Hemoglobinopathies
Kim Smith-Whitley, Janet L. Kwiatkowski
Hemoglobin Disorders
Hemoglobin is a tetramer consisting of 2 pairs of globin chains. Abnormalities in these proteins are referred to as hemoglobinopathies.
More than 800 variant hemoglobins have been described. The most common and useful clinical classification of hemoglobinopathies is based on nomenclature associated with alteration of the involved globin chain. Two hemoglobin (Hb) gene clusters are involved in Hb production and are located at the end of the short arms of chromosomes 16 and 11. Their control is complex, including an upstream locus control region on each respective chromosome and an X-linked control site. On chromosome 16, there are 3 genes within the alpha (α) gene cluster: zeta (ζ), alpha 1 (α1 ), and alpha 2 (α2 ). On chromosome 11, there are 5 genes within the beta (β) gene cluster: epsilon (ε), gamma 1 (γ1 ), gamma 2 (γ2 ), delta (δ), and beta (β).
The order of gene expression within each cluster roughly follows the order of expression during the embryonic period, fetal period, and eventually childhood. After 8 wk of fetal life, the embryonic hemoglobins, Gower-1 (ζ2 ε2 ), Gower-2 (α2 ε2 ), and Portland (ζ2 γ2 ), are formed. At 9 wk of fetal life, the major hemoglobin is HbF (α2 γ2 ). HbA (α2 β2 ) first appears at approximately 1 mo of fetal life, but does not become the dominant hemoglobin until after birth, when HbF levels start to decline. HbA2 (α2 δ2 ) is a minor hemoglobin that appears shortly before birth and remains at a low level after birth. The final hemoglobin distribution pattern that occurs in childhood is not achieved until at least 6 mo of age and sometimes later. The normal hemoglobin pattern is ≥95% HbA, ≤3.5 HbA2 , and <2.5% HbF.
Sickle Cell Disease
Kim Smith-Whitley
Children with sickle cell disease should be followed by experts in the management of this disease, most often by pediatric hematologists. Medical care provided by a pediatric hematologist is also associated with a decreased frequency of emergency department (ED) visits and length of hospitalization when compared to patients who were not seen by a hematologist within the last year.
Pathophysiology
Hemoglobin S (HbS) is the result of a single base-pair change, thymine for adenine, at the 6th codon of the β-globin gene. This change encodes valine instead of glutamine in the 6th residue in the β-globin molecule. Sickle cell anemia (HbSS), homozygous HbSS, occurs when both β-globin alleles have the sickle cell mutation (βs). Sickle cell disease refers not only to patients with sickle cell anemia, but also to compound heterozygotes where one β-globin allele includes the sickle cell mutation and the 2nd β-globin allele includes a gene mutation other than the sickle cell mutation, such as HbC, β-thalassemia, HbD, and HbOArab . In sickle cell anemia, HbS is typically as high as 90% of the total hemoglobin, whereas in sickle cell disease, HbS is >50% of all hemoglobin.
In red blood cells (RBCs), the hemoglobin molecule has a highly specified conformation allowing for the transport of oxygen in the body. In the absence of globin-chain mutations, hemoglobin molecules do not interact with one another. However, the presence of HbS results in a conformational change in the Hb tetramer, and in the deoxygenated state, HbS molecules interact with each other, forming rigid polymers that give the RBC its characteristic “sickled” shape. The lung is the only organ capable of reversing the polymers, and any disease of the lung can be expected to compromise the degree of reversibility.
Intravascular sickling primarily occurs in the postcapillary venules and is a function of both mechanical obstruction by sickled erythrocytes, platelets, and leukocytes and increased adhesion between these elements and the vascular endothelium. Sickle cell disease is also an inflammatory disease based on nonspecific markers of inflammation, including, but not limited to, elevated baseline white blood cell (WBC) count and cytokines. Intraerythrocytic changes lead to a shortened RBC life span and hemolysis. Hemolysis leads to multiple changes, including altered nitric oxide metabolism and oxidant stress that contribute to endothelial dysfunction.
Diagnosis and Epidemiology
Every state in the United States has instituted a mandatory newborn screening program for sickle cell disease. Such programs identify newborns with the disease and provide prompt diagnosis and referral to providers with expertise in sickle cell disease for anticipatory guidance and the initiation of penicillin before 4 mo of age.
The most commonly used procedures for newborn diagnosis include thin-layer/isoelectric focusing (IEF) and high-performance liquid chromatography (HPLC). Some laboratories perform genetic testing on specimens demonstrating abnormal hemoglobins. A confirmatory step is recommended, with all patients who have initial abnormal screens being retested during the first clinical visit. In addition, a complete blood cell count (CBC) and Hb phenotype determination is recommended for both parents to confirm the diagnosis and to provide an opportunity for genetic counseling. Infants who may have HbS-hereditary persistence fetal hemoglobin (HbSHPFH) but do not have full parental studies should have molecular testing for β-globin genotype before 12 mo of age. Table 489.1 correlates the initial hemoglobin phenotype at birth with the type of hemoglobinopathy.
Table 489.1
Various Newborn Sickle Cell Disease Screening Results With Baseline Hemoglobin
| NEWBORN SCREENING RESULTS: SICKLE CELL DISEASE* | POSSIBLE HEMOGLOBIN PHENOTYPE † | BASELINE HEMOGLOBIN RANGE AFTER AGE 5 YR | EXPERTISE IN HEMATOLOGY CARE REQUIRED |
|---|---|---|---|
| FS | SCD-SS | 6-11 g/dL | Yes |
| SCD-S β0 thal | 6-10 g/dL | Yes | |
| SCD-S β+ thal | 9-12 g/dL | Yes | |
| SCD-S δβ− thal | 10-12 g/dL | Yes | |
| S HPFH | 12-14 g/dL | Yes | |
| FSC | SCD-SC | 10-15 g/dL | Yes |
| FSA ‡ | SCD-S β+ thal | 9-12 g/dL | Yes |
| FS other | SCD-S β0 thal | 6-10 g/dL | Yes |
| SCD-SD, SOArab , SCHarlem , SLepore | Variable | Yes | |
| AFS ‡ § | SCD-SS | 6-10 g/dL | Yes |
| SCD-S β+ thal | 6-9 g/dL | Yes | |
| SCD-S β0 thal | 7-13 g/dL variable | Yes |
* Hemoglobins are reported in order of quantity.
† Requires confirmatory hemoglobin analysis after at least 6 mo of age and, if possible, β-globin gene testing or hemoglobin analysis from both parents for accurate diagnosis of hemoglobin phenotype.
‡ Sickle cell trait is another possible diagnosis
§ Impossible to determine the diagnosis because the infant most likely received a blood transfusion before testing.
A, Normal hemoglobin; C, hemoglobin C; F, fetal hemoglobin; HPFH, hereditary persistence of fetal hemoglobin; OArab , hemoglobin OArab ; S, sickle hemoglobin; SC, sickle-hemoglobin C; SCD, sickle cell disease; SS, homozygous sickle cell disease; thal, thalassemia.
In newborn screening programs, the hemoglobin with the greatest quantity is reported first, followed by other hemoglobins in order of decreasing quantity. Some states perform IEF initially on newborn blood samples, then use DNA probes to confirm abnormal hemoglobins found on IEF. In newborns with a hemoglobin analysis result of HbFS , the pattern supports HbSS, HbSHPFH, or HbSβ0 -thalassemia. In a newborn with a hemoglobin analysis of HbFSA , the pattern is supportive of the diagnosis of HbSβ+ -thalassemia. The diagnosis of HbSβ+ -thalassemia is confirmed if at least 50% of the hemoglobin is HbS, HbA is present, and the amount of HbA2 is elevated (typically >3.5%), although HbA2 is not elevated in the newborn period. In newborns with a hemoglobin analysis of HbFSC , the pattern supports a diagnosis of HbSC. In newborns with a hemoglobin analysis of HbFAS , the pattern supports a diagnosis of HbAS (sickle cell trait); however, in this circumstance, care must be taken to confirm that the newborn has not received a red cell transfusion before testing.
A newborn with a hemoglobin analysis of AFS either has been transfused with RBCs before collection of the newborn screen to account for the greater amount of HbA than HbF, or there has been an error. The patient may have either sickle cell disease or sickle cell trait and should be started on penicillin prophylaxis until the final diagnosis can be determined.
Given the implications of a diagnosis of sickle cell disease vs sickle cell trait in a newborn, the importance of repeating the Hb identification analysis in the patient and obtaining a Hb identification analysis and CBC to evaluate the peripheral blood smear and RBC parameters in the parents for genetic counseling cannot be overemphasized. Unintended mistakes do occur in state newborn screening programs. Newborns who have the initial phenotype of HbFS but whose final true phenotype is HbSβ+ -thalassemia have been described as one of the more common errors identified in newborn screening hemoglobinopathy programs. Determining an accurate phenotype is important for appropriate genetic counseling for the parents. In addition, distinguishing HbSS from HbSHPFH in the newborn period usually requires parental or genetic testing. In infants who maintain HbF percentages above 25% after 12 mo of age without evidence of hemolysis should have testing for β-globin gene deletions consistent with HPFH. These children have a much milder clinical course and do not require penicillin prophylaxis or hydroxyurea therapy.
If the parents are tested for sickle cell trait or hemoglobinopathy trait full disclosure to the parents must be provided, and in some circumstances the issue of paternity may be disclosed. For this reason and because of healthcare privacy, common practice is to always seek permission for the genetic testing and to report the hemoglobinopathy trait results back to each parent separately.
In the United States, sickle cell disease is the most common genetic disorder identified through the state-mandated newborn screening program, occurring in 1 : 2,647. In regard to race in the United States, sickle cell disease occurs in African Americans at a rate of 1 : 396 births and in Hispanics at a rate of 1 : 36,000 births. In the United States, an estimated 100,000 people are affected by sickle cell disease, with an ethnic distribution of 90% African American and 10% Hispanic. The U.S. sickle cell disease population represents a fraction of the worldwide burden of the disease, with global estimates of 312,000 neonates born annually with HbSS disease.
Clinical Manifestations and Treatment of Sickle Cell Anemia (HbSS)
For a comprehensive discussion of the clinical management of children and adolescents with sickle cell disease, refer to National Heart, Lung and Blood Institute (NHLBI) 2014 Expert Panel Report on the Evidence-based Management of Sickle Cell Disease (https://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle-cell-disease-report.pdf ).
Fever and Bacteremia
Fever in a child with sickle cell anemia is a medical emergency, requiring prompt medical evaluation and delivery of antibiotics because of the increased risk of bacterial infection and subsequent high mortality rate. As early as 6 mo of age, infants with sickle cell anemia develop abnormal immune function because of splenic dysfunction. By 5 yr of age, most children with sickle cell anemia have complete functional asplenia. Regardless of age, all patients with sickle cell anemia are at increased risk of infection and death from bacterial infection, particularly encapsulated organisms such as Streptococcus pneumoniae , Haemophilus influenzae type b, and Neisseria meningitidis .
The rate of bacteremia in children with sickle cell disease presenting with fever is <1%. Several clinical strategies have been developed to manage children with sickle cell disease who present with fever. These strategies range from hospital admission for intravenous (IV) antimicrobial therapy to administering a third-generation cephalosporin in an ED or outpatient setting to patients without established risk factors for occult bacteremia (Table 489.2 ). Given the observation that the average time for a positive blood culture is <20 hr in children with sickle cell anemia, admission for 24 hr is probably the most prudent strategy for children and families who live out of town or who are identified as high risk for poor follow-up.
Outpatient management of fever without a source should be considered in children with the lowest risk of bacteremia and after appropriate cultures are obtained and IV ceftriaxone or another cephalosporin is given. Observation after antibiotic administration is important, because children who have sickle cell anemia treated with ceftriaxone can develop severe, rapid, and life-threatening immune hemolysis. In the event that Salmonella spp. or Staphylococcus aureus bacteremia occurs, strong consideration should be given to an evaluation for osteomyelitis with a bone scan, given the increased risk of osteomyelitis in children with sickle cell anemia compared to the general population. Screening laboratory and radiologic studies are strongly recommended to identify those at risk for transient red cell aplasia, acute splenic sequestration, and acute chest syndrome (ACS ), because many children with these diagnoses present to acute care settings with isolated fever. Screening children and caregivers for psychosocial factors that could impede their return to the hospital in the case of a positive blood culture is essential.
Aplastic Crisis
Human parvovirus B19 infection poses a unique threat for patients with sickle cell disease because this infection results in temporary red cell aplasia , limiting the production of reticulocytes and causing profound anemia (see Fig. 485.3 in Chapter 485 ). Any child with sickle cell disease, fever, and reticulocytopenia should be presumed to have parvovirus B19 infection until proven otherwise. Reticulocytosis and increased nucleated RBCs may be seen in the recovery phase. Testing for the presence of human parvovirus B19 with PCR testing is superior to using IgM and IgG titers. The acute exacerbation of anemia is treated conservatively using red cell transfusion when the patient becomes hemodynamically symptomatic or has a concurrent illness, such as ACS or acute splenic sequestration. In addition, acute infection with parvovirus B19 is associated with pain, splenic sequestration, ACS, glomerulonephritis, arthropathy, and stroke. Many patients with parvovirus-associated aplastic crisis are contagious, and infection precautions should be taken to avoid nosocomial spread of the infection and to avoid exposure of pregnant caregivers.
Splenic Sequestration
Acute splenic sequestration is a life-threatening complication occurring primarily in infants and young children with sickle cell anemia. The incidence of splenic sequestration has declined from an estimated 30% to 12.6% with early identification by newborn screening and improved parental education. Sequestration can occur as early as 5 wk of age but most often occurs in children between ages 6 mo and 2 yr. Patients with the SC and Sβ+ -thalassemia types of sickle cell disease can have acute splenic sequestration events throughout adolescence and adulthood.
Splenic sequestration is associated with rapid spleen enlargement causing left-sided abdominal pain and Hb decline of at least 2 g/dL from the patient's baseline. Sequestration may lead to signs of hypovolemia as a result of the trapping of blood in the spleen and profound anemia, with total Hb falling below 3 g/dL. A decrease in WBC and platelet count may also be present. Sequestration may be triggered by fever, bacteremia, or viral infections.
Treatment includes early intervention and maintenance of hemodynamic stability using isotonic fluid or blood transfusions. Careful blood transfusions with RBCs are recommended to treat both the sequestration and the resultant anemia. Blood transfusion aborts the RBC trapping in the spleen and allows release of the patient's blood cells that have become sequestered, often raising Hb above baseline values. A reasonable approach is to provide only 5 mL/kg of RBCs and/or a posttransfusion Hb target of 8 g/dL, keeping in mind that the goal of transfusion is to prevent hypovolemia. Blood transfusion that results in Hb levels >10 g/dL may put the patient at risk for hyperviscosity syndrome because blood may be released from the spleen after transfusion.
Repeated episodes of splenic sequestration are common, occurring in two thirds of patients. Most recurrent episodes develop within 6 mo of the previous episode. Prophylactic splenectomy performed after an acute episode has resolved is the only effective strategy for preventing future life-threatening episodes. Although blood transfusion therapy has been used with the goal of preventing subsequent episodes, evidence strongly suggests that this strategy does not reduce the risk of recurrent splenic sequestration compared to no transfusion therapy. However, a short course of regular red cell transfusions can be used until splenectomy is arranged. Children should be appropriately immunized with meningococcal and pneumococcal vaccines before surgery. Penicillin prophylaxis should be prescribed after splenectomy.
Hepatic and Gallbladder Involvement
See Chapters 387 and 393 .
Sickle Cell Pain
Dactylitis , referred to as hand-foot syndrome , is often the first manifestation of pain in infants and young children with sickle cell anemia, occurring in 50% of children by their 2nd yr of life (Fig. 489.1 ). Dactylitis often manifests with symmetric or unilateral swelling of the hands and/or feet. Unilateral dactylitis can be confused with osteomyelitis, and careful evaluation to distinguish the two is important because treatment differs significantly. Dactylitis requires palliation with pain medications, such as hydrocodone or oxycodone, whereas osteomyelitis requires at least 4-6 wk of IV antibiotics. Given the association between genotype and metabolism of codeine, a subgroup of children may not get pain relief from codeine. Therefore, feedback from the parents is needed to determine if therapy was successful in relieving pain.

The cardinal clinical feature of sickle cell disease is acute vasoocclusive pain . Acute sickle cell pain is characterized as unremitting discomfort that can occur in any part of the body but most often occurs in the chest, abdomen, or extremities. These painful episodes are often abrupt and cause disruption of daily life activities and significant stress for children and their caregivers. A patient with sickle cell anemia has approximately 1 painful episode per year that requires medical attention.
The exact etiology of pain is unknown, but the pathogenesis may be initiated when blood flow is disrupted in the microvasculature by sickled red blood cells and other cellular elements, resulting in tissue ischemia. Acute sickle cell pain may be precipitated by physical stress, infection, dehydration, hypoxia, local or systemic acidosis, exposure to cold, and swimming for prolonged periods. However, most pain episodes occur without an identifiable trigger. Successful treatment of these episodes requires education of both caregivers and patients regarding the recognition of symptoms and the optimal management strategy. Given the absence of any reliable objective laboratory or clinical parameter associated with pain, trust between the patient and the treating physician is paramount to successful clinical management. Specific therapy for pain varies greatly but generally includes the use of acetaminophen or a nonsteroidal antiinflammatory drug (NSAID) early in the course of pain, followed by escalation to a combination analgesic regimen using a single-agent short-acting oral opioid, long-acting oral opioid, and continued nonopioid agent.
Some patients require treatment in an acute care setting for administration of IV morphine or derivatives of morphine. The primary goal of treatment in these settings is timely administration of analgesics to provide relief of pain. The incremental increase and decrease in the use of the medication to relieve pain roughly parallels the 8 phases associated with a chronology of pain and comfort in children (Table 489.3 ). When pain requires continued parenteral analgesic administration, hospitalization is required. The average hospital length of stay for children admitted in pain is 4.4 days. The NHLBI clinical guidelines for treating acute and chronic pain in children and adults with sickle cell disease are comprehensive and represent a starting point for treating pain.
Table 489.3
Phases of a Painful Episode in Patients With Sickle Cell Disease
| PHASE | DESCRIPTION AND COMFORT MEASURES |
|---|---|
| Data From Children | |
| I | Baseline |
| No pain and no comfort measures | |
| II | Prepain phase |
| No evidence of pain | |
| Child begins to display some prodromal signs and symptoms of VOE (yellow eyes, fatigue) | |
| No comfort measures used | |
| Caregivers encouraged child to increase fluids to prevent the pain event from occurring | |
| III | Pain starting point |
| Child complained of mild “ache-ish” pain in one specific area, which gradually or rapidly increased or “waxed” | |
| Mild analgesics (ibuprofen and acetaminophen) given | |
| Child maintained normal activities and continued to attend school | |
| Caregivers hoped to prevent an increase in pain intensity | |
| IV | Pain acceleration |
| Pain continued to escalate; intensity increased from mild to moderate; pain appeared in more areas of the body; child was kept home from school; decreased level of activity; differences in behaviors, appearance, and mood | |
| Stronger oral analgesics may be combined with rest, rubbing, heat, distraction, and psychological comfort | |
| V | Peak pain experience |
| Pain continued to escalate | |
| Some children were incapacitated and unable to obtain pain relief | |
| Pain described as “stabbing,” “drilling,” “pounding,” “banging,” “excruciating,” “unbearable,” or “throbbing” | |
| Caregivers sometimes decide to seek help from ED for stronger analgesics and protection from complications such as fever or respiratory distress | |
| Caregivers may be exhausted from caring for the child for several days with little or no rest | |
| All methods of comfort were used around the clock to reduce the pain and avoid going to the hospital | |
| Pain increased despite all efforts | |
| Decision is made to take the child to ED | |
| VI | Pain decrease starting point |
| Pain begins to resolve after the use of IV fluids and analgesics | |
| Analgesics sedate the child and allow the child to sleep for longer periods | |
| Pain described as “slowly decreasing” | |
| Pain is still sharp and throbbing | |
| VII | Steady pain decline |
| Pain decreased slowly or rapidly | |
| Child takes more interest in surroundings, roommates, and visitors | |
| Child is less irritable | |
| Level of activity increased—child may be taken to tub room for warm bath, may watch television, may play games with other children or hospital volunteers | |
| Mobility was improved | |
| Pain levels reported as “just a little” | |
| More animation in behaviors evident | |
| VIII | Pain resolution |
| Pain was at a tolerable level | |
| Child may be discharged from the hospital on mild oral analgesics; child is at or close to baseline conditions, with behavior, appearance, and mood more normal | |
| Caregiver and child attempt to regain, recapture, and catch up with life as it was before the pain event | |
| Data From Adults | |
| I | Evolving/infarctive phase |
| 3 days | |
| ↓ RBC deformability | |
| ↓ Hemoglobin | |
| ↑ % of dense RBCs | |
| ↑ RDW, ↑ HDW | |
| S/S: fear, anorexia, anxiety, ↑ pain | |
| II | Postinfarctive/inflammatory phase |
| 4-5 days | |
| ↓ Hemoglobin | |
| ↑ White blood cells (leukocytosis) | |
| ↑ Acute-phase reactants C-reactive protein | |
| ↑ Reticulocytes, ↑ LDH, ↑ CPK | |
| ↑ % dense RBCs | |
| ↑ RDW, ↑ HDW | |
| S/S: fever, severe steady pain, swelling, tenderness, joint stiffness, joint effusions | |
| III | Resolving/healing/recovery/postcrisis phase |
| ↑ RBC deformability | |
| Hemoglobin returns to precrisis level | |
| Retics return to precrisis levels | |
| ↓ % of dense RBCs | |
| ↓ RDW, ↓ HDW | |
| ↓ ISC | |
| Precursors to relapse that happens in phase III: ↑ platelets, ↑ acute-phase reactants (fibrinogen, α1 -acid glycoprotein, osmomucoid), ↑ viscosity, ↑ ESR | |
| ↑ Retics expressing the ↑ α4 β1 -integrin complex ICAM-1 | |
CPK, Creatinine phosphokinase; ED, emergency department; ESR, erythrocyte sedimentation rate; HDW, hemoglobin distribution width; ICAM, intracellular adhesion molecule; ISC, irreversibly sickled cells; LDH, lactate dehydrogenase; RBC, red blood cell; RDW, red cell distribution width; S/S, signs and symptoms; VOE, vasoocclusive episode.
Adapted from Jacob E: The pain experience of patients with sickle cell anemia, Pain Manage Nurs 2(3):74–83, 2001; with data from Ballas SK, Smith ED: Red blood cell changes during the evolution of the sickle cell painful crisis, Blood 79:2154–2163, 1992; and Beyer JE, Simmons L, Woods GM, Woods PM: A chronology of pain and comfort in children with sickle cell disease, Arch Pediatr Adolesc Med 153:913–920, 1999.
The only measure for degree of pain is the patient. Healthcare providers working with children in pain should use a consistent, validated pain scale (e.g., Wong-Baker FACES Scale) for assessing pain. Although pain scales have proved useful for some children, others require prenegotiated activities to determine when opioid therapy should be initiated and decreased. For example, sleeping through the night might be an indication for decreasing pain medication by 20% the following morning. The majority of painful episodes in patients with sickle cell disease are managed at home with comfort measures, such as heating blanket, relaxation techniques, massage, and oral pain medication.
Several myths have been propagated regarding the treatment of pain in sickle cell disease. The concept that painful episodes in children should be managed without opioids is without foundation and results in unwarranted suffering on the part of the patient. Blood transfusion therapy during an existing painful episode does not decrease the intensity or duration of the painful episode, because tissue necrosis occurs well before the ability to administer the transfusion. IV hydration does not relieve or prevent pain and is appropriate when the patient is dehydrated or unable to drink as a result of the severe pain. Opioid dependency in children with sickle cell disease is rare and should never be used as a reason to withhold pain medication. However, patients with multiple painful episodes requiring hospitalization within 1 yr or with pain episodes that require hospitalization for >7 days should be evaluated for comorbidities and environmental stressors that are contributing to the frequency or duration of pain. Children with chronic pain should be evaluated for other reasons associated with vasoocclusive pain episodes, including, but not limited to, presence of avascular necrosis, leg ulcers, and vertebral body compression fractures. A careful history is warranted to distinguish chronic pain that often is not relieved by opioids vs recurrent acute prolonged vasoocclusive pain episodes.
Skeletal pain (bone or bone marrow infarction) with or without fever must be differentiated from osteomyelitis . Both Salmonella spp. and S. aureus cause osteomyelitis in children with sickle cell disease, often involving the diaphysis of long bones (in contrast to children without sickle cell anemia, in whom osteomyelitis is in the metaphyseal region of the bone). Differentiating osteonecrosis from a vasoocclusive crisis and osteomyelitis is often difficult. Clinical signs and symptoms can be consistent with both osteonecrosis and vasoocclusive crises, as low-grade fever pain, swelling of the affected area, high WBC counts, and elevated C-reactive protein levels can be present in both. Blood cultures, when positive, are helpful. MRI may be useful for locating an area to obtain fluid for culture. MR findings suggestive of osteomyelitis include localized medullary fluid, sequestrum, and cortical defects. Ultimately, aspiration with or with or without biopsy and culture will be needed to differentiate the 2 processes (see Chapter 704 ).
Avascular Necrosis
Avascular necrosis (AVN ) occurs at a higher rate among children with sickle cell disease than in the general population and is a source of both acute and chronic pain. Most often the femoral head is affected. Unfortunately, AVN of the hip may cause limp and leg-length discrepancy. Other sites affected include the humeral head and mandible. Risk factors for AVN include HbSS disease with α-thalassemia trait, frequent vasoocclusive episodes, and elevated hematocrit (for patients with sickle cell anemia). Optimal treatment of AVN has not been determined, and individual management requires consultation with the disease-specific specialist, orthopedic surgeon, physical therapist, hematologist, and primary care physician. Initial management should include referral to a pediatric orthopedist and a physical therapist to address strategies to increase strength and decrease weight-bearing daily activities that may exacerbate the pain associated with AVN. Opioids are often used but usually can be tapered after the acute pain has subsided. Regular blood transfusion therapy has not been demonstrated as an effective therapy to abate the acute and chronic pain associated with AVN.
Priapism
Priapism, defined as an unwanted painful erection of the penis, affects males of all genotypes but most frequently affects males with sickle cell anemia. The mean age of first episode is 15 yr, although priapism has been reported in children as young as 3 yr. The actuarial probability of a patient experiencing priapism is approximately 90% by 20 yr of age.
Priapism occurs in 2 patterns: prolonged , lasting >4 hr, or stuttering , with brief episodes that resolve spontaneously but may occur in clusters and herald a prolonged event. Both types occur from early childhood to adulthood. Most episodes occur between 3 and 9 AM . Priapism in sickle cell disease represents a low-flow state caused by venous stasis from RBC sickling in the corpora cavernosa. Recurrent prolonged episodes of priapism are associated with erectile dysfunction (impotence).
The optimal treatment for acute priapism is unknown. Supportive therapy, such as a hot shower, short aerobic exercise, or pain medication, is often used by patients at home. A prolonged episode lasting >4 hr should be treated by aspiration of blood from the corpora cavernosa, followed by irrigation with dilute epinephrine to produce immediate and sustained detumescence. Urology consultation is required to initiate this procedure, with appropriate input from a hematologist. Simple blood transfusion with exchange transfusion has been proposed for the acute treatment of priapism, but limited evidence supports this strategy as the initial management. If no benefit is obtained from surgical management, transfusion therapy should be considered. However, detumescence may not occur for up to 24 hr (much longer than with urologic aspiration) after transfusion, and transfusion for priapism has been associated with acute neurologic events. Consultation with a hematologist and urologist will help identify therapies to prevent recurrences.
Neurologic Complications
Neurologic complications associated with sickle cell disease are varied and complex, ranging from acute ischemic stroke with focal neurologic deficit to clinically silent abnormalities found on radiologic imaging. Before the development of transcranial Doppler ultrasonography to screen for stroke risk among children with sickle cell anemia, approximately 11% experienced an overt stroke and 20% a silent stroke before age 18 yr. A functional definition of overt stroke is the presence of a focal neurologic deficit lasting for >24 hr and/or abnormal neuroimaging of the brain indicating a cerebral infarct on T2-weighted MRI corresponding to the focal neurologic deficit (Figs. 489.2 and 489.3 ). A silent cerebral infarct lacks focal neurologic findings lasting >24 hr and is diagnosed by abnormal imaging on T2-weighted MRI. Children with other types of sickle cell disease, such as HbSC or HbSβ+ -thalassemia, develop overt or silent cerebral infarcts as well, but at a lower frequency than children with HbSS and HbSβ0 -thalassemia. Other neurologic complications include transient ischemic attacks, headaches that may or may not correlate to degree of anemia, seizures, cerebral venous thrombosis, and posterior reversible encephalopathy syndrome (PRES) . Chiari I malformations can occur in older children with sickle cell disease. Fat embolism syndrome is a rapidly progressive, potentially fatal complication involving pain, respiratory distress, changes in mental status, and multiorgan system failure. When this syndrome is identified early, exchange transfusion therapy has improved patient survival in small case series.

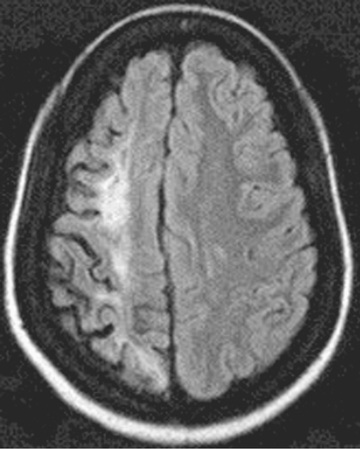
For patients presenting with acute focal neurologic deficit, a prompt pediatric neurologic evaluation and consultation with a pediatric hematologist is recommended. In addition, oxygen administration to keep oxygen saturation (SO 2 ) >96% and simple blood transfusion within 1 hr of presentation, with a goal of increasing Hb to a maximum of 10 g/dL, is warranted. A timely simple blood transfusion is important because this is the most efficient strategy to dramatically increase oxygen content of the blood, if SO 2 is >96%. However, greatly exceeding this posttransfusion Hb limits oxygen delivery to the brain as a result of hyperviscosity by increasing the Hb significantly over the patient's baseline values. Subsequently, prompt treatment with an exchange transfusion should be considered, either manually or with automated erythrocytapheresis, to reduce the HbS percentage to at least <50% and ideally <30%. Exchange transfusion at the time of acute stroke is associated with a decreased risk of 2nd stroke compared to simple transfusion alone. CT of the head to exclude cerebral hemorrhage should be performed as soon as possible, and if available, MRI of the brain with diffusion-weighted imaging to distinguish between ischemic infarcts and PRES. MR venography is useful to evaluate the possibility of cerebral venous thrombosis , a rare but potential cause of focal neurologic deficit in children with sickle cell disease. MR angiography may identify evidence of cerebral vasculopathy; these images are not critical in the initial time management of a child with sickle cell disease presenting with a focal neurologic deficit.
The clinical presentation of PRES or central venous thrombosis can mimic a stroke but would require a different treatment course. For both PRES and cerebral venous thrombosis, the optimal management has not been defined in patients with sickle cell disease, resulting in the need for consultation with both a pediatric neurologist and a pediatric hematologist. The primary approach for prevention of recurrent overt stroke is blood transfusion therapy aimed at keeping the maximum HbS concentration <30%. Despite regular blood transfusion therapy, approximately 20% of patients will have a 2nd stroke and 30% of this group will have a 3rd stroke.
Transcranial Doppler Ultrasonography
Primary prevention of overt stroke can be accomplished using screening transcranial Doppler ultrasonography (TCD) assessment of the blood velocity in the terminal portion of the internal carotid and the proximal portion of the middle cerebral artery. Children with sickle cell anemia with an elevated time-averaged mean maximum (TAMM) blood flow velocity >200 cm/sec are at increased risk for a cerebrovascular event. A TAMM measurement of <200 but ≥180 cm/sec represents a conditional threshold. A repeat measurement is suggested within a few months because of the high rate of conversion to a TCD velocity >200 cm/sec in this group of patients. However a single value ≥220 cm/sec is concerning and does not require repeating before recommending an intervention.
Two distinct methods of measuring TCD velocity are a nonimaging technique and an imaging technique. The nonimaging technique was the method used in the stroke prevention trial sponsored by the National Institutes of Health, whereas most pediatric radiologists in practice use the imaging technique. When compared to each other, the imaging technique produces values that are 10–15% below those of the nonimaging technique. The imaging technique uses the time-averaged mean of the maximum velocity (TAMX), and this measure is believed to be equivalent to the nonimaging calculation of TAMM. A downward adjustment for the transfusion threshold is appropriate for centers using the imaging method to assess TCD velocity. The magnitude of the transfusion threshold in the imaging technique has not been settled, but a transfusion threshold of a TAMX of 185 cm/sec and a conditional threshold of TAMX of 165 cm/sec seem reasonable. Alternatively, some experts recommend using the same thresholds regardless of technique.
Children with TCD values above defined thresholds should begin chronic blood transfusion therapy to maintain HbS levels <30% to decrease the risk of 1st stroke. This strategy results in an 85% reduction in the rate of overt strokes. Once transfusion therapy is initiated, a subset of patients at low risk for the development of increased TCD values, such as those without MRI-confirmed cerebral vasculopathy, may be able to transition from chronic transfusions to long-term hydroxyurea therapy. Acute stroke risk is decreased when hydroxyurea use and chronic transfusions overlap until a robust therapeutic response to hydroxyurea is achieved.
Pulmonary Complications
Lung disease in children with sickle cell disease is the 2nd most common reason for hospital admission and is associated with significant mortality. Acute chest syndrome refers to a life-threatening pulmonary complication of sickle cell disease defined as a new radiodensity on chest radiography plus any 2 of the following: fever, respiratory distress, hypoxia, cough, and chest pain (Fig. 489.4 ). Even in the absence of respiratory symptoms, very young children with fever should receive a chest radiograph to identify evolving ACS, because clinical examination alone is insufficient to identify patients with a new radiographic density. Early detection of ACS may alter clinical management. The radiographic findings in ACS are variable but may include single-lobe involvement, predominantly left lower lobe; multiple lobes, most often both lower lobes; and pleural effusions, either unilateral or bilateral. ACS may progress rapidly from a simple infiltrate to extensive infiltrates and a pleural effusion. Therefore, continued pulse oximetry and frequent clinical exams are required, and repeat chest x-ray films may be indicated for progressive hypoxia, dyspnea, tachypnea, and other signs of respiratory distress.

Most patients with ACS do not have a single identifiable cause. Infection is the most well-known etiology, yet only 30% of ACS episodes will have positive sputum or bronchoalveolar culture, and the most common bacterial pathogens are S. pneumoniae , Mycoplasma pneumoniae, and Chlamydia spp. The most frequent event preceding ACS is a painful episode requiring systemic opioid treatment. Fat emboli have also been implicated as a cause of ACS, arising from infarcted bone marrow, and can be life threatening if large amounts are released to the lungs. Fat emboli can be difficult to diagnose but should be considered in any patient with sickle cell disease presenting with rapid onset of respiratory distress and altered mental status changes. Petechial rash may also occur, but may be difficult to detect if not carefully sought.
Given that the causes of ACS are varied, recommended management is also multimodal (Table 489.4 ). The type of opioid, with morphine being more likely to cause ACS than nalbuphine hydrochloride, is associated with an increase in the risk of ACS in part because of sedation and hypoventilation. However, under no circumstance should opioid administration be limited to prevent ACS; rather, other measures must be taken to prevent ACS from developing. In patients with chest pain, regular use of an incentive spirometer at 10-12 breaths every 2 hr can significantly reduce the frequency of subsequent ACS episodes. Because of the clinical overlap between pneumonia and ACS, all episodes should be treated promptly with antimicrobial therapy, including at least a macrolide and possibly a third-generation cephalosporin. A previous diagnosis of asthma or wheezing with ACS should prompt treatment following standard of care for an asthma exacerbation with bronchodilators. The diagnosis of ACS does not negate the recommended management of a patient with asthma exacerbation. Oxygen should be administered for patients who demonstrate hypoxia. Blood transfusion therapy using either simple or exchange (manual or automated) transfusion is the only method to abort a rapidly progressing ACS episode. The decision when to give blood and whether the transfusion should be a simple or exchange transfusion is less clearly defined. Usually, blood transfusions are given when at least 1 of the following clinical features are present: decreasing SO 2 , increasing work of breathing, rapidly changing respiratory effort either with or without a worsening chest radiograph, a dropping Hb of 2 g/dL below the patient's baseline, or previous history of severe ACS requiring admission to the intensive care unit.
Pulmonary hypertension has been identified as a major risk factor for death in adults with sickle cell anemia. The natural history of pulmonary hypertension in children with sickle cell anemia is unknown. Optimal strategies for screening at risk patients have not been identified (echocardiogram results are not supported by right-sided heart catheterization results demonstrating elevated pulmonary artery pressures), and the best diagnostic methodology carries significant risk of harm. Attempts to identify targeted therapeutic interventions to alter the natural history of pulmonary hypertension in adults have been unsuccessful.
Renal Disease and Enuresis
Renal disease among patients with sickle cell disease is a major comorbid condition that can lead to premature death. Seven sickle cell disease nephropathies have been identified: gross hematuria, papillary necrosis, nephrotic syndrome, renal infarction, hyposthenuria, pyelonephritis, and renal medullary carcinoma. The presentation of these entities is varied but may include hematuria, proteinuria, renal insufficiency, concentrating defects, or hypertension.
The common presence of nocturnal enuresis occurring in children with sickle cell disease is not well defined but is troublesome for affected children and their parents. The overall prevalence of enuresis was 33% in the Cooperative Study of Sickle Cell Disease, with the highest prevalence (42%) among children 6-8 yr old. Furthermore, enuresis may still occur in approximately 9% of older adolescents. Patients with sickle cell disease and nocturnal enuresis should have a systematic evaluation for recurrent urinary tract infections, kidney function, and possibly obstructive sleep apnea syndrome. Unfortunately, most children with nocturnal enuresis do not have an etiology, and targeted therapeutic interventions have been of limited success. However, referrals to pediatric urologists should be considered.
Cognitive and Psychological Complications
Good health maintenance must include routine psychological and social assessment. Ongoing evaluation of the family unit and identification of the resources available to cope with a chronic illness are critical for optimal management. Children and adolescents with sickle cell disease have decreased quality of life, as measured on standardized assessments, compared to their siblings and children with other chronic diseases. Furthermore, children with sickle cell disease are at great risk for academic failure and have a 20% high school graduation rate, possibly because, among other reasons, approximately one third of children with sickle cell anemia have had a cerebral infarct, either silent or an overt stroke. Children with cerebral infarcts require ongoing cognitive and school performance assessment so that education resources can be focused to optimize educational attainment. Participation in relevant support groups and group activities, such as camps for children with sickle cell disease, may be of direct benefit by improving self-esteem and establishing peer relationships.
Other Complications
In addition to the previous organ dysfunctions, patients with sickle cell disease can have other significant complications. These complications include, but are not limited to, sickle cell retinopathy, delayed onset of puberty, and leg ulcers. Optimal treatment for each of these entities has not been determined, and individual management requires consultation with the hematologist and primary care physician.
Therapeutic Considerations
Hydroxyurea
Hydroxyurea, a myelosuppressive agent, is a well-established drug proven effective in reducing the frequency of acute pain episodes. In adults with sickle cell anemia, hydroxyurea decreases the rate of hospitalization for painful episodes by 50% and the rate of ACS and blood transfusion by almost 50%. In addition, adults taking hydroxyurea have shorter hospital stay and require less analgesic medication during hospitalization. In children with sickle cell anemia, a safety feasibility trial demonstrated that hydroxyurea was safe and well tolerated in children >5 yr of age. No clinical adverse events were identified in this study; the primary toxicities were limited to myelosuppression that reversed on cessation of the drug. In addition, infants treated with hydroxyurea experienced fewer episodes of pain, dactylitis, and ACS; were hospitalized less frequently; and less often required a blood transfusion. Despite taking a myelosuppressive agent, the infants treated with hydroxyurea did not experience increased rates of bacteremia or serious infection. Current recommendations are that all children with sickle cell anemia should be offered hydroxyurea beginning at 9 mo of age.
Hydroxyurea may be indicated for other sickle cell–related complications, especially in patients who are unable to tolerate other treatments. For patients who either will not or cannot continue blood transfusion therapy to prevent recurrent stroke, hydroxyurea therapy may be a reasonable alternative. The trial assessing the efficacy of hydroxyurea as an alternative to transfusions to prevent a 2nd stroke was terminated early after the data safety and monitoring found an increased stroke rate in the hydroxyurea arm compared to the transfusion arm (0 vs 7 [10%]). Hydroxyurea alone is inferior to transfusion therapy for secondary stroke prevention in patients who do not have contraindications to ongoing transfusions.
The long-term toxicity associated with initiating hydroxyurea in very young children has not yet been established. However, all evidence to date suggests that the benefits far outweigh the risks. For these reasons, very young children starting hydroxyurea require well-informed parents and medical care by pediatric hematologists, or at least co-management by a physician with expertise in immunosuppressive medications. The typical starting dose of hydroxyurea is 15-20 mg/kg once daily, with an incremental dosage increase every 8 wk of 5 mg/kg, and if no toxicities occur, up to a maximum of 35 mg/kg per dose. The infant hydroxyurea study found young children could safely be started at 20 mg/kg/day without increased toxicity. Achievement of the therapeutic effect of hydroxyurea can require several months, and for this reason, initiating hydroxyurea to address short-term symptom relief is not optimal. We prefer to introduce the concept to parents within the 1st yr of life, preferably by 9 mo; provide literature that describes both the pros and cons of starting hydroxyurea in children with severe symptoms of sickle cell disease; and educate parents on starting hydroxyurea in asymptomatic children as a preventive therapy for repetitive pain and ACS events. Other effects of hydroxyurea that may vary include an increase in the total Hb level and a decrease in the TCD velocity.
The FDA has approved oral L-glutamine, used as an add on to hydroxyurea, for patients ≥5 yr. L-glutamine has been shown to reduce hospitalizations and sickle cell crisis.
Hematopoietic Stem Cell Transplantation
The only cure for sickle cell anemia is transplantation with human leukocyte antigen (HLA)–matched hematopoietic stem cells from a sibling or unrelated donor. Clinical trials are underway to determine whether gene therapy or gene editing therapy is a safe, effective, long-term cure for those with sickle cell anemia. The most common indications for transplant are recurrent ACS, stroke, and abnormal TCD. Sibling-matched stem cell transplantation has a lower risk for graft-versus-host disease than unrelated donors. Surveys suggest that younger children may have lower morbidity and mortality. However, few children have suitable sibling donors. Stem cell transplantation using an unrelated but well-matched donor remains a focus of clinical research. The decision to consider unrelated transplantation should involve appropriate consultation and counseling from physicians with expertise in sickle cell transplantation.
Stem cell transplantation for children with sickle cell disease who have a genetically matched sibling and few complications is not routinely performed. The use of hydroxyurea has dramatically decreased the disease burden for the patient and family, with far fewer hospitalizations for pain or ACS episodes and less use of blood transfusions. Furthermore, the field of stem cell transplantation is progressing so rapidly that nonsibling donor, including haploidentical, transplantation and gene therapy studies are underway. Transplant-related complications caused by conditioning regimens may be decreased by using low-intensity, nonmyeloablative HLA-matched sibling, allogenic stem cell transplantation.
Red Blood Cell Transfusions
RBC transfusions are used frequently both in the treatment of acute complications and to prevent acute or recurrent complications. Typically, short-term transfusions are used to prevent progression of acute complications such as ACS, aplastic crisis, splenic sequestration, and acute stroke, as well as to prevent surgery-related ACS. RBC transfusions are not recommended for uncomplicated acute pain events. Select RBC volumes judiciously to avoid high posttransfusion Hb levels and hyperviscosity. Long-term or chronic transfusion therapy is used to prevent 1st stroke in patients with abnormal TCD or MRI findings (silent stroke), recurrent stroke, or recurrent ACS. Patients with sickle cell disease are at increased risk of developing alloantibodies to less common RBC surface antigens after receiving even a single transfusion. In addition to standard cross matching for major blood group antigens (A, B, O, RhD), more extended matching should be performed to identify donor units that are C-, E-, and Kell-antigen matched. Some centers have begun to perform full RBC antigen phenotyping or genotyping for patients receiving chronic blood transfusions, in order to have the red cell units least likely to result in alloimmunization available for these patients.
Three methods of blood transfusion therapy are used in the management of acute and chronic complications associated with sickle cell disease: automated erythrocytapheresis, manual exchange transfusion (phlebotomy of a set amount of patient's blood followed by rapid administration of donated packed RBCs), and simple transfusion. The decision on which method to use depends on the patient's pretransfusion Hb level, the clinical indication, RBC alloimmunization, and transfusional iron overload. Automated erythrocytapheresis is the preferred method for patients requiring chronic blood transfusion therapy because there is a minimum net iron balance after the procedure, followed by manual exchange transfusion. However, this method requires technical expertise, special machines, and good patient venous access. Manual exchange is more accessible. However, both methods may expose the patient to more red cell units and possible alloimmunization. Simple transfusion therapy may lower donor exposure but may result in higher net iron burden when compared to erythrocytapheresis or exchange transfusion.
Preparation for surgery for children with sickle cell disease requires a coordinated effort from the hematologist, surgeon, anesthesiologist, and primary care provider. Historically, ACS was associated with general anesthesia in patients with sickle cell disease. Blood transfusion prior to surgery for children with sickle cell disease is recommended to raise Hb level preoperatively to no more than 10 g/dL, to avoid ACS development. Because of better general perioperative care and the use of long-term therapies such as hydroxyurea and chronic transfusions, the decision to transfuse before general anesthesia should be made in conjunction with the medical team who provides sickle cell disease–related care for the patient. When preparing a child with sickle cell disease for surgery with a simple blood transfusion, caution must be used not to elevate Hb level beyond 10 g/dL because of the risk of hyperviscosity syndrome. For children with milder forms of sickle cell disease, such as HbSC or HbSβ-thalassemia, a decision must be made on a case-by-case basis as to whether an exchange transfusion is warranted, because a simple transfusion may raise the hemoglobin to an unacceptable level.
Iron Overload
The primary toxic effect of blood transfusion therapy relates to excessive iron stores or iron overload, which can result in organ damage and premature death. Excessive iron stores develop after 100 mL/kg of red cell transfusion, or about 10 transfusions. The assessment of iron overload in children receiving regular blood transfusions is difficult. The most common and least invasive method of estimating total body iron involves serum ferritin levels. Ferritin measurements have significant limitations in their ability to estimate iron stores for several reasons, including, but not limited to, elevation during acute inflammation and poor correlation with excessive iron in specific organs after 2 yr of regular blood transfusion therapy. MRI of the liver has proved to the most effective approach for assessment of iron stores. The imaging strategy is more accurate than serum ferritin in measuring heart and liver iron content. MRI T2* and MRI R2 and R2* sequences are used to estimate iron levels in the heart and liver. The standard for iron assessment previously was liver biopsy, which is an invasive procedure exposing children to the risk of general anesthesia, bleeding, and pain. Liver biopsy alone does not accurately estimate total body iron because iron deposition in the liver is not homogeneous and varies among the affected organs; that is, the amount of iron found in the liver is not equivalent to cardiac tissues. The major advantage of a liver biopsy is that histologic assessment of the parenchyma can be ascertained along with appropriate staging of suspected pathology, particularly cirrhosis.
The primary treatment of transfusion-related iron overload requires iron chelation using medical therapy. In the United States, 3 chelating agents are approved for use in transfusional iron overload. Deferoxamine is administered subcutaneously 5 of 7 nights/wk for 10 hr a night. Deferasirox is taken by mouth daily, and deferiprone is available in tablets taken orally twice a day. The Food and Drug Administration (FDA) approved deferasirox, the newest oral chelator, in 2005 for use in patients age ≥2 yr. A pill formulation of deferasirox is available that does not require mixing before oral administration. Deferiprone is an older oral chelator that has been widely used outside the United States for many years and was approved by the FDA in 2011, but requires weekly CBC monitoring because of neutropenia risk throughout therapy. Transfusion-related excessive iron stores in children with sickle cell disease should be managed by a physician with expertise in chelation therapy because of the risk of significant toxicity from available chelation therapies.
Other Sickle Cell Syndromes
The most common sickle cell syndromes besides HbSS are HbSC, HbSβ0 -thalassemia, and HbSβ+ -thalassemia. The other syndromes—HbSD, HbSOArab , HbSHPFH, HbSE, and other variants—are much less common. Patients with HbSβ0 -thalassemia have a clinical phenotype similar to those with HbSS. In the red cells of patients with HbSC, crystals of HbC interact with membrane ion transport, dehydrating RBCs and inducing sickling. Children who have HbSC disease can experience the same symptoms and complications as those with severe HbSS disease, but less frequently. Children with HbSC have increased incidence of retinopathy, chronic hypersplenism, and acute splenic sequestration over the life span. The natural history of the other sickle cell syndromes is variable and difficult to predict because of the lack of systematic evaluation.
There is no validated model that can predict the clinical course of an individual with sickle cell disease. A patient with HbSC can have a more severe clinical course than a patient with HbSS. Management of end-organ dysfunction in children with sickle cell syndromes requires the same general principles as managing patients with sickle cell anemia; however, each situation should be managed on a case-by-case basis and requires consultation with a pediatric hematologist.
Anticipatory Guidance
Children with sickle cell disease should receive general health maintenance as recommended for all children, with special attention to disease-specific guidance and infection prevention education. In addition to counseling regarding adherence to penicillin and a vaccination schedule, patients, parents and caregivers should be instructed to seek immediate medical attention for all febrile illness. In addition, early detection of acute splenic sequestration has been shown to decrease mortality. Therefore, parents and caregivers should be educated early and repeatedly about the importance of daily penicillin administration and correct palpation of the spleen.
Prophylactic Penicillin
Children with sickle cell anemia should receive prophylactic oral penicillin VK until at least 5 yr of age (125 mg twice daily up to age 3 yr, then 250 mg twice daily thereafter). No established guidelines exist for penicillin prophylaxis beyond 5 yr of age; some clinicians continue penicillin prophylaxis, and others recommend discontinuation. Continuation of penicillin prophylaxis should be continued beyond 5 yr in children with a history of pneumococcal infection because of the increased risk of a recurrent infection. An alternative for children who are allergic to penicillin is erythromycin ethylsuccinate.
Immunizations
In addition to penicillin prophylaxis, routine childhood immunizations, as well as the annual administration of influenza vaccine, are highly recommended. Children with sickle cell disease develop functional asplenia and also require immunizations to protect against encapsulated organisms, including additional pneumococcal and meningococcal vaccinations. The U.S. Centers for Disease Control and Prevention (CDC) provides vaccination guidelines at https://www.cdc.gov/vaccines/hcp/acip-recs/index.html .
Spleen Palpation
Splenomegaly is a common complication of sickle cell disease, and splenic sequestration can be life threatening. Parents and primary caregivers should be taught how to palpate the spleen to determine if the spleen is enlarging starting at the 1st visit, with reinforcement at subsequent visits. Parents should also demonstrate spleen palpation to the provider.
Transcranial Doppler Ultrasound
Primary stroke prevention using TCD has resulted in a decrease in the prevalence of overt stroke among children with sickle cell anemia. Children with HbSS or HbSβ0 -thalassemia should be screened annually with TCD starting at age 2 yr. TCD is best performed when the child is quietly awake and in his or her usual state of health. TCD measurements may be falsely elevated or decreased in the settings of acute anemia, sedation, pain, fever, or immediately after blood transfusions. Screening should occur annually from age 2-16 yr. Abnormal values should be repeated within 2-4 wk to identify patients at greatest risk of overt stroke. Conditional values should be repeated within 3 mo, and normal values should be repeated annually. Routine neuroimaging with MRI in asymptomatic patients requires consultation with a pediatric hematologist or neurologist with expertise in sickle cell disease.
Hydroxyurea
Recommendations provided in the 2014 NHLBI Expert Panel Report include offering hydroxyurea therapy to all children with sickle cell anemia starting at 9 mo of age regardless of clinical symptoms. Monitoring children receiving hydroxyurea is labor intensive. Hydroxyurea is a chemotherapeutic agent, and patients receiving this agent require the same level of nursing and physician oversight as any child with cancer receiving chemotherapy. The parents must be educated about the consequences of therapy, and when ill, children should be promptly evaluated. Starting doses should be approximately 20 mg/kg/day. CBC with differential and reticulocyte count should be checked within 4 wk after initiation of therapy or any dose change to monitor for hematologic toxicity, then every 8-12 wk. Dose escalation should be based on clinical and laboratory parameters. If necessary, dose increases should be in 5 mg/kg/day increments to a maximum of 35 mg/kg/day.
While receiving hydroxyurea, steady-state absolute neutrophil count should be approximately 2,000/µL or higher and platelet count should be 80,000/µL or higher. Younger children may tolerate lower absolute neutrophil counts while receiving hydroxyurea. Holding hydroxyurea and adjusting to lower doses may be required for neutropenia and thrombocytopenia. Hydroxyurea is a pregnancy class D medication, and adolescents should be counseled regarding methods to prevent pregnancy while taking this medication. Close monitoring of the patient requires a commitment by the parents and the patient as well as diligence by a physician to identify toxicity early. Information is scarce regarding the impact of hydroxyurea on fertility, although hydroxyurea has been shown to further reduce sperm count in males with sickle cell disease in several case reports, suggesting that this effect may be reversible once hydroxyurea is discontinued.
Red Cell Transfusion Therapy
At the initiation of blood transfusion therapy, children with sickle cell disease should have testing to identify the presence of alloantibodies and RBC phenotyping or genotyping, which is performed to identify the best matched blood. Red cell units selected should be extended antigen-matched for C, E, and K, when feasible. Goals of transfusion for acute events should be established before initiating therapy, including target posttransfusion Hb level and HbS percentage, or both. For children receiving chronic transfusion therapy, pretransfusion HbS goals should be defined; the most common goal is <30%. Posttransfusion Hb values should be targeted to avoid hyperviscosity. Children, parents, and caregivers should be educated about the symptoms of delayed hemolytic transfusion reactions. Any child with sickle cell disease with a recent history of red cell transfusion who presents with pain, dark urine, increased scleral icterus, or symptoms of worsening anemia should be screened for a delayed hemolytic transfusion reaction after consultation with the blood bank. Children meeting criteria for chronic transfusion therapy should receive annual evaluation for transfusion-transmitted infections, including hepatitis B, hepatitis C, and HIV. After receiving 100 mL/kg RBC transfusions, regular assessments of iron overload should begin, usually including measurements of serum ferritin and assessments for hepatic and cardiac iron every 1-2 yr. For children requiring chelation therapy, an audiogram should be performed annually and monitoring of liver function and pituitary function performed regularly because of iron deposition.
Pulmonary and Asthma Screening
Pulmonary complications of sickle cell disease are common and life threatening. Asthma is common in children with sickle cell disease, and thus evaluation for asthma symptoms and asthma risk factors should be performed routinely, particularly given the high morbidity and mortality. All children should receive annual screening for signs and symptoms of lower airway disease, such as nighttime cough and exercise-induced cough. In children with symptoms consistent with lower airway disease, consultation with an asthma specialist should be considered. Pulse oximetry readings should be performed during well visits to identify children with abnormally low daytime oxygen saturation. For children with snoring, daytime somnolence, and symptoms associated with obstructive sleep apnea syndrome (OSAS), sleep studies should be performed as necessary.
Retinopathy
Effective therapy is available for retinopathy associated with sickle cell disease. Although all patients are at risk for development of retinopathy, those with SC are at very high risk. Patients should receive annual screening by an ophthalmologist to identify vascular changes that would benefit from laser therapy. Although changes may occur earlier, children with sickle cell disease should begin annual screening no later than age 10 yr.
Renal Disease
Sickle cell–associated renal disease starts in infancy and may not become clinically evident until adulthood. Chronic kidney disease is common in adults with sickle cell disease, with high morbidity and mortality. Screening protocols for early signs of sickle nephropathy in children have not been adopted due to lack of data. However, when creatinine elevation, microalbuminuria, or macroalbuminuria is detected, a nephrologist should be consulted to determine next steps for further evaluation and possible treatment. The age to begin screening for proteinuria has not been defined, but some experts recommend screening annually after at least 10 yr, if not sooner. If proteinuria is detected, urine studies should be repeated with an early-morning urine collection; if the protein remains elevated, the patient should be referred to a pediatric nephrologist. Males with sickle cell disease should also receive counseling regarding the diagnosis and treatment of priapism. Because of the high frequency of enuresis beyond early childhood, approximately 9% between 18 and 20 yr of age, parents and caregivers should be educated about the prolonged nature of enuresis in this disease. OSAS is associated with an increased prevalence of enuresis in sickle cell disease. Unfortunately, no evidence-based therapies have been developed to treat enuresis in children and young adults with sickle cell disease. In children with enuresis who have symptoms and clinical features of OSAS, referral to specialists for evaluation is recommended.
Echocardiography
Echocardiography is a screening tool to identify individuals with sickle cell disease who have pulmonary artery hypertension. No evidence currently shows that children with sickle cell disease and elevated tricuspid jet velocity >2.5 cm/sec have an increased rate of mortality. Studies in adults with sickle cell disease have found that echocardiography is insensitive at identifying individuals truly at risk for pulmonary hypertension, although an elevated tricuspid velocity measurement may still be a risk factor for premature death in adults with sickle cell disease. The current recommendation is to refer those with severe cardiopulmonary symptoms from associated pulmonary artery hypertension to a pediatric cardiologist for a more formal evaluation.
Additional Screening
Patients with sickle cell disease are at increased risk for behavioral health issues, including anxiety and depression. Screening should be performed at routine and acute visits. Avascular necrosis of the hips and shoulders is increased in patients with sickle cell disease and may be identified early on routine physical exam. Plain radiographs may not detect early disease; thus, when AVN is suspected and plain films are normal, MRI should be obtained. When AVN is confirmed, patients should be referred promptly to orthopedics and physical therapy.
Bibliography
Abboud MR, Yim E, Musallam KM, et al. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from STOP II. Blood . 2011;118(4):894–898.
Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med . 2017;376(5):429–438.
Ballas SK, Bauserman RL, McCarthy WF, et al. Hydroxyurea and acute painful crises in sickle cell anemia: effects on hospital length of stay and opioid utilization during hospitalization, outpatient acute care contacts, and at home. J Pain Symptom Manage . 2010;40(6):870–882.
Baskin MN, Goh XL, Heeney MM, et al. Bacteremia risk and outpatient management of febrile patients with sickle cell disease. Pediatrics . 2013;131(6):1035–1041.
Belmont AP, Nossair F, Brambilla D, et al. Safety of deep sedation in young children with sickle cell disease: a retrospective cohort study. J Pediatr . 2015;166:1226–1232.
Berger E, Saunders N, Wang L, et al. Sickle cell disease in children. Arch Pediatr Adolesc Med . 2009;163:251255.
Beyer JE, Simmons LE, Woods GM, et al. A chronology of pain and comfort in children with sickle cell disease. Arch Pediatr Adolesc Med . 1999;153(9):913–920.
Bolanos-Meade J, Fuchs EJ, Luznik L, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood . 2012;120(22):4285–4291.
Brandow AM, Zappia KJ, Stucky CL. Sickle cell disease: a natural model of acute and chronic pain. Pain . 2017;158:S79–S84.
Brousse V, Elie C, Benkerrou M, et al. Acute splenic sequestration crisis in sickle cell disease: cohort study of 190 paediatric patients. Br J Haematol . 2012;156(5):643–648.
Brousseau DC, McCarver DG, Drendel AL, et al. The effect of CYP2d6 polymorphisms on the response to pain treatment for pediatric sickle cell pain crisis. J Pediatr . 2007;150(6):623–626.
Brousseau DC, Panepinto JA, Nimmer M, et al. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol . 2010;85(1):77–78.
Buchanan ID, Woodward M, Reed GW. Opioid selection during sickle cell pain crisis and its impact on the development of acute chest syndrome. Pediatr Blood Cancer . 2005;45(5):716–724.
Burnett MW, Bass JW, Cook BA. Etiology of osteomyelitis complicating sickle cell disease. Pediatrics . 1998;101(2):296–297.
Dallas MH, Triplett B, Shook DR, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant . 2013;19(5):820–830.
Davis BA, Allard S, Qureshi A, et al. British committee for standards in haematology: guidelines on red cell transfusion in sickle cell disease. Part I. Principles and laboratory aspects. Br J Haematol . 2017;176:179–191.
DeBaun MR, Gordon M, McKinstry MJ, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med . 2014;371:699–710.
DeBaun MR, Sarnaik SA, Rodeghier MJ, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, gender and relative high systolic blood pressure. Blood . 2012;119:3684–3690.
DeBaun MR, Struck RC. The intersection between asthma and acute chest syndrome in children with sickle-cell anaemia. Lancet . 2016;387:2545–2552.
DeBaun MR, Telfair J. Transition and sickle cell disease. Pediatrics . 2012;130:926–935.
Dever DP, Bal RO, Reinisch A, et al. CRISPR/cas9 β-globin gene targeting in human haematopoietic stem cells. Nature . 2016;539(7629):384–389.
Dowling MM, Quinn CT, Plumb P, et al. Acute silent cerebral ischemia and infarction during acute anemia in children with and without sickle cell disease. Blood . 2012;120:3891–3897.
Ellison AM, Ota KV, McGowan KL, et al. Pneumococcal bacteremia in a vaccinated pediatric sickle cell disease population. Pediatr Infect Dis J . 2012;5:534–536.
Field JJ, Austin PF, An P, et al. Enuresis is a common and persistent problem among children and young adults with sickle cell anemia. Urology . 2008;72:81–84.
Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest . 2007;117:850–858.
Gillis VL, Senthinathan A, Dzingina M, et al. Management of an acute painful sickle cell episode in hospital: summary of NICE guidance. BMJ . 2012;344:e4063.
Gladwin MT. Cardiovascular complications and risk of death in sickle-cell disease. Lancet . 2016;387:2565–2572.
Gluckman E, Cappelli B, Bernaudin F, et al. The pediatric working party of the European society for blood and marrow transplantation, and the center for international blood and marrow transplant research. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood . 2017;129:1548–1556.
Halasa NB, Shankar SM, Talbot TR, et al. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clin Infect Dis . 2007;44:1428–1433.
Hines PC, McKnight TP, Seto W, et al. Central nervous system events in children with sickle cell disease presenting acutely with headache. J Pediatr . 2011;159:472–478.
Hsieh MM, Fithugh CD, Weitzel P, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA . 2014;312:48–56.
Hoppe C. Newborn screening for hemoglobin disorders. Hemoglobin . 2011;35(5/6):556–564.
Huang X, Wang Y, Yan W, et al. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient IPSCs after genome editing of the sickle point mutation. Stem Cells . 2015;33(5):1470–1479.
Hulbert ML, Scothorn DJ, Panepinto JA, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr . 2006;149(5):710–712.
Jain R, Sawhney S, Rizvi SG. Acute bone crises in sickle cell disease: the T1 fat-saturated sequence in differentiation of acute bone infarcts from acute osteomyelitis. Clin Radiol . 2008;63:59–70.
Jordan LC, Casella JF, DeBaun MR. Prospects for primary stroke prevention in children with sickle cell anaemia. Br J Haematol . 2012;157:14–25.
Kassim AA, Sharma D. Hematopoietic stem cell transplantation for sickle cell disease: the changing landscape. Hematol Oncol Stem Cell Ther . 2017;10(4):259–266.
Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest . 2017;127:750–760.
King LGC, Bortolusso-Ali S, Cunningham CA, Reid MEG. Impact of a comprehensive sickle cell center on early childhood mortality in a developing country: the jamaican experience. J Pediatr . 2015;167:702–705.
Knight-Madden JM, Hambleton JR. Inhaled bronchodilators for acute chest syndrome in people with sickle cell disease. Cochrane Database Syst Rev . 2012;(7) [CD003733].
Kuhn JP, Slovis TL, Haller JO. ed 11. Mosby: St Louis; 2008:1087. Caffey's pediatric diagnostic imaging . vol 2.
Kwiatkowski JL, Cohen AR, Garro J, et al. Transfusional iron overload in children with sickle cell anemia on chronic transfusion therapy for secondary stroke prevention. Am J Hematol . 2012;87:221–223.
Lehmann GC, Bell TR, Kirkham FJ, et al. Enuresis associated with sleep disordered breathing in children with sickle cell anemia. J Urol . 2012;188(4 Suppl):1572–1576.
Lettre G, Bauer DE. Feral haemoglobin in sickle-cell disease: from genetic epidemiology to new therapeutic strategies. Lancet . 2016;387:2554–2562.
Madigan C, Malik P. Pathophysiology and therapy for haemoglobinopathies. Part I. Sickle cell disease. Expert Rev Mol Med . 2006;8:1–23.
Mahadeo KM, Oyeku S, Taragin B, et al. Increased prevalence of osteonecrosis of the femoral head in children and adolescents with sickle-cell disease. Am J Hematol . 2011;86(9):806–808.
Mehari A, Gladwin MT, Tian X, et al. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA . 2012;307:1254–1256.
Meier ER, Miller JL. Sickle cell disease in children. Drugs . 2012;72:895–906.
Merritt AL, Haiman C, Henderson SO. Myth: blood transfusion is effective for sickle cell anemia-associated priapism. CJEM . 2006;8:119–122.
Miller AC, Gladwin MT. Pulmonary complications of sickle cell disease. Am J Respir Crit Care Med . 2012;185:1154–1165.
Miller ST, Kim HY, Weiner D, et al. Inpatient management of sickle cell pain: a “snapshot” of current practice. Am J Hematol . 2012;87:333–336.
Naik RP, Derebail VK, Grams ME, et al. Association of sickle cell trait with chronic kidney disease and albuminuria in African Americans. JAMA . 2014;312:2115–2124.
National Heart, Lung and Blood Institute. 2014 Expert Panel report on the evidence-based management of sickle cell disease . www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle-cell-disease-report.pdf .
Nicholson GT, Hsu DT, Colan SD, et al. Coronary artery dilation in sickle cell disease. J Pediatr . 2011;159:789–794.
Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of L-glutamine in sickle cell disease. N Engl J Med . 2018;379(3):226–235.
Nordness ME, Lynn J, Zacharisen MC, et al. Asthma is a risk factor for acute chest syndrome and cerebral vascular accidents in children with sickle cell disease. Clin Mol Allergy . 2005;3:2.
Norris CF, Smith-Whitley K, McGowan KL. Positive blood cultures in sickle cell disease: time to positivity and clinical outcome. J Pediatr Hematol Oncol . 2003;25(5):390–395.
Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood . 1998;91:288–294.
Panepinto JA, Bonner M. Health-related quality of life in sickle cell disease: past, present, and future. Pediatr Blood Cancer . 2012;59:377–385.
Panepinto JA, Brousseau DC, Hillery CA, et al. Variation in hospitalizations and hospital length of stay in children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer . 2005;44:182–186.
Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med . 2011;365:44–52.
Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet . 2013;381:142–151.
Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med . 2017;376(16):1561–1572.
Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: rates and risk factors. N Engl J Med . 1991;325(1):11–16.
Quillen K, Lane C, Hu E, et al. Prevalence of ceftriaxone-induced red blood cell antibodies in pediatric patients with sickle cell disease and human immunodeficiency virus infection. Pediatr Infect Dis J . 2008;27:357–358.
Quinn CT, McKinstry RC, Dowling MM, et al. Acute silent cerebral ischemic events in children with sickle cell anemia. JAMA Neurol . 2013;70:58–65.
Quinn CT, Rogers ZR, McCavit TL, et al. Improved survival of children and adolescents with sickle cell disease. Blood . 2010;115:3447–3452.
Reagan MM, DeBaun MR, Frei-Jones MJ. Multi-modal intervention for the inpatient management of sickle cell pain significantly decreases the rate of acute chest syndrome. Pediatr Blood Cancer . 2011;56:262–266.
Rezende PV, Viana MB, Murao M, et al. Acute splenic sequestration in a cohort of children with sickle cell anemia. J Pediatr (Rio J) . 2009;85:163–169.
Ribeil JA, Hacein-Bay-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med . 2017;376(9):848–855.
Ridha A, Khan A, Al-Abayechi S, et al. Acute compartment syndrome secondary to rhabdomyolysis in a sickle cell trait patient. Lancet . 2014;384:2172.
Rogers ZR. Priapism in sickle cell disease. Hematol Oncol Clin North Am . 2005;19:917–928 [viii].
Scothorn DJ, Price C, Schwartz D, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J Pediatr . 2002;140:348–354.
Smith WR, Bauserman RL, Ballas SK, et al. Climatic and geographic temporal patterns of pain in the multicenter study of hydroxyurea. Pain . 2009;146:91–98.
Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med . 2008;148:94–101.
Smith-Whitley K, Thompson AA. Indications and complications of transfusions in sickle cell disease. Pediatr Blood Cancer . 2012;59:358–364.
Smith-Whitley K, Zhao H, Hodinka RL, et al. Epidemiology of human parvovirus B19 in children with sickle cell disease. Blood . 2004;103:422–427.
Switzer JA, Hess DC, Nichols FT, et al. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol . 2006;5:501–512.
Thornburg CD, Files BA, Luo Z, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood . 2012;120:4304–4310.
Umans H, Haramati N, Flusser G. The diagnostic role of gadolinium enhanced MRI in distinguishing between acute medullary bone infarct and osteomyelitis. Magn Reson Imaging . 2000;18:255–262.
Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood . 1997;89(5):1787–1792.
Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet . 2011;377:1663–1672.
Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia: TCD with transfusions changing to hydroxyurea (TWiTCH): a multicenter, open-label, phase 3, non-inferiority trial. Lancet . 2016;387:661–670.
Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet . 2017;390:311–323.
Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood . 2012;119:3925–3932.
Wilimas JA, Flynn PM, Harris S, et al. A randomized study of outpatient treatment with ceftriaxone for selected febrile children with sickle cell disease. N Engl J Med . 1993;329:472–476.
Yang YM, Shah AK, Watson M, et al. Comparison of costs to the health sector of comprehensive and episodic health care for sickle cell disease patients. Public Health Rep . 1995;110:80–86.
Sickle Cell Trait (Hemoglobin AS)
Kim Smith-Whitley
The prevalence of sickle cell trait varies throughout the world; in the United States the incidence is 7–10% of African Americans. Because all state newborn screening programs include sickle cell disease, for most children, sickle cell trait is first identified on their newborn screen. Communication of sickle cell trait status from infancy to young adulthood for the affected individual, family, and healthcare providers is often inconsistent, and many young adults are unaware of their sickle cell trait status.
The production of HbS is influenced by the number of α-thalassemia genes present and the amount of HbS. By definition, among individuals with sickle cell trait, the HbS level is <50%. The life span of people with sickle cell trait is normal, and serious complications are extremely rare. The CBC is within the normal range (Fig. 489.5B ). Hemoglobin analysis is diagnostic, revealing a predominance of HbA, typically >50%, and HbS <50%. Rare complications of sickle cell trait may exist, but published data do not support this concern, largely because of poorly designed clinical studies. Sickle cell trait is reported to be associated with exertional rhabdomyolysis in military recruits, and possibly with sudden death during rigorous exercise. However, whether these reports establish sickle cell trait as a risk factor that is nonmodifiable by other genetic factors remains unclear. Other complications reported with sickle cell trait include splenic infarction at high altitude, hematuria, hyposthenuria, deep vein thrombosis, and susceptibility to progressive eye injury after hyphema (Table 489.5 ). Renal medullary carcinoma has been reported almost exclusively in individuals with sickle cell trait and occurs predominantly in young people.
Children with sickle cell trait do not require limitations on physical activities. Sudden death in persons with sickle cell trait while exercising under extreme conditions is most likely associated with a 2nd genetic factor and/or environmental factors and not the presence of sickle cell trait itself. However, if exertional rhabdomyolysis is identified, evaluation by metabolism and cardiology should be considered. No causal pathway has been implicated for the presence of sickle cell trait and sudden death. All patients with sickle cell trait who participate in rigorous athletic activities should receive maximum hydration and appropriate rest during exertion, as would be the precautionary steps for all athletes, particularly when participating in hot, humid conditions. The presence of sickle cell trait should never be a reason to exclude a person from athletic participation, but rather should serve as an indication that prudent surveillance is necessary to ensure appropriate hydration and prevention of exhaustion from heat or other strenuous exercise. If athletes are to be screened for sickle cell trait, the appropriate procedure is testing using a hemoglobin electrophoresis followed by genetic counseling, along with the knowledge that genetic information may provide opportunities to challenge paternity. Such situations are typically handled by a pediatrician or hematologist accustomed to providing both a balanced approach to genetic counseling and addressing the challenges about paternity.
Other Hemoglobinopathies
Kim Smith-Whitley
Hemoglobin C
The mutation for HbC is at the same site as in HbS, with substitution of lysine instead of valine for glutamine. In the United States, hemoglobin C trait (HbAC ) occurs in 1 : 40 and homozygous hemoglobin C disease (HbCC ) occurs in 1 : 5,000 African Americans. HbAC is asymptomatic. HbCC can result in mild anemia, splenomegaly, and cholelithiasis; rare cases of spontaneous splenic rupture have been reported. Splenic dysfunction does not occur. This condition is usually diagnosed through newborn screening programs. HbC crystallizes, disrupting the red cell membrane, and HbC crystals may be visible on peripheral smear (see Fig. 489.5C ).
Hemoglobin E
HbE is an abnormal hemoglobin resulting from a qualitative mutation in the β-globin gene and is the 2nd most common globin mutation worldwide. Patients may have asymptomatic hemoglobin E trait (HbAE ) or benign homozygous hemoglobin E disease (HbEE ). Compound heterozygous hemoglobin E/β-thalassemia produces clinical phenotypes ranging from moderate to severe anemia, depending on the β-thalassemia mutation. In California, HbE/β-thalassemia is found almost exclusively in persons of Southeast Asian descent, with a prevalence of 1 : 2,600 births.
Hemoglobin D
At least 16 variants of HbD exist. HbD-Punjab (Los Angeles) is a rare hemoglobin that is seen in 1-3% of Western Indians and in some Europeans with Asian-Indian ancestry and produces symptoms of sickle cell disease when present in combination with HbS. Heterozygous HbD or hemoglobin D trait (HbAD ) is clinically silent. Homozygous hemoglobin D disease (HbDD ) produces a mild to moderate anemia with splenomegaly.
Unstable Hemoglobin Disorders
Kim Smith-Whitley
At least 200 rare unstable hemoglobins have been identified; the most common is Hb Köln . Most patients seem to have de novo mutations rather than inherited hemoglobin disorders. The best-studied unstable hemoglobins are the ones leading to hemoglobin denaturation from mutations affecting heme binding. The denatured hemoglobin can be visualized during severe hemolysis or after splenectomy as Heinz bodies . Unlike the Heinz bodies seen after toxic exposure, in unstable hemoglobins, Heinz bodies are present in reticulocytes and older RBCs (see Fig. 489.5D ). Heterozygotes are asymptomatic.
Children with homozygous gene mutations can present in early childhood with anemia and splenomegaly or with unexplained hemolytic anemia. Hemolysis is increased with febrile illness and with the ingestion of oxidant medications (similar to glucose-6-phosphate dehydrogenase [G6PD] deficiency [see Chapter 490.3 ]) with some unstable hemoglobins. If the spleen is functional, the blood smear can appear almost normal or have only hypochromasia and basophilic stippling. A diagnosis may be made by demonstrating Heinz bodies, Hb instability, or an abnormal Hb analysis (although some unstable hemoglobins have normal mobility and are not detected on Hb analysis).
Treatment is supportive. Transfusion may be required during hemolytic episodes in severe cases. Oxidative drugs should be avoided, and folate supplementation may be helpful if dietary deficiency is a concern. Splenectomy may be considered in patients requiring recurrent transfusion or demonstrating poor growth, but the complications of splenectomy, including bacterial sepsis, risk of thrombosis, and risk of developing pulmonary hypertension, should be considered before surgery.
Abnormal Hemoglobins With Increased Oxygen Affinity
Kim Smith-Whitley
More than 110 high-affinity hemoglobins have been characterized. These mutations affect the state of Hb configuration during oxygenation and deoxygenation. Hemoglobin changes structure when in the oxygenated vs the deoxygenated state. The deoxygenated state is termed the T (tense) state and is stabilized by 2,3-diphosphoglycerate. When fully oxygenated, hemoglobin assumes the R (relaxed) state . The exact molecular interactions between these 2 states are unknown. High-affinity hemoglobins contain mutations that either stabilize the R form or destabilize the T form. The interactions between the R and T forms are complex, and the mechanisms of the mutations are not known. In most cases, the high-affinity hemoglobins can be identified by Hb analysis; approximately 20% must be characterized under controlled conditions where measurements are obtained with the P50 lowered to 9-21 mm Hg (normal: 23-29 mm Hg). The decreased P50 in these hemoglobins leads to an erythrocytosis with Hb levels of 17-20 g/dL. Levels of erythropoietin and 2,3-DPG are normal. Patients are usually asymptomatic and do not need phlebotomy. If phlebotomy is performed, oxygen delivery could be problematic because of the reduced number of Hb molecules to carry oxygen.
Abnormal Hemoglobins Causing Cyanosis
Kim Smith-Whitley
Abnormal hemoglobins causing cyanosis, also called structural methemoglobinemias , are rare. They are referred to as M hemoglobins and represent a group of hemoglobin variants that result from point mutations in one of the globin chains, α, β, or γ, located in the heme pocket; 13 known variants exist. These unstable hemoglobins lead to hemolytic anemia, most pronounced when the β-globin gene is affected. Clinically, these children are cyanotic from birth, without other signs or symptoms of disease, if the mutation is in the α-globin gene (HbM Boston, HbM Iwate, Hb Auckland). Infants with β-globin mutations become cyanotic later in infancy after the fetal hemoglobin switch (HbM Saskatoon, HbM Chile, HbM Milwaukee 1 and 2). The γ-chain mutations (HbF-M Fort Ripley, HbF-M Osaka, HbF Cincinnati, HbF Circleville, HbF Toms River, HbF Viseu) are all transient, presenting with cyanosis at birth, which resolves during the neonatal period after HbF production discontinues.
The abnormal M hemoglobins exhibit autosomal dominant inheritance and are diagnosed by Hb analysis. HbM variants may not be isolated reliably using Hb analysis (HPLC or IEF); consequently, diagnostic confirmation may require DNA sequencing or mass spectrometry. There is no specific treatment, and affected patients do not respond to treatments used for enzyme-deficient methemoglobinemia. Beyond cyanosis, individuals are otherwise asymptomatic and do not require additional monitoring. Children with the β-globin form should avoid oxidant drugs. Individuals with all forms have a normal life expectancy and pregnancy course.
Low-affinity hemoglobins have less cyanosis than the M hemoglobins. The amino acid substitutions destabilize the oxyhemoglobin and lead to decreased oxygen saturation. The best characterized are Hb Kansas, Hb Beth Israel, and Hb Denver. Hb analysis (IEF and HPLC techniques) may be normal in affected individuals. When clinically suspected, oxygen affinity studies reveal a right-shifted dissociation curve, and heat testing demonstrates unstable hemoglobin. Children present with mild cyanosis only.
Hereditary Methemoglobinemia
Kim Smith-Whitley
Hereditary methemoglobinemia is a clinical syndrome caused by an increase in the serum concentration of methemoglobin either as a result of congenital changes in hemoglobin synthesis or of metabolism leading to imbalances in reduction and oxidation of hemoglobin. The iron molecule in hemoglobin is normally in the ferrous state (Fe2+ ), which is essential for oxygen transport. Under physiologic conditions there is a slow, constant loss of electrons to released oxygen, and the ferric (Fe3+ ) form combines with water, producing methemoglobin (MetHb) . The newly formed MetHb has a reduced ability to bind oxygen.
Two pathways for MetHb reduction exist. The physiologic and predominant pathway is a reduced form of nicotinamide adenine dinucleotide (NADH)–dependent reaction catalyzed by cytochrome b5 reductase. This mechanism is >100-fold more efficient than the production of MetHb. The alternate pathway utilizes NAD phosphate generated by G6PD in the hexose monophosphate shunt and requires an extrinsic electron acceptor to be activated (i.e., methylene blue, ascorbic acid, riboflavin). In normal individuals, oxidation of hemoglobin to MetHb occurs at a slow rate, 0.5–3%, which is countered by MetHb reduction to maintain a steady state of 1% MetHb.
MetHb may be increased in the RBC because of exposure to toxic substances or to absence of reductive pathways, such as NADH-cytochrome b5 reductase deficiency. Toxic methemoglobinemia is much more common than hereditary methemoglobinemia (Table 489.6 ). Infants are exceptionally vulnerable to hemoglobin oxidation because their erythrocytes have half the amount of cytochrome b5 reductase seen in adults, fetal hemoglobin is more susceptible to oxidation than hemoglobin A, and the more alkaline infant gastrointestinal tract promotes the growth of nitrite-producing gram-negative bacteria. When MetHb levels are >1.5 g/24 hr, cyanosis is visible (15% MetHb); a level of 70% MetHb is lethal. The MetHb level is usually reported as a percentage of normal hemoglobin, and the toxic level is lower at a lower Hb level. Methemoglobinemia has been described in infants who ingested foods and water high in nitrates , who were exposed to aniline teething gels or other chemicals, and in some infants with severe gastroenteritis and acidosis. Methemoglobin can color the blood brown (Fig. 489.6 ). A patient with significant methemoglobinemia is cyanotic and does not respond to 100% oxygen. Arterial oxygen tension will be normal or elevated despite cyanosis, but blood oxygen saturation determined by multiwavelength co-oximetry will be low. Oxygen saturation calculated from arterial blood gas or pulse oximetry is misleading and inaccurate. Although pulse oximetry is usually lower than normal, it does not reflect the true degree of desaturation.
Hereditary Methemoglobinemia With Deficiency of NADH Cytochrome b5 Reductase
Kim Smith-Whitley
The first reported inherited disorder causing methemoglobinemia resulted from an enzymatic deficiency of NADH cytochrome b5 reductase, which was classified into 2 distinct phenotypes. In type I , the most common form, the deficiency of NADH cytochrome b5 activity is found only in erythrocytes, with other cell types unaffected. In type II the enzyme deficiency is present in all tissues and results in more significant symptoms beginning in infancy with encephalopathy, intellectual impairment, spasticity, microcephaly, and growth retardation, with death most often by 2 yr of age. Both types exhibit an autosomal recessive inheritance pattern.
Cyanosis varies in intensity with season and diet. The time of cyanosis onset also varies, appearing in some patients at birth and others as late as adolescence. Although as much as 50% of the total circulating hemoglobin may be in the form of nonfunctional MetHb, little or no cardiorespiratory distress occurs in these patients, except on exertion.
Daily oral treatment with ascorbic acid (200-500 mg/day in divided doses) gradually reduces the MetHb to approximately 10% of the total pigment and alleviates the cyanosis as long as therapy is continued. Chronic high doses of ascorbic acid have been associated with hyperoxaluria and renal stone formation. Ascorbic acid should not be used to treat toxic methemoglobinemia. When immediately available, poison control should be contacted to verify the most up-to-date therapeutic strategies. As with ascorbic acid, riboflavin uses the alternate pathway of MetHb reduction and is most effective when given in high doses (400 mg once daily). Methylene blue , administered intravenously (1-2 mg/kg initially), is used to treat toxic methemoglobinemia. An oral dose can be administered (100-300 mg/day) as maintenance therapy.
Methylene blue should not be used in patients with G6PD deficiency. This treatment is ineffective and can cause severe oxidative hemolysis. If methylene blue is given to a patient with G6PD deficiency, symptoms will not improve, and marked hemolysis has been reported within 24 hr of administration. Because G6PD deficiency status is rarely known at the time of treatment, a careful history should be elicited. When the history is negative for symptoms of G6PD deficiency, treatment with methylene blue should be initiated judiciously, and the patient should be closely monitored for improvement.
Syndromes of Hereditary Persistence of Fetal Hemoglobin
Kim Smith-Whitley
Hereditary persistence of fetal hemoglobin (HPFH ) syndromes are a form of thalassemia; mutations are associated with a decrease in the production of either or both β- and δ-globins. There is an imbalance in the α:non-α synthetic ratio characteristic of thalassemia. More than 20 variants of HPFH have been described. They are deletional, δβ0 (Black, Ghanaian, Italian), nondeletional (Tunisian, Japanese, Australian), linked to the β-globin–gene cluster (British, Italian-Chinese, Black), or unlinked to the β-globin–gene cluster (Atlanta, Czech, Seattle). The δβ0 forms have deletions of the entire δ- and β-globin gene sequences, and the most common form in the United States is the Black (HPFH 1 ) variant. As a result of the δ and β gene deletions, there is production only of γ-globin and formation of HbF. In the homozygous form, no manifestations of thalassemia are present. There is only HbF with very mild anemia and slight microcytosis. When inherited with other variant hemoglobins, HbF is elevated into the 20–30% range; when inherited with HbS, sickle cell disease is ameliorated, with fewer complications.
Thalassemia Syndromes
Janet L. Kwiatkowski
Thalassemia refers to a group of genetic disorders of globin-chain production in which there is an imbalance between the α-globin and β-globin chain production. β-Thalassemia syndromes result from a decrease in β-globin chains, which results in a relative excess of α-globin chains. β0 -thalassemia refers to the absence of production of the β-globin. When patients are homozygous for the β0 -thalassemia gene, they cannot make any normal β-globin chains (HbA). β+ -thalassemia indicates a mutation that makes decreased amounts of normal β-globin (HbA). β0 -thalassemia syndromes are generally more severe than β+ -thalassemia syndromes, but there is significant variability between the genotype and phenotype. β-thalassemia major , or transfusion-dependent thalassemia, refers to severe β-thalassemia that requires early transfusion therapy. β-thalassemia intermedia (or non–transfusion dependent) is a clinical diagnosis of a patient with a less severe clinical phenotype that usually does not require regular transfusion therapy in childhood. Many of these patients have at least 1 β+ -thalassemia mutation. β-thalassemia syndromes usually require a β-thalassemia mutation in both β-globin genes. Carriers with a single β-globin mutation are generally asymptomatic, except for microcytosis and mild anemia.
In α-thalassemia , there is an absence or reduction in α-globin production usually due to deletions of α-globin genes. Normal individuals have 4 α-globin genes; the more genes affected, the more severe the disease. α0 -thalassemia indicates no α-chains produced from that chromosome (− −/). α+ -thalassemia produces a decreased amount of α-globin chain from that chromosome (-alpha/).
The primary pathology in the thalassemia syndromes stems from the quantity of globin produced, whereas the primary pathology in sickle cell disease is related to the quality of β-globin produced.
Epidemiology
There are >200 different mutations resulting in absent or decreased globin production. Although most are rare, the 20 most common abnormal alleles constitute 80% of the known thalassemias worldwide; 3% of the world's population carries alleles for β-thalassemia, and in Southeast Asia 5–10% of the population carry alleles for α-thalassemia. In a particular region, there are fewer common alleles. In the United States, an estimated 2,000 persons have β-thalassemia major.
Pathophysiology
Two related features contribute to the sequelae of β-thalassemia syndromes : inadequate β-globin gene production leading to decreased levels of normal hemoglobin (HbA) and unbalanced α- and β-globin chain production leading to ineffective erythropoiesis. In β-thalassemia α-globin chains are in excess to non–α-globin chains, and α-globin tetramers (α4 ) are formed and appear as RBC inclusions. The free α-globin chains and inclusions are very unstable, precipitate in RBC precursors, damage the RBC membrane, and shorten RBC survival, leading to anemia and increased erythroid production (Table 489.7 ). This results in a marked increase in erythropoiesis, with early erythroid precursor death in the bone marrow. Clinically, this is characterized by a lack of maturation of erythrocytes and an inappropriately low reticulocyte count. This ineffective erythropoiesis and the compensatory massive marrow expansion with erythroid hyperactivity characterize β-thalassemia. Due to the low or absent production of β-globin, the α-chains combine with γ-chains, resulting in HbF (α2 γ2 ) being the dominant hemoglobin. In addition to the natural survival effect, the γ-globin chains may be produced in increased amounts, regulated by genetic polymorphisms. The δ-chain synthesis is not usually affected in β-thalassemia or β-thalassemia trait, and therefore patients have a relative or absolute increase in HbA2 production (α2 δ2 ).
Table 489.7
| THALASSEMIA | GLOBIN GENOTYPE | RED BLOOD CELL FEATURES | CLINICAL FEATURES | HEMOGLOBIN ANALYSIS |
|---|---|---|---|---|
| α-Thalassemia | ||||
| 1 Gene deletion | −,α/α,α | Normal | Normal | Newborn: Bart: 1–2% |
| 2 Gene deletion (α-thalassemia trait) | −,α/−,α −, −/α,α | Microcytosis, mild hypochromasia | Normal, mild anemia | Newborn: Bart: 5–10% |
| 3 Gene deletion hemoglobin H | −,−/−,α | Microcytosis, hypochromic | Mild anemia, transfusions not required | Newborn: Bart: 20–30% |
| 2 Gene deletion + Constant Spring | −,−/α,αConstant Spring | Microcytosis, hypochromic | Moderate to severe anemia, transfusion, splenectomy. | 2–3% Constant Spring, 10–15% HbH |
| 4 Gene deletion | −,−/−,− | Anisocytosis, poikilocytosis | Hydrops fetalis | Newborn: 89–90% Bart with Gower-1, Gower-2, and Portland |
| Nondeletional | α,α/α,αvariant | Microcytosis, mild anemia | Normal | 1–2% variant hemoglobin |
| β-Thalassemia | ||||
| β0 or β+ heterozygote: trait | β0 /A,β+ /A | Variable microcytosis, mild anemia | Normal | Elevated A2 , variable elevation of F |
| β0 or β+ -Thalassemia severe | β0 /β0 , β+ /β0 , β+ β+ E/β0 | Microcytosis, nucleated RBC | Transfusion dependent | F 98% and A2 2%, E 30–40% (E/β0 ); variably low Hb A with β+ |
| β0 or β+ thalassemia intermedia | Hyopchromic, microcytosis | Mild to moderate anemia, intermittent transfusions | A2 2–5%, F 10–30%, Hb A variably low levels | |
| Dominant (rare) | B0 /A | Microcytosis, abnormal RBCs | Moderately severe anemia, splenomegaly | Elevated F and A2 |
| δ-Thalassemia | A/A | Normal | Normal | A2 absent |
| (δβ)0 -Thalassemia | (δβ)0 /A | Hypochromic | Mild anemia | F 5–20% |
| (δβ)+ -Thalassemia Lepore | βLepore /A | Microcytosis | Mild anemia | Lepore 8–20% |
| Homozygous Hb Lepore | βLepore /βLepore | Microcytic, hypochromic | Thalassemia intermedia | F 80%, Lepore 20% |
| γδβ-Thalassemia | (γA δβ)0/ A | Microcytosis, microcytic, hypochromic | Moderate anemia, splenomegaly, homozygote: thalassemia intermedia | Decreased F and A2 compared with δβ-thalassemia |
| γ-Thalassemia | (γA γG )0 /A | Microcytosis | Insignificant unless homozygote | Decreased F |
In the α-thalassemia syndromes , there is a reduction in α-globin production. Normally, there are 4 α-globin genes (2 from each parent) that control α-globin production. α-thalassemia syndromes vary from complete absence (hydrops fetalis) to only slightly reduced (α-thalassemia silent carrier) α-globin production. In the α-thalassemia syndromes, an excess of β- and γ-globin chains are produced. These excess chains form Bart hemoglobin (γ4 ) in fetal life and HbH (β4 ) after birth. These abnormal tetramers are nonfunctional hemoglobins with very high oxygen affinity. They do not transport oxygen and result in extravascular hemolysis. A fetus with the most severe form of α-thalassemia (hydrops fetalis ) develops in utero anemia and the pregnancy usually results in fetal loss because HbF production requires sufficient amounts of α-globin. In contrast, infants with β-thalassemia major become symptomatic only after birth when HbA predominates and insufficient β-globin production manifests in clinical symptoms.
Homozygous β-Thalassemia (Thalassemia Major, Cooley Anemia)
Clinical Manifestations
If not treated, children with homozygous β0 -thalassemia usually become symptomatic from progressive anemia, with profound weakness and cardiac decompensation during the 2nd 6 mo of life. Depending on the mutation and degree of HbF production, regular transfusions are necessary beginning in the 2nd mo to 2nd yr of life, but rarely later. The decision to transfuse is multifactorial but is not determined solely by the degree of anemia. The presence of signs of ineffective erythropoiesis, such as growth failure, bone deformities secondary to marrow expansion, and hepatosplenomegaly, are important variables in determining transfusion initiation.
The classic presentation of children with severe disease includes thalassemic facies (maxilla hyperplasia, flat nasal bridge, frontal bossing), pathologic bone fractures, marked hepatosplenomegaly, and cachexia and is primarily seen in countries without access to chronic transfusion therapy. Occasionally, patients with moderate anemia develop these features because of severe compensatory, ineffective erythropoiesis.
In nontransfused patients with severe ineffective erythropoiesis, marked splenomegaly can develop with hypersplenism and abdominal symptoms. The features of ineffective erythropoiesis include expanded medullary spaces (with massive expansion of the marrow of the face and skull), extramedullary hematopoiesis, and higher metabolic needs (Fig. 489.7 ). The chronic anemia and increased erythroid drive produce an increase in iron absorption from the gastrointestinal tract and secondary hemosiderosis-induced organ injury.
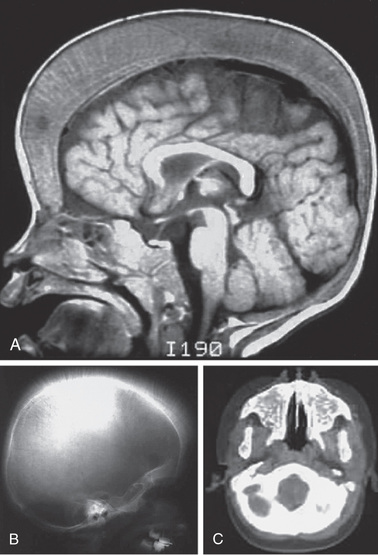
Chronic transfusion therapy dramatically improves the quality of life and reduces the complications of severe thalassemia. Transfusion-induced hemosiderosis becomes the major clinical complication of transfusion-dependent thalassemia. Each mL of packed red cells contains approximately 1 mg of iron. Physiologically, there is no mechanism to eliminate excess body iron. Iron is initially deposited in the liver and is followed by deposition in the endocrine organs and the heart. This leads to a high rate of hypothyroidism, hypogonadotrophic gonadism, growth hormone deficiency, hypoparathyroidism, and diabetes mellitus. Iron deposition in the heart causes heart failure and arrhythmias, and heart disease is the leading cause of death in inadequately chelated patients. Eventually, most patients not receiving adequate iron chelation therapy die from cardiac failure or cardiac arrhythmias secondary to hemosiderosis. Hemosiderosis-induced morbidity can be prevented by adequate iron chelation therapy.
Laboratory Findings
In the United States, some children with β-thalassemia major will be identified on newborn screening as a result of the detection of only HbF on hemoglobin electrophoresis. However, infants with β+ mutations might be missed on newborn screen if small amounts of hemoglobin A are present. A hemoglobin FE pattern can be consistent with hemoglobin E β0 -thalassemia, or the more benign hemoglobin EE disease, and needs to be followed up. The lack of standardized neonatal diagnosis of thalassemia disorders requires close follow-up of newborns with unclear thalassemia mutations and babies from high-risk ethnic groups.
Infants with serious β-thalassemia disorders have a progressive anemia after the newborn period. Microcytosis, hypochromia, and targeting characterize the RBCs. Nucleated RBCs, marked anisopoikilocytosis, and a relative reticulocytopenia are typically seen (see Fig. 489.5E ). The Hb level falls progressively often to <6 g/dL unless transfusions are given. The reticulocyte count is commonly <8% and is inappropriately low compared to the degree of anemia caused by ineffective erythropoiesis. The unconjugated serum bilirubin level is usually elevated, but other chemistries may be initially normal. Even if the child does not receive transfusions, iron eventually accumulates with elevated serum ferritin and transferrin saturation. Evidence of bone marrow hyperplasia can be seen on radiographs (see Fig. 489.7 ).
Early definitive diagnosis is recommended. Newborn screening techniques such as hemoglobin electrophoresis is not definitive. DNA diagnosis of the β-thalassemia mutations, along with testing for common genetic modifiers of the clinical phenotype, is recommended. Co-inheritance of 1 or more α-thalassemia deletions is common, and it decreases the severity of the β-thalassemia disease as it improves the α:β chain imbalance. Some patients' mutations cannot be diagnosed by standard electrophoresis or common DNA probes. Referral of the samples to a tertiary laboratory is indicated, along with parental and family testing. Following the definitive diagnosis, families should undergo detailed counseling.
Management and Treatment of Thalassemia
Transfusion Therapy
β-thalassemia major is a clinical diagnosis that requires the integration of laboratory findings and clinical features. Of patients with homozygous β0 -thalassemia (the most severe mutations), 15–20% may have a clinical course that is phenotypically consistent with thalassemia intermedia. In contrast, 25% of patients with homozygous β+ -thalassemia, typically a more benign genotype, may have transfusion-dependent thalassemia. Transient clinical events, such as a sudden fall in hemoglobin secondary to an episode of parvovirus requiring transfusion, do not necessarily indicate a transfusion-dependent patient. The long-term observation of the clinical characteristics, such as growth, bony changes, and hemoglobin, are necessary to determine chronic transfusion therapy.
Guidelines for Transfusion Therapy.
Patients who require transfusion therapy should have an extended red cell phenotype and/or genotype. Patients should receive RBCs depleted of leukocytes and matched for D, C, c, E, e, and Kell antigens at a minimum. Cytomegalovirus-safe units are indicated in stem cell transplantation candidates. Transfusions should generally be given at intervals of 3-4 wk, with the goal being to maintain a pretransfusion Hb level of 9.5-10.5 g/dL. Ongoing monitoring for transfusion-associated transmitted infections (hepatitis A, B, and C, HIV), alloimmunization, annual blood transfusion requirements, and transfusion reactions is essential.
Iron Overload Monitoring
Excessive iron stores from transfusion cause many of the complications of β-thalassemia major. Accurate assessment of excessive iron stores is essential to optimal therapy. Serial serum ferritin levels provide a useful screening technique in assessing iron balance trends, but results may not accurately predict quantitative iron stores. Undertreatment or overtreatment of presumed excessive iron stores can occur in managing a patient based on serum ferritin alone. Quantitative measurement of liver iron and cardiac iron by magnetic resonance imaging are standard noninvasive methods to measure tissue iron overload; estimation of pancreatic and gonadal iron is being studied. This technology, along with access to multiple chelators, enable targeted chelation therapy for patients with organ-specific hemosiderosis before the onset of overt organ failure. Integration of these imaging technologies with chelation therapy may prevent heart failure, diabetes, and other organ dysfunction.
Quantitative liver iron by approved R2 or R2* MRI is the best indicator of total body iron stores and should be obtained in patients after chronic transfusion therapy has been initiated. The liver iron results will help guide the chelation regimen. Quantitative cardiac iron, determined by T2* MRI cardiac software, is usually obtained starting at 10 yr old, but should be obtained earlier in the setting of severe iron overload or if the transfusion and chelation history is not known. There may be a discrepancy between the liver iron and the heart iron because of different rates of tissue loading and unloading and the differential effects of iron chelators on organ-specific iron removal.
Chelation Therapy
Iron-chelation therapy should start as soon as the patient becomes significantly iron-overloaded. In general, this occurs after 1 yr of transfusion therapy and correlates with the serum ferritin >1,000 ng/mL and/or a liver iron concentration of >5,000 µg/g dry weight. Iron chelation is not currently labeled for use in children <2 yr.
There are 3 available iron chelators (deferoxamine, deferasirox, and deferiprone); each varies in its route of administration, pharmacokinetics, adverse events, and efficacy. Combination chelation therapy may be required for high iron burden. The overall goal is to prevent hemosiderosis-induced tissue injury and avoid chelation toxicity. This requires close monitoring of the patients. In general, chelation toxicity increases as iron stores decrease.
Deferoxamine (Desferal) is the most studied iron chelator; it has an excellent safety and efficacy profile. It requires subcutaneous or IV administration because of its poor oral bioavailability and short half-life of <30 min, necessitating administration as a continuous infusion over at least 8 hr daily, 5-7 days/wk. Deferoxamine is initially started at 25 mg/kg and can be increased to 60 mg/kg in heavily iron-overloaded patients. The major problem with deferoxamine is poor adherence because of the difficult, time-consuming route of administration. Adverse side effects include local skin reactions, ototoxicity, retinal changes, and bone dysplasia with truncal shortening.
The oral iron chelator deferasirox (Exjade, JadeNu) is commercially available in the United States. Of patients treated with deferoxamine, 70% have switched to deferasirox because it is orally available. Deferasirox has a half-life of >16 hr and requires once-daily administration. Two forms of the drug are available, a dispersible tablet that is dissolved in water or juice and a film-coated tablet. A granule form that is sprinkled on soft food and ingested recently was FDA approved. Dosing is different for the different deferasirox formulations. For the dispersible tablet form (Exjade), the initial dose typically is 20 mg/kg/day and can be escalated to as high as 40 mg/kg/day based on the iron burden. The dosing for the film-coated tablet and granule (JadeNu) forms is 30% lower than the dispersible tablet, with a starting dose of 14 mg/kg/day, which can be escalated to a maximum of 28 mg/kg/day. The most common side effects are gastrointestinal (GI) symptoms, which may be lessened with the film-coated tablet form because it does not contain lactose and sodium laureate, which are found in the dispersible tablet and are thought to be responsible for some of the GI symptoms. The most serious side effect of deferasirox is potential kidney damage. Up to 30% of patients have transient increases in creatinine that may require temporary modifications of dosing. This toxicity may occur more commonly in the setting of dehydration. Long-term studies in thousands of patients have not demonstrated progressive renal dysfunction, but isolated cases of renal failure in patients have occurred. In addition, hepatic transaminitis may occur, with an increase to >5 times the upper limit of normal in approximately 8% of patients. All patients require monthly chemistry panels and ongoing monitoring for proteinuria.
Deferiprone (Ferriprox), an oral iron chelator, is approved in the United States for use as a second-line agent. Deferiprone has a half-life of approximately 3 hr and requires dosing 3 times daily. The starting dose is 75 mg/kg/day and can be escalated to 99 mg/kg/day based on the degree of iron overload. Deferiprone, a small molecule, effectively enters cardiac tissue and may be more effective than other chelators in reducing cardiac hemosiderosis. The most serious side effect of deferiprone is transient agranulocytosis, which occurs in 1% of patients and usually in the 1st yr of treatment. It has been associated with rare deaths where patients were not adequately monitored. The use of deferiprone requires frequent blood count monitoring, typically weekly for at least the 1st yr of therapy. Most importantly, the drug should be held and the neutrophil count checked with all febrile illnesses.
As thalassemia patients live longer, the iron chelation goals have changed. Aggressive treatment with combination chelation therapy is often used in heavily iron-overloaded patients to prevent or reverse organ dysfunction. Deferoxamine, in combination with deferiprone, is routinely used in patients with increased cardiac iron. Combination therapy of deferoxamine and deferasirox or deferasirox and deferiprone may also be efficacious in patients with severe iron overload.
Hydroxyurea
Hydroxyurea, a DNA antimetabolite, increases HbF production. It has been most successfully used in sickle cell disease and in some patients with β-thalassemia intermedia. Studies in β-thalassemia major are limited. In many parts of the world, hydroxyurea therapy is used in β-thalassemia intermedia patients. Even though increases in HbF levels are observed, they do not predictively correlate with increase in total Hb in these patients. In general, there appears to be a mean increase in Hb of 1 g/dL (range: 0.1-2.5 g/dL). Hydroxyurea therapy in thalassemia intermedia is associated with a reduced risk of leg ulcers, pulmonary hypertension, and extramedullary hematopoiesis. The initial starting dose for thalassemia intermedia is 10 mg/kg and may be escalated to 20 mg/kg/day. Patients with β-thalassemia are at increased risk of developing cytopenias with hydroxyurea use, which may prevent dose escalation. Close monitoring of the CBC with differential is required.
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation has cured >3,000 patients who had β-thalassemia major. In low-risk HLA-matched sibling patients, there is at least a 90% survival and an 80% event-free survival. In general, myeloablative conditioning regimens have been used to prevent graft rejection and thalassemia recurrence. Most success has been in children <14 yr old without excessive iron stores and hepatomegaly who undergo sibling HLA-matched allogeneic transplantation. All children who have an HLA-matched sibling should be offered the option of bone marrow transplantation. Alternative transplantation regimens for patients without appropriate donors are experimental and have variable success. Gene therapy approaches are under study, and early results with lentiviral vectors have been promising, particularly for patients with β+ - or HbE β-thalassemia genotypes.
Splenectomy
Splenectomy may be required in thalassemia patients who develop hypersplenism. These patients have a falling steady-state Hb level and/or a rising transfusion requirement. However, splenectomy is less frequently used as a therapeutic option; serious adverse effects of splenectomy are increasingly recognized beyond infection risk. In thalassemia intermedia, splenectomized patients have a marked increased risk of venous thrombosis, pulmonary hypertension, leg ulcers, and silent cerebral infarction compared to nonsplenectomized patients. All patients should be fully immunized against encapsulated bacteria and receive appropriate instructions regarding fever management. Prophylactic penicillin should be administered after splenectomy to prevent sepsis, and families need to be educated on the risk of fever and sepsis.
Preventive Monitoring of Thalassemia Patients
Cardiac Disease
Cardiac disease is the major cause of death in thalassemia. Serial echocardiograms should be monitored to evaluate cardiac function and pulmonary artery pressure. Pulmonary hypertension frequently occurs in non-transfusion dependent thalassemia patients and may be an indication for transfusion therapy. After approximately 8 yr of chronic transfusion therapy, cardiac hemosiderosis may occur; consequently, cardiac T2* MRI imaging studies are recommended. Patients with cardiac hemosiderosis and decreasing cardiac ejection fraction require intensive combination chelation therapy. Periodic electrocardiogram studies also are obtained after age 10 yr because of the risk of arrhythmia from cardiac iron overload.
Endocrine Disease
Endocrine function progressively declines with age secondary to hemosiderosis and nutritional deficiencies. Iron deposition in the pituitary and endocrine organs can result in multiple endocrinopathies, including hypothyroidism, growth hormone deficiency, delayed puberty, hypoparathyroidism, diabetes mellitus, osteoporosis, and adrenal insufficiency. Monitoring for endocrine dysfunction starts early, about 5 yr of age, or after at least 3 yr of chronic transfusions. All children require monitoring of their height, weight, pubertal assessment, and sitting height semiannually. Bone density scans should be obtained starting in the 2nd decade of life given the high rate of osteopenia. Nutritional assessments are required. Most patients need vitamin D, vitamin C, and zinc replacement. Fertility is a growing concern among patients and should be assessed routinely.
Psychosocial Support
Thalassemia imposes major disruption in the family unit and significant obstacles to normal development. Culturally sensitive anticipatory counseling is necessary, and the early use of child life services decreases psychological trauma of therapy. Early social service consultation to address financial and social issues is mandatory.
Other β-Thalassemia Syndromes
Non–Transfusion-Dependent Thalassemia: β-Thalassemia Intermedia
The β-thalassemia syndromes are characterized by decreased production of β-globin chains of HbA. There are 200-300 β-thalassemia mutations that have been characterized. These mutations can affect any step in the transcription of β-globin genes. As discussed, β0 -thalassemia is absent production of normal β-chains, and Hb A production of with β+ mutations is decreased. Some β-thalassemia mutations have structural mutations such as HbE. Others, such as δβ-thalassemia or HPFH, are variants of β-thalassemia that have decreased production of β-globin gene with increased compensatory production of HbF. Because phenotypic correlation with genotype is variable, β-thalassemia patients are largely classified by their clinical spectrum. Transfusion-dependent thalassemia, or thalassemia major, is the most severe group. Non–transfusion-dependent thalassemia (thalassemia intermedia) include a spectrum of patients who initially are not chronically transfused in infancy but may be sporadically transfused throughout their lifetime. The major determining characteristic of these patients is less α-β–globin chain imbalance than observed in thalassemia major. Sometimes, genetic modifiers alter the primary mutation severity and improve the globin-chain imbalance. Co-inheritance of α-thalassemia trait or polymorphisms of globin promoters such as BCL11 may lessen disease severity and result in a non-transfusion dependent thalassemia. HbE β-thalassemia is a common cause of both transfusion-dependent and non–transfusion-dependent thalassemia. These secondary genetic modifiers play a role in altering the severity of this disorder. Occasionally, patients with a single β-thalassemia mutation or autosomal dominant β-thalassemia trait have clinical features of thalassemia intermedia, or non–transfusion-dependent thalassemia. Genetic studies of these patients often uncover co-inheritance of genetic modifiers that worsens the condition, such as α-gene triplication or an unstable β-globin mutation.
Thalassemia intermedia patients have significant ineffective erythropoiesis that leads to microcytic anemia with hemoglobin of approximately 7 g/dL (range: 6-10 g/dL). These patients have some of the complications characterized in untransfused thalassemia major patients, but the severity varies depending on the degree of ineffective erythropoiesis. They can develop medullary hyperplasia, hepatosplenomegaly, hematopoietic pseudotumors, pulmonary hypertension, leg ulcers, thrombotic events, and growth failure. Many patients develop hemosiderosis secondary to increased GI absorption of iron requiring chelation. Extramedullary hematopoiesis can occur in the vertebral canal, compressing the spinal cord and causing neurologic symptoms; the latter is a medical emergency requiring immediate local radiation therapy to halt erythropoiesis. Transfusions are indicated in thalassemia intermedia patients with significant clinical morbidity.
Thalassemia trait is often misdiagnosed as iron deficiency in children, because the 2 diagnoses produce similar hematologic abnormalities on CBC. However, iron deficiency is much more prevalent. A short course of iron and reevaluation is all that is required to identify children who will need further evaluation. Children who have β-thalassemia trait have a persistently normal red cell distribution width and low mean corpuscular volume (MCV), whereas patients with iron deficiency develop an elevated red cell distribution width (RDW) with treatment. On Hb analysis, patients with β-thalassemia trait have elevated levels of HbA2 and variably increased Hb F. There are “silent” forms of β-thalassemia trait, and if the family history is suggestive, further studies may be indicated.
α-Thalassemia Syndromes
The same evolutionary pressures that produced β-thalassemia and sickle cell disease produced α-thalassemia. Infants are identified in the newborn period by the increased production of Bart hemoglobin (γ4 ) during fetal life and its presence at birth. The α-thalassemia syndromes occur most frequently in Southeast Asia. Deletion mutations are most common in α-thalassemia. In addition to deletional mutations, there are nondeletional α-globin gene mutations, the most common being Constant Spring (αCS α); these mutations cause a more severe anemia and clinical course than the deletional mutations. Normally, there are 4 α-globin genes. The different phenotypes in α-thalassemia largely result from whether 1 (α+ -thalassemia) or both (α0 -thalassemia) α-globin genes are deleted in each of the 2 loci.
The deletion of 1 α-globin gene (silent trait) is not identifiable hematologically. Specifically, no alterations are noted in the MCV and mean corpuscular hemoglobin (MCH). Persons with this deletion are usually diagnosed after the birth of a child with a 2-gene deletion or HbH (β4 ), but some newborn screening programs report even low concentrations of Hb Bart. During the newborn period, <3% Hb Bart is observed. The deletion of 1 α-globin gene is common in African Americans.
The deletion of 2 α-globin genes results in α-thalassemia trait. The α-globin alleles can be lost in a trans (−α/−α) or cis (α,α/-SEA ) configuration. The trans or cis mutations can combine with other mutations or deletions and lead to HbH or α-thalassemia major. In persons from Africa or of African descent, the most common α-globin deletions are in the trans configuration, whereas in persons from or descended from Asia or the Mediterranean region, cis deletions are most common.
α-Thalassemia trait (2 missing α-globin genes) manifest as a microcytic anemia that can be mistaken for iron-deficiency anemia (see Fig. 489.5F ). The Hb analysis is normal, except during the newborn period, when Hb Bart is typically <8% but >3%. Children with a deletion of 2 α-globin genes are commonly mistaken to have iron deficiency, given the presence of both low MCV and MCH. The simplest approach to distinguish between iron deficiency and α-thalassemia trait is with a good dietary history. Children with iron-deficiency anemia often have a diet that is low in iron and drink significant amount of cow's milk. Alternatively, a brief course of iron supplementation along with monitoring of erythrocyte parameters might confirm the diagnosis of iron deficiency. If both parents of a child diagnosed with α-thalassemia trait are carriers in the cis conformation, they are at risk for a future hydrops fetalis pregnancy. Thus, family screening and genetic counseling are indicated.
The deletion of 3 α-globin genes leads to the diagnosis of HbH disease. A more severe form of HbH disease may be caused by a nondeletional α-globin mutation in combination with 2 gene deletions. HbH Constant Spring (−α/α,αCS ) is the most common type of nondeletional HbH disease.
In California, where a large population of persons of Asian descent resides, approximately 1 : 10,000 of all newborns have HbH disease. The simplest manner of diagnosing HbH disease is during the newborn period, when excess in γ-tetramers are present and Hb Bart is commonly >25%. Obtaining supporting evidence from the parents is helpful. Later in childhood, there is an excess of β-globin chain tetramers that results in HbH. A definitive diagnosis of HbH disease requires DNA analysis. Brilliant cresyl blue can stain HbH, but it is rarely used for diagnosis. Patients with HbH disease have a marked microcytosis, anemia, mild splenomegaly, and, occasionally, scleral icterus or cholelithiasis. Chronic transfusion is not usually required for therapy because the Hb range is 7-11 g/dL, with MCV 51-73 fL, but intermittent transfusions for worsening anemia may be needed. Individuals with non-deletional Hb H disease are more likely to require transfusions than individuals with deletional Hb H disease.
The deletion of all 4 α-globin gene alleles causes profound anemia during fetal life, resulting in hydrops fetalis ; the ζ-globin gene must be present for fetal survival. There are no normal hemoglobins present at birth (primarily Hb Bart, with Hb Gower-1, Gower-2, and Portland). Intrauterine transfusions may rescue the fetus, but congenital abnormalities and neurodevelopmental delay often result. Infants with severe α-thalassemia will have lifelong transfusion dependence, and hematopoietic stem cell transplantation is the only cure.
Treatment of HbH disease requires ongoing monitoring of growth and organ dysfunction. Dietary supplement with folate and multivitamins without iron is indicated. Older patients may develop decreased bone density with calcium and vitamin D deficiency. Vitamin D supplementation is indicated if the level is low, and adequate dietary calcium intake should be encouraged to promote bone health. Iron supplementation should be avoided as patients are at risk of developing iron overload. Intermittent transfusion requirements during intercurrent infection may occur, particularly in nondeletional HbH. Splenectomy is occasionally indicated, and because of the high risk of postsplenectomy thrombosis, aspirin or other anticoagulant therapy following splenectomy should be considered. Hemosiderosis, secondary to GI iron absorption or transfusion exposure, may develop in older patients and require chelation therapy. Because HbH is an unstable hemoglobin sensitive to oxidative injury, oxidative medications should be avoided. At-risk couples for hydrops fetalis should be identified and offered molecular diagnosis on fetal tissue obtained early in pregnancy. Later in pregnancy, intrauterine transfusion can improve fetal survival, but chronic transfusion therapy or bone marrow transplantation for survivors will be required.
Bibliography
Ambati SR, Randolph RE, Mennitt K, et al. Longitudinal monitoring of cardiac siderosis using cardiovascular magnetic resonance (CMR) T2* in patients with thalassemia major on various chelation regimens: a 6 year study. Am J Hematol . 2013;88:652–656.
Ash-Bernal R, Wise R, Wright SM. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) . 2004;83:265–273.
Baksi AJ, Pennell DJ. Randomized controlled trials of iron chelators for the treatment of cardiac siderosis in thalassemia major. Front Pharmacol . 2014;5:217.
Camaschella C. Treating iron overload. N Engl J Med . 2013;368:2325–2327.
Cash C, Arnold DH. Extreme methemoglobinemia after topical benzocaine: recognition by pulse oximetry. J Pediatr . 2017;181:319.
Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood . 2003;101:791–800.
Crowley MA, Mollan TL, Abdulmalik OY, et al. A hemoglobin variant associated with neonatal cyanosis and anemia. N Engl J Med . 2011;364:1837–1842.
Graff DM, Bosse GM, Sullivan J. Case report of methemoglobinemia in a toddler secondary to topical dapsone exposure. Pediatrics . 2016;138(2):e1–e2.
Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassemia. Lancet . 2012;379:373–382.
Hussein AA, Al-Zaben A, Ghatasheh L, et al. Risk adopted allogeneic hematopoietic stem cell transplantation using a reduced intensity regimen for children with thalassemia major. Pediatr Blood Cancer . 2013;60:1345–1349.
Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program . 2010;2010:418–422.
Kwiatkowski JL. Current recommendations for chelation for transfusion-dependent thalassemia. Ann N Y Acad Sci . 2016;1368:107–114.
Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med . 2011;364:710–718.
Martin A, Thompson AA. Thalassemias. Pediatr Clin North Am . 2013;60:1383–1391.
Musallam KM, Taher AT, Cappellini MD, et al. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood . 2013;121:2199–2212 [quiz 372].
Patel K, Livni N, Macdonald D. Renal medullary carcinoma, a rare cause of haematuria in sickle cell trait. Br J Haematol . 2006;132:1.
Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome b(5) reductase deficiency. Br J Haematol . 2008;141:298–308.
Peters M, Heijboer H, Smiers F, et al. Diagnosis and management of thalassaemia. BMJ . 2012;344:40–44.
Songdej D, Babbs C, Higgs DR. BHFS international consortium: an international registry of survivors with hb Bart's hydrops fetalis syndrome. Blood . 2017;9:1251–1259.
Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood . 2010;115(10):1886–1892.
Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood . 2010;115:1886–1892.
Taher AT, Vichinsky E, Musallam KM, et al. Guidelines for the management of non transfusion dependent thalassaemia (NTDT) . Thalassaemia Internation Federation: Nicosia, Cyprus; 2013.
Tsaras G, Owusu-Ansah A, Boateng FO, et al. Complications associated with sickle cell trait: a brief narrative review. Am J Med . 2009;122:507–512.
Vichinsky EP. Clinical manifestations of alpha-thalassemia. Cold Spring Harb Perspect Med . 2013;3:a011742.
Vichinsky E, Levine L. Standard of care guidelines for thalassemia . Children's Hospital: Oakland, CA; 2012.
Vichinsky EP, MacKlin EA, Waye JS, et al. Changes in the epidemiology of thalassemia in north America: a new minority disease. Pediatrics . 2005;116:e818–e825.
Vichinsky E, Cohen A, Thompson AA, et al. Epidemiologic and clinical characteristics of nontransfusion-dependent thalassemia in the United States. Pediatr Blood Cancer . 2018;65(7):e27067.
Wood JC. Impact of iron assessment by MRI. Hematology Am Soc Hematol Educ Program . 2011;2011:443–450.