Motility Disorders and Hirschsprung Disease
Chronic Intestinal Pseudoobstruction
Asim Maqbool, Kristin N. Fiorino, Chris A. Liacouras
Chronic intestinal pseudoobstruction (CIPO) comprises a group of primary and secondary disorders characterized as a motility disorder with the dominant defect of impaired peristalsis; symptoms are consistent with intestinal obstruction in the absence of mechanical obstruction (Table 358.1 ). The natural history of primary pseudoobstruction is that of a progressive disorder, although there are occasional cases of secondary pseudoobstruction caused by conditions that can transiently or permanently alter bowel motility. The most common cause of acute pseudoobstruction is Ogilvie syndrome (acute pseudoobstruction of the colon). Pseudoobstruction represents a wide spectrum of pathologic disorders from abnormal myoelectric activity to abnormalities of the nerves (intestinal neuropathy) or musculature (intestinal myopathy) of the gut. The organs involved can include the entire gastrointestinal tract or be limited to certain components, although almost always include the small bowel. The distinctive pathologic abnormalities are considered together because of their clinical similarities. For these reasons, CIPO may be thought of more as a clinical syndrome at times.
Table 358.1
Causes of Secondary Chronic Intestinal Pseudoobstruction in Children
From Bitton S, Markowitz JF: Ulcerative colitis in children and adolescents. In Wyllie R, Hyams JS, Kay M, editors: Pediatric gastrointestinal and liver disease, 5th ed, Elsevier, 2016, Philadelphia, Box. 44.3, p. 548.
Most congenital forms of primary pseudoobstruction occur sporadically, although autosomal dominant (SOX10), autosomal recessive (RAD2I, SGOL1, TYMP, POLG), X-linked (FLNA, L1CAM), and familial patterns of inheritance have been identified. Patients with autosomal dominant forms of pseudoobstruction have variable expressions of the disease. Patients with mutations in TYMP and POLG genes present with mitochondrial neurogastrointestinal encephalomyopathy syndrome (MNGIE); MELAS syndrome is another mitochondrial disorder associated with CIPO. MNGIE is characterized by intestinal dysmotility, abdominal pain and distention, emesis, cachexia, ptosis, leukoencephalopathy, peripheral neuropathy (paresthesia, pain), and myopathy. Sixty percent have symptoms (often subtle) before age 20 yr (see Chapter 358.2 ). Acquired pseudoobstruction can follow episodes of acute gastroenteritis, presumably resulting in injury to the myenteric plexus.
In congenital pseudoobstruction, abnormalities of the muscle or nerves can be demonstrated in most cases. In myopathies, the smooth muscle is involved, in which the outer longitudinal muscle layer is replaced by fibrous material. These manifestations of visceral myopathies may be primary or secondary phenomenon. The enteric nervous system is usually altered in neuropathies and may involve disorganized ganglia, hypoganglionosis, or hyperganglionosis. Abnormalities in the interstitial cells of Cajal, the intestinal pacemaker, are classified as mesenchymopathies. In others, mitochondrial defects have been identified.
Clinical Manifestations
More than half the children with congenital pseudoobstruction experience symptoms in the first few mo of life (Table 358.2 ). Two-thirds of the infants presenting in the first few days of life are born prematurely, and approximately 40% have malrotation of the intestine. In 75% of all affected children, symptoms occur in the first year of life, while the remainder are usually symptomatic within the next several years. Females present with CIPO more than males do during the first year of life, with equal sex distribution in older children. The most common symptoms are abdominal distention (85–95% of patients) and vomiting (55–90%). Constipation, growth failure, and abdominal pain occur in approximately 60% of patients and diarrhea in 25–30%. The symptoms wax and wane in most patients; poor nutrition, psychologic stress, and intercurrent illness tend to exacerbate symptoms. Urinary tract and bladder involvement occurs in 80% of children with myopathic pseudoobstruction and in 20% of those with neuropathic disease. Symptoms can manifest as recurrent urinary tract infection, megacystis, or obstructive symptoms. Megacystis-microcolon–intestinal hypoperistalsis syndrome is a prenatal or neonatal manifestation of CIPO.
Table 358.2
From Di Nardo G, Di Lorenzo C, Lauro A, et al: Chronic intestinal pseudo-obstruction in children and adults: diagnosis and therapeutic options, Neurogastroenterol Motil 29:e12945, 2017, Table 2.
Diagnosis
The diagnosis of pseudoobstruction is based on the presence of compatible symptoms in the absence of mechanical obstruction (Fig. 358.1 ). Plain abdominal radiographs demonstrate air-fluid levels in the intestine. Neonates with evidence of obstruction at birth may have a microcolon. Contrast studies demonstrate slow passage of barium; water-soluble agents should be considered. Esophageal motility is abnormal in about half the patients. Antroduodenal (small intestinal) motility and gastric emptying studies have abnormal results if the upper gut is involved (Table 358.3 ). The clinical manifestations depend in large part to the areas of the gastrointestinal tract that are involved, with milder forms more common in older children. Although counterintuitive, older children with CIPO may present with both abdominal distention and diarrhea, related to small bowel bacterial overgrowth because of altered motility. Other presentations may include constipation and bilious emesis, as well as failure to thrive, as a consequence of decreased enteral feeding tolerance.

Table 358.3
| GI SEGMENT | FINDINGS* |
|---|---|
| Esophageal motility | Abnormalities in approximately half of CIPO, although in some series up to 85% demonstrate abnormalities |
| Decreased LES pressure | |
| Failure of LES relaxation | |
| Esophageal body: low-amplitude waves, poor propagation, tertiary waves, retrograde peristalsis, occasionally aperistalsis | |
| Gastric emptying | May be delayed |
| EGG | Tachygastria or bradygastria may be seen |
| ADM | Postprandial antral hypomotility is seen and correlates with delayed gastric emptying |
| Myopathic subtype: low-amplitude contractions, <10-20 mm Hg | |
| Neuropathic subtype: contractions are uncoordinated, disorganized | |
| Absence of fed response | |
| Fasting MMC is absent, or MMC is abnormally propagated | |
| Colonic | Absence of gastrocolic reflex because there is no increased motility in response to a meal |
| ARM | Normal rectoanal inhibitory reflex |
* Findings can vary according to the segment(s) of the GI tract that are involved.
ADM, antroduodenal manometry; ARM, anorectal manometry; CIPO, chronic intestinal pseudoobstruction; EGG, electrogastrography; GI, gastrointestinal; LES, lower esophageal sphincter; MMC, migrating motor complex.
From Steffen R: Gastrointestinal motility. In Wyllie R, Hyams JS, Kay M, editors: Pediatric gastrointestinal and liver disease, ed 3, Philadelphia, 2006, WB Saunders, p. 66.
The initial focus is to rule out anatomic obstruction and to assess for bladder involvement, because that is a frequent and significant extraintestinal manifestation of concern. Manometric evidence of a normal migrating motor complex and postprandial activity should redirect the diagnostic evaluation. CIPO due to an intestinal myopathy may demonstrate manometry evidence of low-amplitude contractions, whereas CIPO due to enteric neuropathy demonstrates normal amplitude but poorly organized contractions (nonperistaltic or tonic). Anorectal motility is normal and differentiates pseudoobstruction from Hirschsprung disease. Full-thickness intestinal biopsy might show involvement of the muscle layers or abnormalities of the intrinsic intestinal nervous system.
The differential diagnosis is broad and includes such etiologies as Hirschsprung disease, mitochondrial neurogastrointestinal encephalomyopathy, mechanical obstruction, psychogenic constipation, neurogenic bladder, and superior mesenteric artery syndrome. Secondary causes of ileus or pseudoobstruction that should be considered include medication side effects, infectious etiologies, metabolic disturbances, immunologic disorders, oncologic processes, vasculitides, neuropathies, and myopathies (see Table 358.1 ). Examples include use of opiates hypokalemia, hypothyroidism, hypokalemia, diabetic neuropathy, porphyria, amyloidosis, Chagas disease, scleroderma, hereditary angioedema, mitochondrial disorders, and radiation, and these must be excluded. Other causes of abdominal distention such as small bowel bacterial overgrowth and aerophagia may present similarly and should be considered. Small bowel bacterial overgrowth is a complication of CIPO.
Treatment
Nutritional support is the mainstay of treatment for pseudoobstruction. Thirty to 50% of patients require partial or complete parenteral nutrition. Some patients can be treated with intermittent enteral supplementation, whereas others can maintain themselves on selective oral diets. Prokinetic drugs are generally used, although studies have not shown definitive evidence of their efficacy. Isolated gastroparesis can follow episodes of viral gastroenteritis and spontaneously resolves, usually in 6-24 mo. Erythromycin, a motilin receptor agonist, and cisapride, a serotonin 5-HT4 receptor agonist, may enhance gastric emptying and proximal small bowel motility and may be useful in this select group of patients. Metoclopramide, a prokinetic and antinausea agent, is effective in gastroparesis, although side effects, such as tardive dyskinesia, limit its use. Domperidone, an antidopaminergic agent, is a prokinetic agent that can be considered. Pain management is difficult and requires a multidisciplinary approach.
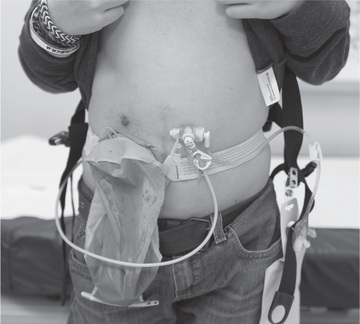
Symptomatic small bowel bacterial overgrowth is usually treated with rotated nonabsorbable oral antibiotics and/or probiotics. Bacterial overgrowth can be associated with steatorrhea and malabsorption. Octreotide, a long-acting somatostatin analog, has been used in low doses to treat small bowel bacterial overgrowth. Patients with acid peptic symptoms are generally treated with acid suppression. Many patients with CIPO benefit from a gastrostomy, and some benefit from decompressive enterostomies (Fig. 358.2 ). Colectomy with ileorectal anastomosis is beneficial if the large bowel is the primary site of the motility abnormality. Bowel transplantation may benefit selected patients with CIPO. The prognosis is better for patients without urinary tract involvement and for those with neuropathic etiologies over myopathic disorders.
Bibliography
Chumpitazi B, Nurko S. Pediatric gastrointestinal motility disorders: challenges and a clinical update. Nat Rev Gastroenterol Hepatol . 2008;4(2):140–148.
Cucchiara S, Borrelli O, Salvia G, et al. A normal gastrointestinal motility excludes chronic intestinal pseudo-obstruction in children. Dig Dis Sci . 2000;45:258–264.
Di Nardo G, Blandizzi C, Volta U, et al. Review article: molecular, pathological and therapeutic features of human enteric neuropathies. Aliment Pharmacol Ther . 2008;28:25–42.
Di Nardo G, Di Lorenzo C, Lauro A, et al. Chronic intestinal pseudo-obstruction in children and adults: diagnosis and therapeutic options. Neurogastroenterol Motil . 2017;29:e12945.
Faure C. Chronic intestinal Pseudo-obstruction syndrome. Walker WA, Goulet O. Pediatric gastrointestinal disease. Pathohysiology, Diagnosis, Management . ed 4. 2004.
Gabbard S, Lacy B. Chronic intestinal pseudo-obstruction. Nutr Clin Pract . 2013;28(3):307–316.
Haas S, Bindl L, Fischer HP. Autoimmune enteric leiomyositis: a rare cause of chronic intestinal pseudo-obstruction with specific morphological features. Human Path . 2005;36:576–580.
Kleinman R, et al. Chronic intestinal pseudo-obstruction . BC Decker: Hamilton, Ontario; 2004:1044–1054.
Knowles CH, Lindberg G, Panza E, De Giorgio R. New perspectives in the diagnosis and management of enteric neuropathies. Nat Rev Gastroenterol Hepatol . 2013;10:206–218.
Lapointe SP, Rivet C, Goulet O, et al. Urological manifestations associated with chronic intestinal pseudo-obstructions in children. J Urol . 2002;168:1768–1770.
Mousa H, Hyman PE, Cocjin J, et al. Long-term outcome of congenital intestinal pseudo-obstruction. Dig Dis Sci . 2002;47:2298–2305.
Nurko S. Motility disorders in children. Pediatr Clin N Am . 2017;64:593–612.
Rahman S. Gastrointestinal and hepatic manifestations of mitochondrial disorders. J Inherit Metab Dis . 2013;36:659–673.
Venkatasubramani N, Sood M. Motility disorders of the gastrointestinal tract. Indian J Pediatr . 2006;73:927–930.
Mitochondrial Neurogastrointestinal Encephalomyopathy
Asim Maqbool, Chris A. Liacouras
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a multisystem autosomal recessive disease that initially presents with severe gastrointestinal disturbances; the neurologic manifestations usually occur later in the illness and may initially be subtle or asymptomatic.
MNGIE is caused by a mutation in the nuclear DNA TYMP gene encoding thymidine phosphorylase that results in abnormalities in intergenomic communication with resulting instability of mitochondrial DNA (some patients have mutations on POLG1 ). There are at least 50 individual mutations with a poor genotype-phenotype correlation and varying manifestations within each family. Consanguinity is present in 30% of families.
MNGIE affects both males and females and is usually diagnosed in the 2nd and 3rd decade (average age: 18 yr; range: 5 mo-35 yr). Onset is usually around age 12 yr, but there is often a 5- to 10-yr delay in the diagnosis.
MNGIE initially presents with gastrointestinal symptoms. Severe intestinal dysmotility and gastroparesis are associated with early satiety, postprandial emesis, episodic pseudoobstruction, diarrhea, constipation, and abdominal pain and cramping, which leads to significant cachexia. Because of the age of onset, emesis, early satiety, and cachexia patients are often misdiagnosed with an eating disorder.
Most often, following the onset of gastrointestinal manifestations, ptosis, progressive external ophthalmoplegia, hearing loss, myopathy, and peripheral neuropathy may develop. The neuropathy is either demyelinating or a mixed axonal demyelinating type and manifests as weakness, decreased or absent deep tendon reflexes, and paresthesias. Leukoencephalopathy is initially asymptomatic and noted on MRI as patchy lesions predominantly in the cortex but also in the basal ganglia and brainstem. Eventually the central nervous system lesions become diffuse and confluent. A small number of patients develop cognitive impairment or dementia.
The diagnosis is suggested by the constellation of gastrointestinal and neurologic symptoms, lactic acidosis, ragged red fibers, and cytochrome C oxidase–deficient fibers seen in most patients on muscle biopsy. Reduced activity of thymidine phosphorylase enzyme and elevated plasma levels of thymidine and deoxyuridine are often diagnostic; genetic testing for the mutation or other genes (POLG1) is recommended.
Treatment is focused on providing sufficient nutritional support and avoidance of infectious complications and of nutritional deficiencies. Domperidone has been used for nausea and emesis, antibiotics for small bowel bacterial overgrowth, amitriptyline or gabapentin for neuropathic pain, and parenteral alimentation for nutritional support. Opiates and any medications that affect intestinal motility or mitochondrial function must be avoided. Stem cell transplantation has been successful in a small number of patients.
Overall the prognosis is poor, with few surviving into the 4th or 5th decade.
Bibliography
Bariş Z, Eminoğlu T, Dalgiç B, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): case report with a new mutation. Eur J Pediatr . 2010;169:1375–1378.
D'Angelo R, Rinaldi R, Carelli V, et al. ITA-MNGIE: an Italian regional and national survey for mitochondrial neuro-gastro-intestinal encephalomyopathy. Neurol Sci . 2016;37:1149–1151.
Garone C, Tadesse S, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain . 2011;134:3326–3332.
Hirano M. Mitochondrial neurogastrointestinal encephalopathy disease. Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews® [internet] . University of Washington, Seattle: Seattle (WA); 2005:1993–2018 [Available from] https://www.ncbi.nlm.nih.gov/books/NBK1179/ [Apr 22; [Updated 2016 Jan 14].
Sivadasan A, Muthusamy K, Kumar A, et al. Pearls and Oy-sters: mitochondrial neurogastrointestinal encephalomyopathy. Neurology . 2016;86(14):e147–e150.
Tang S, Dimberg EL, Milone M, Wong LJC. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol . 2012;259:862–868.
Encopresis and Functional Constipation
Asim Maqbool, Chris A. Liacouras
Constipation is defined as a delay or difficulty in defecation present for >1 month and significant enough to cause distress to the patient. Another approach to the definition is the Rome criteria, outlined in Tables 358.4 and 358.5 . Functional constipation, also known as idiopathic constipation or fecal withholding, can usually be differentiated from constipation secondary to organic causes based on a history and physical examination. Unlike anorectal malformations and Hirschsprung disease, functional constipation typically starts after the neonatal period. Usually, there is an intentional or subconscious withholding of stool. An acute episode usually precedes the chronic course. This acute event could include a social stressor such as initiation of toilet training, birth of a sibling, starting daycare, or abuse. The acute episode may be a dietary change from human milk to cow's milk, secondary to the change in the protein and carbohydrate ratio or an allergy to cow's milk. Although iron has been suspected of causing issues with cow's milk–related constipation, this has not been consistently demonstrated or substantiated. The stool becomes firm, smaller, and difficult to pass, resulting in anal irritation and often an anal fissure. In toddlers, coercive or inappropriately early toilet training is a factor that can initiate a pattern of stool retention. In older children, retentive constipation can develop after entering a situation that makes stooling inconvenient such as school. Because the passage of bowel movements is painful, voluntary withholding of feces to avoid the painful stimulus develops.
Table 358.4
Rome IV Diagnostic Criteria for Defecatory Disorders in Neonates and Toddlers
| FGID | AGE RANGE | CRITERIA REQUIREMENTS | CRITERIA ELEMENTS |
|---|---|---|---|
| Functional constipation | All pediatric age groups |
Must include 1 month of ≥2 of the following in infants up to 4 months of age: In toilet trained children, the following additional criteria may be used: |
FGID, Functional gastrointestinal disorders.
Modified from Benninga MA, Faure C, Hyman PE, et al. Childhood functional gastrointestinal disorders: neonate/toddler, Gastroenterology 150:1443–1455, 2016.
Table 358.5
Rome IV Diagnostic Criteria for Defecatory Disorders in Children and Adolescents
Modified from Hyams JS, Di Lorenzo C, Saps M, et al. Childhood functional gastrointestinal disorders: child/adolescent, Gastroenterology 150:1456–1468, 2016.
Clinical Manifestations
When children have the urge to defecate, typical behaviors include contracting the gluteal muscles by stiffening the legs while lying down, holding onto furniture while standing, or squatting quietly in corners, waiting for the call to stool to pass. The urge to defecate passes as the rectum accommodates to its contents. A vicious cycle of retention develops, as increasingly larger volumes of stool need to be expelled. Caregivers may misinterpret these activities as straining, but it is withholding behavior. There is often a history of blood in the stool noted with the passage of a large bowel movement. Findings suggestive of underlying pathology include failure to thrive, weight loss, abdominal pain, vomiting, or persistent anal fissure or fistula.
In functional constipation, daytime encopresis is common. Encopresis is defined as voluntary or involuntary passage of feces into inappropriate places at least once a mo for 3 consecutive months once a chronologic or developmental age of 4 yr has been reached. Encopresis is not diagnosed when the behavior is exclusively the result of the direct effects of a substance (e.g., laxatives) or a general medical condition (except through a mechanism involving constipation). Subtypes include retentive encopresis (with constipation and overflow incontinence), representing 65–95% of cases, and nonretentive encopresis (without constipation and overflow incontinence). Nonretentive fecal incontinence is defined as no evidence of fecal retention (impaction), ≥1 episodes per week in the previous 1 mo, or defecation in places inappropriate to the social context in a child who has been previously toilet trained and without evidence of anatomic, inflammatory, metabolic, endocrine, or neoplastic process that could explain the symptoms. Encopresis can persist from infancy onward (primary) or can appear after successful toilet training (secondary). The updated Rome criteria (IV) differentiate between infants/toddlers and older children who have been toilet trained versus not toilet trained, for practical assessment purposes.
Diagnosis
The physical examination often demonstrates a large volume of stool palpated in the suprapubic area; rectal examination demonstrates a dilated rectal vault filled with guaiac-negative stool. Children with encopresis often present with reports of underwear soiling, and many parents initially presume that diarrhea, rather than constipation, is the cause. In retentive encopresis , associated complaints of difficulty with defecation, abdominal or rectal pain, impaired appetite with poor growth, and urinary (day and/or night) incontinence are common. Children often have large bowel movements that obstruct the toilet. There may also be retentive posturing or recurrent urinary tract infections. Nonretentive encopresis is more likely to occur as a solitary symptom and have an associated primary underlying psychological etiology. Children with encopresis can present with poor school performance and attendance that is triggered by the scorn and derision from schoolmates because of the child's offensive odor.
The location of the anus relative to perineal anatomic landmarks by sex also needs to be considered. This is expressed as the anogenital index , and it can be calculated when necessary. This is determined by the distance in centimeters from the vagina or scrotum to the anus, divided by the distance from the vagina or scrotum to the coccyx. The normal anogenital index in females is 0.39 ± 0.09, whereas 0.56 ± 0.2 is normal for males. The presence of a hair tuft over the spine or spinal dimple, or failure to elicit a cremasteric reflex or anal wink suggests spinal pathology. A tethered cord is suggested by decreased or absent lower leg reflexes. Spinal cord lesions can occur with overlying skin anomalies. Urinary tract symptoms include recurrent urinary tract infection and enuresis. Children with no evidence of abnormalities on physical examination rarely require radiologic evaluation.
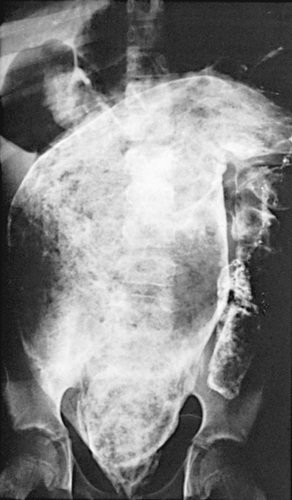
In refractory patients (intractable constipation), specialized testing should be considered to rule out conditions such as hypothyroidism, hypocalcemia, lead toxicity, celiac disease, and disorders of neuromuscular gastrointestinal pathology (Table 358.6 ). Colonic transit studies using radiopaque markers or scintigraphy techniques may be useful. Selected children can benefit from MRI of the spine to identify an intraspinal process, motility studies to identify underlying myopathic or neuropathic bowel abnormalities, or a contrast enema to identify structural abnormalities. In patients with severe functional constipation, water-soluble contrast enema reveals the presence of a mega rectosigmoid (Fig. 358.3 ). Anorectal motility studies can demonstrate a pattern of paradoxical contraction of the external anal sphincter during defecation, which can be treated by behavior modification and biofeedback. Colonic motility can guide therapy in refractory cases, demonstrating segmental problems that might require surgical intervention.
Table 358.6
London Classification of Gastrointestinal Neuromuscular Pathology
|
1.1.1 Aganglionosis* 1.2 Decreased numbers of neurons 1.3 Increased numbers of neurons 1.3.1 Ganglioneuromatosis † 1.3.2 IND, type B ‡ 1.4 Degenerative neuropathy § 1.5.1 Lymphocytic ganglionitis ¶ 1.5.2 Eosinophilic ganglionitis 1.6 Abnormal content in neurons 1.6.1 Intraneuronal nuclear inclusions 1.7 Abnormal neurochemical coding** 1.8 Relative immaturity of neurons 1.9.1 Increased numbers of enteric glia 2.1 Muscularis propria malformations †† 2.2.1 Degenerative leiomyopathy/ ‡‡ 2.2.2 Inflammatory leiomyopathy 2.2.2.1 Lymphocytic leiomyositis 2.2.2.2 Eosinophilic leiomyositis 2.3 Muscle hyperplasia/hypertrophy 2.3.1 Muscularis mucosae hyperplasia 2.4 Abnormal content in myocytes 2.4.1 Filament protein abnormalities 2.4.1.1 Alpha-actin myopathy §§ 2.4.2.3 Megamitochondria ¶¶ 2.5 Abnormal supportive tissue 2.5.1 Atrophic desmosis*** 3. ICC abnormalities (enteric mesenchymopathy) 3.1 Abnormal ICC networks ††† |
* Can include rare cases of non-Hirschsprung disease severe hypoplastic hypoganglionosis with long interganglionic intervals (zonal aganglionosis).
† Although neurons have not been formally quantified, gross increases of disorganized neurons are evident.
‡ Can include retarded neuronal maturation.
§ May occur with or without neuronal loss but is best regarded as a separate entity.
¶ May occur with neuronal degeneration and/or loss; lymphocytic epithelioganglionitis is a variant.
** Includes neurotransmitter loss (e.g., reduced or absent expression) or loss of a neurochemically defined functional subset of nerves (see text).
†† Includes absence, fusion, or additional muscle coats.
‡‡ Hollow visceral myopathy may be diagnosed in familial cases with other characteristic phenotypic features; myopathy with autophagic activity and pink blush myopathy with nuclear crowding are rare variants in which degenerative findings are less overt.
§§ Smooth muscle alpha-actin deficiency is best described, although deficiencies of other proteins related to the contractile apparatus of myocytes have been reported.
¶¶ Mitochondrial neurogastrointestinal encephalomyopathy causes a degenerative appearance predominantly in the longitudinal muscle.
*** Absent connective tissue scaffold has been almost exclusively described in the colon.
††† Generally reduced or absent ICC, although abnormal morphology also reported.
ICC, interstitial cells of Cajal; IND, intestinal neuronal dysplasia.
From Knowles CH, De Giorgio R, Kapur RP, et al: The London classification of gastrointestinal neuromuscular pathology: report on behalf of the Gastro 2009 International Working Group, Gut 59:882–887, 2010, Table 1, p. 883.
Complications of retentive encopresis include day and night urinary incontinence, urinary retention, urinary tract infection, megacystis, and rarely toxic megacolon.
Treatment
Therapy for functional constipation and encopresis includes patient education, relief of impaction, and softening of the stool. Caregivers must understand that soiling associated with overflow incontinence is associated with loss of normal sensation and not a willful act. There needs to be a focus on adherence with regular postprandial toilet sitting and adoption of a balanced diet. In addition, caregivers should be instructed not to respond to soiling with retaliatory or punitive measures, because children are likely to become angry, ashamed, and resistant to intervention. From the outset, parents should be actively encouraged to reward the child for adherence to a healthy bowel regimen and to avoid power struggles.
If an impaction is present on the initial physical examination, an enema is usually required to clear the impaction while stool softeners are started as maintenance medications. Typical regimens include the use of polyethylene glycol preparations, lactulose, or mineral oil (Tables 358.7 and 358.8 ). Prolonged use of stimulants such as senna or bisacodyl should be avoided.
Table 358.7
Suggested Medications and Dosages for Disimpaction
| MEDICATION | AGE | DOSAGE |
|---|---|---|
| RAPID RECTAL DISIMPACTION | ||
| Glycerin suppositories | Infants and toddlers | |
| Phosphate enema | <1 yr | 60 mL |
| >1 yr | 6 mL/kg body weight, up to 135 mL twice | |
| SLOW ORAL DISIMPACTION IN OLDER CHILDREN | ||
| Over 2-3 Days | ||
| Polyethylene glycol with electrolytes | 25 mL/kg body weight/hr, up to 1,000 mL/hr until clear fluid comes from the anus | |
| Over 5-7 Days | ||
| Polyethylene without electrolytes | 1.5 g/kg body weight/day for 3 days | |
| Milk of magnesia | 2 mL/kg body weight twice/day for 7 days | |
| Mineral oil | 3 mL/kg body weight twice/day for 7 days | |
| Lactulose or sorbitol | 2 mL/kg body weight twice/day for 7 days | |
From Loening-Baucke V: Functional constipation with encopresis. In Wyllie R, Hyams JS, Kay M, editors: Pediatric gastrointestinal and liver disease , ed 3, Philadelphia, 2006, WB Saunders, p. 183.
Table 358.8
Suggested Medications and Dosages for Maintenance Therapy of Constipation
| MEDICATION | AGE | DOSE |
|---|---|---|
| TYPICAL DOSES FOR LONG-TERM TREATMENT (YEARS) | ||
| Milk of magnesia | >1 mo | 1-3 mL/kg body weight/day, divided into 1-2 doses |
| Mineral oil | >12 mo | 1-3 mL/kg body weight/day, divided into 1-2 doses |
| Lactulose or sorbitol | >1 mo | 1-3 mL/kg body weight/day, divided into 1-2 doses |
| Polyethylene glycol 3350 (MiraLAX) | >1 yr | 0.7 g/kg body weight/day (max 17.5 g/day) |
| FOR SHORT-TERM TREATMENT (MONTHS) | ||
| Senna (Senokot) syrup, tablets | 1-5 yr | 5 mL (1 tablet) with breakfast, max 15 mL daily |
| 5-15 yr | 2 tablets with breakfast, maximum 3 tablets daily | |
| Glycerin enemas | >10 yr | 20-30 mL/day ( glycerin and
glycerin and  normal saline)
normal saline) |
| Bisacodyl suppositories | >10 yr | 10 mg daily |
From Loening-Baucke V: Functional constipation with encopresis. In Wyllie R, Hyams JS, Kay M, editors: Pediatric gastrointestinal and liver disease , ed 3, Philadelphia, 2006, WB Saunders, p. 185.
Compliance can wane, and failure of this standard treatment approach sometimes requires more intensive intervention. In cases where behavioral or psychiatric problems are evident, involvement of a psychologist or behavioral management (e.g., behavior programs and/or biofeedback) is recommended. Maintenance therapy is generally continued until a regular bowel pattern has been established and the association of pain with the passage of stool is abolished.
For children with chronic diarrhea and/or irritable bowel syndrome where stress and anxiety play a major role, stress reduction and learning effective coping strategies can play an important role in responding to the encopresis. Relaxation training, stress inoculation, assertiveness training, and/or general stress management procedures can be helpful, and the participation of behavioral health specialists is valuable.
Neurostimulation (transcutaneous or sacral implantation) and pelvic physiotherapy are novel approaches used in patients with medication refractory constipation. Children with spinal problems can be successfully managed with low volumes of fluid through a cecostomy or sigmoid tube.
Bibliography
Auth MKH, Vora R, Farrelly P, et al. Childhood constipation. BMJ . 2012;345:38–43.
Bar-Maor JA, Eitan A. Determination of the normal position of the anus (with reference to idiopathic constipation). J Pediatr Gastroenterol Nutr . 1987;6:559–561.
Bekkali NLH, van den Berg MM, Dijkgraaf MGW, et al. Rectal fecal impaction treatment in childhood constipation: enemas versus high doses oral PEG. Pediatrics . 2009;124:e1108–e1115.
Benninga MA, Faure C, Hyman PE, et al. Childhood functional gastrointestinal disorders: Neonate/toddler. Gastroenterology . 2016;150:1443–1455.
Boners ME, Tabbers MM, Benninga MA. Functional nonretentive fecal incontinence in children. J Pediatr Gastroenterol Nutr . 2007;44:5–13.
Burgers R, Levin AD, Di Lorenzo C, et al. Functional defecation disorders in children: comparing the rome II with the rome III criteria. J Pediatr . 2012;161:615–620.
Camilleri M, Kerstens R, Rykx A, et al. A placebo-controlled trial of prucalopride for severe chronic constipation. N Engl J Med . 2008;358:2344–2354.
Candy D, Belsey J. Macrogol (polyethylene glycol) laxatives in children with functional constipation and faecal impaction: a systematic review. Arch Dis Child . 2009;94:156–160.
Carvalho RS, Michail SK, Ashai-Khan F, et al. An update in pediatric gastroenterology and nutrition: a review of some recent advances. Curr Probl Pediatr Adolesc Health Care . 2008;38:197–234.
Emmanuel AV, Kamm MA. Response to a behavioral treatment, biofeedback, in constipated patients is associated with improved gut transit and autonomic innervation. Gut . 2001;49:214–219.
Freedman SB, Rodean J, Hall M, et al. Delayed diagnosis in children with constipation: multicenter retrospective cohort study. J Pediatr . 2017;186:87–94.
Gordon M, MacDonald JK, Parker CE, et al. Osmotic and stimulant laxatives for the management of childhood constipation (review). Cochrane Database Syst Rev . 2016;(8) [CD009118].
Hyams JS, Di Lorenzo C, Saps M, et al. Functional disorders: children and adolescents. Gastroenterology . 2016;150:1456–1468.
Knowles CH, De Giorgio R, Kapur RP, et al. The London classification of gastrointestinal neuromuscular pathology: report on behalf of the gastro 2009 international working group. Gut . 2010;59:882–887.
Koppen IJ, Nurko S, Saps M, et al. The pediatric rome IV criteria: what's new? Expert Rev Gastroenterol Hepatol . 2017;11(3):193–201.
Lu PL, Di Lorenzo C. Neurostimulation of the gastrointestinal tract in children: is it time to shock the gut? Curr Opin Pediatr . 2016;28:631–637.
Masi P, Miele E, Staiano A. Pediatric anorectal disorders. Gastroenterol Clin North Am . 2008;37:709–730.
Pijpers M, Tabbers M, Benninga M, et al. Currently recommended treatments of childhood constipation are not evidence based: a systematic literature review on the effect of laxative treatment and dietary measures. Arch Dis Child . 2009;94:117–131.
Reid H, Bahar RJ. Treatment of encopresis and chronic constipation in young children: clinical results from interactive parent-child guidance. Clin Pediatr (Phila) . 2006;45:157–164.
Rodriguez L, Sood M, Di Lorenzo C, Saps M. An ANMS-NASPGHAN consensus document on anorectal and colonic manometry in children. Neurogastroenterol Motil . 2017;29:e12944.
Rosen R, Buonomo C, Andrade R, et al. Incidence of spinal cord lesions in patients with intractable constipation. J Pediatr . 2004;145:409–411.
Singhal A, Morley R, Abbott R, et al. Clinical safety of iron-fortified formulas. Pediatrics . 2000;105(3):E38.
Stern T, Davis AM. Evaluation and treatment of patients with constipation. JAMA . 2016;315(2):192–193.
The Medical Letter. Plecanatide (Trulance) for chronic idiopathic constipation. Med Lett . 2017;59(1519):66–68.
Van Engelenburg-van Lonkhuyzen ML, Bols EMJ, Benninga MA, et al. Effectiveness of pelvic physiotherapy in children with functional constipation compared with standard medical care. Gastroenterology . 2017;152:82–91.
Voskuijl WP, Reitsma JB, van Ginkel R, et al. Longitudinal follow-up of children with functional nonretentive fecal incontinence. Clin Gastroenterol Hepatol . 2006;4:67–72.
Congenital Aganglionic Megacolon (Hirschsprung Disease)
Asim Maqbool, Chris A. Liacouras
Hirschsprung disease, or congenital aganglionic megacolon, is a developmental disorder (neurocristopathy) of the enteric nervous system, characterized by the absence of ganglion cells in the submucosal and myenteric plexus. It is the most common cause of lower intestinal obstruction in neonates, with an overall incidence of 1 in 5,000 live births. The male:female ratio for Hirschsprung disease is 4 : 1 for short-segment disease and approximately 2 : 1 with total colonic aganglionosis. Prematurity is uncommon.
There is an increased familial incidence in long-segment disease. Hirschsprung disease may be associated with other congenital defects, including trisomy 21, Joubert syndrome, Goldberg-Shprintzen syndrome, Smith-Lemli-Opitz syndrome, Shah-Waardenburg syndrome, cartilage-hair hypoplasia, multiple endocrine neoplasm 2 syndrome, neurofibromatosis, neuroblastoma, congenital hypoventilation (Ondine's curse), and urogenital or cardiovascular abnormalities. Hirschsprung disease has been seen in association with microcephaly, mental retardation, abnormal facies, autism, cleft palate, hydrocephalus, and micrognathia.
Pathology
Hirschsprung disease is the result of an absence of ganglion cells in the bowel wall, extending proximally and continuously from the anus for a variable distance. The absence of neural innervation is a consequence of an arrest of neuroblast migration from the proximal to distal bowel. Without the myenteric and submucosal plexus, there is inadequate relaxation of the bowel wall and bowel wall hypertonicity, which can lead to intestinal obstruction.
Hirschsprung disease is usually sporadic, although dominant and recessive patterns of inheritance have been demonstrated in family groups. Genetic defects have been identified in multiple genes that encode proteins of the RET signaling pathway (RET, GDNF, and NTN ) and involved in the endothelin (EDN) type B receptor pathway (EDNRB, EDN3, and EVE-1 ). Syndromic forms of Hirschsprung disease have been associated with the L1CAM, SOX10, and ZFHX1B (formerly SIP1 ) genes.
The aganglionic segment is limited to the rectosigmoid in 80% of patients. Approximately 10–15% of patients have long-segment disease, defined as disease proximal to the sigmoid colon. Total bowel aganglionosis is rare and accounts for approximately 5% of cases. Observed histologically is an absence of Meissner's and Auerbach's plexuses and hypertrophied nerve bundles with high concentrations of acetylcholinesterase between the muscular layers and in the submucosa.
Clinical Manifestations
Hirschsprung disease is usually diagnosed in the neonatal period secondary to a distended abdomen, failure to pass meconium, and/or bilious emesis or aspirates with feeding intolerance. In 99% of healthy full-term infants, meconium is passed within 48 hr of birth. Hirschsprung disease should be suspected in any full-term infant (the disease is unusual in preterm infants) with delayed passage of stool. Some neonates pass meconium normally but subsequently present with a history of chronic constipation. Failure to thrive with hypoproteinemia from protein-losing enteropathy is a less common presentation because Hirschsprung disease is usually recognized early in the course of the illness but has been known to occur. Breastfed infants might not present as severely as formula-fed infants.
Failure to pass stool leads to dilation of the proximal bowel and abdominal distention. As the bowel dilates, intraluminal pressure increases, resulting in decreased blood flow and deterioration of the mucosal barrier. Stasis allows proliferation of bacteria, which can lead to enterocolitis (Clostridium difficile, Staphylococcus aureus, anaerobes, coliforms) with associated diarrhea, abdominal tenderness, sepsis, and signs of bowel obstruction. Red flags in the neonatal period then include neonatal intestinal obstruction, bowel perforation, delayed passage of meconium, abdominal distention relieved by digital rectal stimulation or enemas, chronic severe constipation, and enterocolitis. Early recognition of Hirschsprung disease before the onset of enterocolitis is essential in reducing morbidity and mortality.
Hirschsprung disease in older patients must be distinguished from other causes of abdominal distention and chronic constipation (see Tables 358.6 and 358.9 and Figs. 358.4 and 358.5 ). The history often reveals constipation starting in infancy that has responded poorly to medical management. Failure to thrive is not uncommon. Fecal incontinence, fecal urgency, and stool-withholding behaviors are usually not present. Significant abdominal distention is unusual in non-Hirschsprung related constipation, as is emesis. The abdomen is tympanitic and distended, with a large fecal mass palpable in the left lower abdomen. Rectal examination demonstrates a normally placed anus that easily allows entry of the finger but feels snug. The rectum is usually empty of feces, and when the finger is removed, there may be an explosive discharge of foul-smelling feces and gas. The stools, when passed, can consist of small pellets, be ribbon-like, or have a fluid consistency, unlike the large stools seen in patients with functional constipation. Intermittent attacks of intestinal obstruction from retained feces may be associated with pain and fever. Urinary retention with enlarged bladder or hydronephrosis can occur secondary to urinary compression.
Table 358.9
Distinguishing Features of Hirschsprung Disease and Functional Constipation
| VARIABLE | FUNCTIONAL | HIRSCHSPRUNG DISEASE |
|---|---|---|
| HISTORY | ||
| Onset of constipation | After 2 yr of age | At birth |
| Encopresis | Common | Very rare |
| Failure to thrive | Uncommon | Possible |
| Enterocolitis | None | Possible |
| Forced bowel training | Usual | None |
| EXAMINATION | ||
| Abdominal distention | Uncommon | Common |
| Poor weight gain | Rare | Common |
| Rectum | Filled with stool | Empty |
| Rectal examination | Stool in rectum | Explosive passage of stool |
| Malnutrition | None | Possible |
| INVESTIGATIONS | ||
| Anorectal manometry | Relaxation of internal anal sphincter | Failure of internal anal sphincter relaxation |
| Rectal biopsy | Normal | No ganglion cells, increased acetylcholinesterase staining |
| Barium enema | Massive amounts of stool, no transition zone | Transition zone, delayed evacuation (>24 hr) |
From Imseis E, Gariepy C: Hirschsprung disease. In Walker WA, Goulet OJ, Kleinman RE, et al, editors: Pediatric gastrointestinal disease , ed 4, Hamilton, ON, 2004, BC Decker, p. 1035.

In neonates, Hirschsprung disease must be differentiated from meconium plug syndrome, meconium ileus, and intestinal atresia. In older patients, the Currarino triad must be considered, which includes anorectal malformations (ectopic anus, anal stenosis, imperforate anus), sacral bone anomalies (hypoplasia, poor segmentation), and presacral anomaly (anterior meningoceles, teratoma, cyst).
Diagnosis
Rectal suction biopsy is the “gold standard” for diagnosing Hirschsprung disease (see Fig. 358.5 ). The biopsy material should contain an adequate amount of submucosa to evaluate for the presence of ganglion cells. To avoid obtaining biopsies in the normal area of hypoganglionosis, which ranges from 3 to 17 mm in length, the suction rectal biopsy should be obtained no closer than 2 cm above the dentate line. The biopsy specimen should be stained for acetylcholinesterase to facilitate interpretation. Patients with aganglionosis demonstrate a large number of hypertrophied nerve bundles that stain positively for acetylcholinesterase with an absence of ganglion cells. Calretinin staining may provide a diagnosis of Hirschsprung disease when acetylcholinesterase staining may not be sufficient.
Anorectal manometry evaluates the internal anal sphincter while a balloon is distended in the rectum. In healthy individuals, rectal distention initiates relaxation of the internal anal sphincter in response to rectal distention (known as the rectoanal inhibitory reflux [RAIR]). In patients with Hirschsprung disease, the internal anal sphincter fails to relax in response to rectal distention, and there is absence of the RAIR. Although the sensitivity and specificity can vary widely, in experienced hands, the test can be quite sensitive. However, the test can be technically difficult to perform in young infants. A normal response in the course of manometric evaluation precludes a diagnosis of Hirschsprung disease; an equivocal or paradoxical response requires a repeat motility or rectal biopsy. The sensitivity and specificity of anorectal manometry are both >90%.
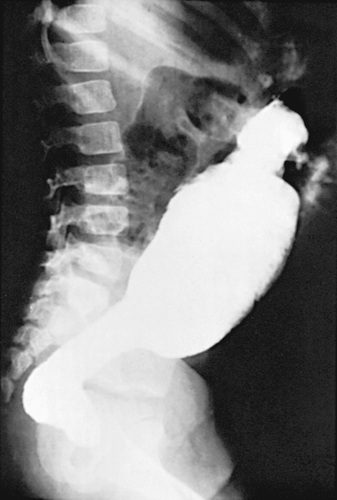
An unprepared contrast enema is most likely to aid in the diagnosis in children older than 1 mo of age because the proximal ganglionic segment might not be significantly dilated in the first few wk of life. Classic findings are based on the presence of an abrupt narrow transition zone between the normal dilated proximal colon and a smaller-caliber obstructed distal aganglionic segment. In the absence of this finding, it is imperative to compare the diameter of the rectum to that of the sigmoid colon, because a rectal diameter that is the same as or smaller than the sigmoid colon suggests Hirschsprung disease. Radiologic evaluation should be performed without prior preparation (i.e., unprepped contrast enema study ) to prevent transient dilation of the aganglionic segment. As many as 10% of newborns with Hirschsprung disease have a normal contrast study. This diagnostic test is most valuable in the disease that involves the distal colon, and specifically, the rectosigmoid. A transition zone may not be readily identifiable in total bowel aganglionosis. Twenty-four-hour delayed films are helpful in showing retained contrast (see Fig. 358.4 ). If significant barium is still present in the colon, it increases the suspicion of Hirschsprung disease even if a transition zone is not identified. Barium enema examination is useful in determining the extent of aganglionosis before surgery and in evaluating other diseases that manifest as lower bowel obstruction in a neonate. The sensitivity (~70%) and specificity (50–80%) of barium enema studies diagnosing Hirschsprung disease is lower than other methodologies. Full-thickness rectal biopsies can be performed at the time of surgery to confirm the diagnosis, level of involvement and to differentiate other disorders (see Fig. 358.5 ).
Treatment
Once the diagnosis is established, the definitive treatment is operative intervention. Previously, a temporary ostomy was placed, and definitive surgery was delayed until the child was older. Currently, many infants undergo a primary pull-through procedure unless there is associated enterocolitis or other complications, when a decompressing ostomy is usually required.
There are essentially three surgical options. The first successful surgical procedure, described by Swenson, was to excise the aganglionic segment and anastomose the normal proximal bowel to the rectum 1-2 cm above the dentate line. The operation is technically difficult and led to the development of two other procedures. Duhamel described a procedure to create a neorectum, bringing down normally innervated bowel behind the aganglionic rectum. The neorectum created in this procedure has an anterior aganglionic segment with normal sensation and a posterior ganglionic segment with normal propulsion. The endorectal pull-through procedure described by Soave involves stripping the mucosa from the aganglionic rectum and bringing normally innervated colon through the residual muscular cuff, thus bypassing the abnormal bowel from within. Advances in techniques have led to successful laparoscopic single-stage endorectal pull-through procedures, which are the treatment of choice.
In ultrashort-segment Hirschsprung disease , also known as anal achalasia , the aganglionic segment is limited to the internal sphincter. The clinical symptoms are similar to those of children with functional constipation. Ganglion cells are present on rectal suction biopsy, but the anorectal manometry is abnormal, with failure of relaxation of the internal anal sphincter in response to rectal distention. Current treatment, although controversial, includes anal botulism injection to relax the anal sphincter and anorectal myectomy if indicated.
Long-segment Hirschsprung disease involving the entire colon and, at times, part of the small bowel presents a difficult problem. Anorectal manometry and rectal suction biopsy demonstrate findings of Hirschsprung disease, but radiologic studies are difficult to interpret because a colonic transition zone cannot be identified. The extent of aganglionosis can be determined accurately by biopsy at the time of laparotomy. When the entire colon is aganglionic, often together with a length of terminal ileum, ileal-anal anastomosis is the treatment of choice, preserving part of the aganglionic colon to facilitate water absorption, which helps the stools to become firm.
The prognosis of surgically treated Hirschsprung disease is generally satisfactory; the great majority of patients achieve fecal continence. Long-term postoperative problems include constipation, recurrent enterocolitis, stricture, prolapse, perianal abscesses, and fecal soiling. Some children require myectomy or a redo pull-through procedure.
Hirschsprung disease–associated enterocolitis can occur at any time prior to or following surgery and is the leading cause of death in these patients. Dysmotility related to partial obstruction, underlying disease, impaired immune function, and the intestinal microbiome may all contribute to this pathophysiologic process. Explosive, foul-smelling and/or bloody diarrhea, abdominal distention, explosive discharge of rectal contents on digital examination, diminished peripheral perfusion, lethargy, and fever are all ominous signs. Management principles include hydration, decompression from above and below (nasogastric Salem Sump, rectal tube, rectal irrigation), and the use of broad-spectrum antibiotics.
Bibliography
Amiel J, Sproat-Emison E, Garcia-Barcelo M, et al. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet . 2008;45:1–14.
Arshad A, Powell C, Tighe MP. Hirschsprung's disease. BMJ . 2012;345:47–49.
Bonnard A, Zeidan S, Degas V, et al. Outcomes of Hirschsprung's disease associated with Mowat-wilson syndrome. J Pediatr Surg . 2009;44:587–591.
Dasgupta R, Langer J. Evaluation and management of persistent problems after surgery for hirschsprung disease in a child. J Pediatr Gastroenterol Nutr . 2008;46(1):13–19.
Dasgupta R, Langer J. Hirschsprung disease. Curr Probl Surg . 2004;41:942–988.
DeLorijn F, Reitsma JB, Voskuijl WP, et al. Diagnosis of Hirschsprung's disease: a prospective comparative accuracy study of common tests. J Pediatr . 2005;146:787–792.
Friedmacher F, Puri P. Classification and diagnostic criteria of variants of Hirschsprung's disease. Pediatr Surg Int . 2013;29:855–872.
Hackam D, Reblock K, Barksdale E, et al. The influence of Down's syndrome on the management and outcome of children with Hirschsprung's disease. J Pediatr Surg . 2003;38:946–949.
Haricharan R, Georgeson K. Hirschsprung disease. Semin Pediatr Surg . 2008;17:266–275.
Heuckeroth RO. Hirschsprung disease. Faure C, et al. Pediatric neurogastroenterology . Springer Science + Business Media: New York; 2013. Gastrointestinal Motility and Functional Disorders in Children, Clinical Gastroenterology . .
Keshtgar AS, Ward HC, Clayden GS, et al. Investigations for incontinence and constipation after surgery for Hirschsprung's disease in children. Pediatr Surg Int . 2003;19:4–8.
Langer JC, Durrant AC, de la Torre L, et al. One-stage transanal soave pull through for hirschsprung disease: a multicenter experience with 141 children. Ann Surg . 2003;238:569–583 [discussion 583–585].
Langer JC. Hirschsprung disease. Curr Opin Pediatr . 2013;25:368–374.
Moore SW. Chromosomal and related mendelian syndromes associated with Hirschsprung's disease. Pediatr Surg Int . 2012;28:1045–1058.
Morris M, Soglio D, Quimet A, et al. A study of calretinin in hirschsprung disease pathology, particularly total colonic hirschsprung disease. J Pediatr Surg . 2013;48(5):1037–1043.
Ozyurek H, Kayacik OE, Gunger O, et al. Rare association of Hirschsprung's disease and joubert syndrome. Eur J Pediatr . 2008;167:475–477.
Pensabene L, Youssef NN, Griffiths JM, et al. Colonic manometry in children with defecatory disorders. Role in diagnosis and management. Am J Gastroenterol . 2003;98:1052–1057.
Prato AP, Musso M, Ceccherini I, et al. Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT). Medicine (Baltimore) . 2009;88:83–90.
Walker WA, Goulet O, Kleinman R, et al. Pediatric gastrointestinal disease . ed 4. BC Decker: Hamilton, Ontario; 2004.
Intestinal Neuronal Dysplasia
Asim Maqbool, Chris A. Liacouras
Intestinal neuronal dysplasia (IND) describes different quantitative (hypoganglionosis or hyperganglionosis) and qualitative (immature or heterotropic ganglion cells) abnormalities of the myenteric and/or submucosal plexus. The typical histology is that of hyperganglionosis and giant ganglia. Type A occurs very rarely and is characterized by congenital aplasia or hypoplasia of the sympathetic innervation. Patients present early in the neonatal period with episodes of intestinal obstruction, diarrhea, and bloody stools. This type B, which accounts for more than 95% of cases, is characterized by malformation of the parasympathetic submucous and myenteric plexus with giant ganglia and thickened nerve fibers, increased acetylcholinesterase staining, and isolated ganglion cells in the lamina propria. IND type B mimics Hirschsprung disease, and patients present with chronic constipation (see Table 358.6 and Fig. 358.5 ). Clinical manifestations include abdominal distention, constipation, and enterocolitis. Various lengths of bowel may be affected from segmental to the entire intestinal tract. IND has been observed in an isolated form and proximal to an aganglionic segment. Other intraintestinal and extraintestinal manifestations are present in patients with IND. It has been reported in all age groups, most commonly in infancy, but is also seen in adults who have had constipation not dating back to childhood.
Associated diseases and conditions include Hirschsprung disease, prematurity, small left colon syndrome, and meconium plug syndrome. Studies have identified a deficiency in substance P in patients with IND. Type A IND may be inherited in a familial, autosomal recessive pattern. Most cases of IND type B are sporadic, with few familial clusters, suggesting autosomal dominant inheritance.
Management includes that for functional constipation, and, if unsuccessful, surgery is indicated.
Bibliography
Hutson J, McNamara J, Shin Y. Review article: slow transit constipation in children. J Paediatr Child Health . 2001;37:426–430.
Kapur R. Neuropathology of paediatric chronic intestinal pseudo-obstruction and related animal models. J Pathol . 2001;194:277–288.
Montedonico S, Acevedo S, Fadda B. Clinical aspects of intestinal neuronal dysplasia. J Pediatr Surg . 2002;37(12):1772–1774.
Neuronal Intestinal Dysplasia. Online Mendelian Inheritance in Man Database . https://www.omim.org/entry/601223?search=intestinal%20neuronal%20dysplasia&highlight=neuronic%20dysplasia%20intestinal%20neuronal%20dysplastic .
Walker WA, Goulet O, Kleinman R, et al. Pediatric gastrointestinal disease . ed 4. BC Decker: Hamilton, Ontario; 2004.
Superior Mesenteric Artery Syndrome (Wilkie Syndrome, Cast Syndrome, Arteriomesenteric Duodenal Compression Syndrome)
Asim Maqbool, Chris A. Liacouras
Superior mesenteric artery syndrome results from compression of the third duodenal segment by the artery against the aorta. Malnutrition or catabolic states may cause mesenteric fat depletion, which collapses the duodenum within a narrowed aortomesenteric angle. Other etiologies include extraabdominal compression (e.g., body cast) and mesenteric tension, as can occur from ileoanal pouch anastomosis. Rapid weight loss and immobilization are risk factors.
Symptoms include intermittent epigastric pain, anorexia, nausea, and vomiting. Risk factors include thin body habitus, prolonged bed rest, abdominal surgery, and exaggerated lumbar lordosis. Onset can be within weeks of a trigger, but some patients have chronic symptoms that evade diagnosis. A classic example is an underweight adolescent who begins vomiting 1-2 wk following scoliosis surgery or spinal fusion. Recognition may be delayed in the context of an eating disorder.
The diagnosis is established radiologically by demonstrating a duodenal cutoff just right of midline along with proximal duodenal dilation, with or without gastric dilation. Although the upper gastrointestinal series remains a mainstay, modalities including CT, MR angiography, or ultrasound may be more appropriate if there is concern for other etiologies such as malignancy. Upper endoscopy should be considered to rule out intraluminal pathology.
Treatment focuses on obstructive relief, nutritional rehabilitation, and correction of associated fluid and electrolyte abnormalities. Lateral or prone positioning can shift the duodenum away from obstructing structures and allow resumption of oral intake. If repositioning is unsuccessful, patients require nasojejunal enteral nutrition past the obstruction or parenteral nutrition if this is not tolerated. This management is successful in the vast majority of cases, with eventual withdrawal of tube feeding once weight has been regained and enteral feeding tolerance orally has been gradually and fully restored. Patients with refractory courses may require surgery to bypass the obstruction.
Acknowledgment
Andrew Chu contributed to the previous version of this chapter.
Bibliography
Biank V, Werlin S. Superior mesenteric artery syndrome in children: a 20-year experience. J Pediatr Gastroenterol Nutr . 2006;42:522–525.
Merrett ND, Wilson RB, Cosman P, et al. Superior mesenteric artery syndrome: diagnosis and treatment strategies. J Gastrointest Surg . 2009;13:287–292.
Welsch T, Büchler MW, Kienle P. Recalling superior mesenteric artery syndrome. Dig Surg . 2007;24(3):149–156 [Epub 2007 Apr 27].