Abnormalities of Lymphatic Vessels
Michael Kelly, Richard L. Tower II, Bruce M. Camitta
Lymphatic Malformations
Lymphatic malformations (LMs) can be isolated, generalized, or associated with syndromes and overgrowth and can occur as combined malformations with other vessel types (see International Society for the Study of Vascular Anomalies (ISSVA ) classification; www.issva.org ). LMs consist of dilated lymphatic channels or cysts lined by lymphatic endothelial cells (LECs). LMs are typically classified by cyst size (macrocystic, microcystic, mixed) and occur more frequently in the head, neck, and axilla.
Generalized lymphatic anomaly (GLA) is defined as a multifocal LM that involves soft tissues, abdominal and thoracic viscera, and often bone. Bone involvement in GLA is usually not progressive and typically spares the cortex. Chylous effusions involving the pleural, pericardial, and peritoneal spaces can occur.
Gorham-Stout disease (GSD; disappearing bone disease) is characterized by an LM involving single or multiple bones and leading to progressive cortical bone loss (also termed vanishing bone syndrome ). LMs often involve soft tissue adjacent to bone and result in effusions in GSD as well.
Central conducting lymphatic anomalies are associated with intestinal dysmotility and, depending on the involved site, may result in chylothorax, pulmonary lymphangiectasia, chylous ascites, protein-losing enteropathy, cutaneous lesions, chylous leakage, and osseous changes from dilated intraosseous lymphatic channels. LMs are also associated with somatic mutations in PIK3CA , which often also produce other vascular malformations and regional tissue overgrowth.
Genetics
Somatic, hyperactivating PIK3CA mutations are present at low frequency (<10 %) in most isolated LMs and LMs that are part of a syndrome. These mutations are the same PIK3CA mutations found in many human cancers. It is not clear how the same somatic mutations cause such phenotypic diversity. However, activation of PIK3CA leads to increased signaling through the AKT/mTOR pathway, likely explaining the sensitivity of most LMs to sirolimus.
Treatment
A decision to treat an LM depends on the anatomic location, involvement of local structures, and symptoms. Referral to a specialized vascular anomalies clinic with the expertise to guide appropriate imaging and treatment decisions is recommended. For localized macrocystic LMs, interventional radiology (IR) with administration of sclerosing agents (OK432, ethanol, bleomycin) is most effective. For lesions involving skin and mucosa, laser treatments may be used. Sirolimus , an inhibitor of mammalian target of rapamycin (mTOR), has been shown to be effective when used alone or in combination with IR for complicated or extensive LMs. Propranolol has been effective in some patients.
Lymphangiectasia
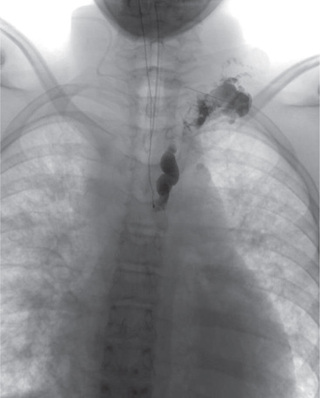
Lymphangiectasia is no longer considered a unique entity and is grouped as either a GLA or channel-type LM according to ISSVA classification. Channel-type LM can result from hypoplasia of the cisternae chylae, lymph nodes, or central collecting ducts, leading to the obstruction of lymph flow from superficial to central collecting channels and resulting in dilation of superficial vessels (Fig. 516.1 ). Symptoms depend on the level of the obstruction and can include protein-losing enteropathy if the mesentery is involved or chylous effusions if obstruction occurs higher.
Lymphedema
Lymphedema is a localized swelling caused by impaired lymphatic flow and can be primary (congenital) or acquired. Primary lymphedemas are grouped as LMs because they result from dysgenesis of lymphatic networks during early development. Primary lymphedema may be found in Turner syndrome, Noonan syndrome, autosomal dominantly inherited Milroy disease, and other chromosomal abnormalities. Mutations in multiple genes, including the vascular endothelial growth factor receptor–3 gene (VEGFR3 ), GJC2 , PTPN14 , and GATA2, are associated with primary lymphedema (www.issva.org ). Autosomal recessive, dominant or de novo mutations of VEGFR3 produce Milroy disease. Mutations in other genes are associated with specific syndromes; CCBE1 (Hennekam), FOXC2 (lymphedema distichiasis), SOX18 (hypotrichosis-telangiectasia-lymphedema), KMT2D/MLL2, and KDM6A (Kabuki). Unilateral or bilateral lower extremity lymphedema in an adolescent may be Meige disease.
Acquired obstruction of the lymphatics can result from tumor, postirradiation fibrosis, and postinflammatory scarring. Filariasis is an important cause of lymphedema in Africa, Asia, and Latin America. One third of the 120 million infected persons (primarily older adolescents and adults) have lymphedema or a hydrocele. Injury to the major lymphatic vessels can cause collection of lymph fluid in the abdomen (chylous ascites) or chest (chylothorax).
Untreated lymphedema can be disabling and is associated with immune dysfunction, inflammation, fibrosis, adipose tissue overgrowth, and lymphangiosarcoma . Current treatment modalities attempt to reduce localized swelling through massage, exercise, and compression. Selenium has been an effective adjuvant to physiotherapy in some adult patients following breast cancer treatment.
Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM ) is characterized by proliferation of lymphatic endothelial cells and smooth muscle cells in the lungs, leading to airway and lymphatic obstruction, cyst formation, pneumothorax, and respiratory failure. It may initially be mistaken for asthma. LAM occurs in young women and is associated with mutations in the tuberous sclerosis tumor-suppressor gene TSC2 in one third of cases. Sirolimus stabilizes lung function, reduces symptoms, and improves life quality. Lung transplantation may be required.
Lymphangitis
Lymphangitis is an inflammation of the lymphatics that drain an area of infection. Tender, erythematous streaks extend proximally from the infected area. Regional nodes may also be tender. Group A streptococci and Staphylococcus aureus are the most common pathogens, and therapy should include antibiotics that treat these organisms.