Disorders Involving Transmembrane Receptors
Julie E. Hoover-Fong, William A. Horton, Jacqueline T. Hecht
Heterozygous mutations of genes encoding FGFR3 (fibroblast growth factor receptor 3) and PTHR1 (parathyroid hormone-1 receptor) result in disorders involving transmembrane receptors. The mutations cause the receptors to become activated in the absence of physiologic ligands, which accentuates normal receptor function of negatively regulating bone growth. The mutations act by gain of negative function. In the FGFR3 mutation group, in which the clinical phenotypes range from severe to mild, the severity appears to correlate with the extent to which the receptor is activated. PTHR1 and especially FGFR3 mutations tend to recur in unrelated individuals (Table 716.1 ).
Table 716.1
| GROUP/NAME OF DISORDER | INHER. | OMIM | GR | ORPHA | GENE |
|---|---|---|---|---|---|
| Thanatophoric dysplasia type I (TD I) | AD | 187600 | 1366 | 1860 | FGFR3 |
| Thanatophoric dysplasia type II (TD II) | AD | 187601 | 1366 | 93274 | FGFR3 |
| Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) | AD | 187600 | 1455 | 85165 | FGFR3 |
| Achondroplasia | AD | 100800 | 1152 | 15 | FGFR3 |
| Hypochondroplasia | AD | 146000 | 1477 | 429 | FGFR3 |
| Camptodactyly, tall stature, and hearing loss syndrome (CATSHL) | AD | 610474 | FGFR3 |
Please also refer to group 33 for craniosynostoses syndromes linked to FGFR3 mutations, as well as LADD syndrome in group 39 for another FGFR3-related phenotype.
OMIM, Online Mendelian Inheritance in Man (omim.org); GR, GeneReviews; ORPHA, Orphanet (orpha.net).
From Campeau P, Schlesinger AE: Skeletal dysplasias. [Updated 2017 Jan 30]. In De Groot LJ, Chrousos G, Dungan K, et al., editors. Endotext [internet] . South Dartmouth, MA, 2000, MDText.com , Inc. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279130/ .
Achondroplasia Group
The achondroplasia group represents a substantial percentage of patients with chondrodysplasias and contains thanatophoric dysplasia (TD), the most common lethal chondrodysplasia, with a birth prevalence of 1 in 35,000 births; achondroplasia, the most common nonlethal chondrodysplasia, with a birth prevalence of 1 in 15,000 to 1 in 40,000 births; and hypochondroplasia. All three have mutations in a small number of locations in the FGFR3 gene. There is a strong correlation between the mutation site and the clinical phenotype.
Thanatophoric Dysplasia
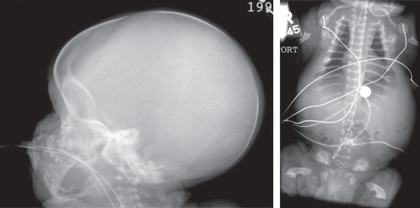
TD (MIM 187600, 187601) manifests before or at birth. In the former situation, ultrasonographic examination in mid-gestation or later reveals a large head and very short limbs; the pregnancy is often accompanied by polyhydramnios and premature delivery. Very short limbs, short neck, long narrow thorax, and large head with midfacial hypoplasia dominate the clinical phenotype at birth (Fig. 716.1 ). The cloverleaf skull deformity known as kleeblattschädel is sometimes found. If the affected fetus survives through pregnancy, the newborn will have severe respiratory distress because of their small thorax. Although this distress can be treated by intense respiratory care, the long-term prognosis is poor.

Skeletal radiographs distinguish two slightly different forms called TD I and TD II. In the more common TD I, radiographs show large calvarium with a small cranial base, marked thinning and flattening of vertebral bodies (platyspondyly) visualized best on lateral view, very short ribs, severe hypoplasia of pelvic bones, and very short and bowed tubular bones with flared metaphyses (Fig. 716.2 ). The femurs are curved and shaped like a telephone receiver. TD II differs mainly in that there are longer and straighter femurs.
The TD II clinical phenotype is associated with mutations that map to codon 650 of FGFR3 , causing the substitution of a glutamic acid for the lysine. This activates the tyrosine kinase activity of a receptor that transmits signals to intracellular pathways. Mutation of lysine 650 to methionine is associated with a clinical phenotype intermediate between TD and achondroplasia, referred to as SADDAN (severe achondroplasia with developmental delay and acanthosis nigricans) . Mutations of the TD I phenotype mainly map to two regions in the extracellular domain of the receptor, where they substitute cysteine residues for other amino acids. Free cysteine residues are thought to form disulfide bonds promoting dimerization of receptor molecules, leading to activation and signal transmission. TD I and TD II represent new mutations in offspring born to unaffected, average stature parents. The recurrence risk is low. Because the mutated codons in TD are mutable for unknown reasons and because of the theoretical risk of germ cell mosaicism, parents are offered prenatal diagnosis for subsequent pregnancies.
Achondroplasia
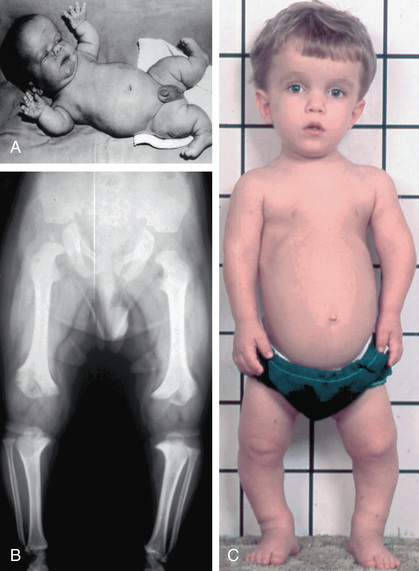
Achondroplasia (MIM 100800) is the prototype chondrodysplasia. It typically manifests at birth with short limbs, a long narrow trunk, and a large head with midfacial hypoplasia and prominent forehead (Fig. 716.3 ). The limb shortening is greatest in the proximal segments, and the fingers often display a trident configuration. Most joints are hyperextensible, but extension is restricted at the elbow. A thoracolumbar gibbus is often found. Birth length may be slightly less than normal but often plots within the low-normal range.
Diagnosis
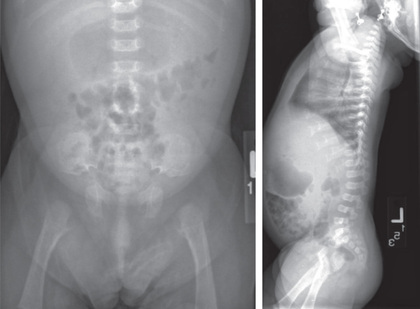
Skeletal radiographs confirm the diagnosis (see Figs. 716.3 and 716.4 ). The calvarial bones are large, whereas the cranial base and facial bones are small. The vertebral pedicles are short throughout the spine as noted on a lateral radiograph. The interpedicular distance, which normally increases from the 1st to the 5th lumbar vertebra, decreases in achondroplasia. The iliac bones are short and round, and the acetabular roofs are flat. The tubular bones are short with mildly irregular and flared metaphyses. The fibula is disproportionately long compared with the tibia, which is often bowed.
Clinical Manifestations
Infants usually exhibit delayed motor milestones, often not walking alone until 18-24 mo. This is because of hypotonia and mechanical difficulty balancing the large head on a normal-sized trunk and short extremities. Intelligence is normal unless central nervous system complications develop. As the child begins to walk, the gibbus usually gives way to an exaggerated lumbar lordosis.
Infants and children with achondroplasia progressively fall below normal standards for length and height. They can be plotted against standards established for achondroplasia. Adult heights typically are 118-145 cm for men and 112-136 cm for women. Surgical limb lengthening and human growth hormone treatment have been used to increase height; however, both are controversial. C-type natriuretic peptide may stimulate bone growth in achondroplasia based on studies in animal models. Clinical trials are underway to study various compounds.
Virtually all infants and children with achondroplasia have large heads, although only a fraction have true hydrocephalus. Head circumference should be carefully monitored using standards developed for achondroplasia, as should neurologic function in general. The spinal canal is stenotic, and spinal cord compression can occur at the foramen magnum and in the lumbar spine. The former usually occurs in infants and small children; it may be associated with hypotonia, failure to thrive, quadriparesis, central and obstructive apnea, and sudden death. Surgical correction may be required for severe stenosis. Lumbar spinal stenosis usually does not occur until early adulthood. Symptoms include paresthesias, numbness, and claudication in the legs. Loss of bladder and bowel control may be late complications. Bowing of the legs is common in patients with achondroplasia and might need to be corrected surgically. Other common problems include dental crowding, articulation difficulties, obesity, and frequent episodes of otitis media, which can contribute to hearing loss.
Genetics
All patients with typical achondroplasia have mutations at FGFR3 codon 380. The mutation maps to the transmembrane domain of the receptor and is thought to stabilize receptor dimers that enhance receptor signals, the consequences of which inhibit linear bone growth. Achondroplasia behaves as an autosomal dominant trait; most cases arise from a new mutation to average stature parents.
Because of the high frequency of achondroplasia among short stature skeletal dysplasias, it is relatively common for adults with achondroplasia to marry. Such couples have a 50% risk of transmitting their condition, heterozygous achondroplasia, to each offspring, as well as a 25% risk of homozygous achondroplasia . The latter condition exhibits intermediate severity between TD and heterozygous achondroplasia and is usually lethal in the newborn period; it is often referred to as “double dominant” inheritance. Prenatal diagnosis is available and has been used to diagnose homozygous achondroplasia. Preimplantation genetic testing can be used to identify double dominant mutations.
Hypochondroplasia
Hypochondroplasia (MIM 146000) resembles achondroplasia but is milder. Usually, it is not apparent until childhood, when mild short stature affecting the limbs becomes evident. Children have a stocky build and slight frontal bossing of the head. Learning disabilities may be more common in this condition. Radiographic changes are mild and consistent with the mild achondroplastic phenotype. Complications are rare; in some patients the condition is never diagnosed. Adult heights range from 116 to 146 cm. An FGFR3 mutation at codon 540 has been found in many patients with hypochondroplasia. Genetic heterogeneity exists in hypochondroplasia; that is, SHOX mutations are associated with a very similar clinical phenotype. Recombinant growth hormone therapy may enhance growth and improve body disproportion.
Jansen Metaphyseal Dysplasia
Jansen metaphyseal chondrodysplasia (MIM 156400) is a rare, dominantly inherited chondrodysplasia characterized by severe shortening of limbs associated with an unusual facial appearance (see Chapter 714 ). Sometimes it is accompanied by clubfoot and hypercalcemia with serum calcium values of 13-15 mg/dL. At birth, a diagnosis can be made from these clinical findings and radiographs that show short tubular bones with characteristic metaphyseal abnormalities that include flaring, irregular mineralization, fragmentation, and widening of the physeal space. The epiphyses are normal. The joints become enlarged and limited in mobility with age. Flexion contractures develop at the knees and hips, producing a bent-over posture. The spine can also be deformed by the irregular growth of vertebrae. Intelligence is normal, although there may be hearing loss.
Jansen metaphyseal chondrodysplasia is caused by activating mutations of PTHR1 . This G-protein–coupled transmembrane receptor serves as a receptor for both parathyroid hormone and parathyroid hormone-related peptide. Signaling through this receptor serves as a brake on the terminal differentiation of cartilage cells at a critical step in bone growth. Because the mutations activate the receptor, they enhance the braking effect and thereby slow bone growth. Loss-of-function mutations of PTHR1 are observed in Blomstrand chondrodysplasia, whose clinical features are the mirror image of Jansen metaphyseal chondrodysplasia.