Chapter 16 NURSING MANAGEMENT: fluid, electrolyte and acid–base imbalances
1. Describe the composition of the major body fluid compartments.
2. Define the following processes involved in the regulation of movement of water and electrolytes between the body fluid compartments: diffusion, osmosis, filtration, hydrostatic pressure, oncotic pressure and osmotic pressure.
3. Describe the aetiology, laboratory diagnostic findings, clinical manifestations, and nursing and collaborative management of the following disorders:
4. Identify the processes that maintain acid–base balance.
5. Discuss the aetiology, laboratory diagnostic findings, clinical manifestations, and nursing and collaborative management of the following acid–base imbalances: metabolic acidosis, metabolic alkalosis, respiratory acidosis and respiratory alkalosis.
6. Describe the composition and indications of common intravenous fluid solutions.
7. Discuss the types and nursing management of commonly used central venous access devices.
Homeostasis
Body fluids and electrolytes play an important role in homeostasis. Homeostasis is the state of equilibrium in the internal environment of the body, which is naturally maintained by adaptive responses that promote healthy survival.1 Maintenance of the composition and volume of body fluids within narrow limits of normal is necessary to maintain homeostasis.2 During normal metabolism the body produces many acids. These acids alter the internal environment of the body, including fluid and electrolyte balances, and must also be regulated to maintain homeostasis. Many diseases and their treatments have the potential to affect fluid and electrolyte balance. For example, a patient with metastatic breast or lung cancer may develop hypercalcaemia as a result of bone destruction from tumour invasion. Chemotherapy that is prescribed to treat the cancer may result in nausea and vomiting and, subsequently, dehydration and acid–base imbalances. Correction of the dehydration with intravenous (IV) fluids must be monitored closely to prevent fluid overload.
It is important for the nurse to anticipate the potential for alterations in fluid and electrolyte balance associated with certain disorders and medical therapies, recognise the signs and symptoms of imbalances, and intervene with the appropriate action. This chapter describes: (1) the normal control of fluids, electrolytes and acid–base balance; (2) aetiologies that disrupt homeostasis and their resultant manifestations; and (3) actions that the nurse can take to prevent or restore fluid, electrolyte and acid–base balance.
Water content of the body
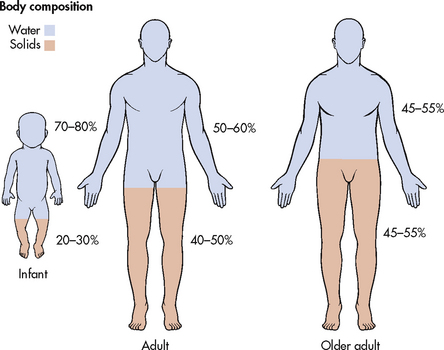
Water is the primary component of the body, accounting for approximately 50–60% of the body weight in the adult. Water is the solvent in which body salts, nutrients and wastes are dissolved and transported. The water content varies with gender, body mass and age (see Fig 16-1). The percentage of body weight that is composed of water is generally greater in men than in women because men tend to have more lean body mass than women. Older adults have less body water than younger individuals for the same reason. Fat cells contain less water than an equivalent volume of lean tissue.3 In the older adult, body water content averages 45–55% of body weight. In the infant, water content is 70–80% of body weight. Thus, infants and the elderly are at a higher risk of fluid-related problems than young adults.
BODY FLUID COMPARTMENTS
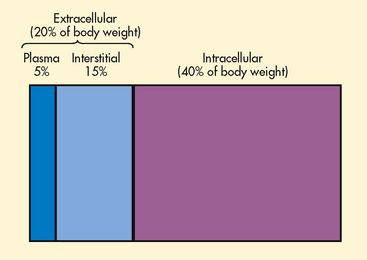
The two major fluid compartments in the body are intracellular space (inside the cells) and extracellular space (outside the cells) (see Fig 16-2). Approximately two-thirds of the body water is located within cells and is termed intracellular fluid (ICF); ICF constitutes approximately 40% of the body weight of an adult. The body of a 70-kg man would contain approximately 42 L of water, of which 28 L would be located within cells. Extracellular fluid (ECF) consists of interstitial fluid, which is composed of the fluid in the interstitium (the space between cells) and lymph, the fluid in blood (plasma), and a very small amount of fluid contained within specialised cavities of the body (cerebrospinal fluid, fluid in the gastrointestinal [GI] tract, and pleural, synovial, peritoneal, intraocular and pericardial fluid). The fluid in the specialised cavities is sometimes referred to as transcellular fluid. The ECF consists of one-third of the body water. This would amount to about 14 L in a 70-kg man. About one-third of the ECF is in the plasma space (3 L in a 70-kg man) and two-thirds is in the interstitial space (10 L in a 70-kg man). The fluid in the specialised cavities totals about 1 L at any given time, but because 3–6 L of fluid is secreted into and reabsorbed from the GI tract every day, loss of this fluid from vomiting or diarrhoea can produce serious fluid and electrolyte imbalances.
FUNCTIONS OF BODY WATER
Body fluids are in constant motion transporting nutrients, electrolytes and oxygen to cells and carrying waste products away from cells. Water is essential in the regulation of body temperature. In addition, it lubricates joints and is a medium for food digestion.3
CALCULATION OF FLUID GAIN OR LOSS
One litre of water weighs 1 kg. Body weight change, especially sudden change, is an excellent indicator of overall fluid volume loss or gain. For example, if a patient drinks 240 mL of fluid, weight gain will be 0.24 kg. A patient receiving diuretic therapy who loses 2 kg in 24 hours has experienced a fluid loss of approximately 2 L. An adult patient who is fasting might lose approximately 0.48–0.96 kg per day. A weight loss exceeding this is likely to be due to loss of body fluid.
Electrolytes
Electrolytes are substances whose molecules dissociate or split into ions when placed in water. Ions are electrically charged particles. Cations are positively charged ions. Examples include sodium (Na+), potassium (K+), calcium (Ca2+) and magnesium (Mg2+) ions. Anions are negatively charged ions. Examples include bicarbonate (HCO3−), chloride (Cl−) and phosphate (PO43−) ions. Most proteins bear a negative charge and are thus anions. The electrical charge of an ion is termed its valence. Cations and anions combine according to their valences. A monovalent ion has the combining power of one hydrogen atom. Definitions of terminology related to body fluid chemistry are presented in Table 16-1.
TABLE 16-1 Terminology related to body fluid chemistry
| Anion | Ion that carries a negative charge |
|---|---|
| Cation | Ion that carries a positive charge |
| Electrolyte | Substance that dissociates in solution into ions (charged particles); a molecule of sodium chloride (NaCl) in solution becomes Na+ and Cl− |
| Monovalent | An ion that has the combining power of one hydrogen atom |
| Non-electrolyte | Substance that does not dissociate into ions in solution; examples include glucose and urea |
| Osmolality | A measure of the total solute concentration per kilogram of solvent |
| Osmolarity | A measure of the total solute concentration per litre of solution |
| Solute | Substance that is dissolved in a solvent |
| Solution | Homogeneous mixture of solutes dissolved in a solvent |
| Solvent | Substance that is capable of dissolving a solute (liquid or gas) |
| Valence | The degree of combining power of an ion |
MEASUREMENT OF ELECTROLYTES
The measurement of electrolytes is important to nurses in evaluating electrolyte balance, as well as in determining the composition of electrolyte preparations. Most hospitals include the normal electrolyte ranges and information on the back of their laboratory forms and this helps nurses with interpretation of results. The concentration of electrolytes can be expressed in milligrams per decilitre (mg/dL), millimoles per litre (mmol/L) or milliequivalents per litre (mEq/L). The international standard for measuring electrolytes is mmol/L. One mole (mol) of a substance is the relative atomic mass of that substance in grams; hence a millimole (mmol) of a substance is the relative atomic mass in milligrams. Sodium’s relative atomic mass is 23; therefore, 23 mg of sodium is 1 mmol of sodium. Sodium and chloride ions are monovalent and therefore will match one to one. One mmol of sodium combines with 1 mmol of chloride. An ion with two electrons, such as calcium, will require two monovalent partners.
In Australia and New Zealand, the unit used to measure electrolytes is the millimole, whereas in the US the unit used is the milliequivalent. The following formula is used to convert milliequivalents to millimoles:
mmol/L = mEq ÷ valence
Electrolytes in body fluids are active chemicals that unite in varying combinations. Routinely in practice it is easier to express their amounts in millimoles where the valence is 1 (e.g. Na+, Cl−); however, when performing more complex electrolyte calculations, the clinician must ensure that multivalent ions (e.g. Ca2+) are accounted for using the above formula—that is, in terms of chemical activity (or milliequivalents) rather than by mass. Ions combine milliequivalent for milliequivalent. For example, 1 mEq (1 mmol) of sodium combines with 1 mEq (1 mmol) of chloride, and 1 mEq (0.5 mmol) of calcium combines with 1 mEq (1 mmol) of chloride. This combining power of electrolytes is important to maintain the balance of positively charged (cation) and negatively charged (anion) ions within body fluids.
ELECTROLYTE COMPOSITION OF FLUID COMPARTMENTS
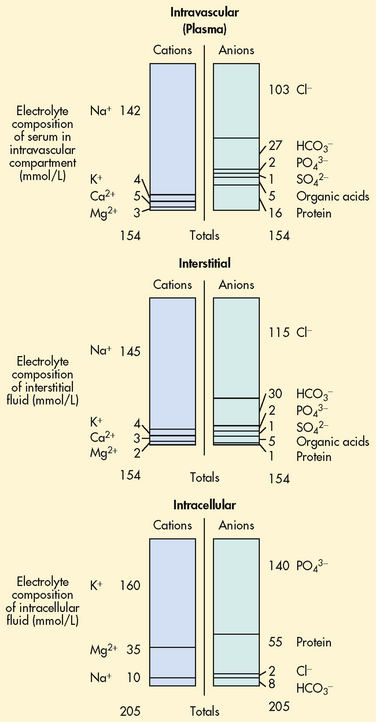
Electrolyte composition varies between the ECF and ICF. The overall concentration of the electrolytes is approximately the same in the two compartments. However, concentrations of specific ions differ greatly (see Fig 16-3). In the ECF, the main cation is sodium with small amounts of potassium, calcium and magnesium. The primary ECF anion is chloride with small amounts of bicarbonate, sulfate and phosphate anions. The plasma has substantial amounts of protein. However, the amount of protein in the plasma is less than in the ICF. There is very little protein in the interstitium under normal conditions. In the ICF the most prevalent cation is potassium with small amounts of magnesium and sodium. The prevalent anion is phosphate with some protein and a small amount of bicarbonate.
Mechanisms controlling fluid and electrolyte movement
Many different processes are involved in the movement of electrolytes and water between the ICF and ECF. Some of the processes include simple diffusion, facilitated diffusion and active transport. Water moves as driven by two forces: hydrostatic pressure and osmotic pressure.
DIFFUSION
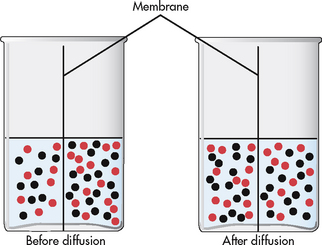
Diffusion is the movement of molecules from an area of high concentration to one of low concentration (see Fig 16-4). It occurs in liquids, gases and solids. Net movement of molecules stops when the concentrations are equal in both areas. The membrane separating the two areas must be permeable to the diffusing substance for the process to occur. Simple diffusion requires no external energy. Gases (e.g. oxygen, nitrogen, carbon dioxide) and substances (e.g. urea) can permeate through cell membranes and are distributed throughout the body.
FACILITATED DIFFUSION
Because of the composition of cellular membranes, some molecules diffuse slowly into the cell. However, when they are combined with a specific carrier molecule, the rate of diffusion accelerates. Like simple diffusion, facilitated diffusion moves molecules from an area of high concentration to one of low concentration. Facilitated diffusion is passive and requires no energy other than that of the concentration gradient. Glucose transport into the cell is an example of facilitated diffusion. A carrier molecule on most cells increases or facilitates the rate of diffusion of glucose into these cells.
ACTIVE TRANSPORT
Active transport is a process in which molecules move against the concentration gradient. External energy is required for this process. The concentrations of sodium and potassium differ greatly intracellularly and extracellularly (see Fig 16-3). By active transport, sodium moves out of the cell and potassium moves into the cell to maintain this concentration difference (see Fig 16-5). This active transport mechanism is referred to as the sodium–potassium pump. The energy source for this mechanism is adenosine triphosphate (ATP), which is produced in the cell’s mitochondria.
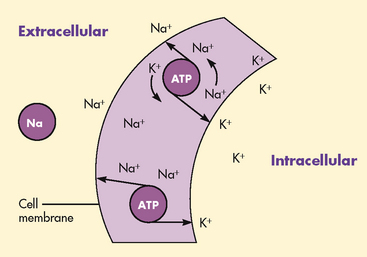
OSMOSIS
Osmosis is the movement of water between two compartments separated by a semipermeable membrane (a membrane permeable to water but not to a solute). Water moves through the membrane from an area of low solute concentration to an area of high solute concentration (see Fig 16-6); that is, water moves from the more dilute compartment (has more water) to the side that is more concentrated (has less water). Osmosis requires no outside energy sources and stops when the concentration differences disappear or when hydrostatic pressure builds and is sufficient to oppose any further movement of water. Diffusion and osmosis are important in maintaining the fluid volume of body cells and the concentration of the solute.
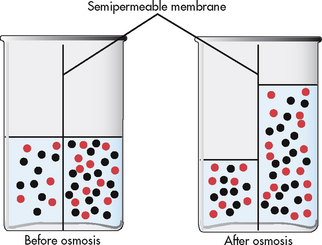
Figure 16-6 Osmosis is the process of water movement through a semipermeable membrane from an area of low solute concentration to an area of high solute concentration.
Osmotic pressure is the amount of pressure required to stop the osmotic flow of water. Osmotic pressure can be understood in terms of imagining a chamber in which two compartments are separated by a semipermeable membrane (see Fig 16-6). Water will move from the less concentrated side to the more concentrated side of the chamber. Osmotic pressure is determined by the concentration of solute in solution. It is measured in milliosmoles and may be expressed as either fluid osmolarity or fluid osmolality. Osmolality measures the osmotic pressure of solute per unit of mass of solvent (mOsm/kg). Osmolarity measures the total milliosmoles of solute per unit of total volume of solution (mOsm/L). Although osmolality and osmolarity are often used interchangeably, osmolality is used to describe fluids inside the body and osmolarity pertains to fluids outside the body.4 Osmolality is the test typically performed to evaluate the concentration of plasma and urine.
Measurement of osmolality
Osmolality is approximately the same in the various body fluid spaces. Determining osmolality is important because it indicates the water balance of the body. To assess the state of the body water balance, one can measure or estimate plasma osmolality. Normal plasma osmolality is between 275 and 295 mOsm/kg. A value greater than 295 mOsm/kg indicates that the concentration of solute is too great or that the water content is too little. This condition is termed water deficit. A value less than 275 mOsm/kg indicates too little solute for the amount of water or too much water for the amount of solute. This condition is termed water excess. Both conditions are clinically significant. Because the major determinants of the plasma osmolality are sodium, glucose and urea, one can calculate the effective plasma osmolality based on the concentrations of these substances. Osmolality of urine can range from 100 to 1300 mOsm/kg, depending on fluid intake and the amount of antidiuretic hormone (ADH) in circulation and the renal response to it.
Osmotic movement of fluids
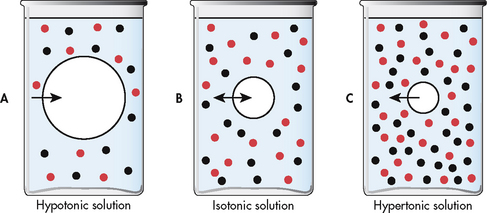
Cells are affected by the osmolality of the fluid that surrounds them. Fluids with the same osmolality as the cell interior are termed isotonic. Solutions in which the solutes are less concentrated than in the cells are termed hypotonic (hypo-osmolar). Those with solutes more concentrated than in cells are termed hypertonic (hyperosmolar).
Normally, the ECF and ICF are isotonic to one another; hence, no net movement of water occurs. In the metabolically active cell there is a constant exchange of substances between the cell and the interstitium but no net gain or loss of water occurs.
If a cell is surrounded by hypotonic fluid, water moves into the cell, causing it to swell and possibly to burst. If a cell is surrounded by hypertonic fluid, water leaves the cell to dilute the ECF; the cell shrinks and may eventually die (see Fig 16-7).
HYDROSTATIC PRESSURE
Hydrostatic pressure is the pressure within a fluid compartment. In the blood vessels, hydrostatic pressure is the blood pressure generated by the contraction of the heart.2 Hydrostatic pressure in the vascular system gradually decreases as the blood moves through the arteries until it is about 40 mmHg (5.33 kPa) at the arterial end of a capillary. Because of the size of the capillary bed and fluid movement into the interstitium, the pressure decreases to about 10 mmHg (1.33 kPa) at the venous end of the capillary. Hydrostatic pressure is the major force that pushes water out of the vascular system at the capillary level.
ONCOTIC PRESSURE
Oncotic pressure (colloidal osmotic pressure) is osmotic pressure exerted by colloids in solution. The major colloid in the vascular system contributing to the total osmotic pressure is protein. Protein molecules attract water, pulling fluid from the tissue space to the vascular space.4 Unlike electrolytes, the large molecular size prevents proteins from leaving the vascular space through pores in capillary walls. Under normal conditions plasma oncotic pressure is approximately 25 mmHg (3.33 kPa). Some proteins are found in the interstitial space; they exert an oncotic pressure of approximately 1 mmHg (0.13 kPa).
Fluid movement in capillaries
There is normal movement of fluid between the capillary and the interstitium. The amount and direction of movement are determined by the interaction of: (1) capillary hydrostatic pressure; (2) plasma oncotic pressure; (3) interstitial hydrostatic pressure; and (4) interstitial oncotic pressure. Capillary hydrostatic pressure and interstitial oncotic pressure cause the movement of water out of the capillary. Plasma oncotic pressure and interstitial hydrostatic pressure cause the movement of fluid into the capillary. At the arterial end of the capillary, capillary hydrostatic pressure exceeds plasma oncotic pressure and fluid is moved into the interstitium. At the venous end of the capillary, the capillary hydrostatic pressure is lower than plasma oncotic pressure and fluid is drawn back into the capillary by the oncotic pressure created by plasma proteins (see Fig 16-8).
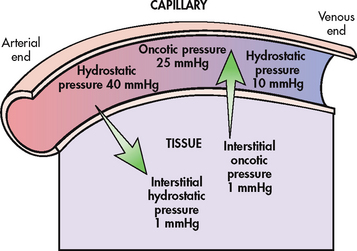
Figure 16-8 Dynamics of fluid exchange between the capillary and the tissue. An equilibrium exists between forces filtering fluid out of the capillary and forces absorbing fluid back into the capillary. Note that the hydrostatic pressure is greater at the arterial end of the capillary than the venous end. The net effect of pressures at the arterial end of the capillary causes a movement of fluid into the tissue. At the venous end of the capillary there is net movement of fluid back into the capillary.
FLUID SHIFTS
If capillary or interstitial pressures are altered, fluid may shift abnormally from one compartment to another, resulting in oedema or dehydration.
Shifts of plasma to interstitial fluid
Accumulation of fluid in the interstitium (oedema) occurs if venous hydrostatic pressure rises, plasma oncotic pressure falls or interstitial oncotic pressure rises. Oedema may also develop if there is an obstruction of lymphatic outflow that causes decreased removal of interstitial fluid.
Elevation of venous hydrostatic pressure
Increasing the pressure at the venous end of the capillary inhibits fluid movement back into the capillary. Causes of increased venous pressure include fluid overload, heart failure, liver failure, obstruction of venous return to the heart (e.g. tourniquets, restrictive clothing, venous thrombosis) and venous insufficiency (e.g. varicose veins).
Decrease in plasma oncotic pressure
Fluid remains in the interstitium if the plasma oncotic pressure is too low to draw fluid back into the capillary. Decreased oncotic pressure is seen when the plasma protein content is low. This can result from excessive protein loss (renal disorders), deficient protein synthesis (liver disease) and deficient protein intake (malnutrition).
Shifts of interstitial fluid to plasma
Fluid is drawn into the plasma space whenever there is an increase in the plasma osmotic or oncotic pressure. This could happen with the administration of colloids, dextran, mannitol or hypertonic solutions. Fluid is drawn from the interstitium. In turn, water is drawn from cells via osmosis, equilibrating the osmolality between ICF and ECF.
Increasing the tissue hydrostatic pressure is another way of causing a shift of fluid into plasma. The wearing of elastic compression gradient stockings to decrease peripheral oedema is a therapeutic application of this effect.
Fluid movement between extracellular fluid and intracellular fluid
Changes in the osmolality of the ECF alter the volume of cells. Increased ECF osmolality (water deficit) pulls water out of cells until the two compartments have a similar osmolality. Water deficit is associated with symptoms that result from cell shrinkage as water is pulled into the vascular system. For example, neurological symptoms are caused by altered central nervous system (CNS) function as brain cells shrink. Decreased ECF osmolality (water excess) develops as the result of gain or retention of excess water. In this case, cells swell. Again, the primary symptoms are neurological as a result of brain cell swelling as water shifts into the cells.
Fluid spacing
Fluid spacing is a term sometimes used to describe the distribution of body water. First spacing describes the normal distribution of fluid in the ICF and ECF compartments. Second spacing refers to an abnormal accumulation of interstitial fluid (i.e. oedema). Third spacing occurs when fluid accumulates in a portion of the body from which it is not easily exchanged with the rest of the ECF. Third-spaced fluid is trapped and essentially unavailable for functional use. Examples of third spacing are ascites, sequestration of fluid in the abdominal cavity with peritonitis and oedema associated with burns.5
Regulation of water balance
HYPOTHALAMIC REGULATION
Water balance is maintained via the finely tuned balance of water intake and excretion. A body fluid deficit or increase in plasma osmolality is sensed by osmoreceptors in the hypothalamus, which in turn stimulates thirst and ADH release. Thirst causes the patient to drink water. ADH (also called vasopressin), which is synthesised in the hypothalamus and stored in the posterior pituitary, acts in the renal distal and collecting tubules, causing water reabsorption. Together these factors result in increased free water in the body and decreased plasma osmolality. If the plasma osmolality is diminished or there is water excess, secretion of ADH is suppressed, resulting in urinary excretion of water.
An intact thirst mechanism is critical because it is the primary protection against the development of hyperosmolality. The patient who cannot recognise or act on the sensation of thirst is at risk of fluid deficit and hyperosmolality. The sensitivity of the thirst mechanism decreases in older adults.
The desire to consume fluids is also affected by social and psychological factors not related to fluid balance. A dry mouth will cause the patient to drink, even when there is no measurable body water deficit. Water ingestion will equal water loss in the individual who has free access to water, a normal thirst and ADH mechanism, and normally functioning kidneys.
PITUITARY REGULATION
Under hypothalamic control, the posterior pituitary releases ADH, which regulates water retention by the kidneys. The distal tubules and collecting ducts in the kidneys respond to ADH by becoming more permeable to water so that water is reabsorbed from the tubular filtrate into the blood and not excreted in urine. Other factors that stimulate ADH release include stress, nausea, nicotine and morphine. These factors usually result in shifts of osmolality within the range of normal values. It is common for the postoperative patient to have a lower serum osmolality after surgery, possibly because of the stress of surgery and opioid analgesia.
A pathological condition seen occasionally is syndrome of inappropriate antidiuretic hormone (SIADH) (see Ch 49). Causes of SIADH include abnormal ADH production in CNS disorders (e.g. brain tumours, brain injury) and certain malignancies (e.g. small cell lung cancer). The inappropriate ADH production causes water retention, which produces a decrease in plasma osmolality below the normal value and a relative increase in urine osmolality, with a decrease in urine volume.
Reduction in the release or action of ADH produces diabetes insipidus (see Ch 49). A copious amount of dilute urine is excreted because the renal tubules and collecting ducts do not appropriately reabsorb water. The patient with diabetes insipidus exhibits extreme polyuria and, if the patient is alert, polydipsia (excessive thirst). Symptoms of dehydration and hypernatraemia develop if the water losses are not adequately replaced.
ADRENAL CORTICAL REGULATION
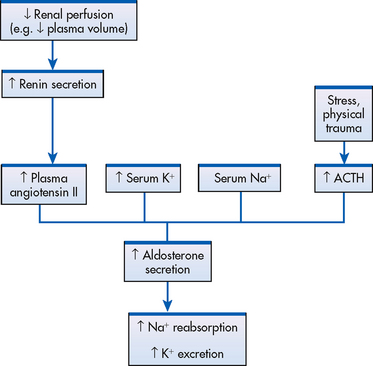
Whereas ADH affects only water reabsorption, glucocorticoids and mineralocorticoids secreted by the adrenal cortex help regulate both water and electrolytes. The glucocorticoids (e.g. cortisol) primarily have an anti-inflammatory effect and increase serum glucose levels, whereas mineralocorticoids (e.g. aldosterone) enhance sodium retention and potassium excretion (see Fig 16-9). When sodium is reabsorbed, water follows due to osmotic changes.
Cortisol is the most abundant glucocorticoid. In large doses, cortisol has both glucocorticoid (glucose-elevating and anti-inflammatory) and mineralocorticoid (sodium-retaining) effects. Cortisol is normally secreted in a diurnal, or circadian, pattern and also in response to increased physical and psychological stress. Many body functions, including fluid and electrolyte balance, are affected by stress (see Fig 16-10).
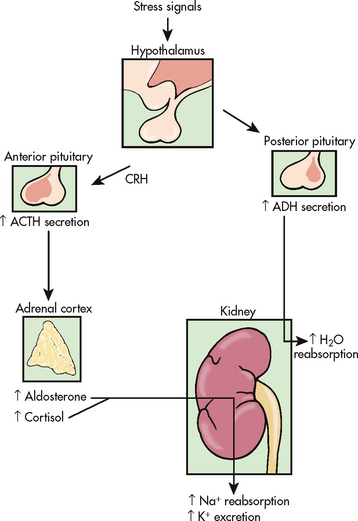
Figure 16-10 Effects of stress on fluid and electrolyte balance. ACTH, adrenocorticotrophic hormone; ADH, antidiuretic hormone; CRH, corticotrophin-releasing hormone.
Aldosterone is a mineralocorticoid with potent sodium-retaining and potassium-excreting capability. Secretion of aldosterone may be stimulated by decreased renal perfusion or decreased sodium delivery to the distal portion of the renal tubule. The kidneys respond by secreting renin into the plasma. Angiotensinogen, produced in the liver and normally found in blood, is acted on by the renin to form angiotensin I, which converts to angiotensin II, which stimulates the adrenal cortex to secrete aldosterone. In addition to the renin–angiotensin–aldosterone system, increased plasma potassium, decreased plasma sodium and adrenocorticotrophic hormone (ACTH) from the anterior pituitary all act directly on the adrenal cortex to stimulate the secretion of aldosterone (see Fig 16-9).
RENAL REGULATION
The primary organs for regulating fluid and electrolyte balance are the kidneys (see Ch 44). The kidneys regulate water balance through adjustments in urine volume. Similarly, urinary excretion of most electrolytes is adjusted so that a balance is maintained between overall intake and output. The total plasma volume is filtered by the kidneys many times each day. In the average adult, the kidney reabsorbs 99% of this filtrate, producing approximately 1.5 L of urine per day. As the filtrate moves through the renal tubules, selective reabsorption of water and electrolytes and secretion of electrolytes result in the production of urine that is greatly different in composition and concentration from the plasma. This process helps maintain normal plasma osmolality, electrolyte balance, blood volume and acid–base balance. The renal tubules are the site for the actions of ADH and aldosterone.
Gerontological considerations: fluid and electrolytes
The older adult experiences normal physiological changes that increase susceptibility to fluid and electrolyte imbalances. Structural changes to the kidneys and a decrease in the renal blood flow lead to a decrease in the glomerular filtration rate, decreased creatinine clearance, the loss of the ability to concentrate urine and conserve water, and narrowed limits for the excretion of water, sodium, potassium and hydrogen ions. Hormonal changes may include a decrease in renin and aldosterone and an increase in ADH and ANP.6 Loss of subcutaneous tissue and thinning of the dermis lead to increased loss of moisture through the skin and an inability to respond to heat or cold quickly. Older adults may also experience a decrease in the thirst mechanism, resulting in decreased fluid intake despite increases in osmolality and serum sodium level. Frail older people, especially if ill, are at increased risk of free-water loss and the subsequent development of hypernatraemia secondary to impairment of the thirst mechanism and barriers to accessible fluids.7
Healthy older adults usually consume adequate fluids to remain well hydrated. However, functional changes may occur that affect the individual’s physical ability to independently obtain fluids. Musculoskeletal changes, such as stiffness of the hands and fingers, can lead to a decreased ability to hold a glass or cup. Mental status changes, such as confusion or disorientation, or changes in ambulation status may lead to a decreased ability to obtain fluids. As a result of incontinent episodes, the older adult may intentionally restrict fluid intake.
To help older patients, the nurse needs to understand the homeostatic changes that occur in older people. It is important to avoid the pitfalls of ageism, wherein an older person’s fluid and electrolyte problems may be inappropriately attributed to the natural processes of ageing. The nurse needs to adapt assessment and nursing implementation to account for these physiological and functional changes. Suggestions for alterations in nursing care for the older adult are presented throughout this chapter and in Chapter 5.
With severely impaired renal function, the kidneys cannot maintain fluid and electrolyte balance. This condition results in oedema, potassium and phosphorus retention, acidosis and other electrolyte imbalances (see Ch 46).
CARDIAC REGULATION
Natriuretic peptides, including atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP), are hormones produced by cardiomyocytes. They are natural antagonists to the renin–angiotensin–aldosterone system. They are produced in response to increased atrial pressure (increased volume such as occurs in heart failure) and high serum sodium levels. They suppress secretion of aldosterone, renin and ADH, and the action of angiotensin II.2 They act on the renal tubules to promote excretion of sodium and water, resulting in a decrease in blood volume and blood pressure.
GASTROINTESTINAL REGULATION
Daily water intake and output are normally between 2000 and 3000 mL (see Table 16-2). Oral intake of fluids accounts for most of the water intake. Water intake also includes water from food metabolism and water present in solid foods. Lean meat is approximately 70% water, whereas the water content of many fruits and vegetables approaches 100%.
TABLE 16-2 Normal fluid balance in the adult
| Intake | |
| Fluids | 1200 ml |
| Solid food | 1000 ml |
| Water from oxidation | 300 ml |
| 2500 ml | |
| Output | |
| Insensible loss (skin and lungs) | 900 ml |
| In faeces | 100 ml |
| Urine | 1500 ml |
| 2500 ml |
In addition to oral intake, the GI tract normally secretes approximately 8000 mL of digestive fluids each day that are reabsorbed. A small amount of the fluid in the GI tract is normally eliminated in faeces, but diarrhoea and vomiting that prevents GI absorption of secretions and fluids can lead to significant fluid and electrolyte loss.
INSENSIBLE WATER LOSS
Insensible water loss, which is invisible vaporisation from the lungs and skin, assists in regulating body temperature. Normally, about 600–900 mL per day is lost. The amount of water loss is increased by accelerated body metabolism, which occurs with increased body temperature and exercise. Water loss through the skin should not be confused with the vaporisation of water excreted by sweat glands. Only water is lost by insensible perspiration. Excessive sweating (sensible perspiration) caused by fever or high environmental temperatures may lead to large losses of water and electrolytes.
FLUID AND ELECTROLYTE IMBALANCES
Fluid and electrolyte imbalances occur to some degree in most patients with a major illness or injury because illness disrupts the normal homeostatic mechanism. Some fluid and electrolyte imbalances are directly caused by illness or disease (e.g. burns, heart failure). At other times, therapeutic measures (e.g. IV fluid replacement, diuretics) cause or contribute to fluid and electrolyte imbalances. Perioperative patients are at risk for the development of fluid and electrolyte imbalances because of restriction of oral intake, gastrointestinal preparation, blood volume loss or fluid shifts.8
The imbalances are commonly classified as deficits or excesses. Each imbalance is discussed separately. (For normal values, see Table 16-3.) In actual clinical situations, more than one imbalance occurring in the same patient is common. For example, a patient with prolonged nasogastric suction will lose sodium, potassium, hydrogen and chloride ions. These imbalances may result in a deficiency of both sodium and potassium, a fluid volume deficit and a metabolic alkalosis due to loss of hydrochloric acid.
TABLE 16-3 Normal serum electrolyte values
| Anions | Normal value |
|---|---|
| Bicarbonate (HCO3−) | 22–26 mmol/L |
| Chloride (Cl−) | 96–106 mmol/L |
| Phosphate (PO43–) | 0.78–1.42 mmol/L |
| Cations | Normal value |
| Potassium (K+) | 3.5–5.0 mmol/L |
| Magnesium (Mg2+) | 0.75–1.25 mmol/L |
| Sodium (Na+) | 135–145 mmol/L |
| Calcium (Ca2+) (total) | 2.15–2.55 mmol/L |
| Calcium (ionised) | 1.16–1.32 mmol/L |
Extracellular fluid volume imbalances
ECF volume deficit (hypovolaemia) and ECF volume excess (hypervolaemia) are commonly occurring clinical conditions. ECF volume imbalances are typically accompanied by one or more electrolyte imbalances, particularly changes in the serum sodium level.
FLUID VOLUME DEFICIT (HYPOVOLAEMIA)
Fluid volume deficit can occur with abnormal loss of body fluids (e.g. diarrhoea, fistula drainage, haemorrhage, polyuria), inadequate intake or a plasma-to-interstitial fluid shift. The term fluid volume deficit should not be used interchangeably with the term dehydration. Dehydration refers to loss of pure water alone without corresponding loss of sodium. The clinical manifestations of fluid volume deficit are listed in Table 16-4.
TABLE 16-4 Extracellular fluid (ECF) imbalances: causes and clinical manifestations
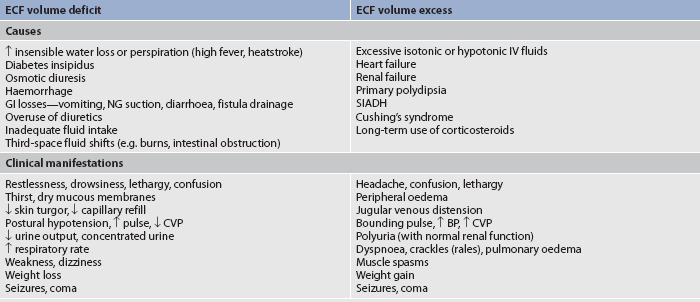
BP, blood pressure; CVP, central venous pressure; GI, gastrointestinal; IV, intravenous; NG, nasogastric; SIADH, syndrome of inappropriate antidiuretic hormone.
The goal of treatment for fluid volume deficit is to correct the underlying cause and to replace both water and any needed electrolytes. Balanced IV solutions, such as lactated Hartmann’s solution, are usually given. Isotonic (0.9%) sodium chloride is used when rapid volume replacement is indicated. Blood is administered when volume loss is due to blood loss.
FLUID VOLUME EXCESS (HYPERVOLAEMIA)
Fluid volume excess may result from excessive intake of fluids, abnormal retention of fluids (e.g. heart failure, renal failure) or an interstitial-to-plasma fluid shift. Although shifts in fluid between the plasma and interstitium do not alter the overall volume of ECF, these shifts do result in changes in the intravascular volume. The clinical manifestations of fluid volume excess are listed in Table 16-4.
The goal of treatment for fluid volume excess is removal of fluid without producing abnormal changes in the electrolyte composition or osmolality of ECF. The primary cause must be identified and treated. Diuretics and fluid restriction are the primary forms of therapy. Restriction of sodium intake may also be indicated. If the fluid excess leads to ascites or pleural effusion, an abdominal paracentesis or thoracentesis may be necessary.
 NURSING MANAGEMENT: EXTRACELLULAR FLUID VOLUME IMBALANCES
NURSING MANAGEMENT: EXTRACELLULAR FLUID VOLUME IMBALANCES
Nursing diagnoses
Nursing diagnoses and collaborative problems for the patient with fluid imbalances include, but are not limited to, the following:
Nursing implementation
Intake and output
The use of 24-hour intake and output records gives valuable information regarding fluid and electrolyte problems. Sources of excessive intake or fluid losses can be identified on an accurately recorded intake-and-output flow sheet. Intake should include oral, IV and tube feedings, and retained irrigants. Output includes urine, excess perspiration, wound or tube drainage, vomitus and diarrhoea. Fluid loss from wounds and perspiration should be estimated. The urine specific gravity should be measured: readings greater than 1.025 indicate concentrated urine, whereas those less than 1.010 indicate dilute urine.
Cardiovascular changes
Monitoring the patient for cardiovascular changes is necessary to prevent or detect complications from fluid and electrolyte imbalances. Signs and symptoms of ECF volume excess and deficit are reflected in changes in blood pressure, pulse force and jugular venous distension. In fluid volume excess, the pulse is full and bounding. Because of the expanded intravascular volume, the pulse is not easily obliterated. Increased volume causes distended neck veins (jugular venous distension) and increased blood pressure.
In mild-to-moderate fluid volume deficit, compensatory mechanisms include sympathetic nervous system stimulation of the heart and peripheral vasoconstriction. Stimulation of the heart increases heart rate and, combined with vasoconstriction, maintains blood pressure within normal limits. A change in position from lying to sitting or standing may elicit a further increase in heart rate or a decrease in blood pressure (orthostatic hypotension). If vasoconstriction and tachycardia provide inadequate compensation, hypotension occurs when the patient is recumbent. Severe fluid volume deficit can cause a weak, thready pulse that is easily obliterated and flattened neck veins. Severe, untreated fluid deficit will result in shock.
Respiratory changes
Both fluid excess and fluid deficit affect respiratory status. ECF excess results in pulmonary congestion and pulmonary oedema as increased hydrostatic pressure in the pulmonary vessels forces fluid into the alveoli. The patient will experience shortness of breath, irritative cough and moist crackles on auscultation.3 The patient with ECF deficit will demonstrate an increased respiratory rate due to decreased tissue perfusion and resultant hypoxia.
Neurological changes
Changes in neurological function may occur with fluid volume excesses or deficits. ECF excess may result in cerebral oedema as a result of increased hydrostatic pressure in cerebral vessels. Alternatively, profound volume depletion may cause an alteration in sensory perception secondary to reduced cerebral tissue perfusion.
Assessment of neurological function includes evaluation of: (1) the level of consciousness, which includes responses to verbal and painful stimuli and the determination of the patient’s orientation to time, place and person; (2) pupillary response to light and equality of pupil size; and (3) voluntary movement of the extremities, degree of muscle strength and reflexes. Nursing care focuses on monitoring and assessing neurological changes using tools such as the Glasgow Coma Scale and ultimately maintaining patient safety.
Daily weights
Accurate daily weights provide the easiest measurement of volume status. An increase of 1 kg is equal to 1000 mL (1 L) fluid retention (provided the patient has maintained usual dietary intake or has not been on nil-by-mouth status). However, weight changes must be obtained under standardised conditions. An accurate weight requires the patient to be weighed at the same time every day, wearing the same garments and on the same carefully calibrated scale. The nurse should remove excess bedding and empty all drainage bags before the weighing. If bulky dressings or tubes are present, which may not necessarily be used every day, a notation regarding these variables should be recorded on the flow sheet or nursing notes. Where patients cannot be moved for effective weighing, the nurse should observe and measure the area potentially affected by oedema, such as abdominal girth measurement for ascites.
Skin assessment and care
Clues to ECF volume deficit and excess can be detected by inspection of the skin. The skin should be examined for turgor and mobility. Normally, a fold of skin, when pinched, will readily move and, on release, will rapidly return to its former position. Skin areas over the sternum, abdomen and anterior forearm are the usual sites for evaluation of tissue turgor (see Fig 16-11). Decreased skin turgor is less predictive of fluid deficit in older people because of the loss of tissue elasticity.6
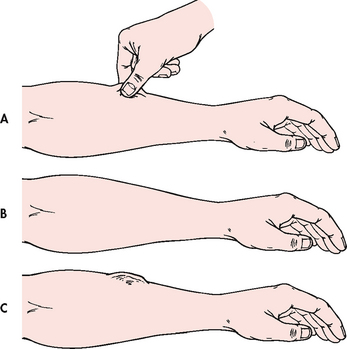
Figure 16-11 Assessment of skin turgor. A and B, When normal skin is pinched, it resumes shape in seconds. C, If the skin remains wrinkled for 20–30 seconds, the patient has poor skin turgor.
In ECF volume deficit, skin turgor is diminished; there is a lag in the pinched skinfold’s return to its original state (referred to as tenting). The skin may be cool and moist if there is vasoconstriction, to compensate for the decreased fluid volume. Mild hypovolaemia usually does not stimulate this compensatory response; consequently, the skin will be warm and dry. Volume deficit may also cause the skin to appear dry and wrinkled. These signs may be difficult to evaluate in the older adult because the patient’s skin may normally be dry, wrinkled and non-elastic. Oral mucous membranes will be dry, the tongue may be furrowed and the individual often complains of thirst. Routine oral care is critical to the comfort of the dehydrated patient and the patient who is fluid restricted for management of fluid volume excess.
Skin that is oedematous may feel cool because of fluid accumulation and a decrease in blood flow secondary to the pressure of the fluid. The fluid can also stretch the skin, causing it to feel taut and hard. Oedema is assessed by pressing with a thumb or forefinger over the oedematous area. A grading scale is used to standardise the description if an indentation (ranging from 1+ [slight, 2 mm indentation] to 4+ [pitting, 8 mm indentation]) remains when pressure is released. The areas to be evaluated for oedema are those where soft tissues overlie a bone. Skin areas over the distal ends of the tibia, fibula and sacrum are the preferred sites.
Good skin care for the patient with ECF volume excess or deficit is important. Oedematous tissues should be protected from extremes of heat and cold, prolonged pressure and trauma. Frequent skin care and changes in position will protect the patient from skin breakdown. Elevation of oedematous extremities helps promote venous return and fluid reabsorption. Dehydrated skin needs ongoing care without the use of soap, preferentially with aqueous solutions. The application of moisturising creams or emollients will increase moisture retention and may aid circulation.
Other nursing measures
The rates of infusion of IV fluid solutions should be monitored carefully. Maintenance of appropriate hydration is essential, particularly when large volumes of fluid or certain electrolytes are involved. This is especially relevant for patients with cardiac, renal or neurological problems. Patients receiving tube feedings need supplementary water added to their enteral formula. The amount of water will depend on the osmolarity of the feeding and the patient’s condition.
The patient with nasogastric suction should not be allowed to drink water because it will result in diffusion of the electrolytes from the mucosal cells into the gastric lumen, which the tube will then suction away. Similarly, the nasogastric tube should always be irrigated with isotonic saline solution and not with water because using water will increase the loss of electrolytes. Occasionally, the patient may be given small amounts of ice to suck.
Nurses in hospitals and nursing homes should encourage and assist older and debilitated patients to maintain adequate oral intake. Attention should be directed to the ability of patients to obtain adequate fluids independently, express thirst and swallow effectively.6 Fluids should be accessible and within easy reach of patients. Patients with physical limitations, such as arthritis, will need assistance to open and hold containers. A variety of types of fluids should be available and individual preferences should be assessed. Room temperature drinks often lack appeal. Therefore, fluids should be served at a temperature that is preferred by the patient. Of the daily intake of fluids, 70–80% should be with meals, with the addition of fluid supplements between meals. Older adults may choose to decrease or eliminate fluids 2 hours before bedtime to decrease nocturia or incontinence. The unconscious or cognitively impaired patient is at increased risk because of an inability to express thirst and respond to it. Therefore, fluid intake and loss must be documented accurately and careful evaluation of adequacy of intake must occur before appropriate fluid intake is administered.9
Sodium imbalances
Sodium is the main cation of the ECF and plays a major role in maintaining the concentration and volume of the ECF, resulting in it being the primary determinant of ECF osmolality. Sodium imbalances are typically associated with parallel changes in osmolality. Because of its impact on osmolality, sodium affects the water distribution between the ECF and the ICF, which has sequelae on the generation and transmission of nerve impulses and the regulation of acid–base balance. The serum sodium level is measured in millimoles per litre.
The GI tract absorbs sodium from foods. Typically, the daily intake of sodium far exceeds the body’s daily requirements. Sodium is removed from the body through urine, sweat and faeces, although the kidneys are the primary regulator of sodium balance. The kidneys regulate the ECF concentration of sodium by excreting or retaining water under the influence of ADH. Aldosterone also plays a role in sodium regulation by promoting sodium reabsorption from the renal tubules. The serum sodium level reflects the ratio of sodium to water, not necessarily the loss or gain of sodium. Thus changes in the serum sodium level may reflect a primary water imbalance, a primary sodium imbalance or a combination of the two. Sodium imbalances are typically associated with imbalances in ECF volume (see Figs 16-12 and 16-13).
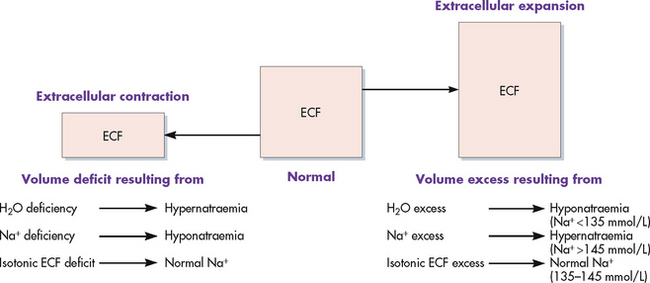
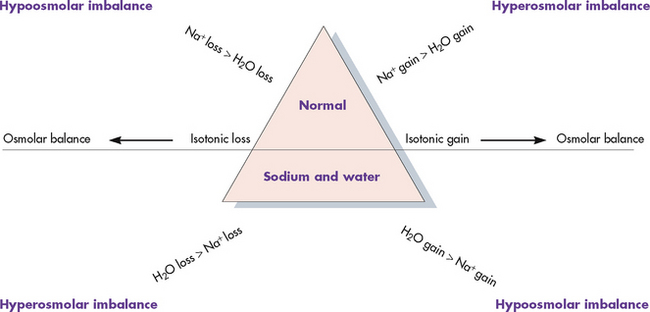
Figure 16-13 Isotonic gains and losses affect mainly the extracellular fluid (ECF) compartment with little or no water movement into the cells. Hypertonic imbalances cause water to move from inside the cell into the ECF to dilute the concentrated sodium, causing cell shrinkage. Hypotonic imbalances cause water to move into the cell, causing cell swelling.
HYPERNATRAEMIA
Common causes of hypernatraemia are listed in Table 16-5. An elevated serum sodium level may occur with water loss or sodium gain; thus, since sodium is the major determinant of the ECF osmolality, hypernatraemia causes hyperosmolality. In turn, ECF hyperosmolality causes a shift of water out of the cells, which leads to cellular dehydration.
TABLE 16-5 Sodium imbalances: causes and clinical manifestations
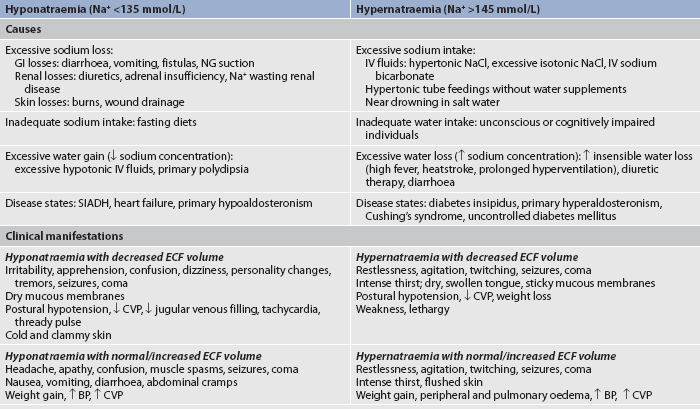
BP, blood pressure; CVP, central venous pressure; ECF, extracellular fluid; GI, gastrointestinal; IV, intravenous; NG, nasogastric; SIADH, syndrome of inappropriate antidiuretic hormone.
As discussed earlier, the primary protection against the development of hyperosmolality is thirst. As the plasma osmolality increases, the thirst centre in the hypothalamus is stimulated and the individual seeks fluids. Hypernatraemia is not a problem in an alert person who has access to water, can sense thirst and is able to swallow. Hypernatraemia secondary to water deficiency is often the result of an impaired level of consciousness or an inability to obtain fluids.
Several clinical states can produce water loss and hypernatraemia. A deficiency in the synthesis or a release of ADH from the posterior pituitary gland (central diabetes insipidus) or a decrease in kidney responsiveness to ADH (nephrogenic diabetes insipidus) can lead to profound diuresis, resulting in a water deficit and hypernatraemia. Hyperosmolality can result from administration of concentrated hyperosmolar tube feedings and osmotic diuretics (mannitol), as well as hyperglycaemia associated with uncontrolled diabetes mellitus. These situations result in osmotic diuresis. Dilute urine is lost, leaving behind a high solute load. Other causes of hypernatraemia include excessive sweating and increased sensible losses from high fever.
Excessive sodium intake with inadequate water intake can also lead to hypernatraemia. Examples of sodium gain include IV administration of hypertonic saline or sodium bicarbonate, use of sodium-containing drugs, concentrated enteral tube feedings, excessive oral intake of sodium (ingestion of sea water) and primary aldosteronism (hypersecretion of aldosterone) caused by a tumour of the adrenal glands.
CLINICAL MANIFESTATIONS
Symptomatic hypernatraemia is rare except in cases where individuals do not have access to water or have an altered thirst mechanism. When symptoms do occur, they are primarily the result of water shifting out of cells into the extracellular fluid with resultant dehydration and shrinkage of cells. Dehydration of brain cells results in neurological manifestations, such as intense thirst, lethargy, agitation, seizures and even coma. Hypernatraemia also has a direct effect on the excitability and conduction of neurons, causing them to be more easily activated. Patients with hypernatraemia will exhibit the symptoms of any accompanying ECF volume deficit, such as postural hypotension, weakness and decreased skin turgor. The clinical manifestations of hypernatraemia are listed in Table 16-5.
NURSING AND COLLABORATIVE MANAGEMENT: HYPERNATRAEMIA
Nursing diagnoses
Nursing diagnoses and consequent problems for the patient with hypernatraemia include, but are not limited to, the following.
Nursing implementation
The goal of treatment in hypernatraemia is to treat the underlying cause. In primary water deficit, the continued water loss must be prevented and water replacement must be provided. If oral fluids cannot be ingested, IV solutions of 5% dextrose in water or hypotonic saline may be given initially. Serum sodium levels must be reduced gradually to prevent too rapid a shift of water back into the cells. Over-rapid correction of hypernatraemia can result in cerebral oedema. The risk is greatest in the patient who has developed hypernatraemia over several days or longer.
The goal of treatment for sodium excess is to dilute the sodium concentration with sodium-free IV fluids, such as 5% dextrose in water, and to promote excretion of the excess sodium by administering diuretics. Dietary sodium intake will also be restricted. (See Ch 49 for specific treatment of diabetes insipidus.)
HYPONATRAEMIA
Hyponatraemia may result from loss of sodium-containing fluids, water excess (dilutional hyponatraemia) or a combination of both. Hyponatraemia causes hypo-osmolality with a shift of water into the cells. A common cause of hyponatraemia caused by water excess is inappropriate use of sodium-free or hypotonic IV fluids. This may occur in patients after surgery or major trauma, during administration of fluids in patients with renal failure or in patients with psychiatric disorders associated with excessive water intake. SIADH will result in dilutional hyponatraemia caused by abnormal retention of water. (See Ch 49 for a discussion of the causes of SIADH.)
Losses of sodium-rich body fluids from the GI tract, kidneys or skin indirectly result in hyponatraemia. Because these fluids are either isotonic or hypotonic, sodium is lost with an equal or greater proportion of water. However, hyponatraemia develops as the body responds to the fluid volume deficit with activation of the thirst mechanism and release of ADH. The resultant retention of water lowers the sodium concentration.3
CLINICAL MANIFESTATIONS
Symptoms of hyponatraemia are related to cellular swelling and are first manifested in the CNS.3 The excess water lowers plasma osmolality, shifting fluid into brain cells, causing irritability, apprehension, confusion, seizures and even coma. The clinical manifestations of hyponatraemia are listed in Table 16-5.
NURSING AND COLLABORATIVE MANAGEMENT: HYPONATRAEMIA
Nursing diagnoses
Nursing diagnoses and consequent problems for the patient with hyponatraemia include, but are not limited to, the following
Nursing implementation
In hyponatraemia that is caused by water excess, fluid restriction is often all that is needed to treat the problem. If severe symptoms (seizures) develop, small amounts of IV hypertonic saline solution (3% NaCl) are given to restore the serum sodium level while the body is returning to a normal water balance. Treatment of hyponatraemia associated with abnormal fluid loss includes fluid replacement with sodium-containing solutions.
Potassium imbalances
Potassium is the major ICF cation with 98% of the body’s potassium being intracellular. For example, potassium concentration within muscle cells is approximately 140 mmol/L; potassium concentration in the ECF is 3.5–5.5 mmol/L. The sodium–potassium pump in cell membranes maintains this concentration difference by pumping potassium into the cell and sodium out, a process that is fuelled by the breakdown of ATP. Adequate intracellular magnesium is necessary for normal functioning of the pump. Because the ratio of ECF potassium to ICF potassium is the major factor in the resting membrane potential of nerve and muscle cells, neuromuscular and cardiac function are commonly affected by potassium imbalances.2
Potassium is critical for many cellular and metabolic functions. In addition to its role in neuromuscular and cardiac function, potassium regulates intracellular osmolality and promotes cellular growth. Potassium moves into cells during the formation of new tissues and leaves the cells during tissue breakdown.3 Potassium also plays a role in acid–base balance (discussed in the section on acid–base regulation later in this chapter).
Diet is the source of potassium. The typical Western diet contains approximately 1950–5460 mg of potassium daily, mainly from fresh fruits, dried fruits and vegetables. Many salt substitutes used in low-sodium diets contain substantial potassium. Patients may receive potassium from parenteral sources, including IV fluids, transfusions of stored, haemolysed blood and medications (e.g. potassium-penicillin).
The kidneys are the primary route for potassium loss and are responsible for excreting approximately 90% of the daily potassium intake, with the remainder lost via faeces and sweat. Impairment of kidney function may lead to the retention of potassium to toxic concentrations. There is an inverse relationship between sodium and potassium reabsorption in the kidneys, where factors that cause sodium retention (e.g. low blood volume, increased aldosterone level) can cause potassium loss in the urine. Large urine volumes can be associated with excess loss of potassium in the urine. The ability of the kidneys to conserve potassium is weak even when body stores are depleted.2
Disruptions in the dynamic equilibrium between ICF and ECF potassium often create clinical problems. Among the factors causing potassium to move from the ECF to the ICF are the following:
• β-adrenergic stimulation (catecholamine release in stress, coronary ischaemia, delirium tremens or administration of β-adrenergic agonist drugs)
• rapid cell building (administration of folic acid or vitamin B12 to patients with megaloblastic anaemia resulting in marked production of red blood cells).
Factors that cause potassium to move from the ICF to the ECF include acidosis, trauma to cells (as in massive soft-tissue damage or tumour lysis) and exercise. Both digoxin-like drugs and β-adrenergic blocking drugs (e.g. propranolol) can impair entry of potassium into cells, resulting in the higher ECF potassium concentration. Causes of potassium imbalance are summarised in Table 16-6.
TABLE 16-6 Potassium imbalances: causes and clinical manifestations
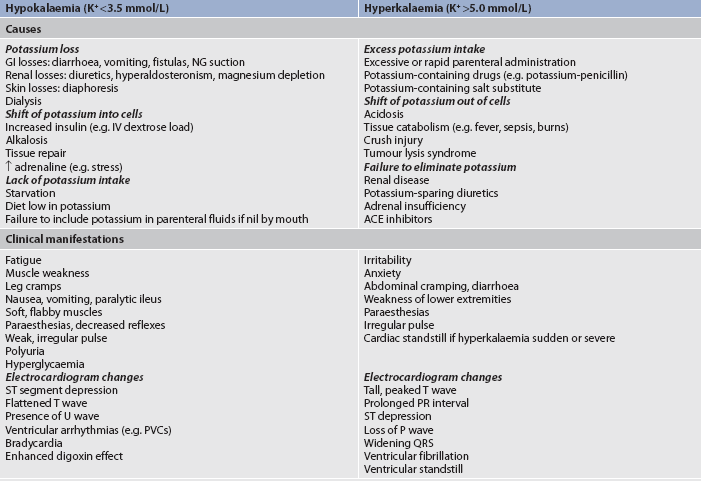
ACE, angiotensin-converting enzyme; GI, gastrointestinal; IV, intravenous; NG, nasogastric; PVC, premature ventricular contraction.
HYPERKALAEMIA
Hyperkalaemia (high serum potassium level) may be caused by a massive intake of potassium, impaired renal excretion, shift of potassium from the ICF to the ECF, or a combination of these factors. The most common cause of hyperkalaemia is renal failure. Hyperkalaemia is also common in patients with massive cell destruction (e.g. burn or crush injury, tumour lysis); rapid transfusion of stored, haemolysed blood; and catabolic states (e.g. severe infections). Metabolic acidosis is associated with a shift of potassium ion from the ICF to the ECF as hydrogen ions move into the cell. Adrenal insufficiency with a subsequent aldosterone deficiency leads to retention of potassium. Certain drugs, such as potassium-sparing diuretics (e.g. spironolactone, triamterene) and angiotensin-converting enzyme (ACE) inhibitors (e.g. enalapril, lisinopril), may contribute to the development of hyperkalaemia. Both of these types of drugs reduce the kidney’s ability to excrete potassium (see Table 16-6).
CLINICAL MANIFESTATIONS
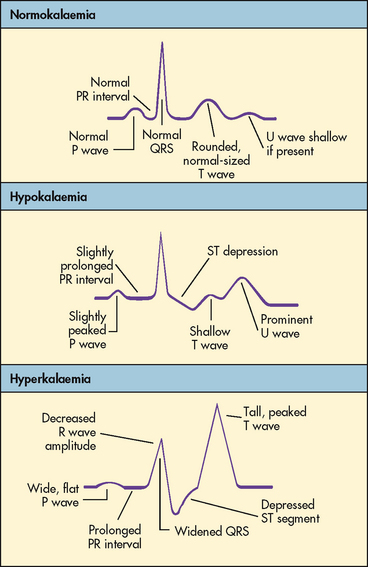
Hyperkalaemia increases the concentration of potassium outside the cell, altering the normal ECF and ICF ratio, resulting in increased cellular excitability. The patient may experience cramping leg pain, followed by weakness or paralysis of skeletal muscles. Leg muscles are affected initially; respiratory muscles are spared. Disturbances in cardiac conduction occur as the potassium level rises.3 Cardiac depolarisation is decreased, leading to flattening of the P wave and widening of the QRS wave. Repolarisation occurs more rapidly, resulting in shortening of the QT interval and causing the T wave to be narrower and more peaked. Ventricular fibrillation or cardiac standstill may occur. Figure 16-14 illustrates the electrocardiographic (ECG) effects of hyperkalaemia. Abdominal cramping and diarrhoea occur from hyperactivity of smooth muscles. Other clinical manifestations are listed in Table 16-6.
NURSING AND COLLABORATIVE MANAGEMENT: HYPERKALAEMIA
Nursing diagnoses
Nursing diagnoses and collaborative problems for the patient with hyperkalaemia include, but are not limited to, the following:
Nursing implementation
Treatment of hyperkalaemia consists of the following:
1. Eliminate oral and parenteral potassium intake (see Box 46-2).
2. Increase elimination of potassium. This is accomplished via diuretics, dialysis and use of ion-exchange resins. Increased fluid intake can enhance renal potassium elimination.
3. Force potassium from the ECF to the ICF. This is accomplished by administration of IV insulin (along with glucose so the patient does not become hypoglycaemic) or via administration of IV sodium bicarbonate in the correction of acidosis. Rarely, a β-adrenergic agonist (e.g. adrenaline) is administered.
4. Reverse the membrane potential effects of the elevated ECF potassium level by administering calcium gluconate intravenously. Calcium ion can immediately reverse the membrane excitability.
In cases where the elevation of potassium is mild and the kidneys are functioning, it may be sufficient to withhold potassium from the diet as well as IV sources and to increase renal elimination by administering fluids and possibly diuretics. All patients with clinically significant hyperkalaemia should be monitored electrocardiographically to detect dysrhythmias and to monitor the effects of therapy. Patients with moderate hyperkalaemia should additionally receive one of the treatments to force potassium into cells, usually IV insulin and glucose. The patient experiencing dangerous cardiac dysrhythmias should receive IV calcium gluconate immediately while the potassium is being eliminated and forced into cells. Haemodialysis is an effective means of removing potassium from the body in the patient with renal failure.
HYPOKALAEMIA
Hypokalaemia (low serum potassium level) can result from abnormal losses of potassium from a shift of potassium from ECF to ICF, or rarely from deficient dietary potassium intake. The most common cause of hypokalaemia is abnormal potassium loss, via either the kidneys or GI tract. Abnormal losses occur when the patient has diuresis, particularly if the patient has an elevated aldosterone level. Aldosterone is released when the circulating blood volume is low; it causes sodium retention in the kidneys but loss of potassium in the urine. Magnesium deficiency may contribute to the development of potassium depletion. Low plasma magnesium levels stimulate renin release and subsequent increased aldosterone levels, which result in potassium excretion.2 GI tract losses of potassium secondary to diarrhoea, laxative abuse, vomiting and ileostomy drainage can cause hypokalaemia.
Metabolic alkalosis can cause a shift of potassium into cells in exchange for hydrogen, thus lowering the potassium in the ECF and causing symptomatic hypokalaemia. Hypokalaemia is sometimes associated with the treatment of diabetic ketoacidosis because of a combination of factors, including an increased urinary potassium loss and a shift of potassium into cells with the administration of insulin and correction of metabolic acidosis. A less common cause of hypokalaemia is the sudden initiation of cell formation; for example, the formation of red blood cells (RBCs) as in treatment of anaemia with vitamin B12, folic acid or erythropoietin.
CLINICAL MANIFESTATIONS
Hypokalaemia alters the resting membrane potential. It most commonly is associated with hyperpolarisation, or increased negative charge within the cell. This causes reduced excitability of cells. The most serious clinical problems are cardiac. Cardiac changes include impaired repolarisation, resulting in a flattening of the T wave and eventually in emergence of a U wave. The P wave may increase in amplitude and may become peaked (see Fig 16-14). The incidence of potentially lethal ventricular dysrhythmias is increased in hypokalaemia. Patients at risk of hypokalaemia and those who are critically ill should have cardiac monitoring to detect cardiac changes related to potassium imbalances. Patients taking digoxin experience increased digoxin toxicity if their serum potassium level is low. Skeletal muscle weakness and paralysis may occur with hypokalaemia. As with hyperkalaemia, symptoms are often observed initially in the legs. Severe hypokalaemia can cause weakness or paralysis of respiratory muscles, leading to shallow respirations and respiratory arrest.
Smooth muscle function is also altered by hypokalaemia. The patient may experience decreased GI motility (e.g. paralytic ileus), decreased airway responsiveness and impaired regulation of arteriolar blood flow, possibly contributing to smooth muscle cell breakdown. Finally, hypokalaemia can impair function in non-muscle tissue. Release of insulin is impaired, leading to hyperglycaemia. Clinical manifestations of hypokalaemia are presented in Table 16-6.
NURSING AND COLLABORATIVE MANAGEMENT: HYPOKALAEMIA
Nursing diagnoses
Nursing diagnoses and collaborative problems for the patient with hypokalaemia include, but are not limited to, the following:
Nursing implementation
Hypokalaemia is treated by giving potassium chloride (KCl) supplements and increasing dietary intake of potassium. KCl supplements can be given orally or intravenously. Except in severe deficiencies, KCl is never given unless there is a urine output of at least 0.5 mL/kg of body weight per hour.
 SAFETY ALERT—KCl
SAFETY ALERT—KClThe preferred level is 40 mmol/L, but stronger concentrations may be given for severe hypokalaemia (up to 80 mmol/L) with continuous cardiac monitoring.10 The rate of IV administration of KCl should not exceed 10–20 mmol per hour and should be administered by infusion pump to ensure correct administration rate. Because KCl is irritating to the vein, assess IV sites at least hourly for phlebitis and infiltration. Infiltration can cause necrosis and sloughing of the surrounding tissue. Central IV lines should be used when rapid correction of hypokalaemia is necessary. Patients should be taught methods to prevent hypokalaemia. Patients at risk should obtain regular serum potassium levels to monitor for hypokalaemia. A teaching guide for the prevention of hypokalaemia is given in Box 16-1 and foods high in potassium are identified in Table 46-9.
BOX 16-1 Prevention of hypokalaemia
PATIENT & FAMILY TEACHING GUIDE
When teaching the patient and/or carer to prevent hypokalaemia, do the following:
2. For patients taking potassium-losing diuretics:
3. For patients taking potassium-sparing diuretics:
4. For patients taking oral potassium supplements:
5. For the patient taking digoxin and others at risk of hypokalaemia:
Calcium imbalances
Calcium is obtained from ingested foods. However, only about 30% of the calcium from foods is absorbed in the GI tract. More than 99% of the body’s calcium is combined with phosphorus and concentrated in the skeletal system. Bones serve as a readily available store of calcium. Thus, wide variations in serum calcium levels are avoided by regulating the movement of calcium into or out of the bone. The functions of calcium include the transmission of nerve impulses, myocardial contractions, blood clotting, the formation of teeth and bone, and muscle contractions.
Calcium is present in the serum in three forms: free or ionised; bound to protein (primarily albumin); and complexed with phosphate, citrate or carbonate. The ionised form is the biologically active form. Approximately half of the total serum calcium is ionised.
Calcium is measured in millimoles per litre. As usually reported, serum calcium levels reflect the total calcium level (all three forms), although ionised calcium levels may be reported separately. The levels listed in Table 16-7 reflect total calcium levels. Changes in serum pH will alter the level of ionised calcium without altering the total calcium level. A decreased plasma pH (acidosis) decreases calcium binding to albumin, leading to more ionised calcium. An increased plasma pH (alkalosis) increases calcium binding, leading to decreased ionised calcium. Alterations in serum albumin levels affect the interpretation of total calcium levels. Low albumin levels result in a drop in the total calcium level, although the level of ionised calcium is not affected.
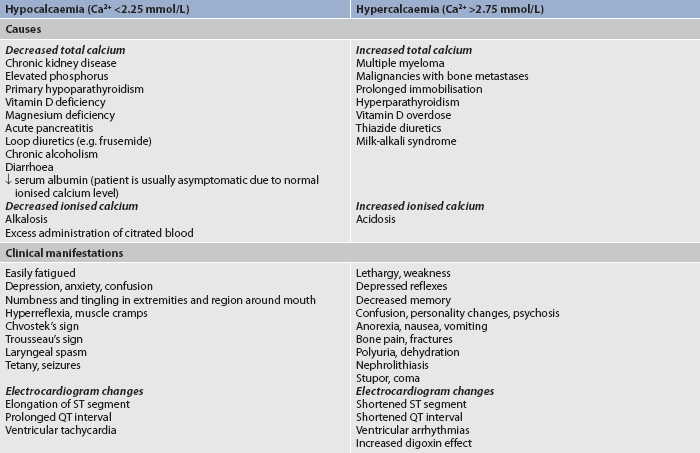
Calcium balance is controlled by parathyroid hormone (PTH), calcitonin and vitamin D. PTH is produced by the parathyroid gland. Its production and release are stimulated by low serum calcium levels. PTH increases bone resorption (movement of calcium out of bones), increases GI absorption of calcium and increases renal tubule reabsorption of calcium. Calcitonin is produced by the thyroid gland and is stimulated by high serum calcium levels. It opposes the action of PTH and thus lowers the serum calcium level by decreasing GI absorption, increasing calcium deposition into bone and promoting renal excretion. Vitamin D is formed through the action of ultraviolet (UV) rays on a precursor found in the skin or is ingested in the diet. Vitamin D is important for absorption of calcium from the GI tract. Causes of calcium imbalances are listed in Table 16-7.
HYPERCALCAEMIA
About two-thirds of hypercalcaemia cases are caused by hyperparathyroidism and one-third of cases are caused by malignancy, especially from breast cancer, lung cancer and multiple myeloma.10 Malignancies lead to hypercalcaemia through bone destruction from tumour invasion or through tumour secretion of a parathyroid-related protein, which stimulates calcium release from bones. Hypercalcaemia is also associated with vitamin D overdose. Prolonged immobilisation results in bone mineral loss and increased plasma calcium concentration. Hypercalcaemia rarely occurs from increased calcium intake (e.g. ingestion of antacids containing calcium, excessive administration during cardiac arrest).
Excess calcium leads to reduced excitability of both muscles and nerves. Manifestations of hypercalcaemia include decreased memory, confusion, disorientation, fatigue, muscle weakness, constipation, cardiac arrhythmias and renal calculi (see Table 16-7).
NURSING AND COLLABORATIVE MANAGEMENT: HYPERCALCAEMIA
Nursing diagnoses
Nursing diagnoses and collaborative problems for the patient with hypercalcaemia include, but are not limited to, the following:
Nursing implementation
The basic treatment of hypercalcaemia is promotion of excretion of calcium in the urine by administration of a loop diuretic (e.g. frusemide) and hydration of the patient with isotonic saline infusions. In hypercalcaemia the patient must drink 3000–4000 mL of fluid daily to promote renal excretion of calcium and to decrease the possibility of kidney stone formation.
Synthetic calcitonin can also be administered to lower serum calcium levels. A diet low in calcium may be prescribed. Mobilisation with weight-bearing activity is encouraged to enhance bone mineralisation. In hypercalcaemia associated with malignancy, the drug of choice is pamidronate, which inhibits the activity of osteoclasts (cells that break down bone and result in calcium release). Pamidronate does not have cytotoxic side effects and it inhibits bone resorption without inhibiting bone formation and mineralisation.
HYPOCALCAEMIA
Any condition that causes a decrease in the production of PTH may result in the development of hypocalcaemia. This may occur with surgical removal of a portion of or injury to the parathyroid glands during thyroid or neck surgery. Acute pancreatitis is another potential cause of hypocalcaemia. Lipolysis, a consequence of pancreatitis, produces fatty acids that combine with calcium ions, decreasing serum calcium levels. The patient who receives multiple blood transfusions can become hypocalcaemic because the citrate used to anticoagulate the blood binds with the calcium. Sudden alkalosis may also result in symptomatic hypocalcaemia despite a normal total serum calcium level. The high pH increases calcium binding to protein, decreasing the amount of ionised calcium. Hypocalcaemia can occur if the diet is low in calcium or if there is increased loss of calcium due to laxative abuse and malabsorption syndromes. (See Table 16-7 for the clinical manifestations and aetiologies of hypocalcaemia.)
Low calcium levels allow sodium to move into excitable cells, decreasing the threshold of action potentials with subsequent depolarisation of the cells. This results in increased nerve excitability and sustained muscle contraction, or tetany. Clinical signs of tetany include Trousseau’s sign and Chvostek’s sign. Trousseau’s sign refers to carpal spasms induced by inflating a blood pressure cuff on the arm (see Fig 16-15). The blood pressure cuff is inflated above the systolic pressure. Carpal spasms become evident within 3 minutes if hypocalcaemia is present. Chvostek’s sign is contraction of facial muscles in response to a tap over the facial nerve in front of the ear (see Fig 16-15), and it also indicates hypocalcaemia with latent tetany. Other manifestations of tetany are laryngeal stridor, dysphagia, and numbness and tingling around the mouth or in the extremities.
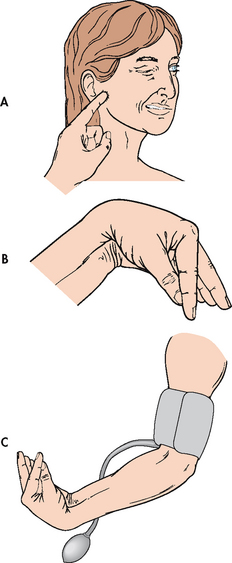
Figure 16-15 Tests for hypocalcaemia. A, Chvostek’s sign is contraction of facial muscles in response to a light tap over the facial nerve in front of the ear. B, Trousseau’s sign is a carpal spasm induced by C, inflating a blood pressure cuff above the systolic pressure for a few minutes.
Cardiac effects of hypocalcaemia include decreased cardiac contractility and ECG changes. A prolonged QT interval may develop into a ventricular tachycardia. Clinical manifestations of hypocalcaemia are listed in Table 16-7.
NURSING AND COLLABORATIVE MANAGEMENT: HYPOCALCAEMIA
Nursing diagnoses
Nursing diagnoses and collaborative problems for the patient with hypocalcaemia include, but are not limited to, the following:
Nursing implementation
The primary goal in the treatment of hypocalcaemia is aimed at treating the cause. Hypocalcaemia can be treated with oral or IV calcium supplements. Calcium is not given intramuscularly because it may cause severe local reactions, such as burning, necrosis and tissue sloughing. IV preparations of calcium, such as calcium gluconate, are administered when severe symptoms of hypocalcaemia are impending or present. A diet high in calcium-rich foods is usually ordered, along with vitamin D supplements. Oral calcium supplements, such as calcium carbonate, may be used when patients are unable to consume enough calcium in the diet, such as those who do not tolerate dairy products. Pain and anxiety must be adequately treated in the patient with suspected hypocalcaemia because hyperventilation-induced respiratory alkalosis can precipitate hypocalcaemic symptoms. Any patient who has had thyroid or neck surgery should be observed closely in the immediate postoperative period for manifestations of hypocalcaemia, because of the proximity of the surgery to the parathyroid glands.
Phosphate imbalances
Phosphorus is a primary anion in the ICF and is essential to the function of muscle, RBCs and the nervous system. It is deposited with calcium for bone and tooth structure. It is also involved in the acid–base buffering system, the mitochondrial energy production of ATP, cellular uptake and use of glucose, and the metabolism of carbohydrates, proteins and fats.
Maintenance of normal phosphate balance requires adequate renal functioning because the kidneys are the major route of phosphate excretion. A small amount is lost in the faeces. A reciprocal relationship exists between phosphorus and calcium in that a high serum phosphate level tends to cause a low calcium concentration in the serum.
HYPERPHOSPHATAEMIA
The major condition that can lead to hyperphosphataemia (high serum phosphate level) is acute or chronic renal failure that results in an altered ability of the kidneys to excrete phosphate. Other causes include chemotherapy for certain malignancies (lymphomas), excessive ingestion of milk or phosphate-containing laxatives and large intakes of vitamin D that increase GI absorption of phosphorus (see Table 16-8).
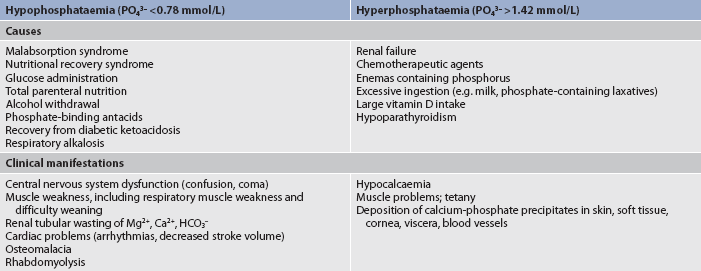
Clinical manifestations of hyperphosphataemia (see Table 16-8) primarily relate to metastatic calcium-phosphate precipitates. Ordinarily, calcium and phosphate are deposited only in bone; however, an increased serum phosphate concentration precipitates readily in conjunction with an increased calcium level, and calcified deposits can occur in soft tissue such as joints, arteries, skin, kidneys and cornea (see Ch 46). Other manifestations of hyperphosphataemia are neuromuscular irritability and tetany, which are related to the low serum calcium levels often associated with high serum phosphate levels.
Management of hyperphosphataemia is aimed at identifying and treating the underlying cause. Ingestion of foods and fluids high in phosphorus (e.g. dairy products) should be restricted. Adequate hydration and correction of hypocalcaemic conditions can enhance the renal excretion of phosphate through the action of PTH. As the serum calcium level increases, it causes the renal excretion of phosphorus. For the patient with renal failure, measures to reduce serum phosphate levels include calcium supplements, phosphate-binding agents or gels and dietary phosphate restrictions (see Ch 46).
HYPOPHOSPHATAEMIA
Hypophosphataemia (low serum phosphate level) is seen in the patient who is malnourished or has a malabsorption syndrome. Other causes include alcohol withdrawal and use of phosphate-binding antacids. Hypophosphataemia may also occur during parenteral nutrition with inadequate phosphorus replacement. Table 16-8 lists causes of phosphorus imbalances.
Most clinical manifestations of hypophosphataemia (Table 16-8) relate to a deficiency of cellular ATP or 2,3-diphosphoglycerate (2,3-DPG), an enzyme in RBCs that facilitates oxygen delivery to the tissues. Because phosphorus is needed for the formation of ATP and 2,3-DPG, its deficit results in impaired cellular energy and oxygen delivery. Mild-to-moderate hypophosphataemia is often asymptomatic and severe hypophosphataemia may be fatal due to decreased cellular function. Acute symptoms include CNS depression, confusion and other mental changes. Other manifestations include muscle weakness and pain, dysrhythmias and cardiomyopathy.
Management of a mild phosphorus deficiency may involve oral supplementation and ingestion of foods high in phosphorus (e.g. dairy products). Severe hypophosphataemia can be serious and may require IV administration of sodium phosphate or potassium phosphate. Frequent monitoring of serum phosphate levels is necessary to guide IV therapy since sudden symptomatic hypocalcaemia, secondary to increased calcium phosphorus binding, is a potential complication of IV phosphorus administration.
Magnesium imbalances
Magnesium is the second most abundant intracellular cation, with approximately 50–60% of the body’s magnesium contained within bone. Magnesium functions as a coenzyme in the metabolism of carbohydrates and protein and is involved in the metabolism of cellular nucleic acids and proteins. Magnesium concentration is regulated by GI absorption and renal excretion;3 the kidneys are able to conserve magnesium in times of need or excrete excesses. Factors that regulate calcium balance (e.g. PTH) appear to similarly influence magnesium balance. Manifestations of magnesium imbalance are often mistaken for calcium imbalances and since magnesium balance is related to calcium and potassium balance, all three cations should be assessed together.11 Magnesium acts directly on the myoneural junction, and neuromuscular excitability is profoundly affected by alterations in serum magnesium levels. Causes of magnesium imbalances are listed in Table 16-9.
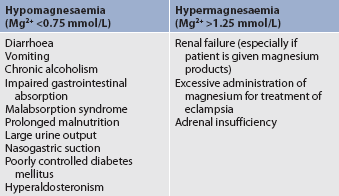
Hypomagnesaemia (low serum magnesium level) produces neuromuscular and CNS hyperirritability, whereas hypermagnesaemia (a high serum magnesium level) depresses neuromuscular and CNS functions. Magnesium is important for normal cardiac function and there is an association between hypomagnesaemia and cardiac dysrhythmias, such as premature ventricular contractions and ventricular fibrillation.
HYPERMAGNESAEMIA
Hypermagnesaemia usually occurs only with an increase in magnesium intake accompanied by renal insufficiency or failure. A patient with chronic renal failure who ingests products containing magnesium (e.g. milk of magnesia) will have a problem with excess magnesium. Magnesium excess could develop in the pregnant woman who receives magnesium sulfate for the management of eclampsia.
Initial clinical manifestations of a mildly elevated serum magnesium concentration include lethargy, drowsiness, and nausea and vomiting. As the levels of serum magnesium increase, deep tendon reflexes are lost, followed by somnolence, and then respiratory and, ultimately, cardiac arrest can occur.
Management of hypermagnesaemia should focus on prevention. Patients with renal failure should not take magnesium-containing drugs and must be cautioned to review all over-the-counter drug labels for magnesium content. The emergency treatment of hypermagnesaemia is IV administration of calcium chloride or calcium gluconate to physiologically oppose the effects of the magnesium on cardiac muscle. Promoting urinary excretion with fluid will decrease serum magnesium levels. The patient with impaired renal function will require dialysis because the kidneys are the major route of excretion for magnesium.
HYPOMAGNESAEMIA
A major cause of magnesium deficiency is prolonged fasting or starvation; chronic alcoholism commonly results in hypomagnesaemia as a result of insufficient food intake. Fluid loss from the GI tract interferes with magnesium absorption. Another potential cause of hypomagnesaemia is prolonged parenteral nutrition without magnesium supplementation. Many diuretics increase the risk of magnesium loss through renal excretion,3 and osmotic diuresis caused by high glucose levels in uncontrolled diabetes mellitus also increases renal excretion of magnesium.
The significant clinical manifestations include confusion, hyperactive deep tendon reflexes, tremors and seizures. Magnesium deficiency also predisposes to cardiac dysrhythmias. Clinically, hypomagnesaemia resembles hypocalcaemia and may contribute to the development of hypocalcaemia as a result of the decreased action of PTH. Hypomagnesaemia may also be associated with hypokalaemia that does not respond well to potassium replacement. This occurs because intracellular magnesium is critical to normal function of the sodium–potassium pump.
Mild magnesium deficiencies can be treated with oral supplements and increased dietary intake of foods high in magnesium (e.g. green vegetables, nuts, bananas, oranges, peanut butter, chocolate). If the condition is severe, controlled parenteral IV or intramuscular (IM) magnesium (e.g. magnesium sulfate) should be administered, since too rapid administration of magnesium can lead to cardiac or respiratory arrest.
Acid–base imbalances
The body normally maintains a steady balance between acids produced during metabolism and bases that neutralise and promote the excretion of the acids. Many health problems may lead to acid–base imbalances, in addition to fluid and electrolyte imbalances. Patients with diabetes mellitus, chronic obstructive pulmonary disease and kidney disease frequently develop acid–base imbalances. Vomiting and diarrhoea may cause loss of acids and bases in addition to fluids and electrolytes. The kidneys are an essential buffer system for acids and, in the older adult, the kidneys are less able to compensate for an acid load. The older adult also has decreased respiratory function, leading to impaired compensation for acid–base imbalances. In addition, tissue hypoxia from any cause may alter acid–base balance. The nurse must always consider the possibility of acid–base imbalance in patients with serious illnesses.
PH AND HYDROGEN ION CONCENTRATION
The acidity or alkalinity of a solution depends on its hydrogen ion (H+) concentration. An increase in H+ concentration leads to acidity; a decrease leads to alkalinity. (Definitions related to acid–base balance are presented in Table 16-10.) Despite the fact that acids are produced by the body daily, the hydrogen ion concentration of body fluids is small (0.0004 mmol/L). This tiny amount is maintained within a narrow range to ensure optimal cellular function. Hydrogen ion concentration is usually expressed as a negative logarithm (symbolised as pH) rather than in millimoles. The use of the negative logarithm means that the lower the pH, the higher the H+ concentration. Relative to a pH of 7, a pH of 8 represents a 10-fold decrease in H+ concentration.
TABLE 16-10 Terms in acid–base physiology
| Acid | Donor of hydrogen ion (H+) by separation of an acid into H+ and its accompanying anion in solution |
|---|---|
| Acidaemia | Signifying an arterial blood pH of less than 7.35 |
| Acidosis | Process that adds acid to or eliminates base from body fluids |
| Alkalaemia | Signifying an arterial blood pH of more than 7.45 |
| Alkalosis | Process that adds base to or eliminates acid from body fluids |
| Anion gap | Reflection of normally unmeasured anions in the plasma; helpful in differential diagnosis of acidosis |
| Base | Acceptor of hydrogen ions; chemical combining of acid and base when hydrogen ions are added to a solution containing a base; bicarbonate (HCO3−) is the most abundant base in body fluids |
| Buffer | Substance that reacts with an acid or a base to prevent a large change in pH |
| pH | Negative logarithm of the H+ concentration |
The pH of a chemical solution may range from 1 to 14. A solution with a pH of 7 is considered neutral. An acid solution has a pH of less than 7, and an alkaline solution has a pH of greater than 7. Blood is slightly alkaline (pH 7.35–7.45); yet if it drops below pH 7.35, the person has acidosis, even though the blood may never become truly acidic. If the blood pH is greater than 7.45, the person has alkalosis (see Fig 16-16).
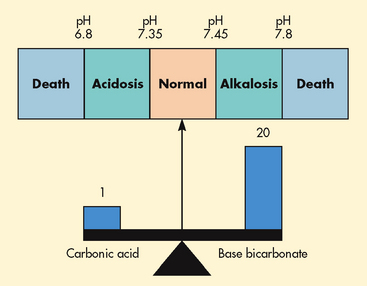
ACID–BASE REGULATION
The body’s metabolic processes constantly produce acids. These acids must be neutralised and excreted to maintain acid–base balance. Normally the body has three mechanisms by which it regulates acid–base balance to maintain the arterial pH between 7.35 and 7.45. These mechanisms are the buffer systems, the respiratory system and the renal system.
The regulatory mechanisms react at different speeds. Buffers react immediately; the respiratory system responds in minutes and reaches maximum effectiveness in hours; the renal response takes 2–3 days to respond maximally, but the kidneys can maintain balance indefinitely in chronic imbalances.
Buffer systems
Buffer systems are the fastest-acting systems and the primary regulators of acid–base balance. Buffers act chemically to change strong acids into weaker acids or to bind acids to neutralise their effect. The buffers in the body include the carbonic acid–bicarbonate, monohydrogen–dihydrogen phosphate, intracellular and plasma protein, and haemoglobin buffers.
A buffer consists of a weakly ionised acid or base and its salt, which functions to minimise the effect of acids on blood pH until they can be excreted from the body. The carbonic acid (H2CO3)–bicarbonate (HCO3−) buffer system neutralises hydrochloric acid (HCl) in the following manner:
In this way, an acid is prevented from making a large change in the blood’s pH, and more H2CO3 is formed. The carbonic acid, in turn, is broken down to water (H2O) and carbon dioxide (CO2). The CO2 is excreted by the lungs, either combined with insensible H2O as carbonic acid or alone as CO2. In this process the buffer system maintains the 20:1 ratio between bicarbonate and carbonic acid as well as the normal pH.
The phosphate buffer system is composed of sodium and other cations in combination with monohydrogen phosphate (HPO42−) or dihydrogen phosphate (H2PO4−). This intracellular buffer system acts in the same manner as the bicarbonate system. Strong acids are neutralised to form sodium chloride (NaCl) and sodium biphosphate (NaH2PO4), a weak acid that can be excreted in the urine. When a strong base such as sodium hydroxide (NaOH) is added to the system, it can be neutralised by sodium dihydrogen phosphate (NaH2PO4) to a weak base (Na2HPO4) and H2O.
Intracellular and extracellular proteins are an effective buffering system throughout the body. The protein buffer system acts like the bicarbonate system. Some of the amino acids of proteins contain free acid radicals, –COOH, which can dissociate into CO2 and H+. Other amino acids have basic radicals, –NH3OH (ammonium hydroxide), which can dissociate into ammonia (NH3) and hydroxide (OH−). The OH− ion can combine with an H+ to form H2O. Haemoglobin is a protein that assists in the regulation of pH by shifting chloride in and out of RBCs in exchange for bicarbonate.
The cell can also act as a buffer by shifting H+ in and out of the cell. With an accumulation of H+ in the ECF, the cells can accept H+ in exchange for another cation (e.g. K+).
The body buffers an acid load better than it neutralises base excess; however, buffers cannot maintain pH without the adequate functioning of the respiratory and renal systems.
Respiratory system
The lungs help maintain a normal pH by excreting CO2 and water, which are by-products of cellular metabolism. When released into circulation, CO2 enters RBCs and combines with H2O to form H2CO3. This carbonic acid dissociates into hydrogen ions and bicarbonate. The free hydrogen ions are buffered by haemoglobin molecules, and the bicarbonate diffuses into the plasma. In the pulmonary capillaries, this process is reversed, and CO2 is formed and excreted by the lungs. The overall reversible reaction is expressed as the following:
CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3−
The amount of CO2 in the blood directly relates to carbonic acid concentration and subsequently to H+ concentration. With increased respirations, more CO2 is expelled and less remains in the blood, which leads to less carbonic acid and thus less H+. With decreased respirations, more CO2 remains in the blood, which leads to increased carbonic acid and thus more H+.
The rate of excretion of CO2 is controlled by the respiratory centre in the medulla in the brainstem. If increased amounts of CO2 or H+ are present, the respiratory centre stimulates an increased rate and depth of breathing; in contrast, respiration rate and depth are inhibited if the centre detects low H+ or CO2 levels.
As a compensatory mechanism the respiratory system acts on the CO2 + H2O side of the reaction by altering the rate and depth of breathing to ‘blow off’ (through hyperventilation) or ‘retain’ (through hypoventilation) CO2. If a respiratory problem is the cause of an acid–base imbalance (e.g. respiratory failure), the respiratory system loses its ability to correct a pH alteration.
Renal system
Under normal conditions the kidneys reabsorb and conserve all of the bicarbonate they filter. The kidneys can generate additional bicarbonate and eliminate excess H+ as compensation for acidosis. The three mechanisms of acid elimination are: (1) secretion of small amounts of free hydrogen ion into the renal tubule; (2) combination of H+ with NH3 to form ammonium (NH4+); and (3) excretion of weak acids.
The body depends on the kidneys to excrete a portion of the acid produced by cellular metabolism. Thus, the kidneys normally excrete acidic urine (average pH equals 6). As a compensatory mechanism, the pH of the urine can decrease to 4 and increase to 8. If the renal system is the cause of an acid–base imbalance (e.g. renal failure), it loses its ability to correct a pH alteration.
ALTERATIONS IN ACID–BASE BALANCE
An acid–base imbalance is produced when the ratio of 1:20 between acid and base content is altered (see Table 16-11). A primary disease or process may alter one side of the ratio (e.g. CO2 retention in pulmonary disease). The compensatory process attempts to maintain the other side of the ratio (e.g. increased renal bicarbonate reabsorption) and when this process fails, an acid–base imbalance results. The compensatory process may be inadequate because either the pathophysiological process is overwhelming or there is insufficient time for the compensatory process to function.
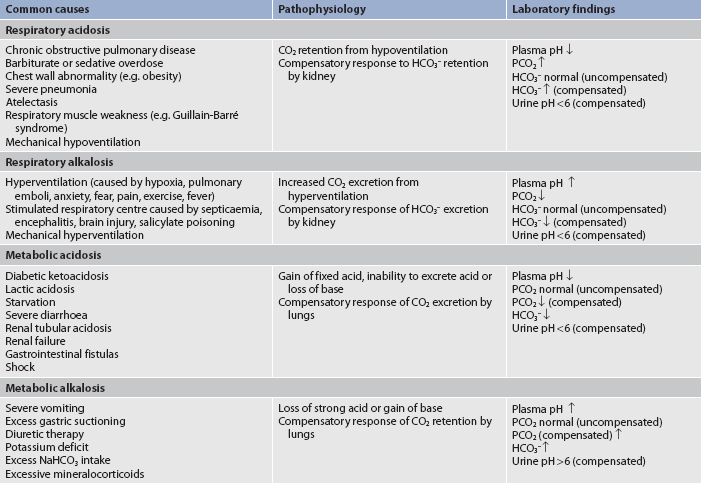
Acid–base imbalances are classified as respiratory or metabolic. Respiratory imbalances affect carbonic acid concentrations; metabolic imbalances affect the base bicarbonate. Therefore, acidosis can be caused by an increase in carbonic acid (respiratory acidosis) or a decrease in bicarbonate (metabolic acidosis). Alkalosis can be caused by a decrease in carbonic acid (respiratory alkalosis) or an increase in bicarbonate (metabolic alkalosis). Imbalances may be further classified as acute or chronic. Chronic imbalances allow greater time for compensatory changes.
Respiratory acidosis
Respiratory acidosis (carbonic acid excess) occurs whenever there is hypoventilation (see Table 16-11). Hypoventilation results in a build-up of CO2; subsequently, carbonic acid accumulates in the blood. Carbonic acid dissociates, liberating H+, and there is a decrease in pH. If CO2 is not eliminated from the blood, acidosis results from the accumulation of carbonic acid (see Fig 16-17, A).
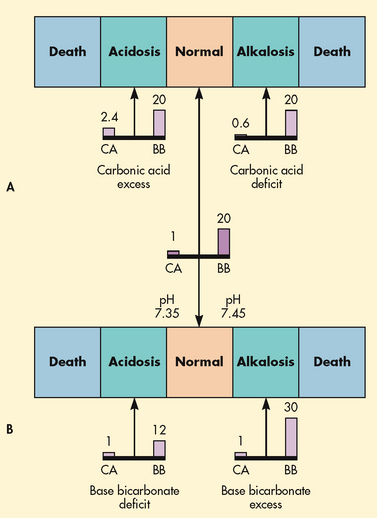
Figure 16-17 Kinds of acid–base imbalances. A, Respiratory imbalances caused by carbonic acid (CA) excess and carbonic acid deficit. B, Metabolic imbalances caused by base bicarbonate (BB) deficit and base bicarbonate excess.
To compensate, the kidneys conserve bicarbonate and secrete increased concentrations of H+ into the urine. In acute respiratory acidosis the renal compensatory mechanisms begin to operate within 24 hours. Until the renal mechanisms have an effect, the serum bicarbonate level will usually be normal.
Respiratory alkalosis
Respiratory alkalosis (carbonic acid deficit) occurs with hyperventilation (see Table 16-11). The primary cause of respiratory alkalosis is hypoxaemia from acute pulmonary disorders. Anxiety, CNS disorders and mechanical overventilation also increase the ventilation rate and decrease the PCO2 level. This leads to decreased carbonic acid and alkalosis (see Fig 16-17, A).
Compensated respiratory alkalosis is rare. In acute respiratory alkalosis, aggressive treatment of the causes of hypoxaemia is essential and usually does not allow time for compensation to occur. However, buffering of acute respiratory alkalosis may occur with shifting of bicarbonate into cells in exchange for Cl−. In chronic respiratory alkalosis that occurs with pulmonary fibrosis or CNS disorders, compensation may include renal excretion of bicarbonate.
Metabolic acidosis
Metabolic acidosis (base bicarbonate deficit) occurs when an acid other than carbonic acid accumulates in the body or when bicarbonate is lost from body fluids (see Table 16-11 and Fig 16-17, B). In both cases a bicarbonate deficit results. Ketoacid accumulation in diabetic ketoacidosis and lactic acid accumulation with shock are examples of accumulation of acids. Severe diarrhoea results in loss of bicarbonate. In renal disease the kidneys lose their ability to reabsorb bicarbonate and secrete hydrogen ions.
The compensatory response to metabolic acidosis is to increase CO2 excretion by the lungs. The patient often develops Kussmaul respiration (deep, rapid breathing). In addition, the kidneys attempt to excrete additional acid.
Metabolic alkalosis
Metabolic alkalosis (base bicarbonate excess) occurs when a loss of acid (prolonged vomiting or gastric suction) or a gain in bicarbonate (ingestion of baking soda) occurs (see Table 16-11 and Fig 16-17, B). The compensatory mechanism is a decreased respiratory rate to increase plasma CO2. Renal excretion of bicarbonate also occurs in response to metabolic alkalosis.
Mixed acid–base disorders
A mixed acid–base disorder occurs when two or more disorders are present at the same time. The pH will depend on the type, severity and acuity of each of the disorders involved and any compensation mechanisms at work. Respiratory acidosis combined with metabolic alkalosis (e.g. chronic obstructive lung disease also treated with a thiazide diuretic) may result in a near-normal pH, while respiratory acidosis combined with metabolic acidosis will cause a greater decrease in pH than either disorder alone. An example of a mixed acidosis appears in a patient in cardiopulmonary arrest. Hypoventilation elevates the CO2 level and anaerobic metabolism due to decreased perfusion produces lactic acid accumulation. An example of a mixed alkalosis is the case of a patient who is hyperventilating because of postoperative pain and is also losing acid secondary to nasogastric suctioning.
CLINICAL MANIFESTATIONS
Clinical manifestations of acidosis and alkalosis are summarised in Tables 16-12 and 16-13. Because a normal pH is vital to all cellular reactions, the clinical manifestations of acid–base imbalances are generalised and non-specific. The compensatory mechanisms also produce some clinical manifestations. For example, the deep, rapid respirations of a patient with metabolic acidosis are an example of respiratory compensation. In alkalosis, hypocalcaemia occurs due to increased calcium binding with albumin, lowering the amount of ionised, biologically active calcium. The hypocalcaemia accounts for many of the clinical manifestations of alkalosis. Determining which patients are at risk for acid–base disturbances includes identifying those patients who have or are at risk for significant electrolyte imbalances, net gain or loss of acids or bases, ventilation abnormalities or abnormal kidney function.12
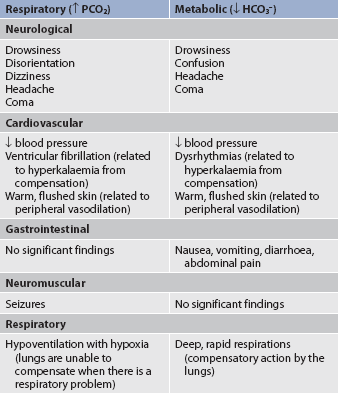
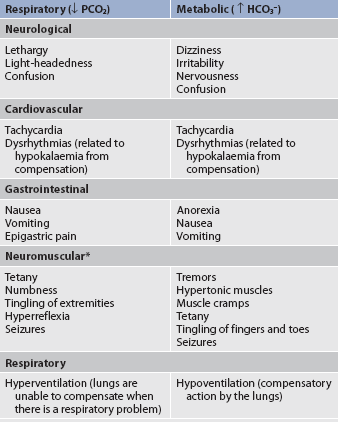
Blood gas values
Arterial blood gas (ABG) values provide valuable information about a patient’s acid–base status, the underlying cause of the imbalance, the body’s ability to regulate pH and the patient’s overall oxygen status. Diagnosis of acid–base disturbances and identification of compensatory processes are done by performing the following six steps:
1. Determine whether the pH is acidotic or alkalotic. Use 7.4 as the starting point. Label values less than 7.4 as acidotic and values greater than 7.4 as alkalotic.
2. Analyse the PCO2 to determine whether the patient has respiratory acidosis or alkalosis. PCO2 is controlled by the lungs and is thus considered the respiratory component of the ABG. Because CO2 forms carbonic acid when dissolved in blood, high CO2 levels indicate acidosis and low CO2 levels indicate alkalosis.
3. Analyse the HCO3− level to determine whether the patient has metabolic acidosis or alkalosis. HCO3−, the metabolic component of the ABG, is controlled primarily by the kidneys. Because HCO3− is a base, high levels of HCO3− result in alkalosis and low levels result in acidosis.
4. At this point, if the CO2 and the HCO3− are within normal limits, the ABGs are normal if the pH is between 7.35 and 7.45.
5. Determine whether the CO2 or the HCO3− matches the acid or base alteration of the pH. For example, if the pH is acidotic and the CO2 is high (respiratory acidosis) but the HCO3− is high (metabolic alkalosis), the CO2 is the parameter that matches the pH alteration. The patient’s acid–base imbalance would be diagnosed as respiratory acidosis.
6. Decide whether the body is attempting to compensate for the pH change. If the parameter that does not match the pH is moving in the opposite direction, the body is attempting to compensate. In step 5, the HCO3− level is alkalotic; this is in the opposite direction of respiratory acidosis and is considered compensation. If compensatory mechanisms are functioning, the pH will return towards 7.40. When the pH is back to normal, the patient has full compensation. The body will not overcompensate for pH changes.
Table 16-14 lists normal ABG values and Table 16-15 provides a sample ABG with interpretation. (Refer to the laboratory findings section of Table 16-11 for the ABG findings of the four major acid–base disturbances.) Table 16.16 shows ROME, a quick memory device (mnemonic) that can be used for understanding acid–base imbalances.
| Parameter | Reference interval |
|---|---|
| pH | 7.35–7.45 |
| PCO2 | 32–48 mmhg |
| Bicarbonate (HCO3−) | 22–26 mmol/l |
| PO2* | 80–100 mmhg (10.67–13.33 kPa) |
| Oxygen saturation | 95% |
| Base excess | ± 2.0 mEq/L |
*Decreases above sea level and with increasing age.
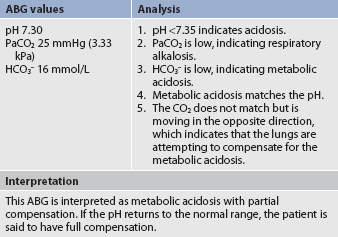
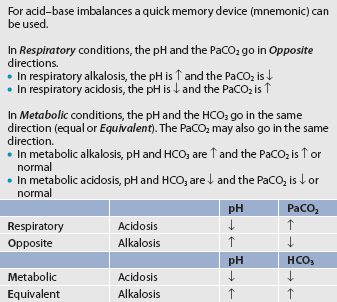
Knowledge of the patient’s clinical situation and the physiological extent of renal and respiratory compensation enables the nurse to identify mixed acid–base disorders as well as the patient’s ability to compensate.
Blood gas analysis will also show the PO2 and oxygen saturation. These values are used to identify hypoxaemia. The values of blood gases differ slightly between arterial and venous samples (see Table 16-14). (Blood gases are discussed further in Ch 25.)
Assessment of fluid, electrolyte and acid–base imbalances
Clinical manifestations for specific fluid, electrolyte and acid–base imbalances have been presented earlier in this chapter. In addition to assessing for those clinical manifestations, subjective and objective data that should be obtained from any patient with suspected fluid, electrolyte or acid–base imbalances are outlined below.
SUBJECTIVE DATA
Important health information
Past health history
The patient should be questioned about any past history of problems involving the kidneys, heart, GI system or lungs that could affect the present fluid, electrolyte and acid–base balance. Information about specific diseases—such as diabetes mellitus, diabetes insipidus, chronic obstructive pulmonary disease, renal failure, ulcerative colitis and Crohn’s disease—should be obtained from the patient. The patient should also be questioned about the incidence of prior fluid, electrolyte or acid–base disorders.
Medications
An assessment of the patient’s current and past use of medications is important. The ingredients in many drugs, especially over-the-counter drugs, are often overlooked as sources of sodium, potassium, calcium, magnesium and other electrolytes. Many prescription drugs, including diuretics, corticosteroids and electrolyte supplements, can cause fluid and electrolyte imbalances.
Functional health patterns
Health perception–health management pattern
If the patient is currently experiencing a problem related to fluid, electrolyte and acid–base balance, a careful description of the illness, including onset, course and treatment, should be obtained. The patient should be questioned about any recent changes in body weight.
Nutritional–metabolic pattern
The patient should be questioned regarding diet and any special dietary practices should be noted. Weight-reduction diets, fad diets or any eating disorders, such as anorexia or bulimia, can lead to fluid and electrolyte problems. If the patient is on a special diet, such as a low-sodium or high-potassium diet, the ability to comply with the dietary prescription should be assessed.
Elimination pattern
Note should be made of the patient’s usual bowel and bladder habits. Any deviations from the expected elimination pattern, such as diarrhoea, oliguria, nocturia, polyuria or incontinence, should be carefully documented.
Activity–exercise pattern
The patient’s exercise pattern is important to determine because excessive perspiration secondary to exercise could result in a fluid and electrolyte problem. Also, the patient’s exposure to extremely high temperatures as a result of leisure or work activity should be determined. The patient should be asked what practices are followed to replace fluid and electrolytes lost through excessive perspiration. An assessment of the patient’s activity level should be done to determine any functional problems that could lead to lack of ability to obtain food or fluids.
Cognitive–perceptual pattern
The patient should be asked about any changes in sensations, such as numbness, tingling, fasciculations (uncoordinated twitching of a single muscle group) or muscle weakness, that could indicate a fluid and electrolyte problem. Additionally, both the patient and the family should be asked whether any changes in mentation or alertness have been noted, such as confusion, memory impairment or lethargy.
OBJECTIVE DATA
Physical examination
There is no specific physical examination to assess fluid, electrolyte and acid–base balance. Common abnormal assessment findings of major body systems offer clues to possible imbalances (see Table 16-17).
Laboratory values
Assessment of serum electrolyte values is a good starting point for identifying fluid and electrolyte imbalance (see Table 16-3). However, serum electrolyte values often provide only limited information. They reflect the concentration of that electrolyte in the ECF but do not necessarily provide information concerning the concentration of the electrolyte in the ICF. For example, the majority of the potassium in the body is found intracellularly. Changes in serum potassium values may be the result of a true deficit or excess of potassium or may reflect the movement of potassium into or out of the cell during acid–base imbalances. An abnormal serum sodium level may reflect a sodium problem or, more likely, a water problem. A reduced haematocrit value could indicate anaemia or it could be caused by fluid volume excess.
Other laboratory tests that help to evaluate the presence of or risk for fluid, electrolyte and acid–base imbalances include serum and urine osmolality, serum glucose, urea and serum creatinine levels, urine specific gravity and urine electrolytes.
In addition to arterial and venous blood gases, serum electrolytes can provide important information about a patient’s acid–base balance. Changes in the serum bicarbonate (often reported as total CO2 or CO2 content) will indicate the presence of metabolic acidosis (low bicarbonate level) or alkalosis (high bicarbonate level). Calculation of the anion gap (serum sodium level minus the sum of the chloride and bicarbonate levels) can help determine the source of metabolic acidosis. The anion gap is increased in metabolic acidosis associated with acid gain (e.g. lactic acidosis, diabetic ketoacidosis) but remains normal (10–14 mmol/L) in metabolic acidosis caused by bicarbonate loss (e.g. diarrhoea).
Oral fluid and electrolyte replacement
In all cases of fluid, electrolyte and acid–base imbalances the treatment is directed towards correction of the underlying cause. The specific diseases or disorders that cause these imbalances are discussed in various chapters throughout this text. Mild fluid and electrolyte deficits can be corrected using oral rehydration solutions containing water, electrolytes and glucose. Glucose not only provides kilojoules but also promotes sodium absorption in the small intestine. Commercial oral rehydration solutions are now available for home use.
Intravenous fluid and electrolyte replacement
IV fluid and electrolyte therapy are commonly used to treat many different fluid and electrolyte imbalances. Many patients need maintenance IV fluid therapy only while they cannot take oral fluids (e.g. during and after surgery). Other patients need corrective or replacement therapy for losses that have already occurred. The amount and type of solution are determined by the normal daily maintenance requirements and by imbalances identified by laboratory results. Table 16-18 provides a list of commonly prescribed IV solutions.
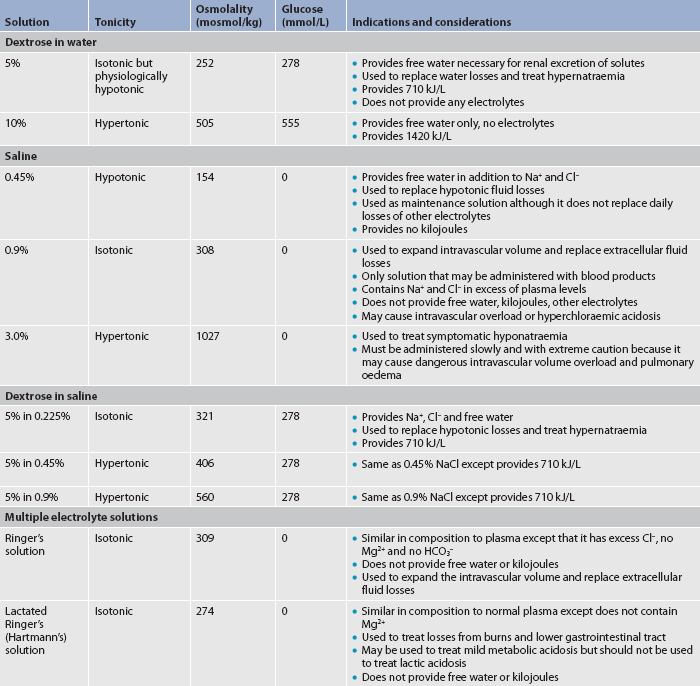
SOLUTIONS
Hypotonic
A hypotonic solution provides more water than electrolytes, diluting the ECF. Osmosis then produces a movement of water from the ECF to the ICF. After osmotic equilibrium has been achieved, the ICF and the ECF have the same osmolality and both compartments have been expanded. Examples of hypotonic fluids are given in Table 16-18. Maintenance fluids are usually hypotonic solutions (e.g. 0.45% NaCl) because normal daily losses are hypotonic. Additional electrolytes (e.g. KCl) may be added to maintain normal levels. Hypotonic solutions have the potential to cause cellular swelling and patients should be monitored for changes in mentation that may indicate cerebral oedema.4,13
Although 5% dextrose in water is considered an isotonic solution, the dextrose is quickly metabolised and the net result is the administration of free water (hypotonic) with proportionately equal expansion of the ECF and the ICF. One litre of a 5% dextrose solution provides 50 g of dextrose, or 710 kJ. Although this amount of dextrose is not enough to meet kilojoule requirements, it helps prevent ketosis associated with starvation. Pure water cannot be administered intravenously because it would cause haemolysis of RBCs.
Isotonic
Administration of an isotonic solution expands only the ECF. There is no net loss or gain from the ICF. An isotonic solution is the ideal fluid replacement for a patient with an ECF volume deficit. Examples of isotonic solutions include Hartmann’s solution and 0.9% NaCl. Hartmann’s solution contains sodium, potassium, chloride, calcium and lactate (the precursor of bicarbonate) in about the same concentrations as those of the ECF. It is contraindicated in the presence of lactic acidosis because of the body’s decreased ability to convert lactate to bicarbonate.
Isotonic saline (0.9% NaCl) has a sodium concentration (154 mmol/L) somewhat higher than plasma (135–145 mmol/L) and a chloride concentration (154 mmol/L) significantly higher than the plasma chloride level (96–106 mmol/L). Thus, excessive administration of isotonic NaCl can result in elevated sodium and chloride levels. Isotonic saline may be used when a patient has experienced both fluid and sodium losses or as vascular fluid replacement in hypovolaemic shock.
Hypertonic
A hypertonic solution initially raises the osmolality of ECF and expands it. It is useful in the treatment of hypovolaemia and hyponatraemia. Examples are listed in Table 16-18. In addition, the higher osmotic pressure draws water out of the cells into the ECF. Hypertonic solutions (e.g. 3% NaCl) require frequent monitoring of blood pressure, lung sounds and serum sodium levels, and should be used with caution because of the risk of intravascular fluid volume excess.13
Although concentrated dextrose and water solutions (10% dextrose or greater) are hypertonic solutions, once the dextrose is metabolised, the net result is the administration of water. The free water provided by these solutions will ultimately expand both the ECF and the ICF. The primary use of these solutions is in the provision of kilojoules. Concentrated dextrose solutions may be combined with amino acid solutions, electrolytes, vitamins and trace elements to provide total parenteral nutrition (see Ch 40). Solutions containing 10% dextrose or less may be administered through a peripheral IV line. Solutions with concentrations greater than 10% must be administered through a central line so that there is adequate dilution to prevent shrinkage of RBCs.
Intravenous additives
In addition to the basic solutions that provide water and a minimum amount of kilojoules and electrolytes, there are additives to replace specific losses. These additives were mentioned previously during the discussion of the particular electrolyte deficiencies. KCl, CaCl2, MgSO4 and HCO3− are common additives to the basic IV solutions.
Plasma expanders
Plasma expanders stay in the vascular space and increase the osmotic pressure. They include colloids, dextran and hetastarch. Colloids are protein solutions such as plasma, albumin and commercial plasmas (e.g. Plasmanate). Albumin is available in 5% and 25% solutions. The 5% solution has an albumin concentration similar to plasma and will expand the intravascular fluid millilitre for millilitre. In contrast, the 25% solution is hypertonic and will draw additional fluid from the interstitium. The 5% concentration is usually given to hypovolaemic patients and the 25% concentration is used if fluid and sodium intake must be minimised.14 Dextran is a complex synthetic sugar. Because dextran is metabolised slowly, it remains in the vascular system for a prolonged period, but not as long as the colloids. It pulls additional fluid into the intravascular space. (Indications for plasma volume expanders are discussed in Ch 66.)
If the patient has lost blood, whole blood or packed RBCs are necessary. Packed RBCs have the advantage of giving the patient primarily RBCs; the blood bank can use the plasma for blood components. Whole blood, with its additional fluid volume, may cause circulatory overload. Although packed RBCs have a decreased plasma volume, they will increase the oncotic pressure and pull fluid into the intravascular space. Loop diuretics may be administered with blood to prevent symptoms of fluid volume excess in anaemic patients who are not volume depleted. (Administration of blood and blood products is discussed in Ch 30.)
CENTRAL VENOUS ACCESS DEVICES
Central venous access devices (CVADs) are catheters that are placed in large blood vessels (e.g. subclavian vein, jugular vein) of patients for whom frequent access to the vascular system is required. In contrast to CVADs, the basic IV catheter is inserted into a peripheral vein in the hand, inside of the arm or antecubital fossa and is used for short-term IV access. Central venous access can be achieved by three different methods: centrally inserted catheters, peripherally inserted central catheters (PICCs) and implanted infusion ports. Centrally inserted catheters and implanted infusion ports must be placed by a doctor, whereas PICCs can be inserted by a nurse with specialised training.
CVADs permit frequent, continuous, rapid or intermittent administration of fluids and medications. They allow for the administration of drugs that are potential vesicants, blood and blood products, and parenteral nutrition. They may also be used for haemodynamic monitoring and venous blood sampling. These devices are indicated for patients who have limited peripheral vascular access or who have a projected need for long-term vascular access. Table 16-19 provides examples of medical conditions where CVADs are used. The Infusion Nurses Society in the US recommends considering a long-term CVAD if the patient will need therapy for more than 1 year.15
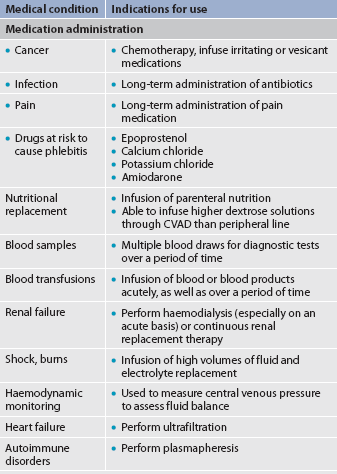
Advantages of CVADs include a reduced need for multiple venipunctures, decreased risk of extravasation injury and immediate access to the central venous system. Although the incidence is decreased, extravasation can still occur if there is displacement of or damage to the device being used. The major disadvantages of CVADs are an increased risk of systemic infection and the invasiveness of the procedure.
CENTRALLY INSERTED CATHETERS
Centrally inserted catheters (also called central venous catheters [CVCs]) are inserted into a vein in the neck or chest (subclavian or jugular) or groin (femoral) with the tip resting in the distal end of the superior vena cava (see Fig 16-18). These catheters are single-, double-, triple- or quad-lumen catheters and they are inserted with the aid of local or general anaesthesia. The other end of the catheter is either non-tunnelled or tunnelled through subcutaneous tissue and exits through a separate incision on the chest or abdominal wall. A Dacron cuff on the catheter serves to stabilise the catheter and may decrease the incidence of infection by impeding bacteria migration along the catheter beyond the cuff. Accurate placement must be verified by chest X-ray before the catheter can be used. Care requirements include injection cap changes, cleansing, flushing and dressing changes. The exact frequency and procedures for these requirements vary and are outlined in the institution’s policy.
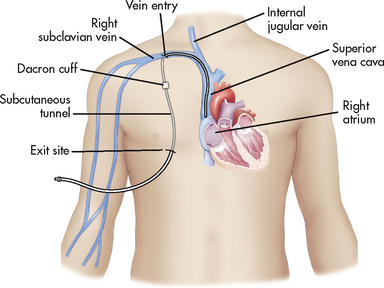
Figure 16-18 Tunnelled centrally inserted catheter. Note the tip of the catheter in the superior vena cava.
Do heparin flushes of central venous catheters decrease occlusions?
EVIDENCE-BASED PRACTICE
Clinical question
For patients with intermittently used central venous catheters (P), do heparin solution flushes (I) versus normal saline flushes (C) decrease catheter occlusions (O)?
Critical appraisal and synthesis of evidence
• Adult patients with a central venous catheter or peripherally inserted central catheter. Excluded patients with implanted ports.
• Outcomes were catheter patency, catheter-related bloodstream infections (CRBSIs) and heparin-induced thrombocytopenia (HIT) rates.
• Weak evidence that heparin flushing reduces catheter occlusions.
• No evidence that heparin flushing reduces bloodstream infections.
Implications for nursing practice
• Maintenance of catheter patency is critical.
• Flushing devices with saline solution may be a safe and effective alternative to heparin flushes for catheter maintenance.
P, patient population of interest; I, intervention or area of interest; C, comparison of interest or comparison group; O, outcome(s) of interest
Specific types of long-term central catheters are Hickman catheters, which require clamps to make sure the valve is closed, and Groshong catheters, which have a valve that opens as fluid is withdrawn or infused and remains closed when not in use.
PERIPHERALLY INSERTED CENTRAL CATHETERS
PICCs are central venous catheters inserted into a vein in the arm rather than a vein in the neck or chest. They are single- or multiple-lumen non-tunnelled catheters that are up to 60 cm in length with gauges ranging from 24 to 16 (see Fig 16-19). PICC lines are inserted at or just above the antecubital fossa (usually cephalic or basilic vein) and advanced to a position with the tip ending in the distal third of the superior vena cava. They are intended for patients who need vascular access for 1 week to 6 months but can be in place for longer periods of time.
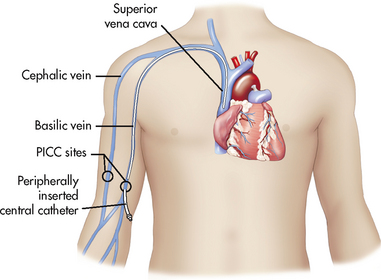
Figure 16-19 Peripherally inserted central catheter (PICC) can be inserted using the basilic or cephalic vein.
The technique for placing a PICC line involves inserting the catheter through a needle with the use of a guide wire or forceps to advance the line. Advantages of PICCs over centrally inserted catheters are lower infection rates, fewer insertion-related complications, decreased cost and insertion at the bedside or outpatient area.
Complications of PICCs include catheter occlusion and phlebitis (see Table 16-20). If phlebitis occurs, it usually appears within 7–10 days following insertion. The arm in which a PICC is in place should not be used for blood pressure readings or blood drawing.16
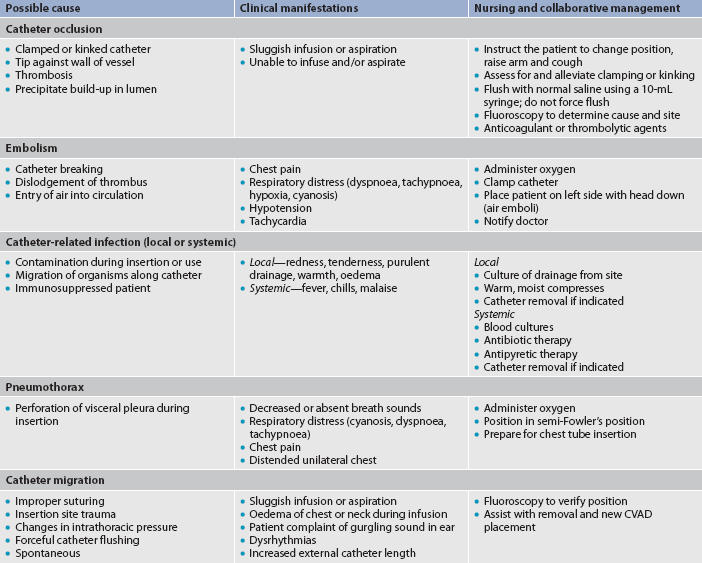
IMPLANTED INFUSION PORTS
Implanted infusion ports consist of a central venous catheter connected to an implanted single or double subcutaneous injection port (see Fig 16-20, A). The catheter is placed into the desired vein and the other end is connected to a port that is surgically implanted in a subcutaneous pocket on the chest wall. The port consists of a metal sheath with a self-sealing silicone septum. Drugs are injected through the skin into the port. After being filled, the reservoir slowly releases the medicine into the bloodstream.
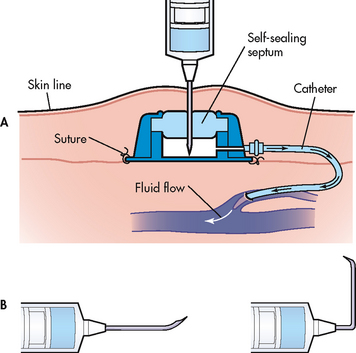
Figure 16-20 A, Cross-section of an implanted infusion port displaying access to the port with a Huber-point needle. Note the deflected point of the Huber-point needle, which prevents coring of the port’s septum. B, Two Huber-point needles used to enter the implanted point. The 90° needle is used for top-entry ports for continuous infusion.
The port is accessed via the septum by means of a special Huber-point needle that has a deflected tip, which prevents damage to the septum that could make the port useless (see Fig 16-20, B).15 Huber-point needles are also available with the tip at a 90° angle for longer infusions. Implanted ports are good for long-term therapy and have a low risk of infection. Because the port is hidden it offers cosmetic advantages.
Implanted ports have been developed that are safe for injections of radio-opaque contrast media at high pressures and controlled rates. For patients who already have poor peripheral venous access, the ability to use the port to inject contrast media decreases discomfort from venipuncture and helps lower the risk for extravasation of vesicant contrast media.17 Care requirements include regular flushing. Formation of ‘sludge’ (accumulation of clotted blood and drug precipitate) may also occur within the port septum.
COMPLICATIONS
The potential for complications associated with CVADs is always present. Astute monitoring and assessment may assist in early identification of potential complications. Table 16-20 lists common possible complications, potential causes, clinical manifestations and interventions.
NURSING MANAGEMENT: CENTRAL VENOUS ACCESS DEVICES
Nursing management of CVADs includes assessment, dressing change and cleansing, injection cap changes and flushing. Although there may be specific institution policies and procedures regarding different types of CVADs, there are some general guidelines to be followed too.
Catheter and insertion site assessment includes inspection of the site for redness, oedema, warmth, drainage and tenderness or pain. Observation of the catheter for misplacement or slippage is important. A comprehensive pain assessment should be performed, particularly noting any complaints of chest or neck discomfort, arm pain or pain at the insertion site.16
Dressing changes and cleansing of the catheter insertion site should be performed according to institution policies and procedures using strict sterile technique. Transparent semipermeable dressings or gauze and tape can be used. If the site is bleeding, a gauze dressing may be preferable; otherwise, transparent dressings have some advantages over gauze and tape. For example, they allow observation of the site without dressing removal and may be left in place for up to 1 week if clean, dry and intact. The dressing should be changed immediately if it becomes damp, loose or visibly soiled.16
The skin around the catheter insertion site should be cleansed according to institution policy. Usually chlorhexidine-based preparations, povidone-iodine or isopropyl alcohol are used. Chlorhexidine persists longer than either povidone-iodine or isopropyl alcohol, offering improved residual killing of bacteria. When using chlorhexidine, the skin should be cleansed using a back-and-forth scrubbing motion, rather than a circular motion, as it is the recommended technique: generating friction during skin preparation is a key to infection prevention.18,19 The area should be allowed to air dry completely before applying the new dressing. Chlorhexidine preparations dry very quickly; povidone-iodine requires a drying time of at least 2 minutes.16 The lumen ports should be secured to the skin above the dressing site. The date and time of the dressing change should be documented as per institution procedure.
Injection caps must be changed at regular intervals using strict sterile technique according to institution policy or when they are damaged from excessive punctures. The patient should be taught to turn the head to the opposite side of the CVAD insertion site during cap change. If the catheter cannot be clamped, the patient should lie flat in bed and perform the Valsalva manoeuvre whenever the catheter is open to air to prevent an air embolism.
Flushing is one of the most effective ways to maintain lumen patency and to prevent occlusion of the CVAD. It also keeps incompatible drugs or fluids from mixing. A normal saline solution in a syringe that has a barrel capacity of 10 mL or more should be used, to avoid excess pressure on the catheter. If resistance is felt, force should not be applied: this could result in a ruptured catheter or create an embolism if a thrombus is present. Because of the risk of contamination and infection, prefilled syringes or single-dose vials are preferred over multiple-dose vials. When flushing, the push–pause method is preferred over a continual even push of saline into the catheter. The push–pause technique creates turbulence within the catheter lumen, promoting the removal of debris that adheres to the lumen. The technique involves injecting the saline with a rapid alternating push–pause motion, instilling 1–2 mL with each push on the syringe plunger.
Removal of CVADs should be done according to institution policy and the nurse’s scope of practice. In many agencies, nurses with demonstrated competency can remove PICCs and non-tunnelled central venous catheters. The procedure involves removing the sutures, if present, and gently withdrawing the catheter while instructing the patient to perform the Valsalva manoeuvre as the last 5–10 cm of the catheter is withdrawn.15 Pressure should be applied to the site immediately with sterile gauze to prevent air from entering and to control bleeding. The catheter tip should be inspected to determine that it is intact. After haemostasis is achieved, an antiseptic ointment and sterile dressing should be applied to the site.
Fluid and electrolyte imbalance
CASE STUDY
Patient profile
Sarah Smith, a 73-year-old female with lung cancer, has been receiving chemotherapy on an outpatient basis. She completed her third treatment 5 days ago and has been experiencing nausea and vomiting for 2 days even though she has been using ondansetron orally as directed. Her daughter brings her to the hospital, where she is admitted to the medical unit. The admitting nurse performs a thorough assessment.
CRITICAL THINKING QUESTIONS
1. Based on her clinical manifestations, what fluid imbalance does this patient have?
2. What additional assessment data should the nurse obtain?
3. What are this patient’s risk factors for fluid and electrolyte imbalances?
4. The nurse takes take blood for a full blood count. What electrolyte imbalances are likely, and why?
5. This patient is at risk for which acid–base imbalance? Describe the changes that would occur in her ABGs with this acid–base imbalance. How would her body compensate?
6. The doctor orders dextrose 5% in 0.45% saline to infuse at 100 mL/h. What type of solution is this and how will it help this patient’s fluid imbalance?
7. What are the priority nursing interventions for this patient?
8. Based on the assessment data presented, this patient is at risk of developing the syndrome of inappropriate antidiuretic hormone. How would the nurse recognise this complication and what is the anticipated treatment?
9. Based on the assessment data presented, write one or more appropriate nursing diagnosis. Are there any collaborative problems?
1. During the postoperative care of a 76-year-old patient, the nurse monitors the patient’s intake and output carefully, knowing that the patient is at risk of fluid and electrolyte imbalances primarily because:
2. If a hypertonic solution is administered, the mechanism involved in equalising the fluid concentration is:
3. An elderly woman was admitted to the medical unit with dehydration. A clinical indication of this problem is:
4. Implementation of nursing care for the patient with hyponatraemia includes:
5. A patient is receiving a loop diuretic. The nurse should be alert for which symptoms?
6. Which patient would be at greatest risk of the potential development of hypermagnesaemia?
7. It is especially important for the nurse to assess for which clinical manifestation(s) in a patient who has just undergone a total thyroidectomy?
8. The nurse anticipates that the patient with hyperphosphataemia secondary to renal failure will require:
9. The lungs act as an acid–base buffer by:
10. A patient has the following arterial blood gas results: pH 7.52; PaCO2 30 mmHg (4 kPa); HCO3− 24 mmol/L. The nurse determines that these results indicate:
11. The typical fluid replacement for the patient with a fluid volume deficit is:
12. The nurse is unable to flush a central venous catheter device and suspects occlusion. The best nursing intervention would be to:
1 Mosby’s dictionary of medicine, nursing, and health professions. 8th edn. St Louis: Mosby; 2008.
2 Huether SE, McCance KL. Understanding pathophysiology, 4th edn. St Louis: Mosby, 2008.
3 Kee JL, Paulanka BJ, Polek C. Handbook of fluid, electrolyte and acid-base imbalances, 3rd edn. Clifton Park, NY: Delmar, 2009.
4 Porth CM, Matfin G. Pathophysiology: concepts of altered health states, 8th edn. Philadelphia: Lippincott, 2009.
5 Holcomb SS. Third-spacing: when body fluid shifts. Nursing. 2008;38:7.
6 Ebersole P, Hess P, Touhy TA, et al. Toward healthy aging, 7th edn. St Louis: Mosby, 2008.
7 Amella EJ. Feeding and hydration issues for older adults with dementia. Nurs Clin North Am. 2004;39:3.
8 Noble KA. Fluid and electrolyte imbalance: a bridge over troubled water. J Perianesth Nurs. 2008;23:4.
9 Scales K, Pilsworth J. The importance of fluid balance in clinical practice. Nurs Stand. 2008;22:47.
10 Wilson BA, Shannon MT, Shields KM. Pearson intravenous drug guide. Upper Saddle River, NJ: Pearson, 2009.
11 Haskal R. Current issues for nurse practitioners: hyponatraemia. J Am Acad Nurse Pract. 2007;19:11.
12 Fournier M. Perfecting your acid-base balancing act: how to detect and correct acid-base disorders. Am Nurse Today. 2009;4:1.
13 Phillips LD. Manual of IV therapeutics, 4th edn. Philadelphia: FA Davis, 2005.
14 Hankins J. The role of albumin in fluid balance. Nursing. 2007;37:12.
15 Ludeman K. Choosing the right vascular access device. Nursing. 2007;37:9.
16 Hadaway LC. Central venous access devices. Nursing. 2008;38:6.
17 Smith LH. Implanted ports, computed tomography, power injectors, and catheter rupture. Clin J Oncol Nurs. 2008;12:5.
18 Rosenthal K. CVAD site prep with pep. Nursing Made Incredibly Easy. 2007;5:6.
19 Weinstein SM. Plumer’s principles and practice of intravenous therapy, 8th edn. Philadelphia: Lippincott Williams & Wilkins, 2007.
