Disorders Involving Defective Bone Resorption
Jacqueline T. Hecht, William A. Horton, David Rodriguez-Buritica
Bone dysplasias displaying increased bone density are rare. Osteopetrosis, which has many subtypes, and pyknodysostosis result from defective bone resorption.
Osteopetrosis
Two main forms of osteopetrosis have been delineated: a severe autosomal recessive form (MIM 259700) with an incidence of ~1/250,000 births and a mild autosomal dominant form (MIM 166600) occurring in ~1/20,000 births. Intrinsic disturbances of osteoclast function due to mutations in genes encoding osteoclast-specific subunits of the vacuolar proton pump TCIRG1 , CLCN7 , OSTM1 , SNX10 , TNFSFR11A, TNFSF11, and PLEKHM1 are found in most patients with the recessive form. Mutations in TNFSFR11A and TNFSF11 produce osteoclast-poor osteopetrosis, due to alteration of the RANK-RANKL interaction. Mutations of CLCN7 are observed in the dominant form of osteopetrosis. All types of mutations lead to disturbances of normal osteoclast function. Mutations in other genes result in additional syndromic presentations like osteopetrosis with renal tubular acidosis due to bi-allelic mutations in CAII.
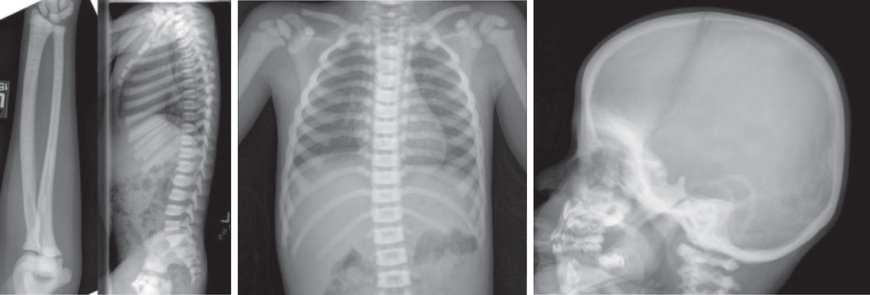
The severe form is usually detected in infancy or earlier because of macrocephaly, hepatosplenomegaly, deafness, blindness, and severe anemia. Radiographs reveal diffuse bone sclerosis and hypocalcemia levels may be detected. Later x-rays show the characteristic bone-within-bone appearance (Fig. 719.1 ). With time, infants typically fail to thrive and show psychomotor delay and worsening of cranial neuropathies and anemia. Dental problems, osteomyelitis of the mandible, and pathologic fractures are common. The most severely affected patients die during infancy; less severely affected patients rarely survive beyond the 2nd decade. Those who survive beyond infancy usually have learning disabilities, but might have normal intelligence despite hearing and vision loss.
Clinical Manifestations
Most of the manifestations are due to failure to remodel growing bones. This leads to narrowing of cranial nerve foramina and encroachment on marrow spaces, which results in secondary complications, such as optic and facial nerve dysfunction, and anemia accompanied by compensatory extramedullary hematopoiesis in the liver and spleen. The unusually dense bones are weak, leading to increased risk of fractures.
The autosomal dominant form of osteopetrosis (Albers-Schönberg disease, osteopetrosis tarda, or marble bone disease) usually manifests during childhood or adolescence with fractures and mild anemia and, less often, as cranial nerve dysfunction, dental abnormalities, or osteomyelitis of the mandible. Skeletal radiographs reveal a generalized increase in bone density and clubbing of metaphyses. Alternating bands of lucent and dense bands produce a sandwich appearance to vertebral bodies. The radiographic changes are sometimes incidental findings in otherwise asymptomatic adolescents and adults.
Treatment
Most of the bone manifestations in severe osteopetrosis due to intrinsic osteoclast defects can be prevented or reversed by hematopoietic stem cell transplantation (HSCT), if carried out before development of irreversible secondary complications, such as visual impairment. RANKL replacement therapy may be useful in patients with RANKL deficiency due to TNFSF11 bi-allelic mutations, who do not benefit from HSCT. Calcitriol and interferon-γ have also been used with equivocal results. Symptomatic care, such as dental care, transfusions for anemia, and antibiotic treatment of infections, is important for patients who survive infancy.
Pyknodysostosis
An autosomal recessive bone dysplasia related to osteopetrosis, pyknodysostosis (MIM 265800) manifests in early childhood with short limbs, characteristic facies, an open anterior fontanel, a large skull with frontal and occipital bossing, and dental abnormalities. The hands and feet are short and broad, and the nails may be dysplastic. The sclerae may be blue. Minimal trauma often leads to fractures. Treatment is symptomatic and focused mainly on the management of dental problems and fractures. The prognosis is generally good, and patients typically reach heights of 130-150 cm.
Skeletal radiographs show a generalized increase in bone density. In contrast to many disorders in this group, the metaphyses are normal. Other changes include wide sutures and wormian bones in the skull, a small mandible, and hypoplasia of the distal phalanges (Fig. 719.2 ).
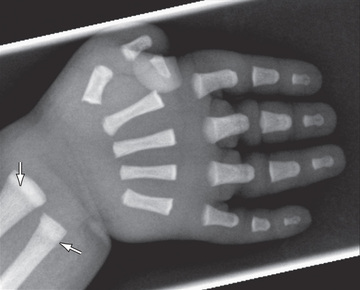
Several mutations have been found in the gene encoding cathepsin K, a cysteine protease that is highly expressed in osteoclasts. The mutations predict loss of enzyme function, suggesting that there is an inability of osteoclasts to degrade bone matrix and remodel bones.