacerbations and remissions. There is also marked heterogeneity in disease manifestation (Table 186.1 ).
Table 186.1
From Koné-Paut I, Shahram F, Darce-Bello M, et al, for PEDBD group: Consensus classification criteria for paediatric Behçet's disease from a prospective observational cohort: PEDBD, Ann Rheum Dis 75:958–964, 2016.
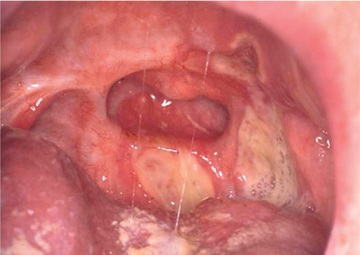
The mean age of the first symptom is between 8 and 12 yr. The most frequent initial symptom is a painful oral ulcer (Fig. 186.1 ). The oral ulcers are often recurrent, may be single or multiple, range from 2-10 mm, and may be in any location in the oral cavity. They are often very painful. The oral ulcers last 3-10 days and heal without scarring. In contrast, the genital ulcers heal with scars. Genital scars are noted in 60–85% of the patients, usually occur after puberty, and are seen on the labia, scrotum, penis, or the anal area.
Another key feature of BD that has significant morbidity is bilateral eye involvement seen in 30–60% of pediatric patients. The main symptoms of anterior uveitis are blurred vision, redness, periorbital or global pain, and photophobia. Although it is often in the form of panuveitis, anterior uveitis may be seen in females. Uveitis in general is more common in males. Vitreitis and retinal vasculitis are the most prominent features of posterior involvement. Complications of uveitis include blindness (unusual with treatment), glaucoma, and cataracts. Retinal vasculitis, retinal detachment, and retrobulbar neuritis (optic neuritis) are less common eye manifestations of BD.
The skin lesions of BD range from erythema nodosum (seen in approximately 50% of patients) to papulopustular acneiform lesions (85%), folliculitis, purpura, and ulcers. Pathergy (seen in 50%) is another skin feature associated with BD and is a pustular reaction occurring 24-48 hr after a sterile needle puncture or saline injection; it is not pathognomonic of BD.
The vasculitis of BD involves both arteries and veins, thrombosis and aneurysm formation, occlusions, or stenosis in arteries of any size. In children, deep venous thrombosis of the lower limbs is the most frequent vasculitic feature. If the hepatic vein is thrombosed, Budd-Chiari syndrome may occur. Pulmonary aneurysms are the most severe feature of pediatric BD, associated with the highest mortality. Coronary artery aneurysms may confuse BD with Kawasaki disease. Microvascular involvement may be noted in the nail bed capillaries.
Central nervous system (CNS) manifestations (approximately 10%) in children include meningoencephalitis (headache, meningismus, cerebrospinal fluid pleocytosis), encephalomyelitis, pseudotumor cerebri, dural sinus thrombosis, and organic psychiatric disorders (psychosis, depression, dementia). Dural sinus thrombosis is the most common CNS manifestation in children.
Gastrointestinal (GI) involvement (seen in 10–30%) manifests with abdominal pain, diarrhea, and intestinal ulcerations, most often in the ileocecal region. Gastrointestinal BD may be difficult to distinguish from inflammatory bowel disease. Oligoarticular arthritis/arthralgia is present in >50% of patients and can be recurrent, but is nondeforming. Other rare manifestations include orchitis, renal vasculitis, glomerulonephritis, or amyloidosis and cardiac involvement.
The International Study Group for Behçet Disease (ISG) criteria are most widely used and require the presence of oral ulcers (at least 3 times per year) along with 2 other major features, including genital ulcers, a positive pathergy test, uveitis, and the characteristic skin lesions (see Table 186.1 ). If only 1 of the criteria is present along with oral ulcerations, the term incomplete or partial Behçet disease is applied.
Classification criteria for children have been suggested by the use of an international prospective observational cohort. According to these criteria, BD is diagnosed when 3 of the following criteria are present: recurrent oral aphthosis, genital ulcers, skin involvement (necrotic folliculitis, acneiform lesions, erythema nodosum), ocular involvement, neurologic involvement, and vascular involvement (venous thrombosis, arterial thrombosis, arterial aneurysm). These criteria performed better than the ISG criteria in the pediatric cohort.
There are no specific laboratory tests. Acute-phase reactants are often mildly elevated. The diagnosis relies on the constellation of symptoms and excluding other causes.
Treatment and Prognosis
Azathioprine is highly recommended to treat inflammatory eye disease. Anti–tumor necrosis factor (TNF) treatment and interferon (IFN)-α should be considered for refractory eye disease. For oral and genital ulcers, topical treatment is recommended (sucralfate, corticosteroids). A placebo-controlled study has shown that apremilast, an oral phosphodiesterase-4 inhibitor, is effective in treating the oral ulcers of Behçet disease. Colchicine is recommended for erythema nodosum or arthritis in males and females and for genital ulcers in females. There is no evidence-based treatment for GI disease, but thalidomide, sulfasalazine, corticosteroids, azathioprine, and anti-TNF agents have been recommended. For CNS disease and vasculitis, corticosteroids, azathioprine, cyclophosphamide, and IFN-α are recommended, and in unresponsive CNS disease, anti-TNF agents. There is no consensus about the benefit of anticoagulation in the management of vein thrombosis in BD.
In patients without major organ involvement, colchicine significantly improves oral and genital ulcers, skin features, and disease activity. In pediatric patients with vascular involvement with venous thrombosis, corticosteroids and azathioprine have been used. In patients with pulmonary arterial or cardiac involvement, cyclophosphamide is typically used initially. Patients treated with anti-TNF drugs have had persistent responses in 90%, 89%, 100%, and 91% of patients with resistant mucocutaneous, ocular, GI, and CNS involvement, respectively.
Mortality in children with BD is low except for the pulmonary aneurysms. However, BD is a chronic disease associated with significant morbidity. Early diagnosis and effective treatment improve the outcome of BD.