Brain Tumors in Childhood
Wafik Zaky, Joann L. Ater, Soumen Khatua
Primary central nervous system (CNS) tumors are a heterogeneous group of diseases that collectively are the most common malignancy in childhood and adolescence. The overall mortality among this group approaches 30%. Patients with CNS tumors have the highest morbidity—primarily neurologic—of all children with malignancies. Outcomes have improved over time with innovations in neurosurgery, radiation therapy (particularly stereotactic conformal radiotherapy), chemotherapy, and immune therapy. The treatment approach for these tumors is multimodal. Surgery with complete resection, if feasible, is the foundation, with radiation therapy and chemotherapy used according to the diagnosis, patient age, and other factors
Etiology
The etiology of pediatric brain tumors is not well defined. A male predominance is noted in the incidence of medulloblastoma and ependymoma. Familial syndromes associated with an increased incidence of brain tumors account for approximately 5% of cases (Table 524.1 ). Cranial exposure to ionizing radiation also is associated with a higher incidence of brain tumors. There are sporadic reports of brain tumors within families without evidence of a heritable syndrome. The molecular events associated with tumorigenesis of pediatric brain tumors are not known.
Table 524.1
Familial Syndromes Associated With Pediatric Brain Tumors
| SYNDROME | CENTRAL NERVOUS SYSTEM MANIFESTATIONS | CHROMOSOME | GENE |
|---|---|---|---|
| Neurofibromatosis type 1 (autosomal dominant) | Optic pathway gliomas, astrocytoma, malignant peripheral nerve sheath tumors, neurofibromas | 17q11 | NF1 |
| Neurofibromatosis type 2 (autosomal dominant) | Vestibular schwannomas, meningiomas, spinal cord ependymoma, spinal cord astrocytoma, hamartomas | 22q12 | NF2 |
| Von Hippel–Lindau (autosomal dominant) | Hemangioblastoma | 3p25-26 | VHL |
| Tuberous sclerosis (autosomal dominant) | Subependymal giant cell astrocytoma, cortical tubers | 9q34 | TSC1 |
| 16q13 | TSC2 | ||
| Li-Fraumeni (autosomal dominant) | Astrocytoma, primitive neuroectodermal tumor | 17q13 | TP53 |
| Cowden (autosomal dominant) | Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) | 10q23 | PTEN |
| Turcot (autosomal dominant) | Medulloblastoma | 5q21 | APC |
| Glioblastoma | 3p21 | hMLH1 | |
| 7p22 | hPSM2 | ||
|
Nevoid basal cell carcinoma Gorlin (autosomal dominant) |
Medulloblastoma | 9q31 | PTCH1 |
Adapted from Kleihues P, Cavenee WK: World Health Organization classification of tumors: pathology and genetics of tumors of the nervous system, Lyon, 2000, IARC Press.
Epidemiology
Approximately 4,600 primary brain tumors are diagnosed each year in children and adolescents in the United States, with an overall annual incidence of approximately 47 cases per 1 million children <20 yr of age. The incidence of CNS tumors is highest in infants and children ≤5 yr old (approximately 52 cases/1 million children).
Pathogenesis
More than 100 histologic categories and subtypes of primary brain tumors are described in the World Health Organization (WHO) classification of CNS tumors. In children 0-14 yr old, the most common tumors are pilocytic astrocytomas (PAs) and medulloblastoma/primitive neuroectodermal tumors (PNETs) . In adolescents (15-19 yr), the most common tumors are pituitary/craniopharyngeal tumors and PAs (Fig. 524.1 ); congenital (neonatal) tumors have a distinct pattern (Table 524.2 ).
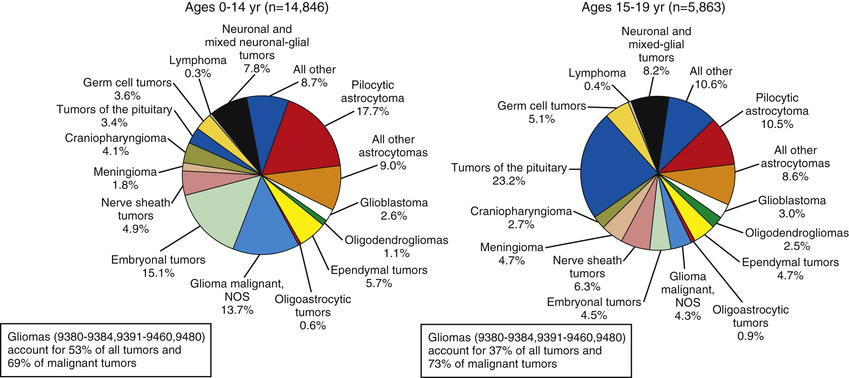
Table 524.2
Congenital and Neonatal Brain Tumors
From Shekdar KV, Schwartz ES: Brain tumors in the neonate, Neuroimag Clin North Am 27:69–83, 2017.
The National Cancer Institute (NCI) Surveillance, Epidemiology, and End Results (SEER) Program reported a slight predominance of infratentorial tumor location (43.2%), followed by the supratentorial region (40.9%), spinal cord (4.9%), and multiple sites (11%) (Fig. 524.2 and Tables 524.3 and 524.4 ). There are age-related differences in primary location of tumor. During the 1st yr of life, supratentorial tumors predominate and most often include choroid plexus complex tumors and teratomas (see Table 524.2 ). In children 1-10 yr old, infratentorial tumors predominate because of the high incidence of juvenile PA and medulloblastoma. After 10 yr of age, supratentorial tumors again predominate, with diffuse astrocytomas most common (see Table 524.4 ). Tumors of the optic pathway and hypothalamus region, the brainstem, and the pineal-midbrain region are more common in children and adolescents than in adults.
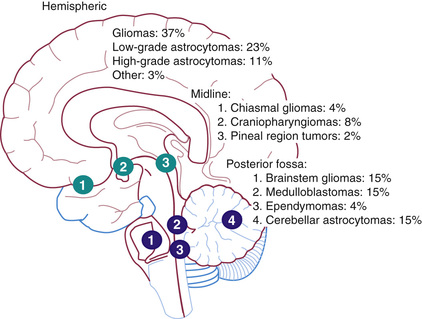
Table 524.3
Posterior Fossa Tumors of Childhood
| TUMOR | RELATIVE INCIDENCE (%) | PRESENTATION | DIAGNOSIS | PROGNOSIS |
|---|---|---|---|---|
| Medulloblastoma | 35-40 | 2-3 mo of headaches, vomiting, truncal ataxia | Heterogeneously or homogeneously enhancing 4th ventricular mass; may be disseminated | 65–85% survival; dependent on stage/type; poorer (20–70%) in infants |
| Cerebellar astrocytoma | 35-40 | 3-6 mo of limb ataxia; secondary headaches, vomiting | Cerebellar hemisphere mass, usually with cystic and solid (mural nodule) components | 90–100% survival in totally resected, pilocytic type |
| Brainstem glioma | 10-15 | 1-4 mo of double vision, unsteadiness, weakness, and cranial nerve dysfunction, including facial weakness, swallowing dysfunction, and oculomotor abnormalities | Diffusely expanded, minimally or partially enhancing mass in 80%; 20% more focal tectal or cervicomedullary lesion | >90% mortality in diffuse tumors; better in localized |
| Ependymoma | 10-15 | 2-5 mo of unsteadiness, headaches, double vision, and facial asymmetry | Usually enhancing, 4th ventricular mass with cerebellopontine predilection | >75% survival in totally resected lesions |
| Atypical teratoid/rhabdoid | >5 (10–15% of infantile malignant tumors) | As in medulloblastoma, but primarily in infants; often associated facial weakness and strabismus | As in medulloblastoma, but often more laterally extended | ≤20% survival in infants |
Adapted from Packer RJ, MacDonald T, Vezina G: Central nervous system tumors, Pediatr Clin North Am 55:121–145, 2008.
Table 524.4
Pediatric Supratentorial Brain Tumors With Key Features on Neuroimaging
| TUMOR | KEY FEATURES |
|---|---|
| GLIAL CELL TUMORS | |
| Pilocytic astrocytoma |
Most common primary tumor in children Excellent prognosis Cystic with enhancing mural nodule or solid mass Lack of significant vasogenic edema |
| Diffuse astrocytoma |
Much less common in children than in adults Relatively ill-defined without contrast enhancement Dedifferentiation rarely seen in children |
| Anaplastic astrocytoma |
Poorly circumscribed margins No hemorrhage or necrosis Usually no contrast enhancement |
| Glioblastoma |
Rare in children Heterogeneous enhancement Necrosis and marked peritumoral edema |
| Subependymal giant cell tumor |
Associated with the tuberous sclerosis complex Avid enhancement Virtually always in a lateral ventricle near foramen of Monro |
| Pleomorphic xanthoastrocytoma |
Almost always supratentorial Solid components show avid enhancement Peripheral location abutting meningeal surface |
| Oligodendroglial tumors |
Relatively well circumscribed, expanded cortex Enhancement and calcification less common than in adults High rCBV often found in low-grade tumors |
| Ependymoma |
Half of supratentorial tumors are parenchymal Higher incidence of cysts than infratentorial ones Calcifications, hemorrhage and inhomogeneous enhancement ADC values usually higher than embryonal tumors |
| Angiocentric glioma |
Superficial cortical lesions T1 hyperintensity is a characteristic but infrequent feature Usually no contrast enhancement |
| NEURONAL AND MIXED NEURONAL-GLIAL TUMORS | |
| Ganglioglioma |
Most common in temporal lobes Mixed cystic and solid masses with avidly enhancing nodule Calcifications are common |
| Desmoplastic infantile tumors |
Very rare, typically 18 mo of age or younger Predominantly cystic with solid nodules located near cortex Solid components may show low ADC values even if benign |
| Dysembryoplastic neuroepithelial tumors |
Cortically based, favor temporal lobes 30% associated with cortical dysplasia May have a characteristic bubbly appearance Can rarely have nodular or ringlike enhancement |
| EMBRYONAL TUMORS | |
| Embryonal tumors not otherwise specified |
Usually children <5 yr of age Large at presentation with little surrounding edema Intense and heterogeneous contrast enhancement Low ADC values |
| Atypical teratoid rhabdoid tumor |
10% of CNS tumors in children <12 mo of age Rare aggressive neoplasms Large and predominantly solid with minimal edema Calcifications, hemorrhage, and cysts are common Moderate to marked enhancement and low ADC values |
ADC, Apparent diffusion coefficient; CNS, central nervous system; rCBV, relative cerebral blood volume.
From Zamora C, Huisman TAGM, Izbudak I: Supratentorial tumors in pediatric patients, Neuroimag Clin North Am 27:39–67, 2017, pp 55-56.
Clinical Manifestations
The clinical presentation of the patient with a brain tumor depends on tumor location, tumor type, and patient age. Signs and symptoms are related to obstruction of cerebrospinal fluid (CSF) drainage paths by the tumor, leading to increased intracranial pressure (ICP) or causing focal brain dysfunction. In young children the diagnosis of a brain tumor may be delayed because the symptoms are similar to more common illnesses, such as gastrointestinal disorders, with associated vomiting. Infants with open cranial sutures may present with signs of increased ICP, such as vomiting, lethargy, and irritability, as well as the later finding of macrocephaly. The classic triad of headache, nausea, and vomiting as well as papilledema is associated with midline or infratentorial tumors . Headaches associated with brain tumors are often of new onset, persistent (but usually <6 mo), associated with either neurologic findings (papilledema, cognitive-behavioral changes, seizures, focal motor deficits), associated with emesis, and occur on awakening or wake the patient from sleep. Disorders of equilibrium, gait, and coordination occur with infratentorial tumors. Torticollis may occur in cases of cerebellar tonsil herniation. Blurred vision, diplopia, and nystagmus also are associated with infratentorial tumors. Tumors of the brainstem region may be associated with gaze palsy, multiple cranial nerve palsies, and upper motor neuron deficits (e.g., hemiparesis, hyperreflexia, clonus).
Supratentorial tumors are more frequently associated with focal motor weakness, focal sensory changes, language disorders, focal seizures, and reflex asymmetry. Infants with supratentorial tumors may present with premature hand preference. Optic pathway tumors manifest as visual and/or afferent oculomotor disturbances, such as decreased visual acuity, Marcus Gunn pupil (afferent pupillary defect), nystagmus, and/or visual field defects. Suprasellar region tumors and 3rd ventricular region tumors may manifest initially as neuroendocrine deficits, such as subacute development of obesity, abnormal linear growth velocity, diabetes insipidus, galactorrhea, precocious puberty, delayed puberty, and hypothyroidism. In fact, signs of endocrine dysfunction preceded symptoms of neuroophthalmologic dysfunction by an average of 1.9 yr, and their recognition as a possible sign of hypothalamic or pituitary neoplasm can hasten diagnosis and improve outcome. The diencephalic syndrome, which manifests as failure to thrive, emaciation despite normal caloric intake, and inappropriately normal or happy affect, occurs in infants and young children with tumors in these regions. Parinaud syndrome is seen with pineal region tumors and is manifested by paresis of upward gaze, pupillary caliber reactive to accommodation but not to light (pseudo–Argyll Robertson pupil), nystagmus to convergence or retraction, and eyelid retraction. Spinal cord tumors and spinal cord dissemination of brain tumors may manifest as long nerve tract motor and/or sensory deficits often localized to below a specific spinal level, bowel and bladder deficits, and back or radicular pain. The signs and symptoms of meningeal metastatic disease from brain tumors or leukemia include head or back pain and symptoms referable to compression of cranial nerves or spinal nerve roots.
Diagnosis
The evaluation of a patient in whom a brain tumor is suspected is an emergency . Initial evaluation should include a complete history, physical (including ophthalmic) examination, and neurologic assessment with neuroimaging. For primary brain tumors, MRI with and without gadolinium is the neuroimaging standard. Tumors in the pituitary/suprasellar region, optic pathway, and infratentorium are better delineated with MRI than with CT. Patients with tumors of the midline and the pituitary/suprasellar/optic chiasmal region should undergo evaluation for neuroendocrine dysfunction . Formal ophthalmologic examination is beneficial in patients with optic path region tumors to document the impact of the disease on oculomotor function, visual acuity, and fields of vision. The suprasellar and pineal regions are preferential sites for germ cell tumors (Fig. 524.3 ). Both serum and CSF measurements of β–human chorionic gonadotropin (β-hCG), α-fetoprotein (AFP), and placental alkaline phosphatase can assist in the diagnosis of germ cell tumors. In tumors with a propensity for spreading to the leptomeninges, such as medulloblastoma/PNET, ependymoma, and germ cell tumors, lumbar puncture (LP) with cytologic analysis of the CSF is indicated; LP is contraindicated in patients with newly diagnosed hydrocephalus secondary to CSF flow obstruction, in those with tumors that cause supratentorial midline shift, and in patients with infratentorial tumors. LP in these patients may lead to brain herniation, resulting in neurologic compromise and death. Therefore, in children with newly diagnosed intracranial tumors and signs of increased ICP, the LP usually is delayed until surgery or shunt placement.

Specific Tumors
Table 524.5 provides a classification of tumors of the central nervous system.
Table 524.5
WHO Classification of Central Nervous System (CNS) Tumors
| DIFFUSE ASTROCYTIC AND OLIGODENDROGLIAL TUMORS | |
| Diffuse astrocytoma, IDH-mutant | 9400/3 |
| Gemistocytic astrocytoma, IDH-mutant | 9411/3 |
| Diffuse astrocytoma, IDH-wild type | 9400/3 |
| Diffuse astrocytoma, NOS | 9400/3 |
| Anaplastic astrocytoma, IDH-mutant | 9401/3 |
| Anaplastic astrocytoma, IDH-wild type | 9401/3 |
| Anaplastic astrocytoma, NOS | 9401/3 |
| Glioblastoma, IDH-wild type | 9440/3 |
| Giant cell glioblastoma | 9441/3 |
| Gliosarcoma | 9442/3 |
| Epithelioid glioblastoma | 9440/3 |
| Glioblastoma, IDH-mutant | 9445/3* |
| Glioblastoma, NOS | 9440/3 |
| Diffuse midline glioma, H3 K27M-mutant | 9385/3* |
| Oligodendroglioma, IDH-mutant and 1p/19q-codeleted | 9450/3 |
| Oligodendroglioma, NOS | 9450/3 |
| Anaplastic oligodendroglioma, IDH-mutant and 1p/19q-codeleted | 9451/3 |
| Anaplastic oligodendroglioma, NOS | 9451/3 |
| Oligoastrocytoma, NOS | 9382/3 |
| Anaplastic oligoastrocytoma, NOS | 9382/3 |
| OTHER ASTROCYTIC TUMORS | |
| Pilocytic astrocytoma | 9421/1 |
| Pilomyxoid astrocytoma | 9425/3 |
| Subependymal giant cell astrocytoma | 9384/1 |
| Pleomorphic xanthoastrocytoma | 9424/3 |
| Anaplastic pleomorphic xanthoastrocytoma | 9424/3 |
| EPENDYMAL TUMORS | |
| Subependymoma | 9383/1 |
| Myxopapillary ependymoma | 9394/1 |
| Ependymoma | 9391/3 |
| Papillary ependymoma | 9393/3 |
| Clear cell ependymoma | 9391/3 |
| Tanycytic ependymoma | 9391/3 |
| Ependymoma, RELA fusion-positive | 9393/3* |
| Anaplastic ependymoma | 9392/3 |
| OTHER GLIOMAS | |
| Chordoid glioma of the third ventricle | 9444/1 |
| Angiocentric glioma | 9431/1 |
| Astroblastoma | 9430/3 |
| CHOROID PLEXUS TUMORS | |
| Choroid plexus papilloma | 9390/0 |
| Atypical choroid plexus papilloma | 9390/1 |
| Choroid plexus carcinoma | 9390/3 |
| NEURONAL AND MIXED NEURONAL-GLIAL TUMORS | |
| Dysembryoplastic neuroepithelial tumor | 9413/0 |
| Gangliocytoma | 9492/0 |
| Ganglioglioma | 9505/1 |
| Anaplastic ganglioglioma | 9505/3 |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | 9493/0 |
| Desmoplastic infantile astrocytoma and ganglioglioma | 9412/1 |
| Papillary glioneuronal tumor | 9509/1 |
| Rosette-forming glioneuronal tumor | 9509/1 |
| Diffuse leptomeningeal glioneuronal tumor | |
| Central neurocytoma | 9506/1 |
| Extraventricular neurocytoma | 9506/1 |
| Cerebellar liponeurocytoma | 9506/1 |
| Paraganglioma | 8693/1 |
| TUMORS OF THE PINEAL REGION | |
| Pineocytoma | 9361/1 |
| Pineal parenchymal tumor of intermediate differentiation | 9362/3 |
| Pineoblastoma | 9362/3 |
| Papillary tumor of the pineal region | 9395/3 |
| EMBRYONAL TUMORS | |
| Medulloblastomas, genetically defined | |
| Medulloblastoma, WNT-activated | 9475/3* |
| Medulloblastoma, SHH-activated and TP53 -mutant | 9476/3* |
| Medulloblastoma, SHH-activated and TP53-wild type | 9471/3 |
| Medulloblastoma, non-WNT/non-SHH | 9477/3* |
| Medulloblastoma, group 3 | |
| Medulloblastoma, group 4 | |
| Medulloblastomas, histologically defined | |
| Medulloblastoma, classic | 9470/3 |
| Medulloblastoma, desmoplastic/nodular | 9471/3 |
| Medulloblastoma, with extensive nodularity | 9471/3 |
| Medulloblastoma, large cell/anaplastic | 9474/3 |
| Medulloblastoma, NOS | 9470/3 |
| Embryonal tumor with multilayered rosettes, C19MC-altered | 9478/3* |
| Embryonal tumor with multilayered rosettes, NOS | 9478/3 |
| Medulloepithelioma | 9501/3 |
| CNS neuroblastoma | 9500/3 |
| CNS ganglioneuroblastoma | 9490/3 |
| CNS embryonal tumor, NOS | 9473/3 |
| Atypical teratoid/rhabdoid tumor | 9508/3 |
| CNS embryonal tumor with rhabdoid features | 9508/3 |
| TUMORS OF THE CRANIAL AND PARASPINAL NERVES | |
| Schwannoma | 9560/0 |
| Cellular schwannoma | 9560/0 |
| Plexiform schwannoma | 9560/0 |
| Melanotic schwannoma | 9560/1 |
| Neurofibroma | 9540/0 |
| Atypical neurofibroma | 9540/0 |
| Plexiform neurofibroma | 9550/0 |
| Perineurioma | 9571/0 |
| Hybrid nerve sheath tumors | |
| Malignant peripheral nerve sheath tumor | 9540/3 |
| Epithelioid MPNST | 9540/3 |
| MPNST with perineurial differentiation | 9540/3 |
| MENINGIOMAS | |
| Meningioma | 9530/0 |
| Meningothelial meningioma | 9531/0 |
| Fibrous meningioma | 9532/0 |
| Transitional meningioma | 9537/0 |
| Psammomatous meningioma | 9533/0 |
| Angiomatous meningioma | 9534/0 |
| Microcystic meningioma | 9530/0 |
| Secretory meningioma | 9530/0 |
| Lymphoplasmacyte-rich meningioma | 9530/0 |
| Metaplastic meningioma | 9530/0 |
| Chordoid meningioma | 9538/1 |
| Clear cell meningioma | 9538/1 |
| Atypical meningioma | 9539/1 |
| Papillary meningioma | 9538/3 |
| Rhabdoid meningioma | 9538/3 |
| Anaplastic (malignant) meningioma | 9530/3 |
| MESENCHYMAL, NONMENINGOTHELIAL TUMORS | |
| Solitary fibrous tumor/hemangiopericytoma** | |
| Grade 1 | 8815/0 |
| Grade 2 | 8815/1 |
| Grade 3 | 8815/3 |
| Hemangioblastoma | 9161/1 |
| Hemangioma | 9120/0 |
| Epithelioid hemangioendothelioma | 9133/3 |
| Angiosarcoma | 9120/3 |
| Kaposi sarcoma | 9140/3 |
| Ewing sarcoma/PNET | 9364/3 |
| Lipoma | 8850/0 |
| Angiolipoma | 8861/0 |
| Hibernoma | 8880/0 |
| Liposarcoma | 8850/3 |
| Desmoid-type fibromatosis | 8821/1 |
| Myofibroblastoma | 8825/0 |
| Inflammatory myofibroblastic tumor | 8825/1 |
| Benign fibrous histiocytoma | 8830/0 |
| Fibrosarcoma | 8810/3 |
| Undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma | 8802/3 |
| Leiomyoma | 8890/0 |
| Leiomyosarcoma | 8890/3 |
| Rhabdomyoma | 8900/0 |
| Rhabdomyosarcoma | 8890/3 |
| Chondroma | 9220/0 |
| Chondrosarcoma | 9220/3 |
| Osteoma | 9180/0 |
| Osteochondroma | 9210/0 |
| Osteosarcoma | 9180/3 |
| MELANOCYTIC TUMORS | |
| Meningeal melanocytosis | 8728/0 |
| Meningeal melanocytoma | 8728/1 |
| Meningeal melanoma | 8720/3 |
| Meningeal melanomatosis | 8728/3 |
| LYMPHOMAS | |
| Diffuse large B-cell lymphoma of the CNS | 9680/3 |
| Immunodeficiency-associated CNS lymphomas | |
| AIDS-related diffuse large B-cell lymphoma | |
| EBV-positive diffuse large B-cell lymphoma, NOS | |
| Lymphomatoid granulomatosis | 9766/1 |
| Low-grade B-cell lymphomas of the CNS | |
| T-cell and NK/T-cell lymphomas of the CNS | |
| Anaplastic large cell lymphoma, ALK-positive | 9714/3 |
| Anaplastic large cell lymphoma, ALK-negative | 9702/3 |
| HISTIOCYTIC TUMORS | |
| Langerhans cell histiocytosis | 9751/3 |
| Erdheim-Chester disease | 9750/1 |
| Rosai-Dorfman disease | |
| Juvenile xanthogranuloma | |
| Histiocytic sarcoma | 9755/3 |
| GERM CELL TUMORS | |
| Germinoma | 9064/3 |
| Embryonal carcinoma | 9070/3 |
| Yolk sac tumor | 9071/3 |
| Choriocarcinoma | 9100/3 |
| Teratoma | 9080/1 |
| Mature teratoma | 9080/0 |
| Immature teratoma | 9080/3 |
| Teratoma with malignant transformation | 9084/3 |
| Mixed germ cell tumor | 9085/3 |
| TUMORS OF THE SELLAR REGION | |
| Craniopharyngioma | 9350/1 |
| Adamantinomatous craniopharyngioma | 9351/1 |
| Papillary craniopharyngioma | 9352/1 |
| Granular cell tumor of the sellar region | 9582/0 |
| Pituicytoma | 9432/1 |
| Spindle cell oncocytoma | 8290/0 |
| METASTATIC TUMORS | |
* These new codes were approved by the IARC/WHO Committee for ICD-O.
** Grading according to the 2013 WHO Classification of Tumors of Soft Tissue and Bone.
The morphology codes are from the International Classification of Diseases for Oncology (ICD-O) (742A). Behavior is coded /0 for benign tumors; /1 for unspecified, borderline, or uncertain behavior; /2 for carcinoma in situ and grade III intraepithelial neoplasia; and /3 for malignant tumors. The classification is modified from the previous WHO classification, taking into account changes in our understanding of these lesions.
The italicized entries are provisional, i.e., the WHO Working Group thought there was insufficient evidence to recognize these as distinct disease entities at this time.
NOS, Not otherwise specified.
Adapted from Louis DN, Ohgaki H, Wiestler OD, Cavenee WK: World Health Organization histological classification of tumours of the central nervous system, 2016, International Agency for Research on Cancer, France.
Astrocytoma
Astrocytomas are a heterogeneous group of tumors that account for approximately 40% of pediatric CNS malignancies. These tumors occur throughout the CNS.
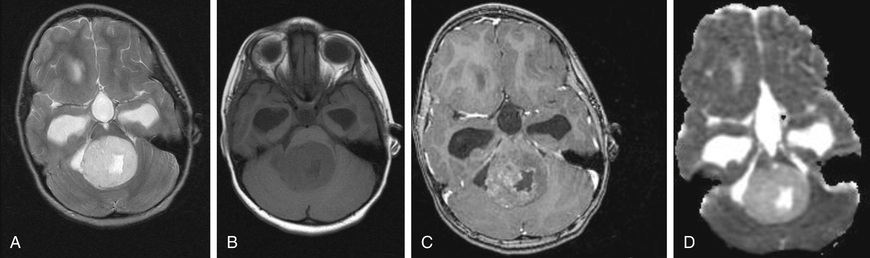
Low-grade astrocytomas (LGAs) , the predominant group of astrocytomas in childhood, are characterized by an indolent clinical course. Pilocytic astrocytoma (PA) is the most common astrocytoma in children, accounting for approximately 20% of all brain tumors. Based on clinicopathologic features using the WHO classification, PA is classified as a grade I tumor. Although PA can occur anywhere in the CNS, the classic sites are the cerebellum and the optic pathway region (Fig. 524.4 ). The classic but not exclusive neuroradiologic finding in PA is the presence of a contrast-enhancing nodule within the wall of a cystic mass. The microscopic findings include the biphasic appearance of bundles of compact fibrillary tissue interspersed with loose, microcystic, spongy areas. The presence of Rosenthal fibers (condensed masses of glial filaments occurring in compact areas) with low mitotic potentials helps establish the diagnosis. A small proportion of these tumors can progress and develop leptomeningeal spread, particularly in the optic pathway region and very rarely transform to higher-grade aggressive type. A PA of the optic nerve and chiasmal region is a relatively common finding in patients with neurofibromatosis type 1 (15% incidence).PA has activation of the MAPK pathway in the form of BRAF fusion or duplication and less often BRAF mutation (V600E ). Other low-grade tumors occurring in the pediatric age-group with clinicopathologic characteristics similar to those of PA include pleomorphic xanthoastrocytoma, pilomyxoid astrocytoma, and subependymal giant cell astrocytoma.
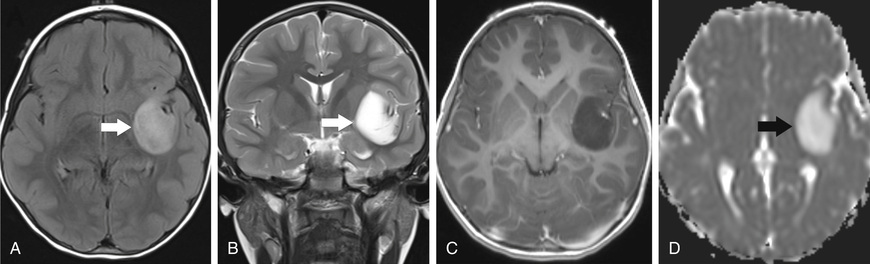
The 2nd most common astrocytoma is diffuse astrocytoma (DA), which consists of a group of tumors characterized by a pattern of diffuse infiltration of tumor cells amidst normal neural tissue. DA accounts for 15% of brain tumors, with the fibrillary type the most common in children. Histologically, these low-grade tumors demonstrate greater cellularity, with few mitotic figures, nuclear pleomorphism, and microcysts. They occur anywhere in the CNS, with a predilection to supratentorial locations (Fig. 524.5 ). The characteristic MRI finding is a lack of enhancement after contrast infusion. Molecular genetic abnormalities found in DA include mutations of P53 and overexpression of platelet-derived growth factor receptor α. Evolution of DA into malignant astrocytoma is associated with cumulative acquisition of multiple molecular abnormalities. Over activation of MAPK pathway was detected in DA in the form of BRAF V600E mutation and FGFR1 duplication.
Pilomyxoid astrocytoma occurs most commonly in the hypothalamic/optic chiasmic region and carries a high risk of local as well as cerebrospinal spread. This astrocytoma affects young children and infants. It is classified as a WHO grade II tumor.
The clinical management of LGAs focuses on a multimodal approach incorporating surgery as the primary treatment, as well as radiation therapy and chemotherapy. With complete surgical resection, overall survival (OS) approaches 80–100%. In patients with partial resection, OS varies from 50–95%, depending on the anatomic location of the tumor. In the patient who has undergone partial tumor resection and has stable neurologic status, the current approach is to follow the patient closely by examination and imaging. With evidence of progression, a 2nd surgical resection should be considered. In patients in whom a 2nd procedure was less than complete or is not feasible, radiation therapy is beneficial. Radiation therapy is delivered to the tumor bed at a total cumulative dose ranging from 50-55 Gy. Modern surgical techniques and innovative radiation therapy methodology, including proton-beam radiation, may have a positive impact on the survival and clinical outcome of these patients. The role of chemotherapy in the management of LGAs is evolving. Because of concerns regarding morbidity from radiation therapy in young children, several chemotherapy approaches have been evaluated, especially in children <10 yr old. Complete response to chemotherapy is uncommon; however, these approaches have yielded durable control of disease in 70–100% of patients. Patients with midline tumors in the hypothalamic/optic chiasmatic region have tended to do less well (Fig. 524.6 ). Taken together, the chemotherapy approaches have permitted delay and, potentially, avoidance of radiation therapy. Chemotherapy agents given singly or in combination for LGA include carboplatin, vincristine, lomustine, procarbazine, temozolomide, and vinblastine. Observation is the primary approach in clinical management of selected patients with LGAs that are biologically indolent (neurofibromatosis type 1 and midbrain astrocytoma). Astrocytomas associated with tuberous sclerosis have responded to everolimus (mammalian target of rapamycin inhibitor).
Malignant astrocytomas are less common in children and adolescents than in adults, accounting for 7–10% of all childhood brain tumors. Among this group, anaplastic astrocytoma (WHO grade III) (Fig. 524.7 ) is more common than glioblastoma multiforme (WHO grade IV) (Fig. 524.8 ). The histopathology of anaplastic astrocytomas demonstrates greater cellularity than that of LGA, with cellular and nuclear atypia, and the presence of mitoses. Characteristic histopathologic findings in glioblastoma multiforme include dense cellularity, high mitotic index, microvascular proliferation, and foci of tumor necrosis. Genome-wide DNA methylation patterns have now identified 5 molecular subgroups of pediatric high-grade glioma (HGG), these subgroups appear to have distinct cellular origins and biologic drivers. Common genetic alterations include gene mutations in histone H3.3 or H3.1 , P53 , and BRAF, in addition to focal amplifications of oncogene (PDGFRA and EGFR ) and deletions of tumor suppressor genes (CDKN2A and CDKN2B ).

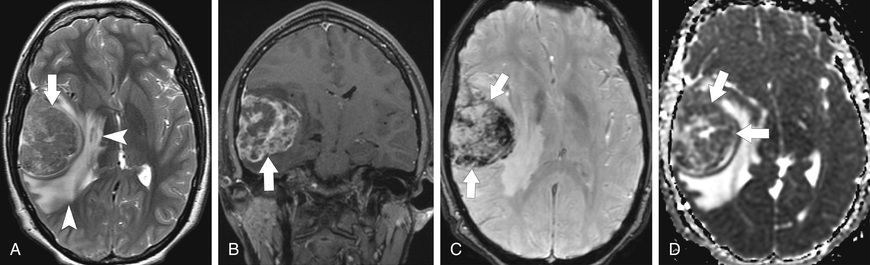
Optimal therapeutic approaches for malignant astrocytomas have yet to be defined. Standard therapy continues to be surgical resection followed by involved-field radiation therapy. A study of adult glioblastoma showed significantly better survival with temozolomide during and after irradiation than with irradiation alone. Current therapeutic approaches incorporate novel chemotherapeutic agents with radiation therapy. Enrollment in a clinical trial may be the best therapeutic option in these tumors for which standard therapy is suboptimal. Immune therapy with chimeric antigen receptor T cells targeting the tumor antigen interleukin-13 receptor α holds promise as a therapy for glioblastomas.
Oligodendrogliomas are uncommon tumors of childhood. These infiltrating tumors occur predominantly in the cerebral cortex and originate in the white matter. Histologically, oligodendrogliomas consist of rounded cells with little cytoplasm and microcalcifications. Observation of a calcified cortical mass on CT in a patient presenting with a seizure is suggestive of oligodendroglioma. Treatment approaches are similar to those for infiltrating astrocytomas.
Ependymal Tumors
Ependymal tumors are derived from the ependymal lining of the ventricular system. Cellular ependymoma (WHO grade II) is the most common of these neoplasms, accounting for 10% of childhood tumors. Approximately 70% of ependymomas in childhood occur in the posterior fossa. The mean age of patients is 6 yr, with approximately 40% of cases occurring in children <4 yr old. The incidence of leptomeningeal spread approaches 10% overall. Clinical presentation can be insidious and often depends on the anatomic location of the tumor. MRI demonstrates a well-circumscribed tumor with variable and complex patterns of gadolinium enhancement, with or without cystic structures (Fig. 524.9 ). These tumors usually are noninvasive, extending into the ventricular lumen and/or displacing normal structures, sometimes leading to significant obstructive hydrocephalus. Histologic characteristics include perivascular pseudorosettes, ependymal rosettes, monomorphic nuclear morphology, and occasional nonpalisading foci of necrosis.
Other histologic subtypes include anaplastic ependymoma (WHO grade III), which is much less common in childhood and is characterized by a high mitotic index and histologic features of microvascular proliferation and pseudopalisading necrosis. Myxopapillary ependymoma (WHO grade I) is a slow-growing tumor arising from the filum terminale and conus medullaris and appears to be a biologically different subtype. Preliminary studies suggest that there are genetically distinct subtypes of ependymoma, exemplified by an association between alterations in the NF2 gene and spinal ependymoma.
Surgery is the primary treatment modality, with extent of surgical resection a major prognostic factor. Two other major prognostic factors are age, with younger children having poorer outcomes, and tumor location, with localization in the posterior fossa, which often is seen in young children, associated with poorer outcomes. Surgery alone is rarely curative. Multimodal therapy incorporating irradiation with surgery has resulted in long-term survival in approximately 40% of patients with ependymoma undergoing gross total resection. Recurrence is predominantly local. The role of chemotherapy in multimodal therapy of ependymoma is still unclear. Current investigations are directed toward identification of optimal radiation dose, surgical questions addressing the use of second-look procedures after chemotherapy, and further evaluation of classic as well as novel chemotherapeutic agents. Genome-wide DNA methylation patterns have identified nine molecular subgroups in these tumors, across 3 anatomic compartments: supratentorial (ST), posterior fossa (PF), and spinal locations. Two subgroups (A and B) of PF ependymoma have been identified with distinct molecular and clinical characteristics, and use of targeted chemotherapy against these subtypes is now being evaluated.
Choroid Plexus Tumors
Choroid plexus tumors account for 2–4% of childhood CNS tumors. They are the most common CNS tumors in children <1 yr old and account for 10–20% of CNS tumors in infants. These tumors are intraventricular epithelial neoplasms arising from the choroid plexus. Children present with signs and symptoms of increased ICP. Infants may present with macrocephaly and focal neurologic deficits. In children, these tumors predominantly occur supratentorially in the lateral ventricles.
The group of choroid plexus tumors comprises choroid plexus papillomas (WHO grade I), atypical choroid plexus papillomas (WHO grade II), and choroid plexus carcinomas (WHO grade III). Choroid plexus papilloma is the most common of this group and is a well-circumscribed lesion with focal calcification on neuroimaging. Choroid plexus carcinoma is a malignant tumor with metastatic potential to seed into the CSF pathways. This malignancy has the histologic characteristics of nuclear pleomorphism, high mitotic index, necrosis, and increased cell density. MRI typically demonstrates a large, hyperdense, contrast-enhancing intraventricular mass with peritumoral edema, hemorrhage, and hydrocephalus. The tumor suppressor p53 is crucially involved in the biology of this cancer and may contribute to aggressive tumor behavior. Molecular data subclassify choroid plexus tumors into 3 distinct subgroups, with different molecular aberrations and clinical outcomes. These tumors are associated with the Li-Fraumeni syndrome .
After complete surgical resection, the frequency of cure for choroid plexus papilloma approaches 100%, whereas that for choroid plexus carcinoma approaches 20–40%. Reports suggest that radiation therapy and/or chemotherapy may lead to better disease control for choroid plexus carcinoma.
Embryonal Tumors
Embryonal tumors or primitive neuroectodermal tumors (PNETs) are the most common group of malignant CNS tumors of childhood, accounting for approximately 20% of pediatric CNS tumors. They have the potential to metastasize to the neuraxis and beyond. The group includes medulloblastoma, supratentorial PNET, ependymoblastoma, medulloepithelioblastoma, and atypical teratoid/rhabdoid tumor, all of which are histologically classified as WHO grade IV tumors.
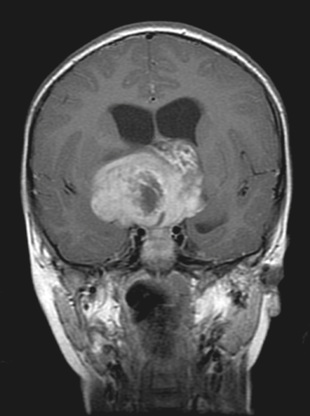
Medulloblastoma accounts for 90% of embryonal CNS tumors and is a cerebellar tumor occurring predominantly in males and at a median age of 5-7 yr (Table 524.6 ). CT and MRI demonstrate a solid, homogeneous, contrast medium–enhancing mass in the posterior fossa causing 4th ventricular obstruction and hydrocephalus (Fig. 524.10 ). Up to 30% of patients with medulloblastoma present with neuroimaging evidence of leptomeningeal spread. Among a variety of diverse histologic patterns of this tumor, the most common is a monomorphic sheet of undifferentiated cells classically noted as small, blue, round cells. Neuronal differentiation is more common among these tumors and is characterized histologically by the presence of Homer Wright rosettes and immunopositivity for synaptophysin. An anaplastic variant is often more aggressive and may be associated with worse prognosis. Patients present with signs and symptoms of increased ICP (i.e., headache, nausea, vomiting, mental status changes, hypertension) and cerebellar dysfunction (i.e., ataxia, poor balance, dysmetria). Standard clinical staging evaluation includes MRI of the brain and spine, both preoperatively and postoperatively, as well as LP after the increased ICP has resolved. The Chang staging system, originally based on surgical information, has been modified to incorporate information from neuroimaging to identify risk categories. Clinical features that have consistently demonstrated prognostic significance include age at diagnosis, extent of disease, and extent of surgical resection. Patients <4 yr old have a poor outcome, partly as the result of a higher incidence of disseminated disease on presentation and past therapeutic approaches that have used less intense therapies. Patients with disseminated disease at diagnosis (M >0), including positive CSF cytologic result alone (M1), have a much poorer outcome than those with no dissemination (M0). Similarly, patients with gross residual disease after surgery have worse outcomes than those in whom surgery achieved gross total resection of disease.
Table 524.6
Summary of the Most Common Integrated Medulloblastoma Diagnoses, With Clinical Correlates
| GENETIC PROFILE | HISTOLOGY | PROGNOSIS |
|---|---|---|
| Medulloblastoma, WNT-activated | Classic | Low-risk tumor; classic morphology found in almost all WNT-activated tumors |
| Large cell/anaplastic (very rare) | Tumor of uncertain clinicopathologic significance | |
| Medulloblastoma, SHH-activated, TP52 -mutant | Classic | Uncommon high-risk tumor |
| Large cell/anaplastic | High-risk tumor; prevalent in children age 7-17 yr | |
| Desmoplastic/nodular (very rare) | Tumor of uncertain clinicopathologic significance | |
| Medulloblastoma, SHH-activated, TP53 -wild type | Classic | Standard-risk tumor |
| Large cell/anaplastic | Tumor of uncertain clinicopathologic significance | |
| Desmoplastic/nodular | Low-risk tumor in infants; prevalent in infants and adults | |
| Extensive nodularity | Low-risk tumor of infancy | |
| Medulloblastoma, non-WNT/non-SHH, group 3 | Classic | Standard-risk tumor |
| Large cell/anaplastic | High-risk tumor | |
| Medulloblastoma, non-WNT/non-SHH, group 4 | Classic | Standard-risk tumor; classic morphology found in almost all group 4 tumors |
| Large cell/anaplastic (rare) | Tumor of uncertain clinicopathologic significance |
Adapted from Louis DN, Ohgaki H, Wiestler OD, Cavenee WK: World Health Organization histological classification of tumours of the central nervous system, 2016, International Agency for Research on Cancer, France.
Cytogenetic and molecular genetic studies have demonstrated multiple abnormalities in medulloblastoma. The most common abnormality involves chromosome 17p deletions, which occur in 30–40% of all cases. These deletions are not associated with P53 mutations. Several signaling pathways have been shown to be active in medulloblastomas, including the sonic hedgehog (SHH) pathway, predominately associated with the desmoplastic variants, and the WNT pathway, which can occur in up to 15% of cases and has been associated with improved survival. Integrative genomic studies have recently identified at least 4 distinct molecular subgroups of medulloblastoma—WNT, SHH, group 3, and group 4—which exhibit highly discriminate transcriptional, cytogenetic, and mutational spectra, in addition to divergent patient demographics and clinical behavior. These prognostic groups still must be validated in larger prospective studies, though the WNT subgroup is known to have the most favorable outcome.
A multimodal treatment approach is pursued in medulloblastoma, with surgery as the starting point of treatment. Medulloblastoma is sensitive to both chemotherapy and radiation therapy. With technologic advances in neurosurgery, neuroradiology, and radiation therapy, as well as identification of chemotherapy as an effective modality, the overall outcome among all patients approaches 60–70%. Standard radiation treatment in standard-risk medulloblastoma incorporates craniospinal radiation at a total cumulative dose of 24 Gy, with a cumulative dose of 50-55 Gy to the tumor bed. Craniospinal radiation at this dose in children <3 yr old results in severe late neurologic sequelae, including microcephaly, learning disabilities, cognitive impairment, neuroendocrine dysfunction (growth failure, hypothyroidism, hypogonadism, absence/delay of puberty), and second malignancies. Similarly, in older children, late sequelae such as learning disabilities, neuroendocrine dysfunction, and second malignancies can occur.
These observations have resulted in stratification of treatment approaches into (1) patients <3 yr old; (2) standard-risk patients >3 yr old with surgical total resection and no disease dissemination (M0); and (3) high-risk patients >3 yr old with disease dissemination (M >0) and/or bulky residual disease after surgery. With the risk-based approach to treatment, children with high-risk medulloblastoma receive high-dose craniospinal radiation (36 Gy) with chemotherapy during and after radiation therapy. Since the dose of radiation depends on the risk stratification, complete staging with MRI of the spine before starting treatment is essential for the best chance of survival.
Approaches in young children (usually <3 yr) incorporate high-dose chemotherapy with peripheral stem cell reinfusion to avoid radiation therapy. OS in children with nonmetastatic medulloblastoma and gross total tumor resection approaches 85%. The presence of bulky residual tumor (56% survival) or metastases (38% survival) confers a poor prognosis. The molecular classification is being evaluated to stratify risk groups and tailor therapy accordingly. The WNT subgroup and nonmetastatic group 4 tumors are recognized as low-risk tumors that may qualify for reduced therapy. High-risk groups were defined as patients with metastatic SHH or group 4 tumors, where intensification of therapy is being profiled.
Supratentorial primitive neuroectodermal tumors (SPNETs) account for 2–3% of childhood brain tumors, primarily in children within the 1st decade of life. These tumors are similar histologically to medulloblastoma and are composed of undifferentiated or poorly differentiated neuroepithelial cells. Historically, patients with SPNETs have had poorer outcomes than those with medulloblastoma after combined-modality therapy. In current clinical trials, children with SPNETs are considered among the high-risk groups and receive dose-intense chemotherapy with craniospinal radiation therapy.
Atypical teratoid/rhabdoid tumor is a very aggressive embryonal malignancy that occurs predominantly in children <5 yr old and can occur at any location in the neuraxis. The histology demonstrates a heterogeneous pattern of cells, including rhabdoid cells that express epithelial membrane antigen and neurofilament antigen. The characteristic cytogenetic pattern is partial or complete deletion of chromosome 22q11.2 that is associated with mutation in the INI1 gene. The relation between this mutation and tumorigenesis is unclear. Though the overall prognosis remains poor, intensive chemotherapy, focal radiation, and high-dose chemotherapy with stem cell rescue has shown a trend toward improved survival. This trend is noted more in patients who undergo complete resection of tumor and focal radiation. Newer data now suggest 3 molecular subtypes within this tumor, and the favorable response seen in some patients reaffirms the molecular intertumor heterogeneity
Pineal Parenchymal Tumors
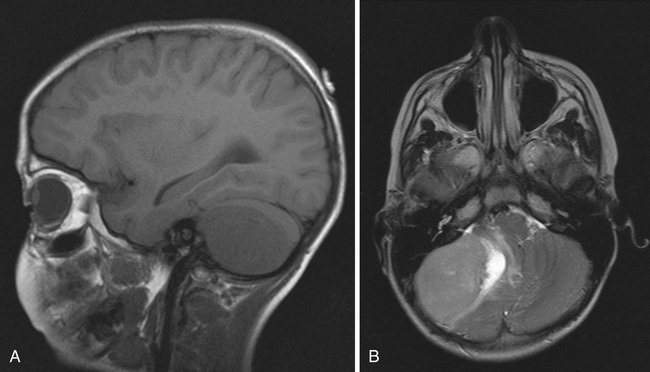
The pineal parenchymal tumors are the most common malignancies after germ cell tumors that occur in the pineal region. These include pineoblastoma (Fig. 524.11 ), occurring predominantly in childhood; pineocytoma ; and the mixed pineal-parenchymal tumors . The therapeutic approach in this group of diseases is multimodal. There was significant concern regarding the location of these masses and the potential complications of surgical intervention. With developments in neurosurgical technique and surgical technology, the morbidity and mortality associated with these approaches have greatly decreased. Stereotactic biopsy of these tumors may be adequate to establish diagnosis; however, consideration should be given to total resection of the lesion before institution of additional therapy. Pineoblastoma, the more malignant variant, is considered a subgroup of childhood PNETs. Chemotherapy regimens incorporate cisplatin, cyclophosphamide (Cytoxan), etoposide (VP-16), and vincristine and/or lomustine. Data have shown that survival outcome of combined chemotherapy and radiation therapy in pineal-region PNETs approaches 70% at 5 yr, similar to that for medulloblastoma. Pineocytoma usually is approached with surgical resection.
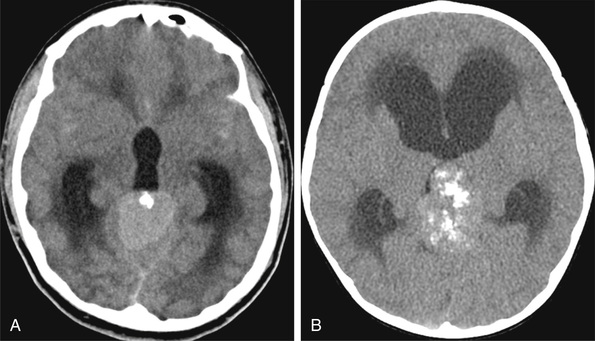
Craniopharyngioma
Craniopharyngioma (CP; WHO grade I) is a common tumor of childhood, accounting for 7–10% of all childhood tumors. Two histologic subtypes have been identified, adamantinomatous CP and papillary CP, each with specific origin and genetic alterations. BRAF V600E mutations were solely found in the papillary CP subgroup, which is a common type in adults, whereas CTNNB1 mutations were exclusively detected in adamantinomatous CP, which is common in children. Children with CP often present with endocrinologic abnormalities (growth failure and delayed sexual maturation) and/or visual changes (decreased acuity or visual field abnormalities). These tumors are often large and heterogeneous, displaying both solid and cystic components, and occur within the suprasellar region. They are minimally invasive, adhere to adjacent brain parenchyma, and engulf normal brain structures. MRI demonstrates the solid tumor with cystic structures containing fluid of intermediate density, and CT may show calcifications (Fig. 524.12 ). Surgery is the primary treatment modality, with gross total resection curative. Controversy exists regarding the relative roles of surgery and radiation therapy in large, complex tumors. Significant morbidity (panhypopituitarism, growth failure, visual loss) is associated with CPs and their therapy because of the anatomic location. There is no role for chemotherapy in CP.

Germ Cell Tumors
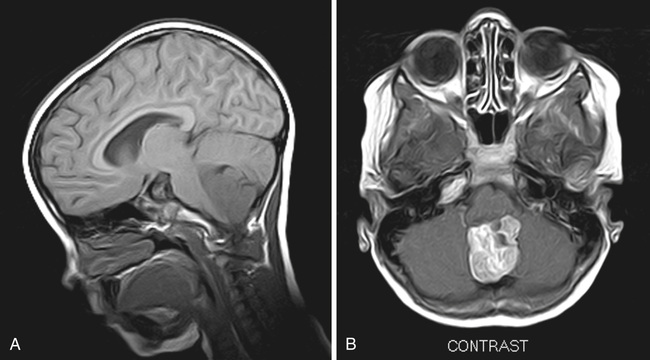
Germ cell tumors of the CNS are a heterogeneous group and primarily tumors of childhood, arising predominantly in midline structures of the pineal and suprasellar regions (see Figs. 524.3 and 524.11 ). They account for 3–5% of pediatric brain tumors. The peak incidence of germ cell tumors occurs in children 10-12 yr old. Overall, there is a male preponderance, although there is a female preponderance for suprasellar tumors. Germ cell tumors occur multifocally in 5–10% of cases. This group of tumors is much more prevalent in Asian than in European populations. Delays in diagnosis can occur because these tumors have a particularly insidious course; the initial presenting symptoms may be subtle. As in peripheral germ cell tumors, the analysis of AFP and β-hCG levels may be useful in establishing the diagnosis and monitoring treatment response. Surgical biopsy is recommended to establish the diagnosis; however, secreting germinomas and nongerminomatous germ cell tumors may be diagnosed by protein marker elevations. Therapeutic approaches to germinomas and nongerminomatous germ cell tumors are different. Survival among patients with pure germinoma exceeds 90%. The postsurgical treatment of pure germinomas is somewhat controversial in defining the relative roles of chemotherapy and radiation therapy. Clinical trials have investigated the use of chemotherapy and reduced radiation and field after surgery in pure germinomas. The therapeutic approach to nongerminomatous germ cell tumors is more aggressive, combining more intense chemotherapy regimens with craniospinal radiation therapy. Survival rates among patients with nongerminomatous germ cell tumors are much lower than in those with germinoma, ranging from 40–70% at 5 yr. Trials have shown the benefit of the use of high doses of chemotherapy with peripheral blood stem cell rescue.
Tumors of the Brainstem
Tumors of the brainstem are a heterogeneous group and account for 10–15% of childhood primary CNS tumors. Outcome depends on tumor location, imaging characteristics, and the patient's clinical status. Patients with these tumors may present with motor weakness, cranial nerve dysfunction, cerebellar dysfunction, and signs of increased ICP. On the basis of MRI evaluation and clinical findings, tumors of the brainstem can be classified into 4 types: focal (5–10% of patients); dorsally exophytic (5–10%); cervicomedullary (5–10%); and diffuse intrinsic pontine glioma (DIPG) (70–85%) (Fig. 524.13 ). Surgical resection is the primary treatment approach for focal and dorsally exophytic tumors and leads to a favorable outcome. Histologically, these 2 groups usually are low-grade gliomas. Because of their location, cervicomedullary tumors may not be amenable to surgical resection but are sensitive to radiation therapy. DIPG, characterized by the diffuse, infiltrating grade II-IV glioma, is associated with a poor outcome independent of histologic diagnosis. These tumors are not amenable to surgical resection. Biopsy in children in whom MRI shows DIPG is controversial and is not recommended unless there are atypical radiographic findings suspicious for another diagnosis, such as infection, vascular malformation, myelination disorder, or metastatic tumor. Recent studies have unraveled the unique genetic makeup of this fatal brain cancer, with almost 80% found to harbor mutations in histone H3.3 or H3.1 (H3-K27M) and 20% with mutations affecting the activin receptor gene (ACVR1). Now, 3 molecularly distinct subgroups have been identified: H3-K27M, silent, and MYCN.

The standard treatment approach has been radiation therapy, and median survival with this treatment is 12 mo, at best. Use of chemotherapy, including high-dose chemotherapy with peripheral blood stem cell rescue, has not yet been of survival benefit in this group of patients. Current approaches include evaluation of investigational agents alone or in combination with radiation therapy, similar to approaches being pursued in patients with malignant gliomas.
Metastatic Tumors
Metastatic spread of other childhood malignancies to the brain is uncommon. Childhood acute lymphoblastic leukemia and non-Hodgkin lymphoma can spread to the leptomeninges, causing symptoms of communicating hydrocephalus. Chloromas are collections of myeloid leukemia cells and can occur throughout the neuraxis. Rarely, brain parenchymal metastases occur from lymphoma, neuroblastoma, rhabdomyosarcoma, Ewing sarcoma, osteosarcoma, and clear cell sarcoma of the kidney. Therapeutic approaches are based on the specific histologic diagnosis and may incorporate radiation therapy, intrathecal chemotherapy, and systemic chemotherapy. Medulloblastoma is the childhood brain tumor that most often metastasizes extraneurally. Less often, extraneural metastases from malignant glioma, PNET, and ependymoma can occur. Ventriculoperitoneal shunts have been known to allow extraneural metastases, primarily within the peritoneal cavity but also systemically.
Complications and Long-Term Management
Data from the NCI SEER Program indicate that >70% of patients with childhood brain tumors will be long-term survivors. At least 50% of these survivors will experience chronic problems as a direct result of their tumors and treatment. These problems include chronic neurologic deficits (e.g., focal motor/sensory abnormalities), seizure disorders, neurocognitive deficits (e.g., developmental delays, learning disabilities), and neuroendocrine deficiencies (e.g., hypothyroidism, growth failure, delay/absence of puberty). These patients are also at significant risk for secondary malignancies. Supportive multidisciplinary interventions for children with brain tumors both during and after therapy may help improve the ultimate outcome. Optimal seizure management, physical therapy, endocrine management with timely growth hormone and thyroid replacement therapy, tailored educational programs, and vocational interventions can enhance the childhood brain tumor survivor's quality of life.