Chapter 49 NURSING MANAGEMENT: endocrine problems
1. Describe the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with an imbalance of hormones produced by the anterior pituitary gland.
2. Explain the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with an imbalance of hormones produced by the posterior pituitary gland.
3. Explore the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with thyroid dysfunction.
4. Examine the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with an imbalance of the hormone produced by the parathyroid glands.
5. Describe the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with an imbalance of hormones produced by the adrenal cortex.
6. Describe the pathophysiology, clinical manifestations, multidisciplinary care and nursing management of the patient with an excess of hormones produced by the adrenal medulla.
7. Evaluate the side effects of corticosteroid therapy.
8. Utilise common nursing assessments, interventions, rationales and expected outcomes for patient teaching in the management of chronic endocrine problems.
Growth hormone excess
AETIOLOGY AND PATHOPHYSIOLOGY
Growth hormone (GH), an anabolic hormone, promotes protein synthesis and mobilises glucose and free fatty acids. GH is produced by the anterior pituitary and stimulates the liver to produce insulin-like growth factor-1 (IGF-1), also known as somatomedin C. IGF-1 stimulates the growth of bones and soft tissues. Normally, IGF-1 signals to the anterior pituitary to reduce GH production. Overproduction of GH is almost always caused by a benign pituitary tumour (adenoma). The pituitary tumour secretes GH despite elevated IGF-1 levels, leading to the unwanted growth of bones and other soft tissues. Overproduction of GH also causes elevation of blood glucose levels through insulin antagonism. Prolonged elevated blood glucose levels associated with an elevation in GH leads to glucose intolerance.
In children, excessive secretion of GH results in gigantism. When the onset of GH excess occurs before closure of the epiphyses, the long bones are still capable of longitudinal growth. The excessive growth is usually proportional. These children may grow as tall as 240 cm and weigh more than 136 kg. In adults, excessive secretion of GH results in acromegaly. Acromegaly is characterised by an overgrowth of the bones and soft tissues. Because the problem develops after epiphyseal closure in adults, the bones are unable to grow longer. Instead, they increase in thickness and width.
The incidence in New Zealand (less than 200 people) and Australia (approximately 1000 people) is extrapolated from US data, where about 3 adults per million are diagnosed with this condition each year, an estimated prevalence of 40–60 per million people.1 Acromegaly is most common in people in their forties and fifties, is equally common in men and women, and is found in all races of people. Although this is a rare condition, the effects are profound and treatment is necessary.
CLINICAL MANIFESTATIONS
Manifestations of acromegaly begin gradually, usually in the third and fourth decades of life. Typically there is an average of 7–9 years between the initial onset of symptoms and final diagnosis. Individuals experience enlargement of the hands and feet. The fingertips develop a tufted or clubbed-like appearance. The enlargement of the bones and cartilage may cause symptoms that range from mild joint pain to deforming, crippling arthritis. Changes in physical appearance occur with thickening and enlargement of bony and soft tissues on the face and head (see Fig 49-1). Enlargement of the mandible causes the jaw to jut forwards. The paranasal and frontal sinuses enlarge, as does the bony tissue of the forehead. Enlargement of soft tissue around the eyes, nose and mouth results in a coarsening of facial features. Enlargement of the tongue results in speech difficulties, and the voice deepens as a result of hypertrophy of the vocal cords.

Sleep apnoea may also occur and is related to upper airway narrowing and obstruction resulting from increased amounts of pharyngeal soft tissues.2 The skin becomes thick, leathery and oily. People with acromegaly may also experience peripheral neuropathy and proximal muscle weakness. Women may develop menstrual disturbances. Individuals with acromegaly are more likely to develop polyps in the colon and colon cancer.
The enlarged pituitary tumour can exert pressure on surrounding structures within the brain, leading to visual disturbances and headaches. Because GH mobilises stored fat for energy, it increases free fatty acid levels in the blood and predisposes the patient to atherosclerosis. The hormone also antagonises the action of insulin and causes hyperglycaemia. Manifestations of diabetes mellitus may occur, including polydipsia and polyuria. Prolonged secretion of GH leads to glucose intolerance.
Acromegaly, when left untreated, can lead to a number of changes in the body. Effects on the cardiovascular system include cardiomegaly, left ventricular hypertrophy, angina pectoris and hypertension. For this reason, disease of the cardiovascular system is associated with increased mortality rates in these individuals. Other systems that undergo changes include the respiratory, gastrointestinal, genitourinary, musculoskeletal and nervous systems.
DIAGNOSTIC STUDIES
In addition to the history and physical examination, a diagnosis of acromegaly requires evaluation of plasma IGF-1 levels, and GH response to an oral glucose challenge. A single measurement of serum GH is of limited value in the diagnosis of acromegaly because GH levels normally fluctuate and can change minute by minute. IGF-1 levels are more constant and thus provide a more reliable measure than GH levels. The definitive test for acromegaly is the oral glucose challenge test. Normally, GH concentration falls during an oral glucose tolerance test. In acromegaly, these levels do not fall below 1 μg/L.3
Magnetic resonance imaging (MRI) is indicated for identifying and determining the extent of spread of the pituitary tumour into surrounding tissue. High-resolution computed tomography (CT) scanning with contrast media may also be used to localise the tumour. A complete ophthalmological examination, including visual fields, is typically done because the tumour (especially a macroadenoma >10 mm) may cause pressure on the optic chiasma or optic nerves.
MULTIDISCIPLINARY CARE
The therapeutic goal in acromegaly is to return the patient’s GH levels to normal. This is accomplished by surgery, radiation or drug therapy, or a combination of these therapies. The biochemical targets for treatment are a GH of <2.5 ng/mL and a normal, age-adjusted IGF-1.2 The prognosis depends on age at onset, age when treatment is initiated and tumour size. Individuals with large pituitary tumours invading the dura, bone or cavernous sinus (80% of patients diagnosed with acromegaly) require multiple therapies. Usually bone growth can be arrested and soft-tissue hypertrophy can be reversed. However, sleep apnoea and diabetic and cardiac complications may persist in spite of treatment.
Surgical therapy
Surgery (hypophysectomy) is the treatment of choice as it offers the best hope for a cure, especially for smaller tumours (microadenomas <10 mm). The majority of surgery done to remove pituitary tumours associated with acromegaly is accomplished with the trans-sphenoidal approach.4 The goal of trans-sphenoidal surgery is to remove only the tumour that is causing GH secretion (see Fig 49-2). This procedure produces an immediate reduction in GH levels followed by a drop in IGF-1 levels within a few weeks.
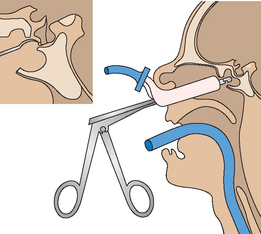
Figure 49-2 Surgery on the pituitary gland is most commonly performed using the trans-sphenoidal approach. An incision is made in the inner aspect of the upper lip and gingiva. The sella turcica is entered through the floor of the nose and sphenoid sinuses.
Although most of these procedures are effective, some patients (especially those with larger tumours or those with GH levels >45 μg/L) do not obtain a cure with the surgery and require adjuvant radiation or drug therapy.5 In some cases, the entire pituitary gland is removed during surgery (hypophysectomy), resulting in a permanent absence of pituitary hormones. Rather than replacing the pituitary (trophic) hormones, which requires parenteral administration, the essential hormones produced by target organs (glucocorticoids, thyroid hormone and sex hormones) can be given orally. Hormone replacement must then be continued throughout life.
Radiation therapy
Radiation therapy is considered when surgery has failed to produce complete remission. External radiation can successfully reduce GH levels in 30–70% of patients, but the primary disadvantage is the long delay (5–10 years) for GH levels to normalise. Because of the length of time it takes to achieve GH reduction, radiation therapy is usually offered in combination with drugs that reduce GH levels. Radiation has also been used to reduce the size of a tumour before surgery. The patient may experience local skin changes, alopecia or oral complications. Hypopituitarism commonly results from radiation therapy and requires hormone replacement therapy.
Stereotactic radiosurgery (gamma surgery, proton beam, linear accelerator [LINAC]) may be used for small, surgically inaccessible pituitary tumours or in place of conventional radiation. This procedure consists of a single dose of radiation delivered to one site from multiple angles. It is used to occlude blood vessels supplying the tumour and results in cell death. Focused radiotherapy may lead to earlier GH reduction than conventional radiotherapy.5
Drug therapy
Three types of drugs are used in the treatment of acromegaly: somatostatin analogues, GH receptor antagonists and dopamine agonists. These drugs reduce GH levels and can be used as initial treatment or as adjunct therapy to surgery or radiation.
The drug most commonly used for acromegaly is octreotide, a somatostatin analogue that reduces GH levels to within the normal range in many patients. Octreotide and lanreotide, which are both long-acting analogues, are given as intramuscular (IM) injection every 2–4 weeks. Octreotide can also be given by a subcutaneous injection 3 times a week. Somatostatin analogues achieve biochemical control in approximately 60% of patients.
Pegvisomant (not currently funded in New Zealand) is a GH receptor antagonist and is considered an alternative to somatostatin analogues. This drug is used for patients who have received surgery or radiation therapy but still have hypersecretion of GH. It is not considered appropriate for primary treatment because it only blocks hormone action and has no effect on tumour mass.
Dopamine agonists (bromocriptine or cabergoline) may also be used in the treatment of acromegaly to suppress GH secretion. Dopamine agonists may be tried first as they are less expensive than the other two classes of drugs, but they are often not effective.5
Somatropin, a recombinant human GH product, is now available for long-term replacement therapy in adults with GH deficiency, given daily as a subcutaneous (SC) injection (preferably in the evening). It is funded in New Zealand for adults who meet criteria as per the New Zealand Adult Growth Hormone Committee. Gradual dosage increases occur at 4–8 week intervals up to a maximum of 0.08 mg/kg/week, based on patient tolerance. Mild-to-moderate side effects include fluid retention and myalgia.
 NURSING MANAGEMENT: GROWTH HORMONE EXCESS
NURSING MANAGEMENT: GROWTH HORMONE EXCESS
Nursing assessment
The nurse needs to assess for signs and symptoms of abnormal tissue growth and evaluate changes in the physical size of each patient. The adult should be questioned about increases in hat, ring, glove and shoe sizes. Photographs are helpful to evaluate any changes. Because physical changes occur slowly and over a long period of time, it is possible that the individual is not even aware of such changes.
Nursing diagnoses
Nursing diagnoses for the patient with GH excess include, but are not limited to, the following:
Planning
The overall goals are that the patient with GH excess will: (1) cope effectively with altered body image; (2) maintain adequate fluid volume; (3) have restful sleep patterns; (4) develop no complications; and (5) obtain long-term follow-up care.
Nursing implementation
Acute intervention
Patients typically have many questions and concerns about surgery. It is important for the nurse to offer reassurance and to provide accurate information regarding this process. The individual treated surgically will need skilled neurosurgical nursing care and must be prepared before surgery for postoperative care. The patient should be instructed to avoid vigorous coughing, sneezing and straining at stool (Valsalva manoeuvre) to prevent cerebrospinal fluid (CSF) leakage from the point at which the sella turcica was entered.
After surgery in which a trans-sphenoidal approach has been used, the head of the patient’s bed should be elevated at a 30° angle at all times. This elevation avoids pressure on the sella turcica and decreases headaches, a frequent postoperative problem. Monitoring neurological status, including pupillary response, should be done in order to detect neurological complications.
Any clear nasal drainage should be sent to the laboratory to be tested for glucose. A glucose level greater than 1.67 mmol/L indicates CSF leakage from an open connection to the brain. If this happens, the patient is at an increased risk of meningitis. Complaints of persistent and severe generalised or supraorbital headache may indicate CSF leakage into the sinuses. A CSF leak usually resolves within 72 hours when treated with head elevation and bed rest. If the leak persists, daily spinal taps may reduce pressure to below-normal levels and allow the fossa to heal. Intravenous (IV) antibiotics are usually administered when there is a CSF leak to prevent meningitis. If the leak does not respond to treatment in 72 hours, surgical intervention may be required.
Mild analgesia is given for headaches. The nurse should perform mouth care every 4 hours to keep the surgical area clean and free of debris and to promote patient comfort. Tooth brushing should be avoided for at least 10 days to prevent disrupting the suture line and to avoid discomfort.
If stereotactic radiosurgery is used, the patient is usually moved from the specialised radiation centre to the neurosurgical nursing unit for overnight observation. The patient will be in a stereotactic head frame. Vital signs, neurological status and fluid volume status must be monitored carefully. Possible complications include increased headaches, seizures, nausea and vomiting. The patient with a history of seizures is at increased risk of seizures for at least 24 hours after the procedure. All staff should be instructed in removing a stereotactic frame in case of an emergency. The patient may experience discomfort at the pin sites. Pin-site care should be done according to institutional policy. Family members can be instructed in pin-site care if the patient is discharged the day after the procedure.
A possible postoperative complication is transient diabetes insipidus (DI). This may occur because of the loss of antidiuretic hormone (ADH), which is stored in the posterior lobe of the pituitary gland, or cerebral oedema related to manipulation of the pituitary during surgery. To assess for DI, urine output and serum and urine osmolarity must be monitored closely. Clinical manifestations and treatment of DI are discussed in more detail on p 1402.
Ambulatory and home care
If a hypophysectomy is performed or the pituitary is damaged, hormone replacement will be necessary. ADH, cortisol and thyroid hormone replacement may be needed. Because these medications need to be taken for life, careful patient teaching is essential when replacement of these hormones is necessary.
Surgery may result in permanent hormone deficiencies and possible decreased fertility, so the patient needs assistance in working through the grieving process associated with these losses. The need for continued drug therapy reduces the patient’s perception of independence and requires considerable emotional adjustment. The nurse must consider the emotional impact of a hypophysectomy when counselling the patient and planning the educational program related to hormone replacement. Serial photographs to show improvement may be helpful. Psychological support from the nurse and family and friends are needed to promote positive mental health outcomes for individuals with acromegaly. The teaching plan includes self-administration of SC injection if prescribed.
Excesses of other trophic hormones
An excess of trophic hormones and the overproduction of a single anterior pituitary hormone usually produce a syndrome related to hormone excess from the target organ. For example, if adrenocorticotrophic hormone (ACTH) is increased, Cushing’s disease results; if thyroid-stimulating hormone (TSH) levels are excessive, hyperthyroidism develops.
Prolactinomas (prolactin-secreting adenomas) are the most frequently occurring pituitary tumour. Common manifestations experienced by women with prolactinomas include galactorrhoea, ovulatory dysfunction (anovulation, infertility), menstrual dysfunction (oligomenorrhoea or amenorrhoea), decreased libido and hirsutism. In men, impotence and decreased libido and sperm density may result. The affected patient may also experience headaches and visual problems. The visual problems are secondary to pressure on the optic chiasm. Because prolactinomas do not typically progress in size, drug therapy is usually the first-line treatment. Dopamine agonists such as bromocriptine, cabergoline or pergolide have successfully been used to treat this disorder. Surgery using the trans-sphenoidal approach (discussed previously) may be considered depending on the size and extent of the tumour. The use of radiation for treatment of prolactinomas has been somewhat limited. It is mainly used in those patients who have failed to respond to medical or surgical therapy.
Hypofunction of the pituitary gland
Hypopituitarism is a rare disorder that involves a decrease in one or more of the pituitary hormones. The anterior pituitary gland secretes ACTH, TSH, follicle-stimulating hormone (FSH), luteinising hormone (LH), GH and prolactin; the posterior pituitary gland secretes ADH and oxytocin. A deficiency of only one pituitary hormone is referred to as selective hypopituitarism. Total failure of the pituitary gland results in deficiency of all pituitary hormones, a condition referred to as panhypopituitarism. The most common hormone deficiencies associated with hypopituitarism involve gonadotrophins (e.g. LH, FSH), GH and ACTH.
AETIOLOGY AND PATHOPHYSIOLOGY
The most common cause of pituitary hypofunction is a pituitary tumour. Autoimmune disorders, infections, pituitary infarction (Sheehan’s syndrome) or destruction of the pituitary gland (as a result of trauma, radiation and surgical procedures) can also cause hypopituitarism. Sheehan’s syndrome is a postpartum condition of pituitary necrosis and hypopituitarism that occurs after circulatory collapse from uterine haemorrhaging.
Hormone deficiencies involving anterior pituitary hormones lead to end-organ failure; thus, the effects of hypopituitarism depend on the specific pituitary hormone or hormones that are lacking. For example, infertility may be the first indication of pituitary hypofunction associated with a pituitary tumour. Deficiencies of TSH and ACTH are life-threatening. ACTH deficiency causes a tendency towards shock and may result in an episode of acute adrenal insufficiency (refractory and life-threatening shock from sodium and water depletion). (Adrenal shock, also known as addisonian crisis, is discussed on p 1424.)
CLINICAL MANIFESTATIONS
The signs and symptoms associated with pituitary hypofunction vary with the degree and speed of onset of pituitary dysfunction and are related to hyposecretion of the target glands and/or a growing pituitary tumour. Common symptoms associated with a space-occupying lesion include headaches, visual changes (decreased peripheral vision or decreased visual acuity), anosmia (loss of the sense of smell) and seizures.
Adults with GH deficiency often have subtle non-specific clinical findings. They have truncal obesity and decreased muscle mass causing reduced strength, decreased energy and reduced exercise capability. They may have a flat affect or appear depressed. Impaired psychological wellbeing is a common finding associated with GH deficiency in adults.
FSH and LH deficiencies in the adult woman are first manifested as menstrual irregularities, diminished libido and changes in secondary sex characteristics (e.g. decreased breast size). Men with FSH and LH deficiencies experience testicular atrophy, diminished spermatogenesis, loss of libido, impotence, and decreased facial hair and muscle mass.
A deficiency of ACTH and cortisol often produces a non-specific clinical picture. Signs and symptoms may include weakness, fatigue, headaches, dry and pale skin, and diminished axillary and pubic hair. Individuals may have postural hypotension, fasting hypoglycaemia, diminished tolerance for stress and poor resistance to infection.
The clinical presentation of an individual with thyroid hormone deficiency associated with hypopituitarism is similar (although usually milder) to what is seen with primary hypothyroidism. Common symptoms include cold intolerance, constipation, fatigue, lethargy and weight gain. (Hypothyroidism is discussed in greater detail on p 1412.)
DIAGNOSTIC STUDIES
In addition to conducting a history and physical examination, diagnostic studies are useful in the diagnosis and treatment of hypopituitarism. Radiological tests such as MRI and CT are used to determine the presence of a pituitary tumour. The laboratory tests for hypopituitarism vary widely but generally involve the direct measurement of pituitary hormones or an indirect determination of the hormone level. Diagnostic tests are also used to evaluate the effectiveness of therapy. See Chapter 47 for more information about diagnostic studies.
MULTIDISCIPLINARY CARE
The treatment for hypopituitarism consists of surgery or radiation for tumour removal, followed by lifelong hormone replacement therapy. Surgery and radiation of pituitary tumours were discussed earlier in this chapter. Hormone replacement therapy is carried out with the appropriate hormone needed (e.g. GH, corticosteroids, thyroid hormone, sex hormones). Hormone replacement therapies for thyroid hormone and corticosteroids are discussed on pages 1413 and 1425, respectively.
Somatrophin is used for GH replacement therapy. Adults with GH deficiency respond well to GH replacement and experience increased energy, increased lean body mass, a feeling of wellbeing and improved body image. The side effects most commonly reported by adults include swelling in the feet and hands, pain in the joints and headache. Somatrophin is given as an SC injection daily or one to two times per month in adults.6 The dose is variable because it is adjusted based on relief of symptoms, IGF-1 levels and the development of adverse effects.
Although gonadal deficiency is not life-threatening, replacement therapy is offered to improve sexual function and general wellbeing. Replacement therapy is contraindicated in individuals with certain medical conditions, such as breast cancer, phlebitis and pulmonary embolism in women, and prostate cancer in men. Oestrogen and progesterone replacement therapy may be indicated for hypogonadal women to treat hot flushes, vaginal dryness and decreased libido. Hormone replacement for women is discussed in greater detail in Chapter 53. Testosterone is used to treat men with gonadotrophin deficiency. The benefits achieved with testosterone therapy include a return of male secondary sex characteristics, improvement in libido and increased muscle mass, bone mass and bone density. Hormone replacement for men is discussed in greater detail in Chapter 54.
NURSING MANAGEMENT: HYPOFUNCTION OF THE PITUITARY GLAND
A primary nursing role in anterior pituitary insufficiency is assessment and recognition of the signs and symptoms associated with hypopituitarism. Nursing management is directed at providing interventions associated with problems that result from hormone deficiency. The nurse also plays a pivotal role in teaching the patient about diagnostic procedures, the disease process and multidisciplinary care options. Because of the need for lifelong hormonal therapy, patient teaching about hormonal administration, side effects and follow-up therapy is important.
DISORDERS ASSOCIATED WITH ANTIDIURETIC HORMONE SECRETION
The two primary conditions associated with ADH secretion result from either overproduction or underproduction of ADH. Overproduction or oversecretion of ADH results in a condition known as syndrome of inappropriate antidiuretic hormone (SIADH), while underproduction or undersecretion of ADH results in a condition referred to as diabetes insipidus (DI). ADH, also referred to as arginine vasopressin (AVP), is synthesised in the hypothalamus and then transported and stored in the posterior pituitary gland. It plays a major role in the regulation of water balance and osmolarity.
Syndrome of inappropriate antidiuretic hormone
AETIOLOGY AND PATHOPHYSIOLOGY
Syndrome of inappropriate antidiuretic hormone occurs when ADH is released despite normal or low plasma osmolarity (see Fig 49-3). SIADH results from an abnormal production or sustained secretion of ADH and is characterised by fluid retention, serum hypoosmolality, dilutional hyponatraemia, hypochloraemia, concentrated urine in the presence of normal or increased intravascular volume, and normal renal function. This syndrome occurs more commonly in older adults. SIADH has various causes (see Box 49-1). The most common causes are malignancy (especially small cell lung cancer), stroke, meningitis and encephalitis. Small cell lung cancerous cells are capable of producing, storing and releasing ADH.7 SIADH tends to be self-limiting when caused by head trauma or drugs but it is chronic in nature when associated with tumours or metabolic diseases.
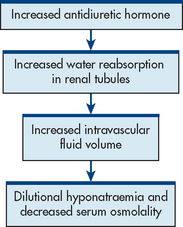
CLINICAL MANIFESTATIONS
Excess ADH increases the permeability of the distal tubule and collecting duct, which leads to the reabsorption of water into the circulation. Consequently, extracellular fluid volume expands, plasma osmolality declines, the glomerular filtration rate increases and sodium levels decline (dilutional hyponatraemia). Hyponatraemia causes muscle cramps and weakness. Initially, thirst, dyspnoea on exertion, fatigue and dulled sensorium may be evident. The patient with SIADH will experience low urinary output and increased body weight.8 As the serum sodium level falls (usually <120 mmol/L), manifestations become more severe and include vomiting, abdominal cramps, muscle twitching and seizures. As plasma osmolality and serum sodium levels continue to decline, cerebral oedema may occur, leading to lethargy, anorexia, confusion, headache, seizures and coma.
DIAGNOSTIC STUDIES
The diagnosis of SIADH is made by simultaneous measurements of urine and serum osmolality. The dilutional hyponatraemia is indicated by a serum sodium level less than 134 mmol/L, serum osmolality less than 280 mOsm/kg (280 mmol/kg) and a urine specific gravity greater than 1.005. A serum osmolality much lower than the urine osmolality indicates the inappropriate excretion of concentrated urine in the presence of dilute serum.
MULTIDISCIPLINARY CARE
Once SIADH is diagnosed, treatment is directed at the underlying cause. Medications that stimulate the release of ADH should be avoided or discontinued (see Box 49-1). The immediate treatment goal is to restore normal fluid volume and osmolality. If symptoms are mild and the serum sodium level is greater than 125 mmol/L, the only treatment may be restriction of fluids to 500–1000 mL per day. This restriction should result in gradual, daily reductions in weight, a progressive rise in serum sodium concentration and osmolality, and symptomatic improvement.
In cases of severe hyponatraemia (<120 mmol/L), especially in the presence of neurological symptoms such as seizures, IV hypertonic saline solution (3–5%) may be administered. Hypertonic saline requires a very slow infusion rate on an infusion pump to avoid too rapid a rise in sodium. A diuretic such as frusemide may be used to promote diuresis, but only if the serum sodium level is at least 125 mmol/L, as it may promote further loss of sodium. Because frusemide increases potassium, calcium and magnesium losses, supplements may be needed. A fluid restriction of 490 mL per day is also indicated for those with severe hyponatraemia.
In chronic SIADH, water restriction of 800–1000 mL per day is recommended. Because this degree of restriction may not be tolerated, demeclocycline and lithium may be administered. These agents block the effect of ADH on the renal tubules, thereby allowing a more dilute urine.
NURSING MANAGEMENT: SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE
An appropriate nursing assessment (see Box 49-2) should be conducted for those at risk and those who have confirmed SIADH. Specifically, the nurse should be alert for low urinary output with a high specific gravity, a sudden weight gain without oedema or a serum sodium level decline. Nursing management of acute onset of SIADH is also presented in Box 49-2.
BOX 49-2 Syndrome of inappropriate antidiuretic hormone
NURSING ASSESSMENT AND MANAGEMENT
Assessment
Management
• Restrict total fluid intake to no more than 1000 mL/day (including that taken with medications)
• Position head of bed flat or with no more than 10° of elevation to enhance venous return to heart and increase left atrial filling pressure, reducing ADH release
• Protect from injury (i.e. assist with ambulation, bed alarm) because of potential alterations in mental status
• Ensure frequent turning, positioning and range-of-motion exercise (if patient is bedridden)
• Follow frequent oral hygiene
• Provide distractions to decrease the discomfort of thirst related to fluid restrictions
• Provide support for the patient and significant others about diagnosis and any mental status changes
When SIADH is chronic, the patient must learn to self-manage their treatment. Fluids are restricted to 800–1000 mL per day. Ice chips or sugarless chewing gum can help decrease thirst. If drinking liquids is an aspect of socialisation, the patient should be assisted in planning fluid intake so liquid allowances are saved for social occasions. The patient may be treated with a diuretic to remove excess fluid volume. The diet should be supplemented with sodium and potassium, especially if diuretics are prescribed. Solutions of these electrolytes must be well diluted to prevent gastrointestinal (GI) irritation or damage. They are best taken at mealtimes to allow mixing with and dilution by food. The patient should be taught the symptoms of fluid and electrolyte imbalances, especially those involving sodium and potassium, so that responses to treatment can be monitored (see Ch 16).
Diabetes insipidus
AETIOLOGY AND PATHOPHYSIOLOGY
Diabetes insipidus is associated with a deficiency of production or secretion of ADH or a decreased renal response to ADH. The decrease in ADH results in fluid and electrolyte imbalances that are caused by increased urinary output and increased plasma osmolality (see Fig 49-4). Depending on the cause, DI may be transient or a chronic lifelong condition.
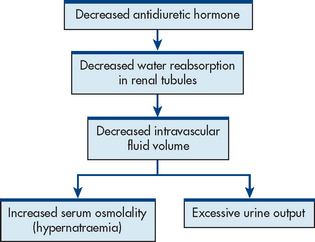
There are several classifications of DI (see Table 49-1).
• Central DI (also known as neurogenic DI) occurs when any organic lesion of the hypothalamus, infundibular stem or posterior pituitary interferes with ADH synthesis, transport or release.
• Nephrogenic DI describes a condition in which there is adequate ADH, but there is a decreased response to ADH in the kidneys. Lithium is one of the most common causes of drug-induced nephrogenic DI. Hypokalaemia and hypercalcaemia may also lead to nephrogenic DI.
• Psychogenic DI, a less common condition, is associated with excessive water intake. This can be caused by a structural lesion in the thirst centre or may be caused by psychiatric problems.
TABLE 49-1 Types and causes of diabetes insipidus
| Types | Causes |
|---|---|
| Central DI (neurogenic) | Problem results from an interference with ADH synthesis or release. Multiple causes include brain tumour, head injury, brain surgery and Cns infections. |
| Nephrogenic DI | Problem results from inadequate renal response to ADH despite presence of adequate ADH. Caused by drug therapy (especially lithium), renal damage or hereditary renal disease. |
| Psychogenic DI | Problem results from excessive water intake. Caused by structural lesion in thirst centre or psychological disorder. |
ADH, antidiuretic hormone; CNS, central nervous system.
CLINICAL MANIFESTATIONS
DI is characterised by increased thirst (polydipsia) and increased urination (polyuria) (see Fig 49-4). The primary characteristic of DI is the excretion of large quantities of urine with osmolality of less than 100 mOsm/kg. Serum osmolality is elevated (usually >295 mOsm/kg) as a result of hypernatraemia due to pure water loss in the kidneys. Most patients compensate for fluid loss by drinking great amounts of water so that serum osmolality is normal or only moderately elevated. The patient may be exhausted from nocturia and may experience generalised weakness.
Central DI usually occurs suddenly with excessive fluid loss. After intracranial surgery, DI usually has a triphasic pattern: the acute phase with abrupt onset of polyuria; an interphase, where urine volume apparently normalises; and a third phase, where central DI is permanent. The third phase is usually apparent within 10–14 days postoperatively. Central DI that results from head trauma is usually self-limiting and improves with treatment of the underlying problem. DI following cranial surgery is more likely to be permanent. Although the clinical manifestations of nephrogenic DI are similar, the onset and amount of fluid losses are less dramatic than with central DI.
If oral fluid intake cannot keep up with urinary losses, severe fluid volume deficit results. This deficit is manifested by weight loss, constipation, poor tissue turgor, hypotension, tachycardia and shock. In addition, the patient shows central nervous system (CNS) manifestations, ranging from irritability and mental dullness to coma. These manifestations are related to increasing serum osmolality and hypernatraemia. Because of the polyuria, severe dehydration and hypovolaemic shock may occur.
DIAGNOSTIC STUDIES
Because DI may be central, nephrogenic or psychogenic in origin, identification of the cause is the initial step. A complete history and physical examination is undertaken. Psychogenic DI is associated with overhydration and hypervolaemia rather than with dehydration and hypovolaemia, which are seen in other forms of DI. A water deprivation test is usually carried out to confirm the diagnosis of central DI. Before the test is done, the patient’s baseline weight, pulse, urine and plasma osmolalities, and blood pressure (BP) are obtained. All fluids are withheld for 8–16 hours. The patient may be anxious and should be reassured that the test will be stopped if fluid volume deficit becomes severe. The patient should be observed throughout the test because of the craving to drink. During the test, the patient’s BP, weight and urine osmolality are assessed hourly. The test continues until urine osmolalities stabilise (hourly increase <30 mOsm/kg in 3 consecutive hours) or body weight declines by 3% or orthostatic hypotension develops. ADH is then given, and urine osmolality is measured 1 and 2 hours later. In central DI, the rise in urinary osmolality after vasopressin exceeds 9%. Individuals with nephrogenic DI will have no response.9
MULTIDISCIPLINARY CARE
Determining and treating the primary cause is central to the collaborative management of DI. The therapeutic goal is maintenance of fluid and electrolyte balance.
For central DI, fluid and hormonal replacement is the cornerstone of treatment. In acute DI, hypotonic saline or dextrose 5% in water is administered intravenously and titrated to replace urinary output. Hormone replacement is necessary because of the lack of ADH production or secretion. Desmopressin acetate (DDAVP), an analogue of ADH, is the hormone replacement of choice for central DI. DDAVP can be administered orally, intravenously or as a nasal spray. Several other drugs are available for ADH replacement, including aqueous vasopressin, vasopressin tannate and lysine vasopressin. Several drugs can be used for the treatment of partial central DI, including carbamazepine. Hormone replacement has little effect in the treatment of nephrogenic DI because the kidneys are unable to respond to ADH. Instead, the treatment revolves around dietary measures (low-sodium diet) and thiazide diuretics. Limiting sodium intake to no more than 3 g per day is thought to help decrease urine output. Thiazide diuretics (e.g. hydrochlorothiazide, chlorothiazide) are able to slow the glomerular filtration rate and allow the kidneys to reabsorb more water in the loop of Henle and distal tubules. When a low-sodium diet and thiazide drug use are not effective, indomethacin may be prescribed. Indomethacin, a non-steroidal anti-inflammatory agent, helps increase renal responsiveness to ADH.
NURSING MANAGEMENT: DIABETES INSIPIDUS
Nursing management of the patient with DI includes early detection, maintenance of adequate hydration and patient teaching for long-term management.
During acute central DI, the nurse administers fluids and hormone replacement. Fluids are replaced orally or intravenously, depending on the patient’s condition and ability to drink copious amounts of fluids. Adequate fluids should be kept at the bedside. If IV glucose solutions are used, the serum glucose level should be monitored, as hyperglycaemia and glycosuria can lead to osmotic diuresis, which increases the fluid volume deficit. Accurate records of intake and output, urine specific gravity and daily weights are mandatory in the assessment of fluid volume status.
Nursing interventions also include the administration of DDAVP. The patient should be assessed for weight gain, headaches, restlessness, and signs of hyponatraemia and water intoxication. The adequacy of treatment is assessed by monitoring fluid intake and output. The healthcare provider should be notified immediately if the patient develops increased urine volume, as this indicates the need for increased dosing of DDAVP.
The patient with chronic DI who requires long-term ADH replacement needs instruction in self-management. DDAVP can be taken orally or intranasally. Nasal irritation may occur due to nasal administration. Headaches, nausea and other signs of hyponatraemia may indicate overdosage. Failure to improve may indicate underdosage. The patient should be instructed to report any of these symptoms. Increases in weight may indicate fluid retention. The need for close follow-up, including laboratory studies, is an essential part of the teaching plan.
DISORDERS OF THE THYROID GLAND
The thyroid hormones, thyroxine (T4) and triiodothyronine (T3), regulate energy metabolism and growth and development. Disorders of the thyroid gland include enlargement, benign and malignant nodules, inflammation, and hyper- and hypo-functioning (see Fig 49-5).
Thyroid enlargement
Goitre is hypertrophy and enlargement of the thyroid gland due to excess TSH stimulation, which in turn can be caused by inadequate circulating thyroid hormones. Goitre may also be caused by growth-stimulating immunoglobulins and other growth factors. Goitrogens (foods or drugs that contain thyroid-inhibiting substances) can cause goitre (see Box 49-3) but usually only in individuals who live in iodine-deficient areas (endemic goitre). A goitre is also commonly found in patients with Graves’ disease (see Fig 49-6).

TSH and T4 levels are measured to determine whether goitre is associated with hyperthyroidism, hypothyroidism or normal thyroid function. Thyroid antibodies are measured to assess for thyroiditis. Treatment with thyroid hormone may prevent further thyroid enlargement. Surgery to remove large goitres may be necessary.
Thyroid nodules
Thyroid nodules are common clinically (prevalence about 5%). They result in a palpable deformity of the thyroid gland and may be benign or malignant. Benign nodules are usually not dangerous but they can cause tracheal compression if they become too large. About 5% of thyroid nodules are malignant.10 The major sign of thyroid cancer is the presence of a hard, painless nodule or nodules on an enlarged thyroid gland.
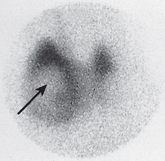
Nodular enlargement of the thyroid gland or palpation of a mass usually requires radiological evaluation. Ultrasound is often the first radiological test used in the diagnostic examination of a thyroid nodule. CT, MRI and ultrasound-guided fine-needle aspiration (FNA) are other diagnostic options. FNA is indicated when a tissue sample for pathological examination is necessary. FNA is considered one of the most effective methods to identify malignancy.11 A thyroid scan may also be undertaken to evaluate for possible malignancy. The scan shows whether nodules on the thyroid are ‘hot’ or ‘cold’. Thyroid tumours may or may not take up radioactive iodine. Tumours that take up the radioactive iodine are called ‘hot’ nodules and are nearly always benign. If the nodule does not take up the radioactive iodine, it appears as ‘cold’ and has a higher risk of being malignant (see Fig 49-7). An increase in the level of serum calcitonin may also be helpful in diagnosis because increased levels are associated with a certain type (medullary) of thyroid cancer.
Surgical removal of the tumour is usually indicated in the treatment of thyroid cancer. Surgical procedures may range from unilateral total lobectomy with removal of the isthmus to total thyroidectomy with bilateral lobectomy. Many thyroid cancers are TSH dependent, and thyroid hormone in hyperphysiological doses is often prescribed to inhibit pituitary secretion of TSH. Radiation therapy may be indicated to prolong survival.
Nursing care for the patient with thyroid tumours is similar to care for the patient who has undergone thyroidectomy and also includes general nursing measures for the patient with cancer (see Ch 15).
Thyroiditis
Thyroiditis is an inflammatory process in the thyroid that can have several causes. Subacute granulomatous thyroiditis (de Quervain’s thyroiditis), which causes thyrotoxicosis, is thought to be caused by a viral infection. Acute thyroiditis is due to bacterial or fungal infection. Subacute and acute forms of thyroiditis have an abrupt onset and the thyroid gland is painful. Chronic autoimmune thyroiditis (Hashimoto’s thyroiditis) can lead to hypothyroidism. Hashimoto’s thyroiditis is a chronic autoimmune disease in which thyroid tissue is replaced by lymphocytes and fibrous tissue. It a common cause of goitrous hypothyroidism. Silent painless thyroiditis is a form of lymphocytic thyroiditis with a variable onset. In women, this condition may occur in the postpartum period and usually resolves within 3–12 months. It is believed to be an autoimmune disease and may be early Hashimoto’s thyroiditis.11
T4 and T3 are initially elevated in subacute, acute and silent thyroiditis but may become depressed with time. TSH levels are low and then elevated. Thyroid hormone levels are usually low in chronic Hashimoto’s thyroiditis and TSH is high. Suppression of radioactive iodine uptake (RAIU) is seen in subacute and silent thyroiditis. Antithyroid antibodies are present in Hashimoto’s thyroiditis.
Recovery from thyroiditis may be complete in weeks or months without treatment. If the condition is bacterial in origin, treatment may include specific antibiotics or surgical drainage. In the subacute and acute forms, salicylates and non-steroidal anti-inflammatory drugs are used. If there is no response to these drugs in 48 hours, corticosteroids are given. Propranolol or atenolol may be used for cardiovascular symptoms from a hyperthyroid condition. Thyroid hormone replacement is indicated if the patient is hypothyroid.
Nursing care of the patient with thyroiditis depends, in part, on the therapeutic management. Education regarding treatment and encouraging compliance are important for all types of thyroiditis. The patient should be instructed to remain under close healthcare supervision so that their progress can be monitored and to report any change in symptoms to their healthcare provider.
The patient with thyroiditis of an autoimmune origin may be susceptible to other autoimmune diseases, such as Addison’s disease, pernicious anaemia, premature gonadal failure or Graves’ disease. The patient should be taught the signs and symptoms of these disorders, particularly Addison’s disease. A patient receiving thyroid hormone replacement must be taught the expected side effects of these drugs and measures to manage them. The patient treated surgically needs care similar to that given to the patient undergoing thyroidectomy.
Hyperthyroidism
Hyperthyroidism is hyperactivity of the thyroid gland with sustained increase in synthesis and release of thyroid hormones. The term thyrotoxicosis refers to the physiological effects or clinical syndrome of hypermetabolism that result from excess circulating levels of T4 or T3, or both. Hyperthyroidism and thyrotoxicosis usually occur together, as in Graves’ disease. However, in some forms of thyroiditis, thyrotoxicosis may occur without hyperthyroidism.12
Hyperthyroidism occurs in women more than men, with the highest frequency in people between 20 and 40 years of age. The most common cause of hyperthyroidism is Graves’ disease. Other causes include toxic nodular goitre, thyroiditis, exogenous iodine excess, pituitary tumours and thyroid cancer.
AETIOLOGY AND PATHOPHYSIOLOGY
Graves’ disease is an autoimmune disease of unknown aetiology marked by diffuse thyroid enlargement and excessive thyroid hormone secretion. Precipitating factors such as insufficient iodine supply, infection and stressful life events may interact with genetic factors to cause the disease. Graves’ disease is the most common cause of hyperthyroidism in Australia and New Zealand, accounting for about 75% of cases. The patient develops antibodies to the TSH receptor. These antibodies attach to the receptors and stimulate the thyroid gland to release T3 or T4, or both. The excessive release of thyroid hormones leads to the clinical manifestations associated with thyrotoxicosis. The disease is characterised by remissions and exacerbations, with or without treatment. It may progress to destruction of the thyroid tissue, causing hypothyroidism.
Nodular goitres are thyroid hormone-secreting nodules that function independently of TSH stimulation. If these nodules are associated with hyperthyroidism, they are termed toxic nodular goitre. There may be multiple nodules (multinodular goitre) or a single nodule (solitary autonomous nodule). The nodules are usually benign follicular adenomas. Toxic nodular goitres occur equally in men and women. Although they can appear at any age, the frequency of toxic multinodular goitre is greatest in people over 40 years of age. Small solitary autonomous nodules do not usually secrete enough thyroid hormone to cause clinical thyrotoxicosis. However, larger nodules (>3 cm) may result in clinical disease.
CLINICAL MANIFESTATIONS
The clinical manifestations of hyperthyroidism are related to the effect of thyroid hormone excess. Excess circulating thyroid hormone directly increases metabolism. It also increases tissue sensitivity to stimulation by the sympathetic nervous system.
Palpation of the thyroid gland may reveal goitre. When the thyroid gland is excessively large, goitre may be noted on inspection. Auscultation of the thyroid gland may reveal bruits, a reflection of increased blood supply. Another common finding associated with hyperthyroidism is ophthalmopathy, a term used to describe abnormal eye appearance or function. A classic finding in Graves’ disease is exophthalmos, a protrusion of the eyeballs from the orbits (see Fig 49-6). Exophthalmos is a type of infiltrative ophthalmopathy that is due to impaired venous drainage from the orbit, which causes increased fat deposits and fluid (oedema) in the retro-orbital tissues. Due to increased pressure, the eyeballs are forced outwards and protrude. This sign is seen in 20–40% of patients with Graves’ disease. It is usually bilateral but can be unilateral or asymmetrical. In non-infiltrative ophthalmopathy, the upper lids are usually retracted and elevated, with the sclera visible above the iris. When the eyelids do not close completely, the exposed corneal surfaces become dry and irritated. Serious consequences, such as corneal ulcers and eventual loss of vision, can occur.
Other manifestations of thyroid hyperfunction are listed in Table 49-2. A patient with advanced disease may exhibit many of the manifestations, including acropachy (see Fig 49-8), whereas a patient in the early stages of hyperthyroidism may exhibit only weight loss and increased nervousness. Symptoms in the elderly patient with this disorder can be very different (referred to as apathetic hyperthyroidism) and may include anorexia, apathy, lassitude, depression, weight loss, atrial fibrillation and confusion.13 Table 49-3 compares features of hyperthyroidism in younger and older adult patients.
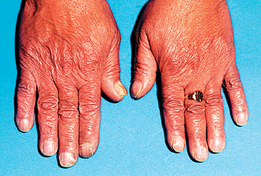
TABLE 49-3 Comparison of hyperthyroidism in younger and older adults
| Younger adult | Older adult | |
|---|---|---|
| Common causes | Graves’ disease in >90% of cases | Graves’ disease or toxic nodular goitre |
| Common symptoms | Nervousness, irritability, weight loss, heat intolerance, warm, moist skin | Anorexia, weight loss, apathy, lassitude, depression, confusion |
| Goitre | Present in >90% of cases | Present in about 50% of cases |
| Ophthalmopathy | Exophthalmos present in 20–40% of cases | Exophthalmos less common |
| Cardiac features | Tachycardia and palpitations common but without heart failure | Angina, arrhythmia, congestive heart failure may occur |
COMPLICATIONS
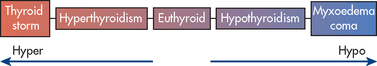
Thyrotoxic crisis (also called thyroid storm) is an acute, rare condition in which all hyperthyroid manifestations are heightened. Although it is considered a life-threatening emergency, death is rare when treatment is vigorous and initiated early. The physiological factor(s) that initiate thyrotoxic crisis are unknown. The cause is thought to be stressors (e.g. infection, trauma, surgery) in a patient with pre-existing hyperthyroidism, either diagnosed or undiagnosed. Heart and nerve tissues become more sensitive to catecholamines due to the presence of more binding sites for adrenaline and noradrenaline.
Manifestations include severe tachycardia, heart failure, shock, hyperthermia (up to 40.7°C), restlessness, agitation, seizures, abdominal pain, nausea, vomiting, diarrhoea, delirium and coma. Treatment is aimed at reducing circulating thyroid hormone levels and the clinical manifestations of this disorder by appropriate drug therapy. Supportive therapy is directed at managing respiratory distress, fever reduction, fluid replacement, and elimination or management of the initiating stressor(s).
DIAGNOSTIC STUDIES
The two primary laboratory findings used to confirm the diagnosis of hyperthyroidism are decreased TSH levels and elevated free thyroxine (FT4) levels. Total T3 and T4 may also be assessed but these are not as useful. Measurements of total T3 and T4 measure both free and bound (to protein) hormone levels. The free hormone is the only form of the hormone that is biologically active.
The RAIU test is used to differentiate Graves’ disease from other forms of thyroiditis. The patient with Graves’ disease will show a diffuse, homogeneous uptake of 35–95%, whereas the patient with thyroiditis will show an uptake of less than 2%. The person with a nodular goitre will have an uptake in the high–normal range (see Box 49-4).
MULTIDISCIPLINARY CARE
The overall goal in the treatment of hyperthyroidism is to block the adverse effects of thyroid hormones and stop their oversecretion. The three primary treatment options for the patient with hyperthyroidism are antithyroid medications, radioactive iodine (RAI) therapy and subtotal thyroidectomy (see Box 49-4). In general, the treatment of choice in non-pregnant adults in New Zealand and Australia is RAI therapy. However, the choice of treatment is influenced by the patient’s age and preferences, the severity of the disorder and complicating features (including pregnancy). If surgery is to be performed, the patient is usually given antithyroid drugs and iodine to produce a euthyroid state and possibly β-adrenergic blockers to relieve symptoms preoperatively.
Drug therapy
Drugs used in the treatment of hyperthyroidism include antithyroid drugs, iodine and β-adrenergic blockers. These drugs are useful in the treatment of thyrotoxic states, but they are not considered curative. Radiation therapy or surgery may ultimately be required.
Antithyroid drugs
The first-line antithyroid drugs are propylthiouracil (PTU) and carbimazole. These drugs inhibit the synthesis of thyroid hormones. PTU also blocks peripheral conversion of T4 to T3. Although there is individual variation, improvement usually begins 1–2 weeks after the start of therapy. Good results are usually seen within 4–8 weeks. Therapy is continued for 6–15 months to allow for spontaneous remission, which occurs in 20–40% of individuals with hyperthyroidism. These drugs are not curative. The major disadvantages of these drugs are patient non-compliance and a high rate of recurrence of hyperthyroidism when the drugs are discontinued. PTU lowers hormone levels more quickly but requires a dose three times a day. Carbimazole is administered in a single daily dose of 20–40 mg. Indications for the use of antithyroid drugs include Graves’ disease in the young patient, hyperthyroidism during pregnancy and the need to achieve a euthyroid state before surgery or radiation therapy.
Iodine
Iodine is used with other antithyroid drugs to prepare the patient for thyroidectomy or for treatment of thyrotoxic crisis. The administration of iodine in large doses rapidly inhibits synthesis of T3 and T4 and blocks the release of these hormones into circulation. It also decreases the vascularity of the thyroid gland, making surgery safer and easier. The maximal effect of iodine is usually seen within 1–2 weeks. Because there is a reduction in the therapeutic effect, long-term iodine therapy is not effective in controlling hyperthyroidism. Iodine is available in the form of saturated solution of potassium iodine (SSKI) and Lugol’s solution.
β-adrenergic blockers
β-adrenergic blockers are used for symptomatic relief of thyrotoxicosis that results from increased β-adrenergic receptor stimulation caused by excess thyroid hormones. Propranolol is usually administered with other antithyroid agents and rapidly provides symptomatic relief. Atenolol is the preferred β-adrenergic blocker for use in the hyperthyroid patient with asthma or heart disease.
Radioactive iodine therapy
RAI therapy is the treatment of choice for most non-pregnant adults. (A pregnancy test is done on all women who experience menstrual cycles before initiation of therapy.) RAI therapy damages or destroys thyroid tissue, thus limiting thyroid hormone secretion. RAI therapy has a delayed response and the maximum effect may not be seen for 2–3 months. For this reason, the patient is usually treated with antithyroid drugs before and during the first 3 months after the initiation of RAI therapy until the effects of radiation become apparent. Although RAI therapy is usually effective, there is a high incidence of post-treatment hypothyroidism (80% of adequately treated individuals), resulting in the need for lifelong thyroid hormone replacement.
Surgical therapy
Thyroidectomy is indicated for individuals who have been unresponsive to antithyroid therapy, for individuals with very large goitres causing tracheal compression and for individuals with a possible malignancy. Additionally, this surgery may be done when an individual is not a good candidate for RAI therapy. One advantage that thyroidectomy has over RAI therapy is a more rapid reduction in T3 and T4 levels. A subtotal thyroidectomy is the preferred surgical procedure and involves the removal of a significant portion of the thyroid gland. For subtotal thyroidectomy to be effective, approximately 90% of thyroid tissue must be removed. If too much tissue is taken, the gland will not regenerate after surgery and hypothyroidism will result.
Endoscopic thyroidectomy is a minimally invasive procedure. It is an appropriate procedure for patients with small nodules (<3 cm) where there is no evidence of malignancy. The advantages of endoscopic thyroidectomy over open thyroidectomy are less scarring, less pain and a faster return to normal activity.
Before surgery, antithyroid drugs, iodine and β-adrenergic blockers may be administered to achieve a euthyroid state and to control symptoms. Iodine reduces vascularisation of the gland, reducing the risk of haemorrhage. Postoperative complications include hypothyroidism, damage to or inadvertent removal of the parathyroid glands causing hypoparathyroidism and hypocalcaemia, haemorrhage, injury to the recurrent or superior laryngeal nerve, thyrotoxic crisis and infection.
Nutritional therapy
The potential for nutritional deficits is high when an increased metabolic rate is present. A high-energy diet (16,800–21,000 kJ/day) may be ordered to satisfy hunger and prevent tissue breakdown. This can be accomplished with six full meals a day and snacks high in protein, carbohydrates, minerals and vitamins, particularly vitamin A, thiamine, vitamin B6 and vitamin C. The protein content should be 1–2 g/kg of ideal body weight. Increased carbohydrates should compensate for an altered metabolism, while providing energy and lessening the use of body stored protein. Highly seasoned and high-fibre foods should be avoided because they stimulate the already hyperactive GI tract. Substitutes should be provided for caffeine-containing liquids, such as coffee, tea and cola, because the stimulating effects of these fluids increase restlessness and sleep disturbances. A dietician should be consulted for guidance in meeting the nutritional needs of patients with hyperthyroidism.
NURSING MANAGEMENT: HYPERTHYROIDISM
Nursing assessment
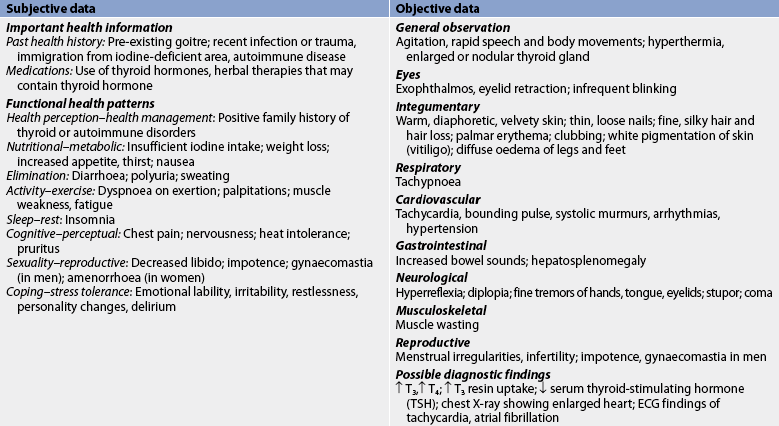
Subjective and objective data that should be obtained from an individual with hyperthyroidism are presented in Table 49-4.
Nursing diagnoses
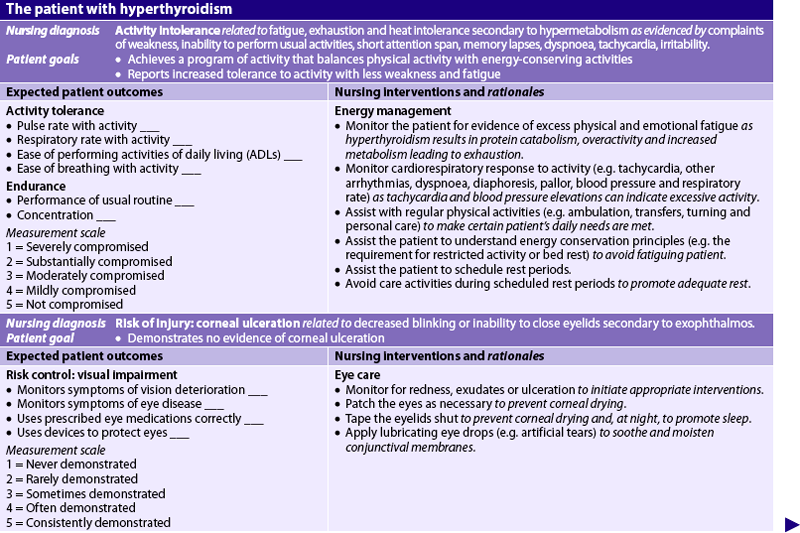
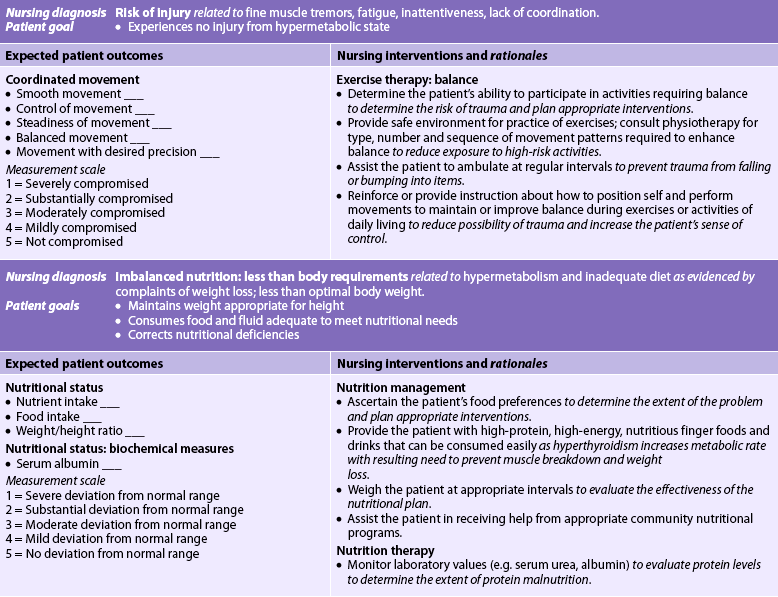
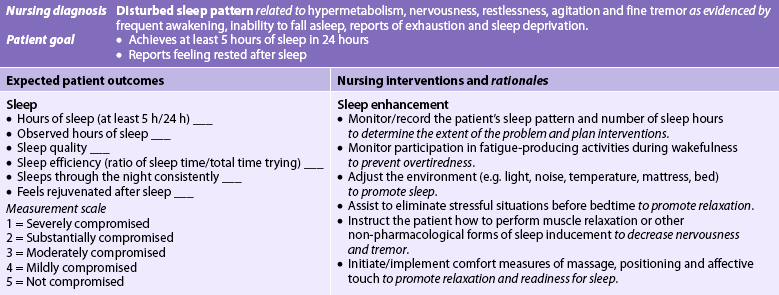
Nursing diagnoses for the patient with hyperthyroidism include, but are not limited to, those presented in NCP 49-1.

Planning
The overall goals are that the patient with hyperthyroidism will: (1) experience relief of symptoms; (2) have no serious complications related to the disease or treatment; (3) maintain nutritional balance; and (4) cooperate with the therapeutic plan.
Nursing implementation
Acute intervention
Individuals who have hyperthyroidism are usually treated in an outpatient setting. However, patients who develop acute thyrotoxicosis (thyroid storm) or those who undergo thyroidectomy require hospitalisation and acute care.
Acute thyrotoxicosis
Acute thyrotoxicosis is a systemic syndrome that requires aggressive treatment, often in an intensive care unit. The nurse needs to administer medications (previously discussed) that block thyroid hormone production. Nursing management also includes provisions for supportive therapy. Having an understanding of the major organ response to the hypermetabolic state is a critical aspect of nursing management. Supportive therapy includes monitoring for cardiac arrhythmias and decompensation, ensuring adequate oxygenation and administering IV fluids to replace fluid and electrolyte losses. This is especially important in patients who develop vomiting and diarrhoea.
A calm, quiet room should be provided because increased metabolism causes sleep disturbances. Providing for adequate rest may be a challenge because of the patient’s irritability and restlessness. Specific interventions may include: (1) placing the patient in a cool room, away from very ill patients and noisy, high-traffic areas; (2) using light bed coverings and changing the linen frequently if the patient is diaphoretic; (3) encouraging and assisting with exercise involving large muscle groups (tremors can interfere with small-muscle coordination) to allow the release of nervous tension and restlessness; and (4) establishing a supportive, trusting relationship to help the patient cope with aggravating events and to lessen anxiety.
If exophthalmos is present, there is a potential for corneal injury related to irritation and dryness. The patient may also have orbital pain. Nursing interventions to relieve eye discomfort and prevent corneal ulceration include applying artificial tears to soothe and moisten conjunctival membranes. Salt restriction may help reduce periorbital oedema. Elevation of the patient’s head promotes fluid drainage from the periorbital area; the patient should sit upright as much as possible. Dark glasses reduce glare and prevent irritation from smoke, air currents, dust and dirt. If the eyelids cannot be closed, they should be lightly taped shut for sleep. To maintain flexibility, the patient should be taught to exercise the intraocular muscles several times a day by turning the eyes in the complete range of motion. Good grooming can be helpful in reducing the loss of self-esteem that can result from an altered body image. If exophthalmos is severe, corticosteroids, radiation of retro-orbital tissues, orbital decompression or corrective lid or muscle surgery may be used.
Thyroid surgery
When subtotal thyroidectomy is the treatment of choice, the patient must be adequately prepared to avoid postoperative complications. The signs and symptoms of thyrotoxicosis must be alleviated as much as possible, and cardiac problems must be controlled before surgery. If iodine is used to relieve hyperthyroid symptoms, it should be mixed with water or juice, sipped through a straw and administered after meals. The patient must be assessed for signs of iodine toxicity, such as swelling of buccal mucosa and other mucous membranes, excessive salivation, nausea and vomiting, and skin reactions. If toxicity occurs, iodine administration should be discontinued and the doctor notified.
Preoperative teaching should include comfort and safety measures in which the patient can participate. Coughing, deep breathing and leg exercises should be practised and their importance explained. The patient should be taught how to support the head manually while turning in bed because this manoeuvre minimises stress on the suture line after surgery. Range-of-motion exercises of the neck should be practised. The nurse should explain routine postoperative care, such as IV infusions. The patient should be told that talking is likely to be difficult for a short time after surgery.
The hospital room must be prepared before the patient’s return from surgery. Oxygen, suction equipment and a tracheostomy tray should be readily available. A tracheostomy tray is required in case airway obstruction occurs. Although this rarely occurs, it is an emergency situation the nurse must be prepared for. Recurrent laryngeal nerve damage leads to vocal cord paralysis. If there is paralysis of both cords, spastic airway obstruction will occur, requiring an immediate tracheostomy.
Respiration may also become difficult because of excess swelling of the neck tissues, haemorrhage, haematoma formation and laryngeal stridor. Laryngeal stridor (harsh, vibratory sound) may occur during inspiration and expiration as a result of oedema of the laryngeal nerve. Laryngeal stridor may also be related to tetany (a condition of neuromuscular hyperexcitability associated with a sudden decrease in calcium levels), which occurs if the parathyroid glands are removed or damaged during surgery leading to hypocalcaemia. To treat tetany, IV calcium salts, such as calcium gluconate or calcium gluceptate, should be available.
After a thyroidectomy the nurse should:
1. assess the patient every 2 hours for 24 hours for signs of haemorrhage or tracheal compression, such as irregular breathing, neck swelling, frequent swallowing, sensations of fullness at the incision site, choking, and blood on the anterior or posterior dressings
2. place the patient in a semi-Fowler position and support the patient’s head with pillows, and avoid flexion of the neck and any tension on the suture lines
3. monitor the patient’s vital signs—the initial assessment should be completed by checking for signs of tetany secondary to hypoparathyroidism (e.g. tingling in toes, fingers or around the mouth; muscular twitching; apprehension) and by evaluating difficulty in speaking and hoarseness; Trousseau’s sign and Chvostek’s sign should be monitored for 72 hours; some hoarseness is to be expected for 3–4 days after surgery because of oedema
If postoperative recovery is uneventful, the patient is ambulated within hours after surgery, is permitted to take fluid as soon as tolerated and eats a soft diet the day after surgery.
The appearance of the incision may be highly distressing to the patient. The patient can be reassured that the scar will fade in colour and eventually look like a normal neck wrinkle. A scarf, jewellery, high collar or other covering can effectively camouflage the scar.
Ambulatory and home care
Postoperative care
Discharge teaching for the patient following surgery is an important aspect of nursing care. The patient and family need to be aware that thyroid hormone balance should be monitored periodically to ensure that normal function has returned. Most patients experience a period of relative hypothyroidism soon after surgery because of the substantial reduction in the size of the thyroid. However, the remaining tissue usually hypertrophies, recovering the capacity to produce the hormone needed by the body, although this takes time. The administration of thyroid hormone is avoided because exogenous hormone inhibits pituitary production of TSH and delays or prevents the restoration of normal gland function and thyroid tissue regeneration.
Energy intake must be reduced substantially below the amount that was required before surgery to prevent weight gain. Adequate iodine is necessary to promote thyroid function but excesses can inhibit the thyroid. Seafood once or twice a week or normal use of iodised salt should provide sufficient intake. Regular exercise helps stimulate the thyroid gland and should be encouraged. High environmental temperature should be avoided because it inhibits thyroid regeneration.
Regular follow-up care is necessary. The patient should be seen postoperatively after 4–6 weeks and then assessed six-monthly in the first year for the development of hypothyroidism. Once the patient is stable and on appropriate thyroid replacement therapy, if necessary, they may be managed by their doctor. If a complete thyroidectomy has been performed, the patient needs instruction in lifelong thyroid replacement. Failure of thyroid function is considered the end stage of Graves’ disease. The patient should be taught the signs and symptoms of progressive thyroid failure and instructed to seek medical care if these develop. Hypothyroidism is relatively easy to manage with oral administration of thyroid replacement.
Radioactive iodine therapy
Radioactive iodine therapy is administered on an outpatient basis and is the therapy of choice for the non-pregnant adult. Because the therapeutic dose of radioactive iodine is low, no radiation safety precautions are necessary. The patient should be instructed that radiation thyroiditis and parotiditis are possible and may cause dryness and irritation of the mouth and throat. Relief may be obtained with frequent sips of water, ice chips or a salt and soda gargle three or four times per day. This gargle is made by dissolving 1 teaspoon of salt and 1 teaspoon of baking soda in 2 cups of warm water. The discomfort should subside in 3–4 days. Because of the high frequency of hypothyroidism after radioactive iodine therapy, the patient and family should be taught the symptoms of hypothyroidism and instructed to seek medical help if these symptoms occur.
Hypothyroidism
AETIOLOGY AND PATHOPHYSIOLOGY
Hypothyroidism results from insufficient circulating thyroid hormone as a result of a variety of abnormalities and occurs in about 5% of the adult population.12 The most common cause of hypothyroidism in Australia is autoimmune chronic lymphocytic thyroiditis, characterised by raised circulating levels of thyroid peroxidase antibody.12 Hypothyroidism can be primary (related to destruction of thyroid tissue or defective hormone synthesis) or secondary (related to pituitary disease with decreased TSH secretion or hypothalamic dysfunction with decreased thyrotrophin-releasing hormone [TRH] secretion). It may also be transient, related to thyroiditis or discontinuance of thyroid hormone therapy.
Iodine deficiency is the most common cause of hypothyroidism worldwide and is most prevalent in iodine-deficient areas of the world. In areas where iodine intake is adequate the most common cause of primary hypothyroidism in the adult is atrophy of the thyroid gland. This atrophy is the end result of Hashimoto’s thyroiditis and Graves’ disease. These autoimmune diseases destroy the thyroid gland. Hypothyroidism also may develop due to treatment for hyperthyroidism, specifically the surgical removal of the thyroid glands or RAI therapy. Drugs such as amiodarone (contains iodine) and lithium (blocks hormone production) are known to produce hypothyroidism.
Hypothyroidism that develops in infancy, known as congenital hypothyroidism (formally termed cretinism), is caused by thyroid hormone deficiencies during fetal or early neonatal life. All infants in Australia and New Zealand are screened for decreased thyroid function at birth; however, there is continuing concern in both countries about borderline iodine insufficiency in school-aged children due to low iodine levels in the modern diet.14,15
CLINICAL MANIFESTATIONS
All hypothyroid states have certain features in common, regardless of the cause. Manifestations vary depending on the severity and duration of thyroid deficiency, as well as the patient’s age at onset of the deficiency.
Hypothyroidism has systemic effects characterised by an insidious and non-specific slowing of body processes. The clinical presentation can range from a patient with no symptoms to a patient with classic symptoms and physical changes easily detected on examination. Unless hypothyroidism occurs after thyroidectomy or thyroid ablation, or during treatment with antithyroid drugs, the onset of symptoms may occur over months to years. The severity of symptoms depends on the degree of thyroid hormone deficiency and the long-term physiological effects of thyroid hormone deficiency. Long-term effects may involve any body system but are more pronounced in the neurological, cardiovascular, GI, reproductive and haematological systems.
The adult with hypothyroidism often is exhausted and lethargic, and experiences personality and mental changes including impaired memory, slowed speech, decreased initiative and somnolence. Many individuals with hypothyroidism appear depressed. Although patients with hypothyroidism sleep for long periods of time, the stages of sleep are altered.
Hypothyroidism is associated with decreased cardiac output and decreased cardiac contractility. Thus, the patient may experience low exercise tolerance and shortness of breath on exertion. In the patient with a pre-existing cardiovascular condition, hypothyroidism may cause significant haemodynamic compromise.
Anaemia is a common feature of hypothyroidism. Erythropoietin levels may be low or normal. Oxygen demand is decreased, and there is a hypocellular bone marrow. The result is a low haematocrit. Other haematological problems are related to vitamin B12, iron and folate deficiencies. The patient may bruise easily. Increased serum cholesterol and triglyceride levels and the accumulation of mucopolysaccharides in the intima of small blood vessels can result in coronary atherosclerosis. This accumulation is seldom symptomatic (i.e. characterised by angina) because of the decreased myocardial oxygen consumption that has been observed in hypothyroidism.
GI motility is also decreased, and achlorhydria (absence or decrease of hydrochloric acid) is common. Constipation, which is a common complaint, may progress to obstipation (discussed in Ch 42) and, rarely, to intestinal obstruction. The underlying metabolic disease makes the individual a high-risk candidate for intestinal surgery.
Other physical changes include cold intolerance, hair loss, dry and coarse skin, brittle nails, hoarseness, muscle weakness and swelling, and weight gain. Weight gain is most likely a result of a decreased metabolic rate.
Patients with severe longstanding hypothyroidism may display myxoedema, which is the accumulation of hydrophilic mucopolysaccharides in the dermis and other tissues (see Fig 49-9). This mucinous oedema causes the characteristic facies of hypothyroidism (i.e. puffiness, periorbital oedema and mask-like affect). Individuals with hypothyroidism may describe an impaired self-image in regard to their disabilities and altered appearance.
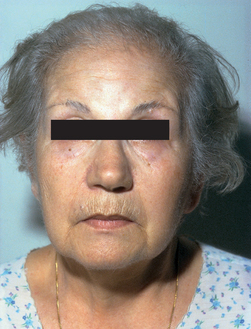
Figure 49-9 Common features of myxoedema. Dull, puffy skin; coarse, sparse hair; periorbital oedema; and prominent tongue.
Women with hypothyroidism frequently complain of menorrhagia. Some affected individuals have been treated for menorrhagia for years and may have undergone hysterectomy before the hypothyroidism was diagnosed. In addition, anovulatory cycles with subsequent infertility may occur.
In the older adult, the typical manifestations of hypothyroidism (including fatigue, cold and dry skin, hoarseness, hair loss, constipation and cold intolerance) may be attributed to normal ageing. For this reason, the patient’s symptoms may not raise suspicion of an underlying condition. Older adults who have confusion, lethargy and depression should be evaluated for thyroid disease.
COMPLICATIONS
The mental sluggishness, drowsiness and lethargy of hypothyroidism may progress gradually or suddenly to a notable impairment of consciousness or coma. This situation, termed myxoedema coma, constitutes a medical emergency. Myxoedema coma can be precipitated by infection, drugs (especially opioids, tranquillisers and barbiturates), exposure to cold and trauma. It is characterised by subnormal temperature, hypotension and hypoventilation. For the patient to survive, vital functions must be supported and IV thyroid hormone replacement must be administered.
DIAGNOSTIC STUDIES
The most common and reliable laboratory tests used to evaluate thyroid function are those that measure TSH and FT4.16 These values, correlated with symptoms gathered from the history and physical examination, confirm the diagnosis. Serum TSH levels help determine the cause of hypothyroidism. Serum TSH is high when the defect is in the thyroid and low when it is in the pituitary or hypothalamus. An increase in TSH after TRH injection suggests hypothalamic dysfunction, whereas no change suggests anterior pituitary dysfunction (see Box 49-5). Other abnormal laboratory findings are elevated cholesterol and triglyceride levels, anaemia and increased creatinine kinase.
Collaborative therapy
Thyroid hormone replacement (e.g. levothyroxine)
Monitor thyroid hormone levels and adjust dosage (if needed)
Nutritional therapy to promote weight loss
Patient and family teaching (see Box 49-6)
TRH, thyrotrophin-releasing hormone; TSH, thyroid-stimulating hormone.
MULTIDISCIPLINARY CARE
The overall treatment in a patient with hypothyroidism is restoration of a euthyroid state as safely and rapidly as possible with hormone replacement therapy. A low-kilojoule diet is indicated to promote weight loss.
Levothyroxine is the drug of choice to treat hypothyroidism. In the young and otherwise healthy patient, the maintenance replacement dose is adjusted according to the patient’s response and laboratory findings. In the older adult patient and the person with compromised cardiac status, a smaller initial dose is recommended because the usual dose may increase myocardial oxygen demand. The increased oxygen demand may cause angina and cardiac arrhythmias. Any chest pain experienced by a patient starting thyroid replacement should be reported immediately, and an electrocardiogram (ECG) and serum cardiac enzyme tests must be performed. In the patient without side effects the dose is increased at 4–6 week intervals. It is important that the patient take replacement medication regularly. Lifelong thyroid replacement therapy is usually required.
A number of levothyroxine preparations are currently available. Controversy exists regarding the relative amount of thyroid hormone from brand to brand. Individuals using levothyroxine should be cautioned by the healthcare team to have serum TSH levels checked 4–6 weeks after changing the levothyroxine preparation.
NURSING MANAGEMENT: HYPOTHYROIDISM
Nursing assessment
Careful assessment may reveal early and subtle changes that indicate dysfunction. Assessment of the patient who is suspected of having hypothyroidism should include questions about weight gain, mental changes, fatigue, slowed and slurred speech, cold intolerance, skin changes such as increased dryness or thickening, constipation and dyspnoea. In addition, the nurse should question the patient about the recent introduction of iodine-containing medications. The patient should be assessed for bradycardia; distended abdomen; dry, thick, cold skin; thick, brittle nails; paraesthesias; and muscular aches and pains.
Nursing diagnoses
Nursing diagnoses for the patient with hypothyroidism may include, but are not limited to, those presented in NCP 49-2.
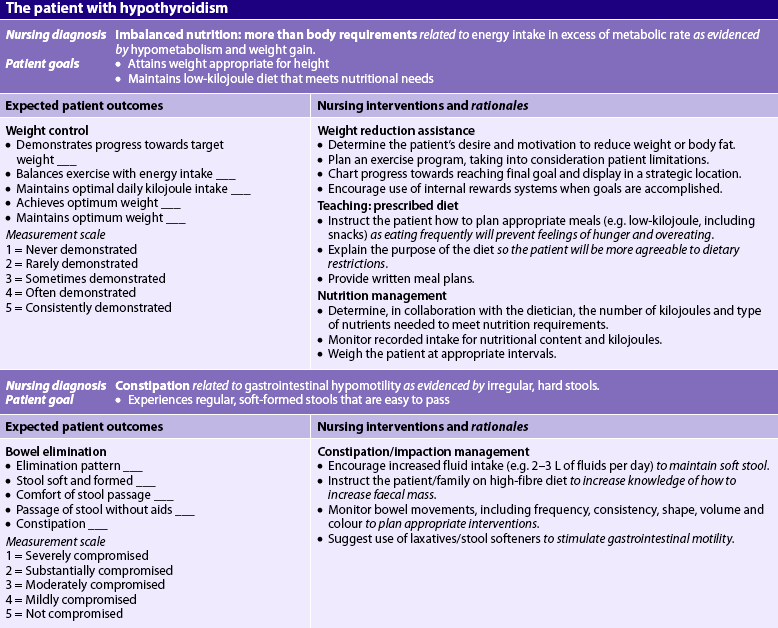
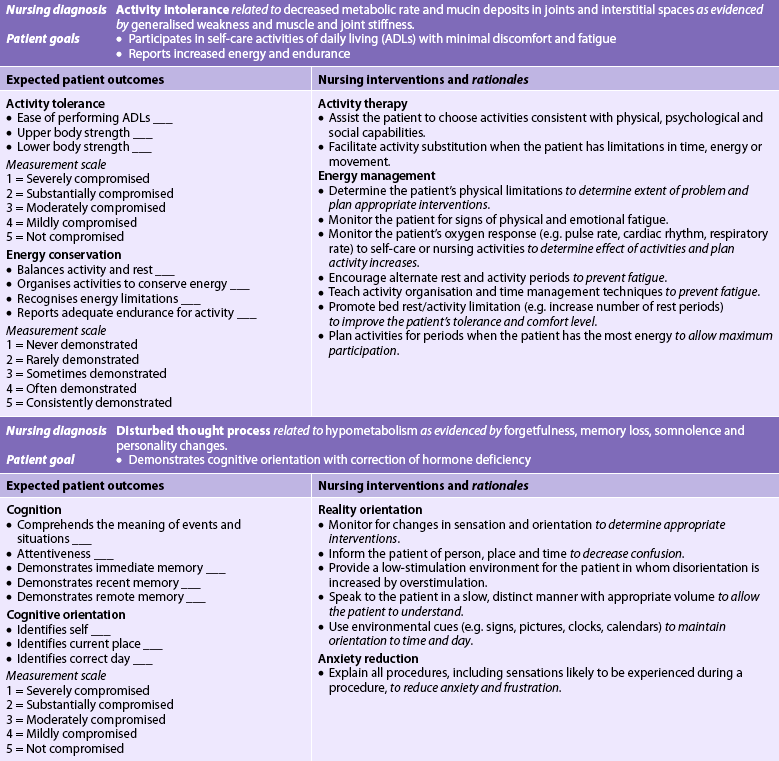
Planning
The overall goals are that the patient with hypothyroidism will: (1) experience relief of symptoms; (2) maintain a euthyroid state; (3) maintain a positive self-image; and (4) comply with lifelong thyroid replacement therapy.
Nursing implementation
Health promotion
There is currently no consensus about thyroid-function screening. Although hypothyroidism is relatively common, particularly among women over the age of 49, there does not appear to be strong justification to screen the general population.17 Research suggests that high-risk populations should be screened for subclinical (asymptomatic) thyroid disease.17 High-risk individuals include those with a family history of thyroid disease, those with a history of neck radiation, women over 49 years of age and postpartum women.
Acute intervention
Most individuals with hypothyroidism do not require acute nursing care because most are managed on an outpatient basis. The patient who develops myxoedema coma requires acute nursing care, often in an intensive care setting. Mechanical respiratory support and cardiac monitoring are frequently necessary. The nurse will administer thyroid hormone replacement therapy and all other medications intravenously because the paralytic ileus associated with myxoedema coma causes unreliable absorption of oral medications. If the patient is hyponatraemic, hypertonic saline may be administered until the serum sodium level reaches at least 130 mmol/L. The nurse should monitor core temperature because the patient with myxoedema coma is often hypothermic.
For assessment of the patient’s progress, vital signs, body weight, fluid intake and output, and visible oedema should be monitored. Cardiac assessment is especially important because the cardiovascular response to the hormone determines the medication regimen. Energy level and mental alertness should be noted. These should increase within 2–14 days and continue to rise steadily to normal levels.
Ambulatory and home care
Patient teaching is imperative for the patient with hypothyroidism. A patient and family teaching guide is provided in Box 49-6. Initially the hypothyroid patient needs more time to comprehend all of the necessary information. It is important to provide written instructions, repeat the information often and assess the patient’s comprehension level.
PATIENT & FAMILY TEACHING GUIDE
1. The patient and family must understand the importance of thyroid replacement therapy. It is especially important to emphasise the need (a) for lifelong replacement, (b) to continually take the medication and (c) for regular follow-up care. Self-care practices to prevent complications should also be emphasised.
2. Emphasise the need for a comfortable, warm environment because of intolerance to cold.
3. Teach measures to prevent skin breakdown. Soap should be used sparingly and lotion applied to skin.
4. Caution the patient, especially the older adult, to avoid sedatives. If they must be used, suggest that the lowest dose be used. Family members should closely monitor mental status, level of consciousness and respirations.
5. Discuss with the patient measures to minimise constipation. Suggestions should include a gradual increase in activity and exercise, increased fibre in the diet, use of stool softeners and maintenance of a regular bowel elimination time. Use of enemas should be avoided because they produce vagal stimulation, which can be hazardous if cardiac disease is present.
The need for lifelong drug therapy must be stressed. Patients should be instructed in expected and unexpected side effects. Specifically, the signs and symptoms of hypothyroidism or hyperthyroidism that indicate hormone imbalance should be included in the teaching plan. Toxic symptoms should be clearly defined. Table 49-2 lists signs of hyperthyroidism that are the same as the toxic symptoms of thyroid hormone replacement.
Patients must be taught to contact their healthcare provider immediately if signs of overdose, such as orthopnoea, dyspnoea, rapid pulse, palpitations, nervousness or insomnia, appear. Patients with diabetes mellitus should test their capillary blood glucose at least daily because return to the euthyroid state frequently increases insulin requirements. In addition, thyroid preparations potentiate the effects of anticoagulants and decrease the effect of digoxin compounds. Thus, patients should be taught the toxic signs and symptoms of these medications and should remain under close medical observation until stable.
It is sometimes difficult for patients to recognise signs of overdosage or underdosage of drug therapy. Therefore, a family member or friend should be included in the instruction process. Patient handouts should be written in understandable language and should accompany verbal instructions. The handouts should be reviewed with the patient and family to assess understanding, and information should be clarified when necessary.
With treatment, striking transformations occur in both appearance and mental function. Most adults return to a normal state. Cardiovascular conditions and (occasionally) psychosis may persist despite corrections of the hormonal imbalance. Relapses occur if treatment is interrupted.
Hyperparathyroidism
AETIOLOGY AND PATHOPHYSIOLOGY
Hyperparathyroidism is a condition involving an increased secretion of parathyroid hormone (PTH). PTH helps regulate calcium and phosphate levels by stimulating bone resorption of calcium, renal tubular reabsorption of calcium and activation of vitamin D. Thus, oversecretion of PTH is associated with increased serum calcium levels. Hyperparathyroidism affects approximately 1% of the general population and is more common in women than men.18,19
Hyperparathyroidism is classified as primary, secondary or tertiary:
• Primary hyperparathyroidism is due to an increased secretion of PTH leading to disorders of calcium, phosphate and bone metabolism. The most common cause is a benign tumour (adenoma) in the parathyroid gland. Primary hyperparathyroidism usually occurs between 30 and 70 years of age. Peak incidence is in the fifth and sixth decades of life. Patients who have previously undergone head and neck radiation may have an increased risk of developing a parathyroid adenoma.
• Secondary hyperparathyroidism appears to be a compensatory response to conditions that induce or cause hypocalcaemia, the main stimulus of PTH secretion. Disease conditions associated with secondary hyperparathyroidism include vitamin D deficiencies, malabsorption, chronic renal failure and hyperphosphataemia.
• Tertiary hyperparathyroidism occurs when there is hyperplasia of the parathyroid glands and a loss of negative feedback from circulating calcium levels. Thus, there is autonomous secretion of PTH, even with normal calcium levels. This condition is observed in the patient who has had a kidney transplant after a long period of dialysis treatment for chronic kidney disease (see Ch 46).
CLINICAL PRACTICE
Situation
The nurse is caring for a woman with thyroid disease for whom thyroid replacement therapy is the planned treatment. The patient regularly takes traditional Chinese medicine and her alternative therapist has suggested she begin a herbal regimen instead of taking the medication prescribed. Should the nurse intervene?
Important points for consideration
• Culturally competent nursing care should incorporate the patient’s values and beliefs.
• Patient autonomy, the patient’s right to choose a treatment plan, should be respected.
• Having adequate, understandable information about available treatment options and their possible consequences facilitates an informed choice.
CRITICAL THINKING QUESTIONS
1. What information should the nurse obtain from the patient? What information should the nurse provide for the patient? What should the nurse discuss with the doctor?
2. How should the nurse proceed? Should the nurse try to incorporate the therapist’s herbal regimen into the plan of care while attempting to persuade the patient of the need for thyroid replacement therapy?
Excessive levels of circulating PTH usually lead to hypercalcaemia and hypophosphataemia, creating a multisystem effect (see Table 49-5). In the bones, decreased bone density, cyst formation and general weakness can occur as a result of the effect of PTH on osteoclastic (bone resorption) and osteoblastic (bone formation) activity. In the kidneys, the excess calcium cannot be reabsorbed, leading to increased levels of calcium in the urine (hypercalciuria). This urinary calcium, along with a large amount of urinary phosphate, can lead to calculi formation.20 In addition, PTH stimulates the synthesis of a biologically active form of vitamin D, a potent stimulator of calcium transport in the intestines. In this way, PTH indirectly increases GI absorption of calcium, which further contributes to the high serum calcium levels.
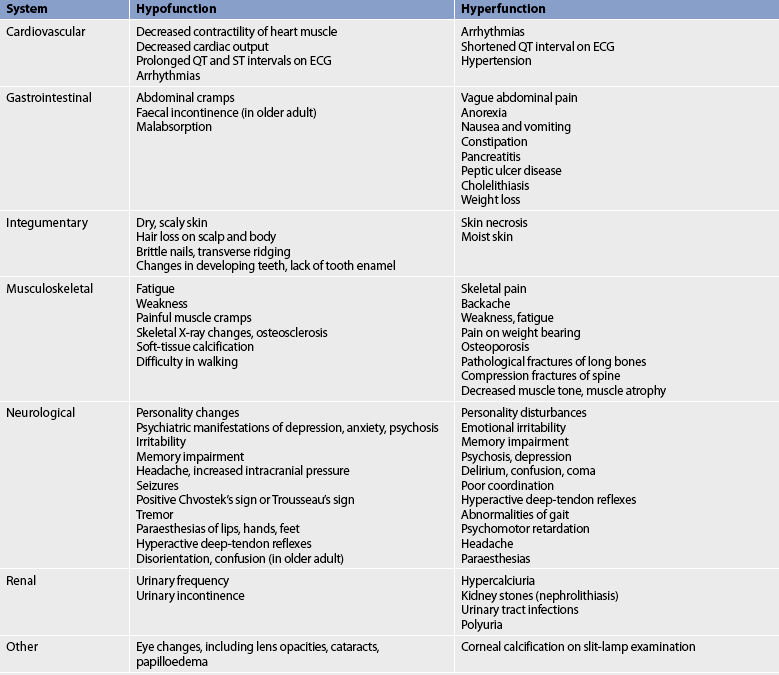
CLINICAL MANIFESTATIONS
Clinical manifestations of hyperparathyroidism range from the asymptomatic individual (who is diagnosed through testing for unrelated problems) to the patient with overt symptoms. Clinical manifestations are associated with hypercalcaemia and are shown in Table 49-5. The major manifestations include weakness, loss of appetite, constipation, increased need for sleep, emotional disorders and shortened attention span. Major signs include loss of calcium from bones (osteoporosis), fractures and kidney stones (nephrolithiasis). Neuromuscular abnormalities are characterised by muscle weakness, particularly in the proximal muscles of the lower extremities. Asymptomatic cases are often identified with routine calcium screening.
COMPLICATIONS
Serious complications of hyperparathyroidism are renal failure, pancreatitis, cardiac changes, and long bone, rib and vertebral fractures.
DIAGNOSTIC STUDIES
PTH, as measured by radioimmunoassay, is elevated in hyperparathyroidism. Serum calcium levels usually exceed 2.5 mmol/L. Because of its inverse relationship with calcium, the serum phosphorus level is usually below 0.1 mmol/L. Elevations in other laboratory tests include urine calcium, serum chloride, uric acid, creatinine, amylase (if pancreatitis is present) and alkaline phosphatase (in the presence of bone disease). Bone density measurements may also be used to detect bone loss. Conversely, individuals found to have bone loss on a screening dual-energy X-ray absorptiometry (DEXA) scan should be tested for hypercalcaemia.19 A 24-hour urine collection for calcium and creatinine excretion may be part of the diagnostic examination to rule out benign familial hypercalcaemia hypocalciuria. An MRI, CT scan and/or ultrasound may be used for localisation of the adenoma.
MULTIDISCIPLINARY CARE
The treatment objectives are to relieve symptoms and prevent complications caused by excess PTH. The choice of therapy depends on the urgency of the clinical situation, the degree of hypercalcaemia and the underlying cause of the disorder.
Surgical therapy
The most effective treatment of primary and secondary hyperparathyroidism is surgical intervention. Parathyroidectomy leads to a rapid reduction of chronically high calcium levels. Criteria for surgery include serum calcium levels greater than 3.0 mmol/L, hypercalciuria (>400 mg/day), markedly reduced bone mineral density, overt symptoms (e.g. neuromuscular effects, nephrolithiasis) or age under 49 years. Surgery involves partial or complete removal of the parathyroid glands. The procedure that is most commonly used involves an endoscope and is done on an outpatient basis. Successful removal of the parathyroid glands is facilitated by intraoperative PTH assay, nuclear scanning with sestamibi and a radio-guided probe.21
Autotransplantation of normal parathyroid tissue in the forearm or near the sternocleidomastoid muscle is usually done. This allows PTH secretion to continue with normalisation of calcium levels. If autotransplantation is not possible, or if it fails, the patient will need to take calcium supplements for life.
Non-surgical therapy
If the patient does not meet the criteria for surgical intervention, or if the patient is elderly or at increased surgical risk from other health problems, a conservative management approach is used. This includes an annual examination with tests for serum PTH, calcium, phosphorus and alkaline phosphatase levels; renal function assessment; X-rays to assess for metabolic bone loss; and measurement of urinary calcium excretion. Continued ambulation and the avoidance of immobility are critical aspects of management. Dietary measures include maintaining a high fluid intake and a moderate calcium intake.
Phosphorus is usually supplemented unless contraindicated by an increased risk of urinary calculi formation. Several drugs currently used in the treatment of hyperparathyroidism are helpful in lowering calcium levels, but do not, in themselves, treat the underlying problem. Bisphosphonates (e.g. alendronate) inhibit osteoclastic bone resorption and rapidly normalise serum calcium levels. Oestrogen or progestin therapy can reduce serum and urinary calcium levels in postmenopausal women and may retard demineralisation of the skeleton. Oral phosphate may be used to inhibit the calcium-absorbing effects of vitamin D in the intestine. Phosphates should be used only if the patient has normal renal function and low serum phosphate levels. Diuretics may be given to increase the urinary excretion of calcium.
Calcimimetic agents (e.g. cinacalcet) are a new class of drugs that increase the sensitivity of the calcium receptor on the parathyroid gland, resulting in decreased PTH secretion and calcium blood levels, and thus sparing calcium stores in the bone. Drugs in this class are currently approved for secondary hyperparathyroidism in patients with chronic kidney disease on dialysis and patients with parathyroid cancer.22 Cinacalcet is under investigation for use in primary hyperparathyroidism.
NURSING MANAGEMENT: HYPERPARATHYROIDISM
Nursing care for the patient after parathyroidectomy is similar to that for a patient after thyroidectomy. The major postoperative complications are associated with haemorrhage and fluid and electrolyte disturbances. Tetany is another concern; it is usually apparent early in the postoperative period but may develop over several days. Mild tetany, characterised by unpleasant tingling of the hands and around the mouth, may be present but should decrease over time. If tetany becomes more severe (e.g. muscular spasms or laryngospasms develop), IV calcium may be given. IV calcium gluconate or calcium gluceptate should be readily available for patients following parathyroidectomy in the event that acute tetany occurs.
Intake and output are monitored to evaluate fluid status. Calcium, potassium, phosphate and magnesium levels are assessed frequently, as well as Chvostek’s sign and Trousseau’s sign. Mobility is encouraged to promote bone calcification.
If surgery is not performed, treatment to relieve symptoms and prevent complications is initiated. The nurse can assist the patient to adapt the meal plan to their lifestyle—a referral to a dietician may be useful. Because immobility can aggravate bone loss, the nurse also needs to stress the importance of an exercise program. The patient should be encouraged to keep the regular appointments, and the tests being performed should be explained. The patient should also be instructed in the symptoms of hypocalcaemia or hypercalcaemia and to report these should they occur. Hypocalcaemia and hypercalcaemia are discussed in Chapter 16.
Hypoparathyroidism
AETIOLOGY AND PATHOPHYSIOLOGY
Hypoparathyroidism, a condition associated with inadequate circulating PTH, is uncommon. It is characterised by hypocalcaemia resulting from a lack of PTH to maintain serum calcium levels. PTH resistance at the cellular level may also occur (pseudohypoparathyroidism). This is caused by a genetic defect resulting in hypocalcaemia in spite of normal or high PTH levels and is often associated with hypothyroidism and hypogonadism.
The most common cause of hypoparathyroidism is iatrogenic. This may include accidental removal of the parathyroid glands or damage to the vascular supply of the glands during neck surgery (e.g. thyroidectomy, radical neck surgery). Idiopathic hypoparathyroidism resulting from the absence, fatty replacement or atrophy of the glands is a rare disease that usually occurs early in life and may be associated with other endocrine disorders. Affected patients may have antiparathyroid antibodies. Severe hypomagnesaemia also leads to suppression of PTH secretion.23
CLINICAL MANIFESTATIONS
The clinical features of acute hypoparathyroidism are due to hypocalcaemia (see Table 49-5). Sudden decreases in calcium concentration cause tetany. This state is characterised by tingling of the lips, fingertips and occasionally feet, and increased muscle tension leading to paraesthesias and stiffness. Painful tonic spasms of smooth and skeletal muscles (particularly of the extremities and face), dysphagia, a constricted feeling in the throat and laryngospasms are also present. Chvostek’s sign and Trousseau’s sign are usually positive. Respiratory function may be severely compromised by accessory muscle spasm and laryngospasm-induced airway obstruction. Patients are usually anxious and apprehensive. Abnormal laboratory findings include decreased serum calcium and PTH levels and increased serum phosphate levels. Other causes of chronic hypocalcaemia include chronic kidney disease, vitamin D deficiency and hypomagnesaemia.
NURSING AND COLLABORATIVE MANAGEMENT: HYPOPARATHYROIDISM
The primary management objectives for the patient with hypoparathyroidism are to treat acute complications such as tetany, maintain normal serum calcium levels and prevent long-term complications. Emergency treatment of tetany requires the administration of IV calcium. IV calcium chloride, calcium gluconate or calcium gluceptate should be given slowly. Calcium must be infused slowly because high blood levels can cause hypotension, serious cardiac arrhythmias or cardiac arrest. Thus, ECG monitoring is indicated when calcium is administered. The patient who takes digoxin is particularly vulnerable. IV calcium can cause venous irritation and inflammation. Extravasation may cause cellulitis, necrosis and tissue sloughing. IV patency should be assessed before administration.
Rebreathing may partially alleviate acute neuromuscular symptoms associated with hypocalcaemia, such as generalised muscle cramps or mild tetany. The patient who can cooperate should be instructed to breathe in and out of a paper bag or breathing mask. This reduces carbon dioxide excretion from the lungs, increases carbonic acid levels in the blood and lowers the pH. A lower pH (acidic environment) enhances the degree of ionisation of calcium, causing an increase in the proportion of total body calcium available in the active form. This will then temporarily relieve the manifestations of hypocalcaemia.
The patient with hypoparathyroidism needs instruction in the management of long-term drug therapy and nutrition. PTH replacement is not a recommended drug therapy because of the expense and the need for parenteral administration. Oral calcium supplements of at least 1.5–3 g per day in divided doses are usually prescribed.
Vitamin D is used in chronic and resistant hypocalcaemia to enhance intestinal calcium absorption and bone resorption. Preferred preparations are dihydrotachysterol and 1,25-dihydroxycholecalciferol (calcitriol). These drugs raise calcium levels rapidly and are quickly metabolised. Rapid metabolism is desired because vitamin D is a fat-soluble vitamin and toxicity can cause irreversible renal impairment. Ergocalciferol, the least expensive of the vitamin D preparations, may also be prescribed. A high-calcium meal plan includes foods such as dark-green vegetables, soybeans and tofu. The patient should be told that foods containing oxalic acid (e.g. spinach, rhubarb), phytic acid (e.g. bran, wholegrains) and phosphorus reduce calcium absorption. The patient should be instructed about the need for lifelong treatment and follow-up care, including the monitoring of calcium levels three to four times a year.
DISORDERS OF THE ADRENAL CORTEX
There are three main classifications of adrenal cortex steroid hormones: glucocorticoid, mineralocorticoid and androgen. Glucocorticoids regulate metabolism, increase blood glucose levels and are critical in the physiological stress response. In humans the primary glucocorticoid is cortisol. Mineralocorticoids regulate sodium and potassium balance. The primary mineralocorticoid is aldosterone. Androgens contribute to growth and development in both genders and to sexual activity in adult women. The term corticosteroid refers to any one of these three types of hormones produced by the adrenal cortex.
Cushing’s syndrome
AETIOLOGY AND PATHOPHYSIOLOGY
Cushing’s syndrome is a rare condition affecting about 2 people per million a year. It is a spectrum of clinical abnormalities caused by an excess of corticosteroids, particularly glucocorticoids. Several conditions can cause Cushing’s syndrome (see Box 49-7). The most common cause is iatrogenic administration of exogenous corticosteroids (e.g. prednisone). Approximately 85% of the cases of endogenous Cushing’s syndrome are due to an ACTH-secreting pituitary tumour (Cushing’s disease). Other causes of Cushing’s syndrome include adrenal tumours and ectopic ACTH production by tumours (usually of the lung or pancreas) outside the hypothalamic–pituitary–adrenal axis. Cushing’s disease and primary adrenal tumours are more common in women in the 20–40-year age group; ectopic ACTH production is more common in men.
BOX 49-7 Causes of Cushing’s syndrome
• Prolonged administration of high doses of corticosteroids
• ACTH-secreting pituitary tumour (Cushing’s disease)
• Cortisol-secreting neoplasm within the adrenal cortex that can be either carcinoma or adenoma
• Excess secretion of ACTH from carcinoma of the lung or other malignant growth outside the pituitary or adrenal glands
CLINICAL MANIFESTATIONS
The clinical manifestations of Cushing’s syndrome can be seen in most body systems and are related to excess levels of corticosteroids (see Table 49-6). Although manifestations of glucocorticoid excess usually predominate, symptoms of mineralocorticoid and androgen excess may also be seen.
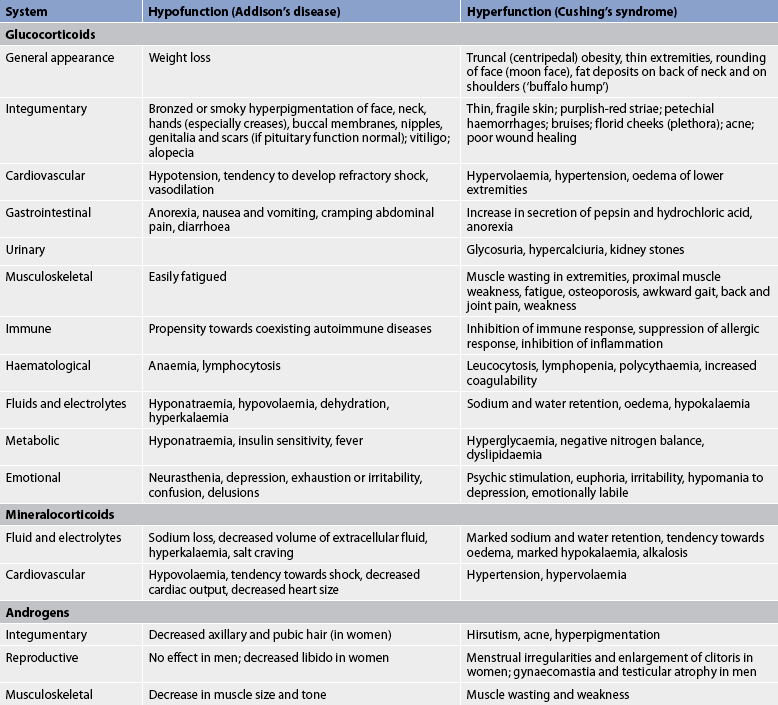
Corticosteroid excess causes pronounced changes in physical appearance (see Fig 49-10). Weight gain, the most common feature, results from the accumulation of adipose tissue in the trunk, face and cervical area (see Fig 49-11).24 Transient weight gain from sodium and water retention may be present because of the mineralocorticoid effects of cortisol. Hyperglycaemia occurs because of glucose intolerance (associated with cortisol-induced insulin resistance) and increased gluconeogenesis by the liver.
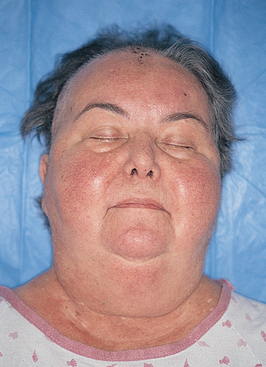
Figure 49-10 Cushing’s syndrome. Facies include a rounded face (‘moon face’) with thin, reddened skin. Hirsutism may also be present.
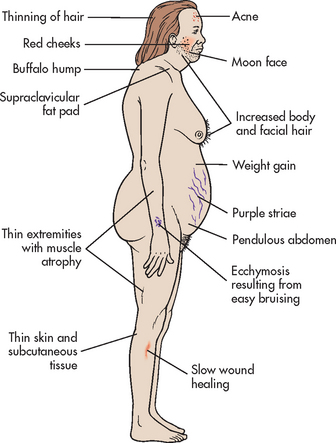
Protein wasting is caused by the catabolic effects of cortisol on peripheral tissue. Muscle wasting leads to muscle weakness, especially in the extremities. A loss of protein matrix in the bone leads to osteoporosis with subsequent pathological fractures (e.g. vertebral compression fractures) and bone and back pain. The loss of collagen makes the skin weaker and thinner, and therefore more easily bruised. Catabolic processes predominate and wound healing is delayed. Mood disturbances (irritability, anxiety, euphoria), insomnia, irrationality and occasionally psychosis may occur.
Mineralocorticoid excess may cause hypertension (secondary to fluid retention), whereas adrenal androgen excess may cause pronounced acne, virilisation in women and feminisation in men. Menstrual disorders and hirsutism in women and gynaecomastia and impotence in men are seen more commonly in adrenal carcinomas. The clinical presentation is the first indication of Cushing’s syndrome. Of particular importance are: (1) centripedal (truncal) obesity or generalised obesity; (2) ‘moon facies’ (fullness of the face) with facial plethora; (3) purplish red striae, which are usually depressed below the skin surface, on the abdomen, breast or buttocks (see Fig 49-12); (4) hirsutism in women; (5) menstrual disorders in women; (6) hypertension; and (7) unexplained hypokalaemia.
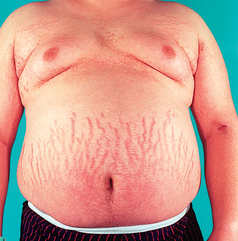
DIAGNOSTIC STUDIES
When Cushing’s syndrome is suspected, a 24-hour urine collection for free cortisol is carried out. Urine cortisol levels of 49–100 μg per day in adults indicate Cushing’s syndrome.25 If the results are borderline, a high-dose dexamethasone suppression test is done. False-positive results can occur in patients with depression, those who are under acute stress and those who are actively alcoholics. Plasma cortisol (the primary glucocorticoid) levels may be elevated, with loss of diurnal variation. CT scanning and MRI of the pituitary and adrenal glands may be used.
Plasma ACTH levels may be low, normal or elevated depending on the underlying problem. High or normal levels indicate ACTH-dependent Cushing’s disease, whereas low or undetectable levels indicate an adrenal or exogenous aetiology. Other findings on diagnostic tests associated with, but not diagnostic of, Cushing’s syndrome include granulocytosis, lymphopenia, eosinopenia, hyperglycaemia, glycosuria, hypercalciuria and osteoporosis. Hypokalaemia and alkalosis are seen in ectopic ACTH syndrome and adrenal carcinoma.
MULTIDISCIPLINARY CARE
The primary goal of treatment for Cushing’s syndrome is to normalise hormone secretion. The specific treatment is dependent on the underlying cause (see Box 49-8). If the underlying cause is a pituitary adenoma, the standard treatment is surgical removal of the pituitary tumour using the trans-sphenoidal approach.26 Patients with Cushing’s disease usually have small tumours that are less than 1 cm in diameter (microadenomas) and are surgically cured about 70–90% of the time.24 In patients with larger tumours, cure rates are lower (<65% in most centres).24 It has been shown that the success of the surgery is dependent on the amount of experience that the surgeon has at performing pituitary operations. Therefore, to achieve the best rate of cure and have fewer complications, patients with Cushing’s disease should be referred to a neurosurgeon with a special interest in pituitary surgery.24
MULTIDISCIPLINARY CARE
*Treatment is based on underlying cause.
Radiation to the pituitary adenoma may be necessary if surgical outcomes are not optimal or if the patient is not a good surgical candidate. Adrenalectomy is indicated for Cushing’s syndrome caused by adrenal tumours or hyperplasia. Occasionally, bilateral adrenalectomy is necessary. Laparoscopic adrenalectomy is used unless a known or suspected malignant adrenal tumour is present. An open surgical adrenalectomy is used for adrenal cancer. Patients with ectopic ACTH-secreting tumours are managed by treating the primary neoplasm.
Drug therapy is used when surgery is contraindicated or as an adjunct to surgery. The goal of drug therapy is the inhibition of adrenal function. Metyrapone, ketoconazole and aminoglutethimide are used to inhibit cortisol synthesis. Common side effects of these agents include anorexia, nausea and vomiting, GI bleeding, depression, vertigo, skin rashes and diplopia. The GI side effects may be minimised by administering drugs with meals and with a bedtime snack.
If Cushing’s syndrome has developed during the course of prolonged administration of corticosteroids (e.g. prednisone), one or more of the following alternatives may be tried: (1) gradual discontinuance of corticosteroid therapy; (2) reduction of the corticosteroid dose; and (3) conversion to an alternate-day regimen.27 Gradual tapering of the corticosteroids is necessary to avoid potentially life-threatening adrenal insufficiency. An alternate-day regimen is one in which twice the daily dosage of a shorter-acting corticosteroid is given every other morning to minimise hypothalamic–pituitary–adrenal suppression, growth suppression and altered appearance. This regimen is not used when the corticosteroids are given as endocrine replacement therapy.
NURSING MANAGEMENT: CUSHING’S SYNDROME
Nursing assessment
Subjective and objective data that should be obtained from the patient with Cushing’s syndrome are presented in Table 49-7.
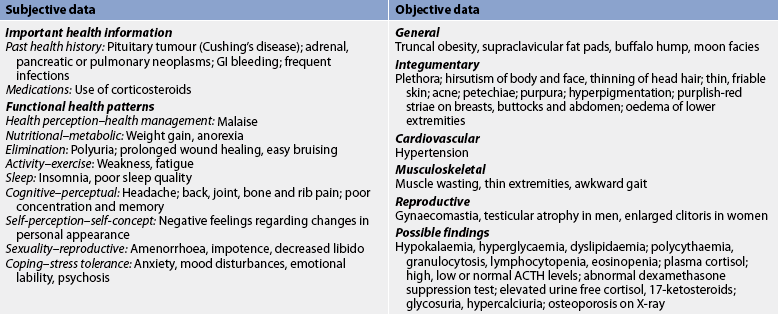
Nursing diagnoses
Nursing diagnoses for the patient with Cushing’s syndrome may include, but are not limited to:
• risk of infection related to lowered resistance to stress and suppression of the immune system
• imbalanced nutrition, more than body requirements, related to increased appetite, high kilojoule content of foods and inactivity
• disturbed self-esteem related to altered body image and emotional lability
• impaired skin integrity related to excess corticosteroids, immobility and altered skin fragility.
Planning
The overall goals are that the patient with Cushing’s syndrome will: (1) experience relief of symptoms; (2) have no serious complications; (3) maintain a positive self-image; and (4) actively participate in the therapeutic plan.
Nursing implementation
Health promotion
Health promotion is focused on identifying patients at risk of Cushing’s syndrome. Patients receiving long-term exogenous cortisol for a variety of diseases are at risk. Patient teaching related to medication use and monitoring of side effects is an important preventative measure.
Acute intervention
The patient with Cushing’s syndrome is seriously ill. Because the therapy has many side effects, the focus of assessment is on signs and symptoms of hormone and drug toxicity and complicating conditions (e.g. cardiovascular disease, diabetes mellitus, infection). Nursing assessment should include monitoring of vital signs, daily weight, blood glucose levels, possible infection (especially pain, loss of function and purulent drainage, because other signs and symptoms of inflammation such as fever and redness may be minimal or absent) and signs and symptoms of abnormal thromboembolic phenomena, such as sudden chest pain, dyspnoea or tachypnoea.
Another important focus of nursing care is emotional support. Changes in appearance, such as centripedal obesity, multiple bruises, hirsutism in women and gynaecomastia in men, can be distressing. The patient may feel unattractive, repulsive or unwanted. The nurse can help by remaining sensitive to the patient’s feelings and offering respect and unconditional acceptance. The patient can be reassured that the physical changes and many of the emotional effects will resolve when hormone levels return to normal.
If treatment involves surgical removal of a pituitary adenoma, an adrenal tumour or one or both adrenal glands, nursing care will additionally focus on preoperative and postoperative care.
Preoperative care
Before surgery the patient should be brought to optimal physical condition. Hypertension and hyperglycaemia must be controlled, and hypokalaemia must be corrected with diet and potassium supplements. A high-protein diet helps correct protein depletion. Preoperative teaching will depend on the type of surgical approach planned (hypophysectomy or adrenalectomy), but should include information regarding the postoperative care the patient should anticipate. In the postoperative period (for both open and laparoscopic adrenalectomy), patients will probably have a nasogastric tube, urinary catheter, IV therapy, central venous pressure monitoring and leg sequential compression devices to prevent emboli.
Postoperative care
Surgery on the adrenal glands poses risks beyond those of other types of operations. Because these glands are highly vascular, the risk of haemorrhage is increased. The manipulation of glandular tissue during surgery may release large amounts of hormone into the circulation, producing marked fluctuations in the metabolic processes affected by these hormones. Postoperatively, BP, fluid balance and electrolyte levels tend to be unstable because of these hormone fluctuations.
High doses of corticosteroids (e.g. hydrocortisone) are administered intravenously during surgery and for several days afterwards to ensure adequate responses to the stress of the procedure. If large amounts of endogenous hormone have been released into the systemic circulation during surgery, the patient is likely to develop hypertension, increasing the risk of haemorrhage. High levels of corticosteroids also increase susceptibility to infection and delay wound healing.
Any rapid or significant changes in BP, respirations or heart rate should be reported. Fluid intake and output are monitored carefully and assessed for potential imbalances. The critical period for circulatory instability ranges from 24 to 48 hours after surgery. IV corticosteroids are given, and the dose and rate of flow are adjusted to the patient’s clinical manifestations and fluid and electrolyte balance. Oral doses are given as tolerated. The IV line may be kept in place after IV corticosteroids are withdrawn to keep a line open for quick administration of corticosteroids or vasopressors. Morning urine levels of cortisol (obtained at the same time each morning) are measured to evaluate the effectiveness of the surgery.
If corticosteroid dosage is tapered too rapidly after surgery, acute adrenal insufficiency may develop. Vomiting, increased weakness, dehydration and hypotension may indicate hypocortisolism. In addition, the patient may complain of painful joints, pruritus or peeling skin and may experience severe emotional disturbances. These signs and symptoms should be reported so that drug doses can be adjusted as necessary. The nurse must constantly be alert for signs of corticosteroid imbalance. After surgery the patient is usually maintained on bed rest until their BP stabilises. The nurse must be alert for subtle signs of postoperative infections because the usual inflammatory responses are suppressed. Meticulous care must be taken when changing the dressing and during any other procedures that necessitate access to body cavities, circulation or areas under the skin, so that infection is prevented.
Ambulatory and home care
Discharge instructions relate to the patient’s lack of endogenous corticosteroids and resulting inability to react to stressors physiologically. Referral to the community nurse should be considered, especially for older adults, because of the need for ongoing evaluation and education. Patients should wear a medical alert bracelet at all times and carry medical identification and instructions in their wallet or purse. Exposure to extremes of temperature, infections and emotional disturbances should be avoided as much as possible. Stress may produce or precipitate acute adrenal insufficiency because the remaining adrenal tissue cannot meet an increased hormonal demand. Many patients can be taught to adjust their corticosteroid replacement therapy in accordance with their stress levels. The nurse should consult with the patient’s medical practitioner to determine the parameters for dosage changes if this plan is feasible. If the patient cannot adjust their own medication, or if weakness, fainting, fever or nausea and vomiting occur, the patient should contact their healthcare provider for a possible adjustment in corticosteroid dosage. Many patients require lifetime replacement therapy, but it may take several months to adjust the hormone dose satisfactorily and patients should be prepared for this.
Adrenocortical insufficiency
AETIOLOGY AND PATHOPHYSIOLOGY
Adrenocortical insufficiency (hypofunction of the adrenal cortex) may be from a primary cause (known as Addison’s disease) or a secondary cause (lack of pituitary ACTH secretion). In Addison’s disease, all three classes of adrenal corticosteroids (glucocorticoids, mineralocorticoids and androgens) are reduced. In secondary adrenocortical insufficiency, corticosteroids and androgens are deficient but mineralocorticoids rarely are. ACTH deficiency may be caused by pituitary disease or suppression of the hypothalamic–pituitary axis as a result of the administration of exogenous corticosteroids.
The most common cause of Addison’s disease in industrialised nations is an autoimmune response. Adrenal tissue is destroyed by antibodies against the patient’s own adrenal cortex. Susceptibility genes for Addison’s disease are beginning to be identified.28 Often, other endocrine conditions are present and Addison’s disease is considered a component of polyendocrine deficiency syndrome. Tuberculosis causes Addison’s disease worldwide, but this is rare in developed countries. Other causes include infarction, fungal infections (e.g. histoplasmosis), acquired immunodeficiency syndrome (AIDS) and metastatic cancer. Iatrogenic Addison’s disease may be due to adrenal haemorrhage, often related to anticoagulant therapy, antineoplastic chemotherapy, ketoconazole therapy for AIDS or bilateral adrenalectomy. Adrenal insufficiency most often occurs in adults under 60 years of age and affects both genders equally. Addison’s disease, if caused by an autoimmune response, is most common in females of European descent.
CLINICAL MANIFESTATIONS
Manifestations do not tend to become evident until 90% of the adrenal cortex is destroyed, so the disease is often advanced before it is diagnosed. The manifestations have a very slow (insidious) onset and include progressive weakness, fatigue, weight loss and anorexia as primary features. Skin hyperpigmentation, a striking feature, is seen primarily in sun-exposed areas of the body, at pressure points, over joints and in creases, especially palmar creases (see Fig 49-13). It is most likely due to increased secretion of β-lipotrophin (which contains melanocyte-stimulating hormone [MSH]) or ACTH. These trophic hormones are increased because of decreased negative feedback and subsequent low corticosteroid levels. Other frequent manifestations are orthostatic hypotension, hyponatraemia, hyperkalaemia, nausea and vomiting, and diarrhoea. Irritability and depression may also occur in primary adrenal hypofunction.
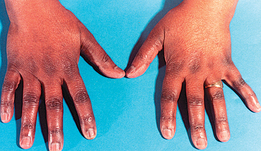
Patients with secondary adrenocortical hypofunction may have many signs and symptoms in common with patients with Addison’s disease but are characteristically not hyperpigmented because ACTH and related peptide levels are low.
COMPLICATIONS
Patients with adrenocortical insufficiency are at risk of an acute adrenal insufficiency (addisonian crisis), which is a life-threatening emergency caused by insufficient adrenocortical hormones or a sudden sharp decrease in these hormones. Addisonian crisis is triggered by stress (e.g. from infection, surgery, trauma, haemorrhage or psychological distress), the sudden withdrawal of corticosteroid hormone replacement therapy (which is often done by a patient who lacks knowledge of the importance of replacement therapy), after adrenal surgery or following sudden pituitary gland destruction.
During acute adrenal insufficiency, severe manifestations of glucocorticoid and mineralocorticoid deficiencies are exhibited, including hypotension (particularly postural), tachycardia, dehydration, hyponatraemia, hyperkalaemia, hypoglycaemia, fever, weakness and confusion. Hypotension may lead to shock. Circulatory collapse associated with adrenal insufficiency is often unresponsive to the usual treatment (vasopressors and fluid replacement). GI manifestations include severe vomiting, diarrhoea and pain in the abdomen. Pain may also occur in the lower back or legs.
DIAGNOSTIC STUDIES
In addition to clinical features, a diagnosis of Addison’s disease can be made when cortisol levels are subnormal or fail to rise over basal levels with an ACTH-stimulation test. A failure of cortisol levels to rise in response to ACTH stimulation indicates primary adrenal disease. A positive response to ACTH stimulation indicates a functioning adrenal gland and points to a probable diagnosis of pituitary disease (see Table 47-6).
Other abnormal laboratory findings include hyperkalaemia, hypochloraemia, hyponatraemia, hypoglycaemia, anaemia and increased blood urea nitrogen levels. Urine levels of free cortisol are low. An ECG may show low voltage and peaked T waves caused by hyperkalaemia. CT scans and MRI are used to localise tumours or to identify adrenal calcifications or enlargement (see Box 49-9).
MULTIDISCIPLINARY CARE
Collaborative therapy
Daily glucocorticoid replacement (two-thirds on awakening in morning, one-third in late afternoon)*
Daily mineralocorticoid in morning*
Salt additives for excess heat or humidity
Increased doses of cortisol for stressful situations (e.g. surgery, hospitalisation)
ACTH, adrenocorticotrophic hormone; CT, computed tomography; MRI, magnetic resonance imaging.
*For conditions of normal daily stress in individuals with usual daytime activity.
MULTIDISCIPLINARY CARE
Treatment of adrenocortical insufficiency is focused on management of the underlying cause when possible. The mainstay of treatment for adrenocortical insufficiency is replacement therapy (see Box 49-9). Hydrocortisone, the most commonly used form of replacement therapy, has both glucocorticoid and mineralocorticoid properties. During situations associated with physiological stress, glucocorticoid dosage must be increased to prevent addisonian crisis. Mineralocorticoid replacement with fludrocortisone acetate is administered daily with increased salt in the diet.
Addisonian crisis is a life-threatening emergency requiring aggressive management. Treatment must be directed towards shock management and high-dose hydrocortisone replacement. Large volumes of 0.9% saline solution and 5% dextrose are administered intravenously to reverse hypotension and electrolyte imbalances until BP returns to normal.
NURSING MANAGEMENT: ADDISON’S DISEASE
Nursing implementation
Acute intervention
When the patient with Addison’s disease is hospitalised, whether for diagnosis, an acute crisis or some other health problem, frequent nursing assessment is necessary. Vital signs and signs of fluid volume deficit and electrolyte imbalance should be assessed every 30 minutes to 4 hours for the first 24 hours depending on the patient’s instability. In addition, daily weighing, diligent corticosteroid administration, protection against exposure to infection and complete assistance with daily hygiene should be practised. The patient should be protected from noise, light and environmental temperature extremes as they cannot cope with these stresses because of the inability to produce corticosteroids.
If the hospitalisation was due to adrenal crisis, the patient usually responds by the second day and can start oral corticosteroid replacement. Because discharge frequently occurs before the maintenance dose of corticosteroids is reached, the patient should be instructed on the importance of keeping scheduled follow-up appointments.
Ambulatory and home care
The nurse has an important role in the long-term management of Addison’s disease. The serious nature of the disease and the need for lifelong replacement therapy necessitate a well-organised and carefully presented teaching plan. Box 49-10 outlines the major areas that must be included in the teaching plan.
PATIENT & FAMILY TEACHING GUIDE
The following should be included in a teaching plan for the patient and family.
3. Symptoms of overdose and underdose
4. Conditions requiring increased medication (e.g. trauma, infection, surgery, emotional crisis)
5. Course of action to take relative to changes in medication
6. Prevention of infection and need for prompt and vigorous treatment of existing infections
7. Need for lifelong replacement therapy
Glucocorticoids are usually given in divided doses, two-thirds in the morning and one-third in the afternoon. Mineralocorticoids are given once daily, preferably in the morning. This dosage schedule reflects normal circadian rhythm in endogenous hormone secretion and decreases the side effects associated with corticosteroid replacement therapy. Because the aim of replacement therapy is a return to normal hormone levels, nursing care is focused on helping the patient to maintain hormone balance while managing the medication regimen.
Patients are unable to tolerate physical or emotional stress without additional exogenous corticosteroids, so long-term care revolves around recognising the need for extra medication and techniques for stress management. The need for corticosteroid hormone is proportional to stress levels. Examples of situations requiring corticosteroid adjustment are fever, influenza, teeth extraction and rigorous physical activity, such as playing tennis on a hot day or running a marathon. Doses of glucocorticoids are usually doubled when minor stress occurs (e.g. a respiratory infection, dental work) and tripled when major stress occurs (e.g. divorce, loss of parent). When in doubt, it is better to err on the side of overreplacement. If vomiting or diarrhoea occurs, as may happen with influenza, the healthcare provider must be notified immediately because electrolyte replacement may be necessary. In addition, these manifestations may be early indicators of crisis. Overall, patients who take their medications consistently can anticipate a normal life expectancy.
Patients must be taught the signs and symptoms of corticosteroid deficiency and excess (Cushing’s) and to report these signs to their healthcare provider so that the dose can be adjusted to their own needs. It is critical that patients wear an identification bracelet (medical alert) and carry a card in their wallet or purse stating that they have Addison’s disease so that appropriate therapy can be initiated in case of an unexpected stressful event. Patients should be instructed and given handouts regarding other medications that can cause a need to increase glucocorticoid dosage (e.g. phenytoin, barbiturates, rifampicin and antacids). In addition, oestrogen inhibits steroid metabolism. Patients using mineralocorticoid therapy should be instructed how to take their BP and increase their salt intake, and be given parameters to report to their healthcare provider. Changes may indicate a need for dosage adjustment.
Patients should carry an emergency kit at all times. The kit should consist of 100 mg of IM hydrocortisone, syringes and instructions for use. The patient and significant others should be instructed in how to give an IM injection in case the replacement therapy cannot be taken orally. The patient should verbalise instructions, practise IM injections with saline and have written instructions as to when to alter the dose.29
Corticosteroid therapy
Cortisol and related glucocorticoids are used to relieve the signs and symptoms associated with many diseases (see Box 49-11). The long-term administration of corticosteroids in therapeutic doses often leads to serious complications and side effects (see Box 49-12). For this reason, corticosteroid therapy is not recommended for minor chronic conditions. Therapy should be reserved for diseases in which there is a risk of death or permanent loss of function, and conditions in which short-term therapy is likely to produce remission or recovery. The potential benefits of treatment must always be weighed against the risks.
BOX 49-11 Diseases and disorders treated with corticosteroids
DRUG THERAPY
BOX 49-12 Side effects of corticosteroids
DRUG THERAPY
• Predisposition to peptic ulcer disease.
• Skeletal muscle atrophy and weakness occurs.
• Mood and behaviour changes may be observed.
• Glucose intolerance predisposes to diabetes mellitus.
• Fat from extremities is redistributed to trunk and face.
• Hypocalcaemia related to anti-vitamin D effect may occur.
• Healing is delayed. An increased risk of wound dehiscence.
• Susceptibility to infection is increased. Infection develops more rapidly and spreads more widely.
• Suppression of pituitary ACTH synthesis occurs. Corticosteroid deficiency is likely if hormones are withdrawn abruptly. Taper corticosteroid doses.
• Increased blood pressure occurs because of excess blood volume and potentiation of vasoconstrictor effects. Hypertension predisposes to heart failure.
• Protein depletion decreases bone formation, density and strength. Predisposes to pathological fractures, especially compression fractures of the vertebrae (osteoporosis).
EFFECTS OF CORTICOSTEROID THERAPY
There are multiple effects of corticosteroid therapy. Although these actions can prove to be beneficial and therapeutic in some situations, they can also contribute to adverse effects as well. The expected effects of corticosteroid therapy include the following:
1. Anti-inflammatory action. Corticosteroids decrease the number of circulating lymphocytes, monocytes and eosinophils. They enhance the release of polymorphonuclear leucocytes from bone marrow, inhibit the accumulation of leucocytes at the site of inflammation and inhibit the release of substances involved in the inflammatory response (e.g. kinins, prostaglandins, histamine) from the leucocytes. As a result, manifestations of inflammation, including redness, tenderness, heat, swelling and local oedema, are suppressed.
2. Immunosuppression. Corticosteroids cause atrophy of lymphoid tissue, suppress the cell-mediated immune responses and decrease the production of antibodies.
3. Maintenance of normal BP. Corticosteroids potentiate the vasoconstrictor effect of noradrenaline and act on the renal tubules to increase sodium reabsorption and enhance potassium and hydrogen excretion. Retention of sodium (and subsequently water) increases blood volume and helps maintain BP. Mineralocorticoids have a direct effect on sodium reabsorption in the distal tubule of the kidneys and as a result increase sodium and water retention.
4. Carbohydrate and protein metabolism. Corticosteroids antagonise the effects of insulin and can induce glucose intolerance by increasing hepatic glycogenolysis and insulin resistance. They also stimulate the breakdown of protein for gluconeogenesis, which can lead to skeletal muscle wasting. Although corticosteroids mobilise free fatty acids and redistribute fat in cushingoid patterns, the mechanism for this process is unknown.
COMPLICATIONS ASSOCIATED WITH CORTICOSTEROID THERAPY
A beneficial effect in one situation may be a harmful one in another. For example, the vasopressive effect of the hormone is critical in enabling the body to function in stressful situations but can produce hypertension when used for drug therapy. Suppression of inflammation and the immune response may help save the lives of the victim of anaphylaxis and the transplant recipient, but it can cause reactivation of latent tuberculosis and greatly reduces resistance to other infections and cancers. In addition, corticosteroids inhibit the antibody response to vaccines. Specific side effects related to corticosteroid therapy are listed in Box 49-12.
NURSING AND COLLABORATIVE MANAGEMENT: CORTICOSTEROID THERAPY
Many patients receive corticosteroid therapy, in particular glucocorticoid therapy, for non-endocrine reasons (see Box 49-11). Thorough instruction is necessary to ensure patient compliance. When corticosteroids are used as non-replacement therapies, they are taken once daily or once every other day. They should be taken early in the morning with food to decrease gastric irritation. Exogenous corticosteroid administration may suppress endogenous ACTH and therefore endogenous cortisol (suppression is time and dose dependent), so the danger of abrupt cessation of corticosteroid therapy must be emphasised to patients and significant others. Steroids taken longer than 1 week will suppress adrenal production and oral steroids should be tapered. Nurses must ensure that increased doses of steroid are prescribed in acute care or home care situations with increased physical or emotional stress.
Patients often receive corticosteroid treatment for prolonged periods of time (>3 months), so corticosteroid-induced osteoporosis is an important concern.30 Therapies to reduce the resorption of bone may include increased calcium intake, vitamin D supplementation, bisphosphonates (e.g. alendronate) and the institution of a low-impact exercise program. Further instructions and interventions to minimise the side effects and complications of corticosteroid therapy are shown in Box 49-13.
BOX 49-13 Corticosteroid therapy
PATIENT & FAMILY TEACHING GUIDE
The nurse needs to teach the patient and family the following:
1. Plan a diet high in protein, calcium (at least 1500 mg per day) and potassium but low in fat and concentrated simple carbohydrates, such as sugar, honey, syrups and boiled lollies.
2. Identify measures to ensure adequate rest and sleep, such as daily naps and avoidance of caffeine late in the day.
3. Develop and maintain an exercise program to help maintain bone integrity.
4. Recognise oedema and ways to restrict sodium intake to less than 2000 mg per day if oedema occurs.
5. Monitor glucose levels and recognise symptoms and signs of hyperglycaemia (e.g. polydipsia, polyuria, blurred vision) and glycosuria (glucose in the urine). The patient should be instructed to report hyperglycaemic symptoms or capillary glucose levels greater than 10 mmol/L or urine positive for glucose.
6. Notify the healthcare provider if experiencing postprandial heartburn or epigastric pain that is not relieved by antacids.
7. See an eye specialist yearly to assess possible development of cataracts.
8. Use safety measures, such as getting up slowly from the bed or a chair, and use good lighting to avoid accidental injury.
9. Maintain good hygiene practices and avoid contact with people with colds or other contagious illnesses to avoid infection.
10. Inform all healthcare providers about long-term corticosteroid use.
11. May need increased doses of corticosteroids in times of physical and emotional stress.
12. Never abruptly stop the corticosteroids as this could lead to Addisonian crisis and possible death.
Hyperaldosteronism
AETIOLOGY AND PATHOPHYSIOLOGY
Hyperaldosteronism is characterised by excessive aldosterone secretion. The main effects of aldosterone are sodium retention and potassium and hydrogen ion excretion. Thus, the hallmark of this disease is hypertension with hypokalaemic alkalosis. Primary hyperaldosteronism (PA) is most commonly caused by a small solitary adrenocortical adenoma. Occasionally, multiple lesions are involved and are associated with bilateral adrenal hyperplasia. PA affects both sexes equally and occurs most frequently between 30 and 49 years of age. It is estimated that approximately 1% of cases of hypertension are caused by PA. Secondary hyperaldosteronism occurs in response to a non-adrenal cause of elevated aldosterone levels, such as renal artery stenosis, renin-secreting tumours and chronic kidney disease.
CLINICAL MANIFESTATIONS
Elevated levels of aldosterone are associated with sodium retention and elimination of potassium. Sodium retention leads to hypernatraemia, hypertension and headache. Oedema does not usually occur because the rate of sodium excretion increases, which prevents more severe sodium retention. The potassium wasting leads to hypokalaemia, which causes generalised muscle weakness, fatigue, cardiac arrhythmias, glucose intolerance and metabolic alkalosis, which may lead to tetany.
DIAGNOSTIC STUDIES
The diagnosis of hyperaldosteronism should be suspected in all hypertensive patients with hypokalaemia who are not being treated with diuretics. PA is associated with elevated plasma aldosterone levels, elevated sodium levels, decreased serum potassium levels and decreased plasma renin activity. Adenomas are localised by means of a CT scan or MRI. For more accurate localisation and confirmation of a functioning adenoma, adrenal vein sampling is undertaken as a vascular radiological procedure.
NURSING AND COLLABORATIVE MANAGEMENT: PRIMARY HYPERALDOSTERONISM
The preferred treatment for PA is surgical removal of the adenoma (adrenalectomy). This surgery can be performed as an open procedure; however, laparoscopic adrenalectomy is increasingly performed because of the benefits this minimally invasive surgery offers.31 Before surgery, patients should be treated with a low-sodium diet, potassium-sparing diuretics (spironolactone, eplerenone) and antihypertensive agents to normalise serum potassium levels and BP. Spironolactone and eplerenone block the binding of aldosterone to the mineralocorticoid receptor in the terminal distal tubules and collecting ducts of the kidneys, thus increasing the excretion of sodium and water and the retention of potassium. Eplerenone is the first agent of a new class of drugs known as selective aldosterone receptor antagonists. Oral potassium supplements and sodium restrictions are also necessary. Potassium supplementation and a potassium-sparing diuretic should not be started simultaneously because of the danger of hyperkalaemia.
Patients with bilateral adrenal hyperplasia are treated with spironolactone, amiloride (another potassium-sparing diuretic) or aminoglutethimide (which blocks aldosterone synthesis). Calcium channel blockers may also be used to control BP, and dexamethasone may be used to decrease the hyperplasia.
Nursing care includes careful assessment for signs of fluid and electrolyte balance (especially potassium) and cardiovascular status. BP should be monitored frequently before and after surgery because unilateral adrenalectomy is successful in controlling hypertension in only 80% of patients with adenoma. Patients receiving maintenance therapy with spironolactone or amiloride need instruction about the possible side effects of gynaecomastia, impotence and menstrual disorders, as well as knowledge about the signs and symptoms of hypokalaemia and hyperkalaemia. Patients should be taught how to monitor their own BP and about the need for frequent monitoring. The need for continued health supervision should be stressed.
Phaeochromocytoma
AETIOLOGY AND PATHOPHYSIOLOGY
Phaeochromocytoma is a rare condition characterised by a tumour of the adrenal medulla that produces excessive catecholamines (adrenaline, noradrenaline).32 It can occur at any age and in either sex, but it is found most commonly in young to middle-aged adults. In most cases affecting adults, the tumour is benign, encapsulated, unilateral and solitary. Occasionally, bilateral tumours are found. The secretion of excessive catecholamines results in severe hypertension. If undiagnosed and untreated, phaeochromocytoma may lead to diabetes mellitus, cardiomyopathy and death.32
CLINICAL MANIFESTATIONS
The most striking clinical features of phaeochromocytoma include severe, episodic hypertension accompanied by the classic triad of severe pounding headache, tachycardia with palpitations and profuse sweating, along with unexplained abdominal or chest pain. Attacks of episodic hypertension are due to sympathetic nervous system stimulation and are often accompanied by anxiety. Attacks may be provoked by many medications, including antihypertensives, opioids, radiological contrast media and tricyclic antidepressants. The duration of an attack may vary from a few minutes to several hours.
DIAGNOSTIC STUDIES
Although phaeochromocytoma is associated with a number of symptoms, the diagnosis is often missed. Phaeochromocytoma is an uncommon cause of hypertension, accounting for 0.1% of all cases. This condition should be considered in patients who do not respond to traditional hypertensive treatments. Phaeochromocytoma might be rare, but making the diagnosis is critical because the malignancy rate is 10%, it may be associated with a familial syndrome, it may precipitate life-threatening hypertension and the patient may be cured completely after tumour removal.33
The measurement of plasma metanephrines, followed by 24-hour urine collection for metanephrines, is the simplest and most reliable test. Values are elevated in at least 95% of patients with phaeochromocytoma. CT scans and MRI are used for tumour localisation.
NURSING AND COLLABORATIVE MANAGEMENT: PHAEOCHROMOCYTOMA
The primary treatment consists of surgical removal of the tumour. Preoperatively, calcium channel blockers such as nicardipine are used to control BP and other excess catecholamine symptoms. Sympathetic blocking agents, such as phenoxybenzamine and prazosin, may also be administered along with the calcium channel blockers to reduce BP and alleviate other symptoms of catecholamine excess. Sympathetic blocking agents may result in orthostatic hypotension, so the patient must be advised to make postural changes cautiously. β-adrenergic blockers are used (e.g. propranolol) to decrease tachycardia and other arrhythmias.32,33
Surgery is more commonly carried out via laparoscopic adrenalectomy than via open abdominal incision. Complete removal of the adrenal tumour cures the hypertension in the majority of individuals but hypertension persists in about 10–30% of patients. For these individuals, BP management involves standard antihypertensive drug therapy. If surgery is not an option, metyrosine is used to diminish catecholamine production by the tumour and simplify chronic management.
Case finding is an important nursing function. Any patient with hypertension accompanied by symptoms of sympathoadrenal discharge should be referred to a specialist for definitive diagnosis. An important part of the nursing assessment is observation of the patient for the classic triad of symptoms of phaeochromocytoma (severe pounding headache, tachycardia and profuse sweating). BP should be monitored immediately if the patient is experiencing an attack. The nurse should be prepared to check BP when any of the drugs that might precipitate an attack are given.
The nurse should attempt to make the patient as comfortable as possible. All diagnostic samples should be collected appropriately. Capillary blood glucose levels should be monitored to assess for diabetes mellitus. Patients need rest, nourishing food and emotional support during this period.34
Preoperative and postoperative care is similar to that for any patient undergoing adrenalectomy except that BP fluctuations from catecholamine excesses tend to be severe and must be monitored carefully. Because hypertension may persist even when the tumour is removed, the nurse should stress the importance of follow-up care and routine BP monitoring. If metyrosine is being used, the patient should be instructed to rise slowly and hold on to a secure object, because this medication can cause orthostatic hypotension.
The patient with Graves’ disease
CASE STUDY
Patient profile
Elizabeth Minton, a 43-year-old woman, admitted to hospital with a high fever. Following an endocrine diagnostic examination, she was diagnosed as having Graves’ disease.
CRITICAL THINKING QUESTIONS
1. What is the aetiology of this patient’s symptoms?
2. What diagnostic studies were probably ordered? What would the results have been to establish the diagnosis of Graves’ disease?
4. What is the purpose of drug therapy for this patient?
5. What are this patient’s immediate learning needs and her learning needs preoperatively and postoperatively? What teaching strategies would the nurse use if this patient could not read?
6. What are the nursing interventions for successful long-term management of this patient after the subtotal thyroidectomy? How would nursing interventions change if this patient was 70 years old?
7. Based on the assessment data presented, write one or more appropriate nursing diagnoses pertinent to this patient while hospitalised. Are there any collaborative problems?
1. Following a hypophysectomy for acromegaly, postoperative nursing care should focus on:
2. A patient with a head injury develops syndrome of inappropriate antidiuretic hormone. Symptoms the nurse would expect to find include:
3. The medical practitioner prescribes levothyroxine for a patient with hypothyroidism. Following teaching regarding this medication, the nurse determines that further instruction is needed when the patient says:
4. Following thyroid surgery, the nurse suspects damage to or removal of the parathyroid glands when the patient develops:
5. An important nursing intervention when caring for a patient with Cushing’s syndrome is to:
6. After an adrenalectomy for phaeochromocytoma, the patient is most likely to experience:
7. To control the side effects of corticosteroid therapy, the nurse teaches the patient who is taking corticosteroids to:
8. The nurse teaches the patient that the best time to take corticosteroids for replacement purposes is:
1 Australian Pituitary Foundation. Acromegaly. Available at www.pituitary.asn.au/ThePituitaryGland/ConditionsIntroduction/Acromegaly.aspx, Updated July 2006. accessed 14 January 2011.
2 Burt MG, Ho KK. Newer options in the management of acromegaly. Intern Med J. 2006;36(7):437–444.
3 Cook DM, Yuen KCJ, Biller BMK, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for growth hormone use in growth hormone-deficient adults and transition patients: 2009 update. Endocrine Prac 2009: 15(2):1–29. Available at www.aace.com/pub/pdf/guidelines/GrowthHormoneGuidelines.pdf. accessed 14 January 2011.
4 Aron DC, Findling JW, Tyrrell JB. Hypothalamus and pituitary gland. In Greenspan FS, Gardner DG, eds.: Basic and clinical endocrinology, 8th edn., New York: McGraw-Hill, 2010.
5 Robinson A, Verbalis J, et al. Posterior pituitary. In Kronenberg HM, Melmed S, Polonsky KS, et al, eds.: Williams textbook of endocrinology, 8th edn., Philadelphia: Saunders, 2008.
6 Mulinda JM. Hypopituitarism (panhypopituitarism): treatment & medication. eMedicine. Available at http://emedicine.medscape.com/article/122287-treatment, Updated April 2010. accessed 14 January 2011.
7 Nakayama S, Yokote T, Kobayashi K, et al. Syndrome of inappropriate antidiuretic hormone secretion associated with acute myeloid leukemia with multilineage dyplasia. Endocrine. 2009;35(3):290–292.
8 Bora K, Chaudhry M. Syndrome of inappropriate antidiuretic hormone secretion. eMedicine. Available at http://emedicine.medscape.com/article/768380-overview, Updated October 2009. accessed 14 January 2011.
9 Robinson A. Posterior pituitary—diabetes insipidus. In Goldman L, Ausiello D, eds.: Cecil textbook of medicine, 23rd edn., St Louis: Saunders, 2008.
10 Mackenzie EJ, Mortimer RH. Thyroid nodules and thyroid cancer, Med J Aust. 2004;180(5):242–247. Available at eMJA www.mja.com.au/public/issues/180_05_010304/mac10351_fm.html accessed 14 January 2011.
11 Shomon M. Managing thyroid disease during and after pregnancy: guidelines. Available at http://thyroid.about.com/od/hormonepregnantmenopause1/ss/pregnancyguide.htm, Updated March 2009. accessed 14 January 2011.
12 Best Practice New Zealand. Management of thyroid dysfunction in adults. Available at www.bpac.org.nz/magazine/2010/december/thyroid.asp, 2010. accessed 14 January 2011.
13 Jett K. Chronic diseases in late life. In Ebersole P, Hess P, Touchy TA, et al, eds.: Toward healthy aging: human needs and nursing responses, 7th edn., Philadelphia: Mosby, 2008.
14 Gordon RC, Rose MC, Skeaff SA, et al. Iron supplementation improves cognition in mildly iron-deficient children. Am J Clin Nutr. 2009;90(9):1264–1271.
15 Gallego G, Goodall S, Eastman CJ. Iodine deficiency in Australia: is iodine supplementation for pregnant and lactating women warranted? Med J Aust 2010; 192:461–463. Available at www.thyroidfoundation.com.au/files/media_releases/Viewpoint.pdf. accessed 14 January 2011.
16 Carson M. Assessment and management of patients with hypothyroidism. Nurs Stand. 2009;23:18.
17 Gibbons V, Conaglen JV, Lillis S, et al. Epidemiology of thyroid disease in Hamilton (New Zealand) general practice. Aust NZ J Pub Health. 2008;32(5):421–423.
18 Vaidya B, Pearce S. Management of hypothyroidism in adults. BMJ. 2008;337:a801.
19 Evans RA, Benson RE, Wyndham N. Primary hyperparathyroidism. Aust NZ J Surg. 2008;40:348–351.
20 Grey A, Bolland M. Evaluation and treatment of primary hyperparathyroidism. JAMA. 2005;294(21):2699–2700.
21 Owens B. A review of primary hyperparathyroidism. J Infus Nurs. 2009;32:87.
22 New drugs. Aust Prescr 2008; 31:108–111. Available at www.australianprescriber.com/magazine/31/4/108/11/. accessed 14 January 2011.
23 Chew S, Leslie D. Clinical endocrinology and diabetes. London: Churchill Livingstone, 2006.
24 Jung C, Inder W. Cushing’s syndrome 2009. Australian Pituitary Foundation; Updated 2009. Available at www.pituitary.asn.au/ThePituitaryGland/ConditionsIntroduction/Cushingssyndrome.aspx accessed 14 January 2011.
25 Neiman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1526–1540.
26 Aron DC, Findling MD, Tyrrell JB. Glucocorticoids and adrenal androgens. In Greenspan FS, Gardner DG, eds.: Basic and clinical endocrinology, 7th edn., New York: McGraw-Hill, 2010.
27 Chrousos GP, Lafferty A. Glucocorticoid therapy and Cushing syndrome: treatment & medication. eMedicine. Available at http://emedicine.medscape.com/article/921086-treatment. accessed 14 January 2011.
28 Stewart P. The adrenal cortex. In Kronenberg HM, Melmed S, Polonsky KS, et al, eds.: Williams textbook of endocrinology, 8th edn., Philadelphia: Saunders, 2008.
29 National Institute of Diabetes and Digestive and Kidney Diseases. Addison’s disease: adrenal insufficiency. Available at http://endocrine.niddk.nih.gov/pubs/addison/addison.htm#emergency. accessed 14 January 2011.
30 Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–1723.
31 McKenzie RJ, Lillegard JB, Young WF, et al, eds. Aldosteronomas—state of the art. Surg Clin North Am. 2009;89:1241.
32 Sweeny A, Blake MA. Pheochromocytoma. eMedicine. Available at http://emedicine.medscape.com/article/124059-overview, Updated April 2010. accessed 14 January 2011.
33 Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium, October 2005. Nat Clin Pract Endocrinol Metab. 2007;3(2):92–102.
Australian Addison’s Disease Association. www.addisons.org.au
Australian and New Zealand Bone and Mineral Society. www.anzbms.org.au
Australian Endocrine Surgeons. www.endocrinesurgeons.org.au
Australian Institute of Health & Welfare. www.aihw.gov.au
Australian Pituitary Foundation. www.pituitary.asn.au
Australian Thyroid Foundation. www.thyroidfoundation.com.au
Bone Health for Life. www.bonehealthforlife.org.au
Endocrine Nurses’ Society of Australasia. www.ensa.org.au
Endocrine Society of Australia. www.endocrinesociety.org.au
MAGIC NZ (Major Aspects of Growth in Children). www.magicfoundation.org
New Zealand Addison’s Network. www.addisons.org.nz
Pituitary Network Association. www.pituitary.org
Thyroid Australia. www.thyroid.org.au
Thyroid Federation International. www.thyroid-fed.org
