Hypoparathyroidism
Daniel A. Doyle
Etiology
Hypocalcemia is common in neonates between 12 and 72 hr of life, especially in premature infants, in infants with asphyxia, and in infants of diabetic mothers (early neonatal hypocalcemia ; see Chapter 119.4 ; Table 589.1 and Fig. 589.1 ). After the 2nd to 3rd day and during the 1st wk of life, the type of feeding also is a determinant of the level of serum calcium (late neonatal hypocalcemia ). The role played by the parathyroid glands in these hypocalcemic infants is unclear, although functional immaturity of the parathyroid glands is invoked as 1 pathogenetic factor. In a group of infants with transient idiopathic hypocalcemia (1-8 wk of age), serum levels of parathyroid hormone (PTH) are significantly lower than those in normal infants. It is possible that the functional immaturity is a manifestation of a delay in development of the enzymes that convert glandular PTH to secreted PTH; other mechanisms are possible.
Table 589.1
HDR, hypoparathyroidism, sensorineural deafness, and renal anomaly; HRD, hypoparathyroidism, retardation, dysmorphism; MELAS, mitochondrial encephalomyopathy with lactic acidosis and stroke-like episode; PTH, parathyroid hormone.
Modified from Root AW, Diamond Jr FB: Disorders of mineral homeostasis in children and adolescents. In Sperling MA, editor: Pediatric endocrinology , ed 4, Philadelphia, 2014, Elsevier, Table 18.2A.
Aplasia or Hypoplasia of the Parathyroid Glands
Aplasia or hypoplasia of the parathyroid glands is often associated with the DiGeorge/velocardiofacial syndrome . This syndrome occurs in 1 in 4,000 newborns. In 90% of patients, the condition is caused by a deletion of chromosome 22q11.2. Approximately 25% of these patients inherit the chromosomal abnormality from a parent. Neonatal hypocalcemia occurs in 60% of affected patients, but it is transitory in the majority; hypocalcemia can recur or can have its onset later in life. Associated abnormalities of the 3rd and 4th pharyngeal pouches are common; these include conotruncal defects of the heart in 25%, velopharyngeal insufficiency in 32%, cleft palate in 9%, renal anomalies in 35%, and aplasia of the thymus with severe immunodeficiency in 1%. This syndrome has also been reported in a small number of patients with a deletion of chromosome 10p13, in infants of diabetic mothers, and in infants born to mothers treated with retinoic acid for acne early in pregnancy.
X-Linked Recessive Hypoparathyroidism
Familial clusters of hypoparathyroidism with various patterns of transmission have been described. In 2 large North American pedigrees, this disorder appears to be transmitted by an X-linked recessive gene located on Xq26-q27. In these families, the onset of afebrile seizures characteristically occurs in infants from 2 wk to 6 mo of age. The absence of parathyroid tissue after detailed examination of a boy with this condition suggests a defect in embryogenesis.
Autosomal Recessive Hypoparathyroidism With Dysmorphic Features
Autosomal recessive hypoparathyroidism with dysmorphic features has been described in Middle Eastern children. Parental consanguinity occurred for almost all of several dozen affected patients. Profound hypocalcemia occurs early in life, and dysmorphic features include microcephaly, deep-set eyes, beaked nose, micrognathia, and large floppy ears. Intrauterine and postnatal growth restriction are severe, and cognitive impairment is common. The putative gene is on chromosome 1q42-43. The autosomal recessive form of hypoparathyroidism that occurs with type I polyglandular autoimmune disease is described subsequently. In a few patients with autosomal recessive inheritance of isolated hypoparathyroidism, mutations of the PTH gene have been found.
Hypoparathyroidism, Sensorineural Deafness, and Renal Anomaly Syndrome
Hypoparathyroidism, sensorineural deafness, and renal anomaly occur owing to mutations of the GATA3 gene. The protein encoded by this gene is essential in the development of the parathyroids, auditory system, and kidneys. The GATA3 gene is located at chromosome 10p14 and is nonoverlapping with the DiGeorge critical region at 10p13 (see Fig. 588.1 ). Congenital ichthyosis and HDR have also been reported.
Suppression of Neonatal Parathyroid Hormone Secretion Because of Maternal Hyperparathyroidism
Neonatal PTH secretion can be suppressed by maternal hyperparathyroidism, resulting in transient hypocalcemia in the newborn infant. It appears that neonatal hypocalcemia results from suppression of the fetal parathyroid glands by exposure to elevated levels of calcium in maternal and hence fetal serum. Tetany usually develops within 3 wk but may be delayed by 1 mo or more if the infant is breastfed. Hypocalcemia can persist for weeks or months. When the cause of hypocalcemia in an infant is unknown, measurements of calcium, phosphorus, and PTH should be obtained from the mother. Most affected mothers are asymptomatic, and the cause of their hyperparathyroidism is usually a parathyroid adenoma.
Autosomal Dominant Hypoparathyroidism
Patients with autosomal dominant hypoparathyroidism have an activating (gain-of-function) mutation of the Ca2+ -sensing receptor, forcing the receptor to an on state with subsequent depression of PTH secretion even during hypocalcemia. The patients have hypercalciuria. The hypocalcemia is usually mild and might not require treatment beyond childhood (see Fig. 588.1 ).
Hypoparathyroidism Associated With Mitochondrial Disorders
Mitochondrial DNA mutations in Kearns-Sayre syndrome, MELAS (myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) syndrome, and in mitochondrial trifunctional protein–deficiency syndrome is associated with hypoparathyroidism. A diagnosis of mitochondrial cytopathy should be considered in patients with unexplained symptoms, such as ophthalmoplegia, sensorineural hearing loss, cardiac conduction disturbances, and tetany (see Fig. 588.1 ).
Surgical Hypoparathyroidism
Removal or damage of the parathyroid glands can complicate thyroidectomy. Hypoparathyroidism has developed even when the parathyroid glands have been identified and left undisturbed at the time of operation. This may be the result of interference with the blood supply or of postoperative edema and fibrosis. Symptoms of tetany can occur abruptly postoperatively and may be temporary or permanent. In some instances, symptoms develop insidiously and go undetected until months after thyroidectomy. Occasionally, the first evidence of surgical hypoparathyroidism may be the development of cataract. The status of parathyroid function should be carefully monitored in all patients undergoing thyroidectomy.
Deposition of iron pigment or of copper in the parathyroid glands (thalassemia, Wilson disease) can also produce hypoparathyroidism.
Autoimmune Hypoparathyroidism
An autoimmune mechanism for hypoparathyroidism is strongly suggested by the finding of parathyroid antibodies and by its frequent association with other autoimmune disorders or organ-specific antibodies. Autoimmune hypoparathyroidism is often associated with Addison disease and chronic mucocutaneous candidiasis. The association of at least 2 of these 3 conditions has been classified as autoimmune polyglandular disease type I (see Chapter 586 ). It is also known as autoimmune polyendocrinopathy, candidiasis, and ectodermal dystrophy (APECED). This syndrome is inherited in an autosomal recessive fashion and is not related to any single human leukocyte antigen–associated haplotype. One-third of patients with this syndrome have all 3 components; 66% have only 2 of 3 conditions. The candidiasis almost always precedes the other disorders (70% of cases occur in children younger than 5 yr of age); the hypoparathyroidism (90% of cases occur after 3 yr of age) usually occurs before Addison disease (90% of cases occur after 6 yr of age). A variety of other disorders, including alopecia areata or totalis, malabsorption disorder, pernicious anemia, gonadal failure, chronic active hepatitis, vitiligo, and insulin-dependent diabetes, occur at various times. Some of these associations might not appear until adult life. Autoimmune thyroid disease is a rare concomitant finding.
Affected siblings can have the same or different constellations of disorders (hypoparathyroidism, Addison disease). The disorder is exceptionally prevalent among Finns and Iranian Jews. The gene for this disorder is designated AIRE (autoimmune regulator); it is located on chromosome 21q22. It appears to be a transcription factor that plays an essential role in the development of immunologic tolerance. Patients with Addison disease as part of polyendocrinopathy syndrome type I have demonstrated adrenal-specific autoantibody reactivity directed against the side-chain cleavage enzyme.
Idiopathic Hypoparathyroidism
The term idiopathic hypoparathyroidism should be reserved for the small residuum of children with hypoparathyroidism for whom no causative mechanism can be defined. Most children in whom onset of hypoparathyroidism occurs after the 1st few years of life have an autoimmune condition. Autoantibodies to the extracellular domain of the calcium-sensing receptor have been identified in some patients with acquired hypoparathyroidism. One should always consider incomplete forms of DiGeorge syndrome or an activating calcium-sensing receptor mutation in the differential diagnosis.
Clinical Manifestations
There is a spectrum of parathyroid deficiencies with clinical manifestations varying from no symptoms to those of complete and long-standing deficiency. Mild deficiency may be revealed only by appropriate laboratory studies. Muscular pain and cramps are early manifestations; they progress to numbness, stiffness, and tingling of the hands and feet. There may be only a positive Chvostek or Trousseau sign or laryngeal and carpopedal spasms. Convulsions with or without loss of consciousness can occur at intervals of days, weeks, or months. These episodes can begin with abdominal pain, followed by tonic rigidity, retraction of the head, and cyanosis. Hypoparathyroidism is often mistaken for epilepsy. Headache, vomiting, increased intracranial pressure, and papilledema may be associated with convulsions and might suggest a brain tumor.
In patients with long-standing hypocalcemia, the teeth erupt late and irregularly. Enamel formation is irregular, and the teeth may be unusually soft. The skin may be dry and scaly, and the nails might have horizontal lines. Mucocutaneous candidiasis, when present, antedates the development of hypoparathyroidism; the candidal infection most often involves the nails, the oral mucosa, the angles of the mouth, and less often, the skin; it is difficult to treat.
Cataracts in patients with long-standing untreated disease are a direct consequence of hypoparathyroidism; other autoimmune ocular disorders such as keratoconjunctivitis can also occur. Manifestations of Addison disease, lymphocytic thyroiditis, pernicious anemia, alopecia areata or totalis, hepatitis, and primary gonadal insufficiency may also be associated with those of hypoparathyroidism.
Permanent physical and mental deterioration occurs if initiation of treatment is long delayed.
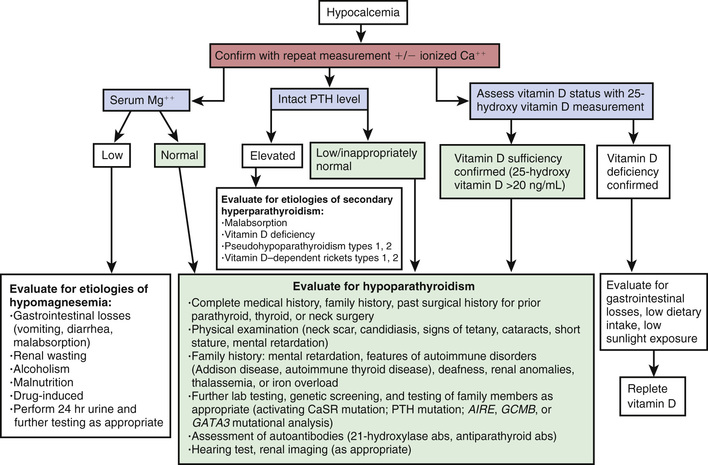
Laboratory Findings
The serum calcium level is low (5-7 mg/dL), and the phosphorus level is elevated (7-12 mg/dL). Blood levels of ionized calcium (usually approximately 45% of the total) more nearly reflect physiologic adequacy but also are low. The serum level of alkaline phosphatase is normal or low, and the level of 1,25(OH)2 D3 is usually low, but high levels have been found in some children with severe hypocalcemia. The level of magnesium is normal but should always be checked in hypocalcemic patients. Levels of PTH are low when measured by immunometric assay. Radiographs of the bones occasionally reveal an increased density limited to the metaphyses, suggesting heavy metal poisoning, or an increased density of the lamina dura. Radiographs or CT scans of the skull can reveal calcifications in the basal ganglia. There is a prolongation of the QT interval on the electrocardiogram, which disappears when the hypocalcemia is corrected. The electroencephalogram usually reveals widespread slow activity; the tracing returns to normal after the serum calcium concentration has been within the normal range for a few weeks, unless irreversible brain damage has occurred or unless the parathyroid insufficiency is associated with epilepsy. When hypoparathyroidism occurs concurrently with Addison disease, the serum level of calcium may be normal, but hypocalcemia appears after effective treatment of the adrenal insufficiency.
Treatment
Emergency treatment of neonatal tetany consists of intravenous injections of 5-10 mL or 1-3 mg/kg of a 10% solution of calcium gluconate (elemental calcium 9.3 mg/mL) at the rate of 0.5-1.0 mL/min while the heart rate is monitored and a total dose not to exceed 20 mg of elemental calcium/kg. Additionally, 1,25-dihydroxycholecalciferol (calcitriol) should be given. The initial dosage is 0.25 µg/24 hr; the maintenance dosage ranges from 0.01-0.10 µg/kg/24 hr to a maximum of 1-2 µg/24 hr. Calcitriol has a short half-life and should be given in 2 equal divided doses; it has the advantages of rapid onset of effect (1-4 days) and rapid reversal of hypercalcemia after discontinuation in the event of overdosage (calcium levels begin to fall in 3-4 days). Calcitriol is supplied as an oral solution.
An adequate intake of calcium should be ensured. Supplemental calcium can be given in the form of calcium gluconate or calcium glubionate to provide 800 mg of elemental calcium daily, but it is rarely essential. Foods with high phosphorus content such as milk, eggs, and cheese should be reduced in the diet.
Clinical evaluation of the patient and frequent determinations of the serum calcium levels are indicated in the early stages of treatment to determine the requirement for calcitriol or vitamin D2 . If hypercalcemia occurs, therapy should be discontinued and resumed at a lower dose after the serum calcium level has returned to normal. In long-standing cases of hypercalcemia, repair of cerebral and dental changes is not likely. Pigmentation, lowering of blood pressure, or weight loss can indicate adrenal insufficiency, which requires specific treatment. Patients with autosomal dominant hypocalcemic hypercalciuria can develop nephrocalcinosis and renal impairment if treated with vitamin D.
Differential Diagnosis
Magnesium deficiency must be considered in patients with unexplained hypocalcemia. Concentrations of serum magnesium <1.5 mg/dL (1.2 mEq/L) are usually abnormal. Familial hypomagnesemia with secondary hypocalcemia has been reported in approximately 50 patients, most of whom developed tetany and seizures at 2-6 wk of age. Administration of calcium is ineffective, but administration of magnesium promptly corrects both calcium and magnesium levels. Oral supplements of magnesium are necessary to maintain levels of magnesium in the normal range. Two genetic forms have been described. One is caused by an autosomal recessive gene on chromosome 9, resulting in a specific defect in absorption of magnesium. The other is caused by an autosomal dominant gene on chromosome 11q23, resulting in renal loss of magnesium.
Hypomagnesemia also occurs in malabsorption syndromes such as Crohn disease and cystic fibrosis. Patients with autoimmune polyglandular disease type I and hypoparathyroidism can also have concurrent steatorrhea and low magnesium levels. Therapy with aminoglycosides causes hypomagnesemia by increasing urinary losses.
It is not clear how low levels of magnesium lead to hypocalcemia. Evidence suggests that hypomagnesemia impairs release of PTH and induces resistance to the effects of the hormone, but other mechanisms also may be operative.
Poisoning with inorganic phosphate leads to hypocalcemia and tetany. Infants administered large doses of inorganic phosphates, either as laxatives or as sodium phosphate enemas, have had sudden onset of tetany, with serum calcium levels <5 mg/dL and markedly elevated levels of phosphate. Symptoms are quickly relieved by intravenous administration of calcium. The mechanism of the hypocalcemia is not clear (see Chapter 68.6 ).
Hypocalcemia can occur early in the course of treatment of acute lymphoblastic leukemia. Hypocalcemia is usually associated with hyperphosphatemia resulting from destruction of lymphoblasts.
Episodic symptomatic hypocalcemia occurs in the Kenny-Caffey syndrome, which is characterized by medullary stenosis of the long bones, short stature, delayed closure of the fontanel, delayed bone age, and eye abnormalities. Idiopathic hypoparathyroidism and abnormal PTH levels have been found. Autosomal dominant and autosomal recessive modes of inheritance have been reported. Mutations of the TBCE gene (1q43-44) perturb microtubule organization in diseased cells.