Hypophosphatasia
Linda A. DiMeglio
Hypophosphatasia is a rare inborn error of metabolism in which tissue-nonspecific (liver, bone, kidney) alkaline phosphatase isoenzyme (TNSALP) activity is deficient, although activity of the intestinal and placental isoenzymes is normal. ALPL gene mutations reduce the TNSALP enzyme activity essential for normal skeletal mineralization.
Most of the >100 ALPL mutations gene identified to date are missense mutations, although splice-site mutations, small deletions, and frameshift mutations also have been found. Some patients have a regulatory defect involving this enzyme rather than a mutation. There is considerable heterogeneity in disease severity related to the degree of enzyme activity impairment. A nosology that describes seven forms of the condition, ranging from neonatal lethal disease to odontohypophosphatasia , which only affects teeth, is employed. The lethal and infantile forms are autosomal recessive; milder forms can be either autosomal recessive or dominant.
The most severe perinatal hypophosphatasia cases are lethal in utero or shortly after birth. Affected infants have profound skeletal hypomineralization with short bones and may also have anemia with hemorrhage, pyridoxine-dependent seizures, and hypoplastic lungs (Fig. 724.1A ). Infantile hypophosphatasia is next on the continuum. These infants present prior to 6 months of age with hypercalcemia/ hypercalciuria (leading to nephrocalcinosis), premature cranial suture fusion (that can lead to increased intracranial pressure), irritability, and failure to thrive. X-rays reveal irregular ossification, punched-out areas, and metaphyseal cupping. Prior to the availability of enzyme replacement therapy with asfotase alfa , mortality was estimated at 50%; survivors had significant disability. There is also a benign prenatal form of hypophosphatasia, seen in newborns who have skeletal abnormalities in utero or at birth that improve spontaneously over time.
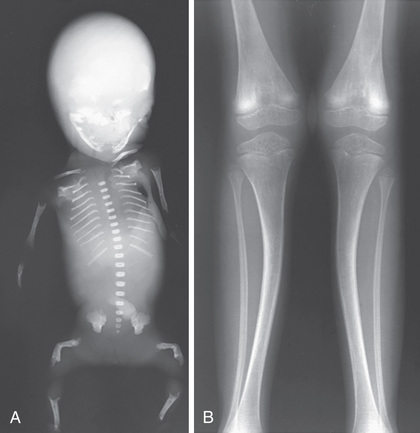
The next form of hypophosphatasia is recognized in childhood (after 6 months of life) or late adolescence (hypophosphatasia tarda) (see Fig. 724.1B ). These children present with premature exfoliation of primary teeth (with the root intact due to poorly mineralized dental cementum), mild skeletal deformities, fracture, and variable short stature. Some children have skeletal pain and muscle weakness. Long bones can have characteristic “tongues” of radiolucency.
An adult hypophosphatasia form manifests in middle age (although some patients can recount a history of early deciduous tooth loss or rickets). This form is characterized by osteopenia/osteoporosis, recurrent metaphyseal stress fractures (particularly of the metatarsals and tibiae), and femoral pseudofractures. Affected individuals can also have psychiatric symptoms (depression/anxiety) chondrocalcinosis, osteoarthritis, myopathy, nephrocalcinosis, and permanent tooth loss between 40 and 60 years of age.
In hypophosphatasia, large quantities of phosphoethanolamine (PEA) are found in the urine because this compound cannot be degraded in the absence of TNSALP activity. Plasma inorganic pyrophosphate and pyridoxal-5′-phosphate (PLP) levels are elevated for the same reason. Pyridoxal-5-phosphate levels tend to be lower than normal in most other bone diseases and hence can aid in the differential diagnosis of hypophosphatasia. Seizures in patients with the lethal and infantile forms of the disease are related to impaired pyridoxine metabolism.
The clinical course of this condition often improves spontaneously as an affected child matures, although early death from renal failure or flail chest leading to pneumonia can occur in the severe infantile form of the disorder. Enzyme replacement therapy with recombinant human TNSALP improves skeletal healing and mineral content, pulmonary status, and overall physical activity.
Rarely patients presenting with identical clinical and radiographic patterns have normal serum alkaline phosphatase activity but increased concentrations of phosphoethanolamine, inorganic phosphate, and pyridoxal-5′-phosphate. Their disease has been labeled pseudohypophosphatasia and might represent the presence of a mutant alkaline phosphatase isoenzyme that reacts to artificial substrates in an alkaline environment (in a test tube), but not in vivo with natural substrates.
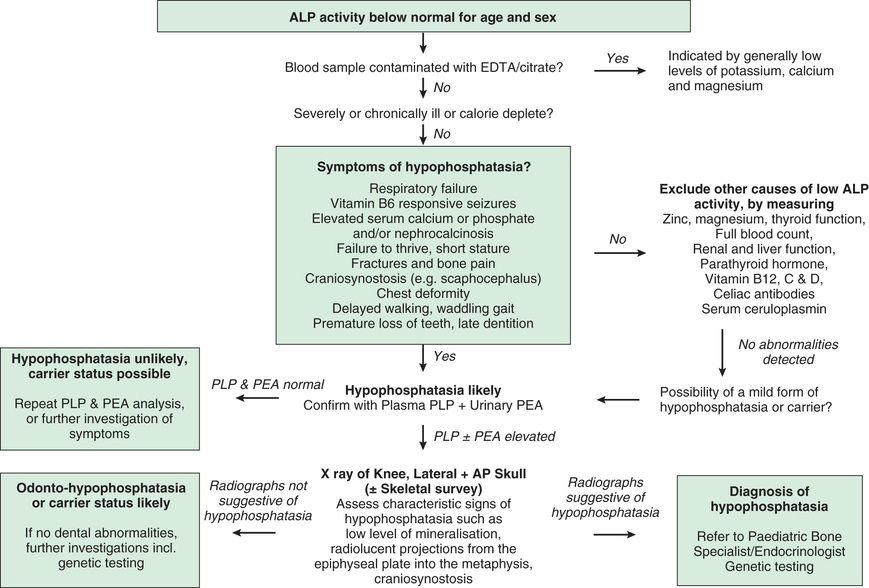
An approach to low ALP levels is noted in Fig. 724.2 .