15
Right Ventricular Failure
Joshua Sternbach, Francois Haddad, John E. Arbo, and Anne-Sophie Beraud
BACKGROUND
Accurate and rapid assessment of right ventricular failure (RVF) is challenging. The thin-walled right ventricle (RV), which serves as a conduit to the typically high-flow low-pressure pulmonary circulation, is less tolerant of increases in afterload and wall stress. Right ventricular hemodynamic compromise can occur as a result of a variety of clinical insults and can benefit from an equally diverse group of therapies. This chapter presents the clinical approach to the evaluation of the failing right heart, while providing evidence-based strategies for effective therapeutic intervention.
CLASSIFICATION AND EPIDEMIOLOGY
RVF is a complex clinical process defined by the inability to provide adequate blood flow through the pulmonary circulation at a normal central venous pressure (CVP).1,2 Although heart failure is often ascribed to either the left or the right ventricle, the interdependence of these structures renders this division somewhat artificial. Isolated left ventricular failure can create an unhealthy milieu for an otherwise well-functioning RV, just as a struggling RV can hinder the performance of a normal LV. In the latter case, RV distension and diminished contractility may lead to paradoxical interventricular septal motion, producing an acute deficit in LV filling and therefore decreased cardiac output and oxygen delivery.
PATHOPHYSIOLOGY
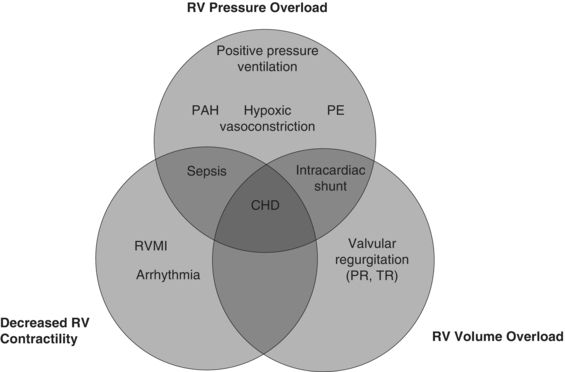
RV dysfunction or failure occurs as the result of one or more of three pathophysiologic processes: RV pressure overload, volume overload, or reduced contractility (see Fig. 15.1). RV pressure overload may occur in conditions such as pulmonary embolism (PE), pulmonary arterial hypertension (PAH) (with or without associated lung disease), and positive pressure ventilation. RV volume overload occurs in the setting of valvular (tricuspid or pulmonic) regurgitation and has particularly deleterious effects on LV systolic function. Finally, depressed RV contractility may manifest as a result of myocardial ischemia, arrhythmia, or sepsis.
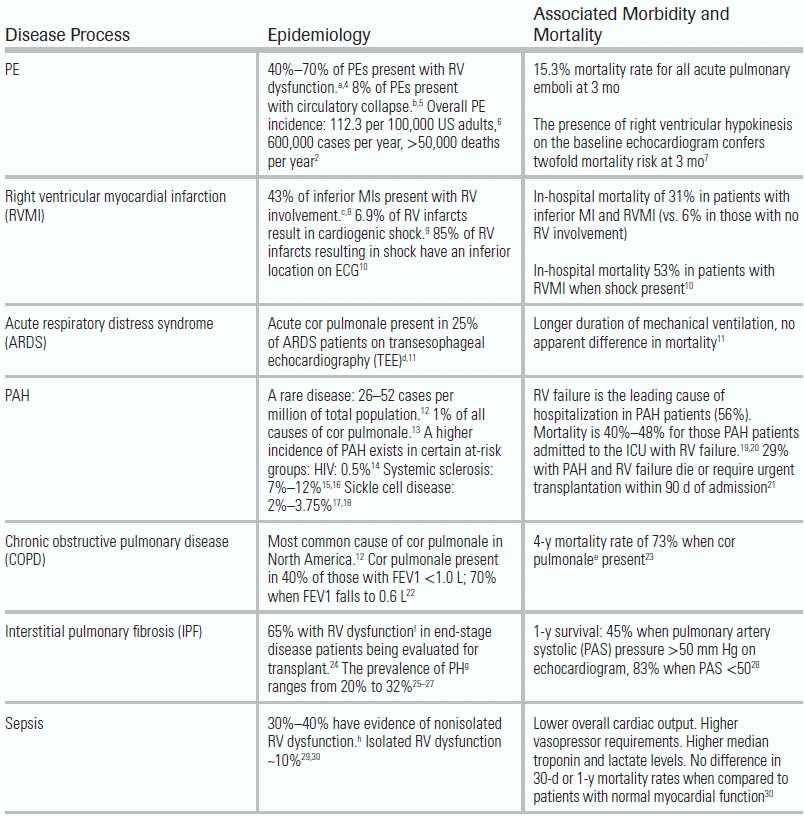
Specific etiologies of RV dysfunction include intrinsic pulmonary or pulmonary vascular conditions (also termed cor pulmonale) or cardiac disease. Data regarding the incidence and associated morbidity and mortality of various causes are presented in Table 15.1. Patients requiring ICU admission for RV failure frequently experience high mortality rates and require prolonged medical intensive care.31,32 Other conditions not mentioned in Table 15.1 that can result in RV failure include adult congenital heart disease, sleep-disordered breathing, any disorder associated with pulmonary hypertension (PH)—including chronic thromboembolic disease—and connective tissue disorders such as scleroderma (Table 15.2).
TABLE 15.1 Epidemiology and Associated Morbidity and Mortality of RV Dysfunction
Reproduced with the permission of Stanford University School of Medicine.
TABLE 15.2 World Health Organization Classification of PH

Adapted from Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classifications of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43.33
HISTORY AND PHYSICAL EXAM
In patients with mild to moderate chronic right heart disease, reports of dyspnea, fatigue, lethargy, light-headedness, angina, and exertional syncope or presyncope will predominate.3 Patients with a more severe disease may initially present with lower extremity edema or ascites, prompting complaints of abdominal pain or fullness due to hepatic congestion.
In a patient with established RV dysfunction, a careful review of medications is an essential part of the initial patient history, as any missed doses or lapses in compliance can result in a dramatic clinical decline. Patients with chronic PH treated with vasodilatory agents—or patients with chronic lung diseases treated with daily inhalers or immunomodulators—may develop advanced symptoms, despite seemingly minimal changes in their prescribed routine. Likewise, recreational or illicit drug use may trigger new, or exacerbate chronic, RV dysfunction. In particular, amphetamines, cocaine, and dietary supplements containing fenfluramine constitute a common cohort of agents known to cause PH and subsequent cor pulmonale.34,46
The initial physical exam centers on a determination of CVP and signs of low cardiac output or hemodynamic compromise. In the absence of significant tricuspid valve stenosis, CVP provides a reasonable estimate of the filling pressure of the RV, in much the same way that the pulmonary capillary wedge pressure can estimate LV diastolic pressure.1 Jugular venous pressure (JVP) is a surrogate for CVP, but it can be measured by direct visualization of the internal jugular vein. To perform the exam, position the patient at a 45-degree angle and measure at the vertical height of the jugular vein, just above the sternal angle (or angle of Louis).36 Calculate the JVP by adding 5 cm to this height (the distance from the middle of the right atrium to the sternal angle). CVP can also be estimated using ultrasound and examining the inferior vena cava (IVC) diameter (see Echocardiography below). A JVP >8 cm is suggestive of elevated right-sided filling pressures.
Additional physical exam signs suggestive of RV failure include an increase in intensity of the pulmonic component of S2, a narrowly split S2, a holosystolic murmur at the left lower sternal border (usually compatible with tricuspid regurgitation), a prominence of the “A” wave in the jugular venous pulse (corresponding to atrial contraction), and either a left parasternal heave or a downward subxiphoid thrust. In the patient presenting with severe decompensated disease, systemic signs of cardiogenic shock may manifest as cool extremities, diminished peripheral pulses, and decreased urine output.37
DIFFERENTIAL DIAGNOSIS
Identifying conditions that mimic RV failure requires consideration of disease processes that cause either a real or perceived elevation in CVP. For example, constrictive pericarditis produces elevated right-sided filling pressures with elevations in CVP as well as signs and symptoms of acute heart failure. Superior vena cava syndrome, as might be seen in patients with a history of central venous catheters, device placement, or known malignancies, can present with an increase in jugular venous distension, or height, without a true elevation in right atrial pressure.
Because lower extremity edema, abdominal ascites, and even hepatic engorgement can result from RVF, other syndromes marked by anasarca—or total body fluid excess—must also be excluded. Cirrhosis, nephrotic syndrome, or disorders disruptive of the hepatic circulation (such as Budd-Chiari or hepatic venoocclusive disease) may produce a pattern of edema identical to that found in states of RVF.
DIAGNOSTIC EVALUATION
Electrocardiogram
The initial evaluation of a patient with suspected RV dysfunction should begin with an assessment using simple and noninvasive diagnostic modalities. An electrocardiogram (ECG) can offer significant insight into the presence or absence of RV strain. Significant ECG findings include an S1Q3T3 pattern, right axis deviation, or signs of RV hypertrophy such as a dominant R wave in V1 and V2 with a prominent S wave in V5 and V6.38 A low threshold for performing a right-sided ECG (providing the additional leads of V3R to V6R) can improve the detection of RV infarction and inferior wall ischemia.8 In a prospective study of patients with acute inferior myocardial infarction (MI) complicated by RV involvement (evidenced by ST-segment elevation in V4R), in-hospital mortality rate was significantly higher when compared to those with inferior MI in the absence of RV ischemia (31% vs. 6%, respectively).39 Posterior MI, especially in patients with a right dominant coronary circulation, represents another potential insult to RV perfusion. This can manifest with subtle ECG findings, including prominent R wave amplitude and ST depressions in V1 to V3. However, more demonstrative ST-segment elevations can become evident with the use of posterior or the so-called esophageal ECG leads (V7 to V9).40
Chest Radiography
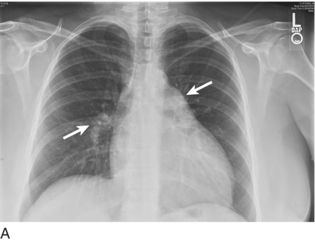
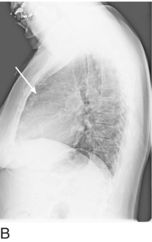
Appreciating signs of RV enlargement on a single- or two-view chest x-ray (CXR) requires close attention to subtle findings. PH and RV hypertrophy may produce enlargement of the proximal pulmonary arteries or a reduction in retrosternal air space (Fig. 15.2). Classic CXR findings in PE include Westermark sign (focal oligemia) and Hampton hump (peripheral area of opacification representing pulmonary infarction); note that these findings lack in sensitivity and specificity.41 The absence of pulmonary edema in the setting of elevated JVP is considered to be the most specific sign for isolated RVF.1,2 Once conventional imaging studies such as CXR and computed tomography demonstrate evidence for RV dysfunction, however, the disease process is likely already advanced.
Echocardiography
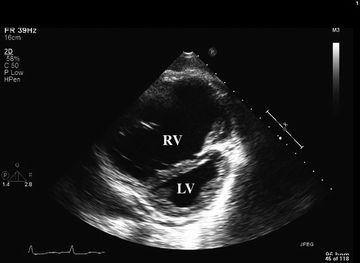
Bedside portable ultrasound and echocardiography has gained widespread popularity and can provide direct assessment of cardiac function. Findings indicative of RV dysfunction include tricuspid regurgitation, RV hypokinesis or dilatation (Fig. 15.3), right atrial enlargement, and paradoxical septal motion.12,37 CVP can be evaluated echocardiographically by measurement of the degree of IVC collapse with inspiration.42 RV failure due to acute PE—or occasionally MI—may present with diffuse hypokinesis of the RV free wall sparing the apex (McConnell sign).3 Echocardiographic findings in RV failure are discussed in further detail in Chapter 6.
Laboratory Investigation
Poor end-organ perfusion as a result of diminished right heart function may be suggested by elevations in serum lactate or evidenced by markers of organ-specific injury such as serum creatinine (kidney) or hepatic function tests and bilirubin (liver).37 Troponin and brain natriuretic peptide (BNP)—two biomarkers of cardiac injury—also have predictive value in the setting of RV dysfunction due to PE or acute exacerbations of PH. In acute PE, an elevated troponin T was found to be associated with in-hospital and 30-day mortality, prolonged hypotension, cardiogenic shock, and need for resuscitation.43,44 BNP has also been shown to have significant predictive value for outcomes in PE. In one study of 79 patients with acute PE, those who had an uncomplicated clinical course (16.5%) all had normal BNP values, whereas all in-hospital deaths and serious events occurred in the group with elevated measures. BNP also serves as a reliable marker of RV strain and correlates well with echocardiographic measurements of RV/LV ratio and IVC dimension.45 In patients with PAH requiring hospitalization for ICU management of acute right heart failure, elevations in admission BNP, C-reactive protein, and creatinine were found to negatively correlate with survival.20
MONITORING AND SUPPORTIVE CARE
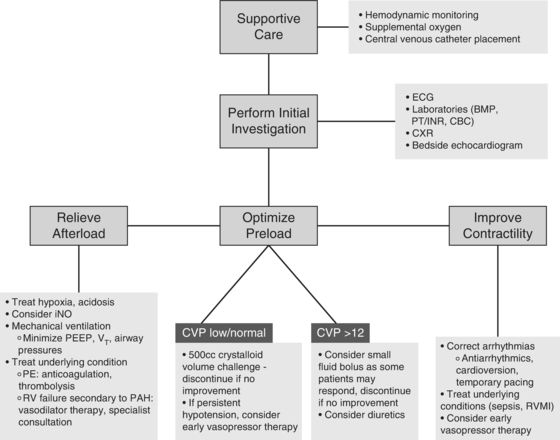
Initial treatment of RVF requires the basic supportive care necessary for all critically ill patients. Monitoring of blood pressure, heart rate, and pulse oximetry—whether performed noninvasively or with the assistance of an intra-arterial catheter—is essential. Central venous access, in addition to enabling complex pharmacotherapy, offers the additional benefit of regular CVP measurement and central venous oxygen saturation (ScvO2), data that can assist in the assessment of response to vasoactive medications and fluid therapy.
Supplemental oxygen administration to maintain oxygen saturation >92% can reverse one of the more injurious influences on RV afterload, namely, hypoxic vasoconstriction (see below). Basic lab work, including a complete blood count, coagulation studies, and a comprehensive metabolic panel, permits identification of easily correctable disorders such as anemia, electrolyte or acid/base abnormalities, as well as renal or hepatic dysfunction.
Once advanced monitoring is in place, and initial diagnostic studies performed, targeted interventions should be implemented to relieve RV strain. Prior to initiating medical therapy, however, consideration of the following unique characteristics of RV failure can help avoid unintentional exacerbation of cardiopulmonary function.
Afterload Reduction
Administration of RV afterload-reducing agents such as inhaled nitric oxide can precipitate acute pulmonary edema if the RV failure is secondary to a left-sided cardiac process (e.g., LV systolic or diastolic failure, mitral stenosis). This occurs as a result of decreased pulmonary vascular resistance in the face of a relatively fixed left-sided obstruction or defect.46
Inotropic Support
In the case of RV failure presenting with systemic hypotension, initiation of certain vasopressor therapies may become necessary. There is no evidence to suggest that one inotrope or vasopressor has greater success than another at maintaining circulatory support in RVF. However, careful consideration should precede the use of strong alpha-1 agonists as they can increase pulmonary vascular tone and further impinge on RV function.
Volume Resuscitation
Although a trial of intravenous volume administration may be appropriate in hypotensive patients without an increased CVP or evidence of pulmonary edema (as in the case of right ventricular MI), signs of RV volume overload including a CVP of >12 to 15 mm Hg should preclude this therapy. In these instances, initiation of vasopressors or inotropes may be preferable; if uncertainty exists, aggressive fluid therapy should be avoided. A trial bolus of 500 cc of crystalloid, with careful monitoring of clinical response, should be used instead.3,47
MANAGEMENT GUIDELINES
Oxygen
Hypoxic pulmonary vasoconstriction—although normally a protective physiologic response against V/Q mismatching when pO2 levels drop below 50 to 60 mm Hg—requires a reversal in instances of life-threatening RV failure. Application of supplemental oxygen via nasal cannula, Venturi mask, simple face mask, nonrebreathing face mask, or other device depending on the degree of hypoxia can reduce pulmonary vascular resistance.48 Additionally, systemic acidosis, if present, can worsen hypoxic vasoconstriction and should be corrected.49
Nitric Oxide
Available in the inhaled form (iNO), nitric oxide can diffuse rapidly across the alveolocapillary membrane into adjacent smooth muscle of pulmonary vessels to increase cyclic guanosine monophosphate, leading to vasodilatation. In right heart failure (RHF), iNO is an attractive therapeutic option given its ability to preferentially improve perfusion to well-ventilated pulmonary segments while avoiding unwanted systemic hypotension (prevented by the scavenging of NO by native hemoglobin). Initial dosing begins at 20 parts per million or less; higher concentrations provide little additional hemodynamic benefit.50 Disadvantages of iNO include the risk of developing methemoglobinemia as well as rebound PH upon discontinuation.51,52
Vasopressors and Inotropes
Improvement of a poorly contractile RV requires augmentation of ventricular perfusion, RV ejection fraction, and ultimately stroke volume. Various agents have been employed for this purpose, and each agent comes with different risks and benefits depending on the underlying etiology of the dysfunction (Table 15.3). Definitive evidence to guide choice of vasopressor or inotrope in patients with acute RHF is lacking. Experimental studies suggest that dobutamine may be more beneficial than norepinephrine.47,54 A recent extensive literature review gave weak evidence-based recommendations for the use of norepinephrine, vasopressin, dobutamine, and levosimendan. Strong recommendations were made for the use of PDE3 inhibitors (e.g., milrinone) for improvement in RV performance and reduction in pulmonary vascular resistance (PVR). A strong recommendation advised against the use of dopamine in cardiogenic shock secondary to an increase in tachyarrhythmias.55
TABLE 15.3 Common Agents Used in the Treatment of RV Failure
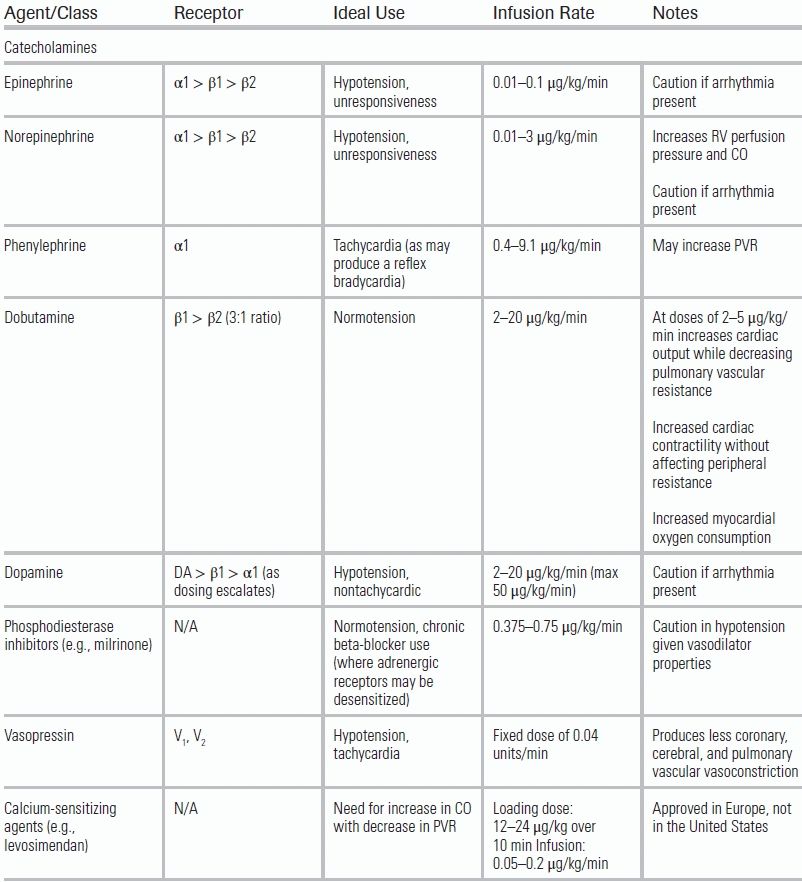
Adapted from Overgaard CB, Dzavík V. Inotropes and vasopressors: review of physiology and clinical use in cardiovascular disease. Circulation. 2008;118(10):1047–1056.53
Diuretics
Although a key therapy for the volume-overloaded RV, achieving clinically meaningful diuresis in the emergency department may be impractical and, at times, detrimental depending upon the patient's hemodynamic stability. Inpatient therapy in the intensive care unit may include salt and fluid restriction, intermittent or continuous administration of loop diuretics, or more rigorous approaches such as continuous or intermittent renal replacement therapy.56 In the emergency setting, however, a more reasonable approach may be to pursue diuresis or natriuresis by augmenting RV contractility, cardiac output, and, by extension, renal perfusion.
Antiarrhythmics
The importance of maintaining sinus rhythm and heart rate control during an episode of acute right heart failure cannot be overstated. Atrial tachyarrhythmias, such as atrial fibrillation or flutter, are the most common ectopic rhythms observed in RV failure and can increase morbidity and mortality.47 Although no randomized, controlled data exist to guide the management of unstable atrial arrhythmias, consensus guidelines characterize unstable patients as those with ventricular rates >150 accompanied by ongoing chest pain or poor perfusion (a systolic blood pressure <90, heart failure, or reduced level of consciousness).57,58 Patients with hemodynamic instability due to a tachyarrhythmia, regardless of the presence of RV strain, should receive direct current cardioversion for restoration of sinus rhythm. For those deemed appropriate for rate control or pharmacologic therapy, evidence for specific treatment in RVF is once again sparse. Digoxin, although not a first-line agent in those who can tolerate calcium channel blockers or beta-blockers, may be useful in decompensated heart failure given its modest inotropic properties and ability to slow conduction velocity. PH patients with evidence of RV dysfunction in sinus rhythm have demonstrated improvements in cardiac output following the administration of this agent.59 Figure 15.4 summarizes a therapeutic approach to the patient with RV failure.
SPECIAL CONSIDERATIONS
Sepsis
The current accepted definition of sepsis-induced myocardial dysfunction requires only a reduced LV ejection fraction—in the absence of cardiac disease—that demonstrates reversibility on the correction of the septic state.30 Sepsis-induced cardiomyopathy affects both ventricles and causes dilation, diminished ejection fraction, and poor response to fluid resuscitation and catecholamines. The underlying pathophysiologic mechanisms are myriad; however, increased concentrations of myocardial suppressant substances such as bacterial toxins, cytokines, tumor necrosis factor–alpha, interleukin-1, and nitric oxide have been implicated. Key treatment strategies involve correction of the underlying infectious disease while providing adequate fluid resuscitation and vasopressor support so as to optimize mean arterial pressure and organ perfusion.60
Severe Pulmonary Hypertension
Whether idiopathic or originating from a known cause, this disease group may require consideration of specialized therapies such as prostacyclin derivatives (e.g., epoprostenol, iloprost), endothelin receptor antagonists (e.g., bosentan), and phosphodiesterase inhibitors (e.g., sildenafil) due to marked elevations in pulmonary vascular resistance. However, such advanced pharmacologic interventions can usually be delayed until clinical stability and transfer to an intensive care setting has occurred. Enlisting specialist consultation early in the clinical course should always be considered.
Massive Pulmonary Embolism
The defining characteristics of massive PE include arterial hypotension (systolic arterial pressure <90 mm Hg or a drop in systolic arterial pressure of at least 40 mm Hg for at least 15 minutes) and cardiogenic shock.61 Diagnosis is suggested by RV strain demonstrated on ECG as well as RV dilation on echocardiogram. Following diagnosis, aggressive interventions such as thrombolysis or thrombectomy (performed via surgical or interventional radiologic methods) in addition to systemic anticoagulation may become necessary. A detailed discussion of massive PE is provided in Chapter 11.
Mechanical Ventilation
In the event that intubation and mechanical ventilation become necessary, the physiologic characteristics of RV dysfunction should be carefully considered. High tidal volumes (VT) and positive end-expiratory pressures (PEEP) can increase pulmonary arterial and right atrial pressures, worsen tricuspid regurgitation, increase RV afterload, and decrease preload.55 This contrasts with the influence of positive pressure ventilation on the failing left ventricle, where reductions in preload and afterload can be a welcome effect.62 As such, the lowest VT and PEEP settings should be sought in order to help preserve adequate oxygenation and ventilation. It is equally important to avoid excessive hypercapnia, which can exacerbate pulmonary vasoconstriction and lead to an increase in RV afterload. This can be attenuated with some measure of hyperventilation, although care must be taken to avoid air trapping (especially in those with a history of obstructive lung disease) that can lead to elevated pleural and pericardial pressures and impaired diastolic filling.3
Pregnancy
Maternal and fetal mortality risk is increased in the presence of cardiac disease. Pregnancy in the setting of PH, for example, is associated with a combined mortality rate that approaches 50%.63 In general, the goals of treatment in pregnant patients are similar to those in nonpregnant patients. Periods of greatest risk include the second trimester and active labor and delivery.47 Consultation with a cardiologist, and if available, a maternal–fetal medicine specialist, is warranted early in the clinical course.
Advanced Mechanical Support
In more severe cases of circulatory collapse, advanced mechanical support devices, including ventricular assist devices or extracorporeal membrane oxygenation, may have clinical utility. These interventions as well as measures such as intraaortic balloon counterpulsation and atrial septostomy have been implemented successfully in cases of severe PH and massive PE and as a bridge to lung transplantation.64–66 Although only low-grade evidence exists for their use, such salvage therapies merit consideration, especially in centers with advanced cardiothoracic surgical capability.
CONCLUSION
Recognizing and successfully treating acute right heart failure require the consideration of a unique collection of illnesses and thoughtful integration of critical care resources. Although some of the available strategies and pharmacotherapies differ from those used in left ventricular disease, the underlying treatment principles are much the same. All clinical efforts should be aimed at preserving and aiding myocardial function, while maintaining focus on correction of precipitating illness and contributing systemic comorbidities. Historically, right ventricular dysfunction has been the subject of less academic study than left ventricular disorders. Future investigation, from both the clinical and basic science realms, will be needed to delineate the optimal therapeutic approach. At present, clinicians faced with the acutely failing RV may still draw upon a broad armamentarium to achieve hemodynamic and respiratory stability.
LITERATURE TABLE
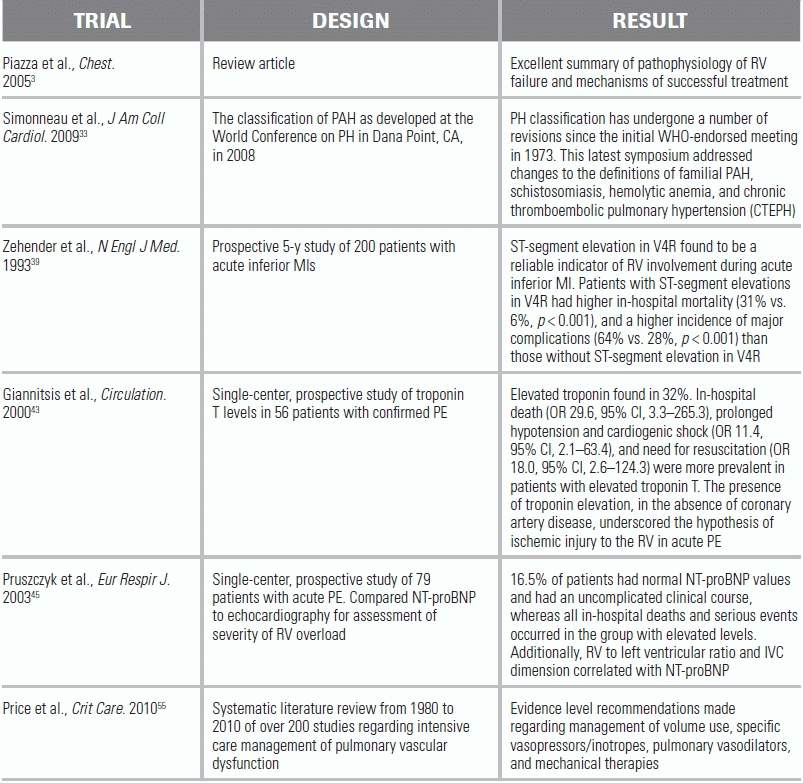
CI, confidence interval; OR, odds ratio.
REFERENCES
1.Greyson CR. Pathophysiology of right ventricular failure. Crit Care Med. 2008;36(suppl 1):S57–S65.
2.Greyson CR. Right heart failure in the intensive care unit. Curr Opin Crit Care. 2012;18(5):424–431.
3.Piazza G, Goldhaber SZ. The acutely decompensated right ventricle, pathways for diagnosis and management. Chest. 2005;128:1836–1852.
4.ten Wolde M, Söhne M, Quak E, et al. Prognostic value of echocardiographically assessed right ventricular dysfunction in patients with pulmonary embolism. Arch Intern Med. 2004;164(15):1685–1689.
5.Stein PD, Beemath A, Matta F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med. 2007;120(10):871–879.
6.Wiener RS, Schwartz LM, Woloshin S. Time trends in pulmonary embolism in the United States: evidence of overdiagnosis. Arch Intern Med. 2011;171(9):831.
7.Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER). Lancet. 1999;353(9162):1386–1389.
8.Braat SH, Brugada P, de Zwaan C, et al. Value of electrocardiogram in diagnosing right ventricular involvement in patients with an acute inferior wall myocardial infarction. Br Heart J. 1983;49(4):368.
9.Mehta SR, Eikelboom JW, Natarajan MK, et al. Impact of right ventricular involvement on mortality and morbidity in patients with inferior myocardial infarction. J Am Coll Cardiol. 2001;37:37–43.
10.Jacobs AK, Leopold JA, et al. Cardiogenic shock caused by right ventricular infarction: a report from the SHOCK registry. J Am Coll Cardiol. 2003;41:1273–1279.
11.Vieillard-Baron A, et al. Acute cor pulmonale in acute respiratory distress syndrome submitted to protective ventilation: incidence, clinical implications, and prognosis. Crit Care Med. 2001;29(8):1551–1555.
12.Peacock AJ, Murphy NF, McMurray JJV, et al. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30:104–109.
13.Budev MM, Arroliga AC, Wiedemann HP, et al. Cor Pulmonale: an overview. Semin Respir Crit Care Med. 2003;24(3):233–244.
14.Sitbon O, Lascoux-Combe C, Delfraissy JF, et al. Prevalence of HIV-related pulmonary arterial hypertension in the current antiretroviral therapy era. Am J Respir Crit Care Med. 2008;177:108–113.
15.Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a french nationwide prospective multicenter study. Arthritis Rheum. 2005;52:3792–3800.
16.Mukerjee D, St George D, Coleiro B, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2003;62:1088–1093.
17.Machado RF, Gladwin MT. Pulmonary hypertension in hemolytic disorders: pulmonary vascular disease: the global perspective. Chest. 2010;137:30S–38S.
18.Fonseca GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by right heart catheterization in sickle cell disease. Eur Respir J. 2012;39(1):112–118.
19.Campo A, Mathai SC, Le Pavec J, et al. Outcomes of hospitalisation for right heart failure in pulmonary arterial hypertension. Eur Respir J. 2011;38(2):359–367.
20.Sztrymf B, Souza R, Bertoletti L, et al. Prognostic factors of acute heart failure in patients with pulmonary arterial hypertension. Eur Respir J. 2010;35:1286–1293.
21.Haddad F, Peterson T, Fuh E, et al. Characteristics and outcome after hospitalization for acute right heart failure in patients with pulmonary arterial hypertension. Circ Heart Fail. 2011;4(6):692–699.
22.Macnee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1994;150(3):833–852.
23.Renzetti AD, McClement JG, Litt BD. The Veterans administration cooperative study of pulmonary function. Am J Med. 1966;41:115–129.
24.Vizza CD, Lynch JP, Ochoa LL, et al. Right and left ventricular dysfunction in patients with severe pulmonary disease. Chest. 1998;113:576–583.
25.Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–752.
26.Patel NM, Lederer DJ, Borczuk AC, et al. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132(3):998.
27.Shorr AF, Davies DB, Nathan SD. Outcomes for patients with sarcoidosis awaiting lung transplantation. Chest. 2002;122:233–238.
28.Strange C, Highland KB. Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med. 2005;11(5):452–455.
29.Redl G, Germann P, Plattner H, et al. Right ventricular function in early septic shock states. Intensive Care Med. 1993;19(1):3–7.
30.Pulido JN, Afessa B, Masaki M, et al. Clinical spectrum, frequency, and significance of myocardial dysfunction in severe sepsis and septic shock. Mayo Clin Proc. 2012;87(7):620–628.
31.Green EM, Givertz MM. Management of acute right ventricular failure in the intensive care unit. Curr Heart Fail Rep. 2012;9(3):228–235.
32.Nieminen MS, Brutsaert D, Dickstein K, et al. EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J. 2006;27:2725–2736.
33.Simonneau G, Robbins IM, Beghetti M, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2009;54:S43.
34.Brenot F, Herve P, Petitpretz P, et al. Primary pulmonary hypertension and fenfluramine use. Br Heart J. 1993;70(6):537.
35.Albertson TE, Walby WF, Derlet RW. Stimulant-induced pulmonary toxicity. Chest. 1995;108(4):1140.
36.Cook DJ, Simel DL. The rational clinical examination. Does this patient have abnormal central venous pressure? JAMA. 1996;275(8):630–637.
37.Matthews JC, McLaughlin V. Acute right ventricular failure in the setting of acute pulmonary embolism or chronic pulmonary hypertension: a detailed review of the pathophysiology, diagnosis, and management. Curr Cardiol Rev. 2008;4(1):49–59.
38.Harrigan RA, Jones K. ABC of clinical electrocardiography. Conditions affecting the right side of the heart. BMJ. 2002;324(7347):1201–1204.
39.Zehender M, Kasper W, Kauder E, et al. Right ventricular infarction as an independent predictor of prognosis after acute inferior myocardial infarction. N Engl J Med. 1993;328(14):981–988.
40.Casas RE, Marriott HJ, Glancy DL. Value of leads V7-V9 in diagnosing posterior wall acute myocardial infarction and other causes of tall R waves in V1-V2. Am J Cardiol. 1997;80(4):508.
41.Stein PD, Terrin ML, Hales CA, et al. Clinical, laboratory, roentgenographic, and electrocardiographic findings in patients with acute pulmonary embolism and no pre-existing cardiac or pulmonary disease. Chest. 1991;100(3):598.
42.Blehar DJ, Dickman E, Gaspari R. Identification of congestive heart failure via respiratory variation of inferior vena cava diameter. Am J Emerg Med. 2009;27:71–75.
43.Giannitsis E, Müller-Bardorff M, Kurowski V, et al. Independent prognostic value of cardiac troponin T in patients with confirmed pulmonary embolism. Circulation. 2000;102(2):211–217.
44.Becattini C, Vedovati MC, Agnelli G. Prognostic value of troponins in acute pulmonary embolism: a meta-analysis. Circulation. 2007;116:427–433.
45.Pruszczyk P, Kostrubiec M, Bochowicz A, et al. N-terminal pro-brain natriuretic peptide in patients with acute pulmonary embolism. Eur Respir J. 2003;22(4):649–653.
46.Bocchi EA, Bacal F, Auler JO Jr, et al. Inhaled nitric oxide leading to pulmonary edema in stable severe heart failure. Am J Cardiol. 1994;74:70–72.
47.Haddad F, Doyle R, Murphy DJ, et al. Right ventricular function in cardiovascular disease, part II: pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117(13):1717–1731.
48.Forfia PR, Vaidya A, Wiegers SE. Pulmonary heart disease: the heart-lung interaction and its impact on patient phenotypes. Pulm Circ. 2013;3(1):5–19.
49.Lejeune P, Brimioulle S, Leeman M, et al. Enhancement of hypoxic pulmonary vasoconstriction by metabolic acidosis in dogs. Anesthesiology. 1990;73(2):256–264.
50.Ichinose F, Roberts JD, Zapol WM. Inhaled nitric oxide: a selective pulmonary vasodilator current uses and therapeutic potential. Circulation. 2004;109(25):3106–3111.
51.Weinberger B, Laskin DL, Heck DE, et al. The toxicology of inhaled nitric oxide. Toxicol Sci. 2001;59(1):5–16.
52.Christenson J, Lavoie A, O'Connor M, et al. The incidence and pathogenesis of cardiopulmonary deterioration after abrupt withdrawal of inhaled nitric oxide. Am J Respir Crit Care Med. 2000;161(5):1443.
53.Overgaard CB, Dzavík V. Inotropes and vasopressors: review of physiology and clinical use in cardiovascular disease. Circulation. 2008;118(10):1047–1056.
54.Kerbaul F, Rondelet B, Motte S, et al. Effects of norepinephrine and dobutamine on pressure load-induced right ventricular failure. Crit Care Med. 2004;32:1035–1040.
55.Price, LC, Wort SJ, et al. Pulmonary vascular and right ventricular dysfunction in adult critical care: current and emerging options for management: a systemic literature review. Crit Care. 2010;14:R169.
56.Lahm T, McCaslin CA, Wozniak TC, et al. Medical and surgical treatment of acute right ventricular failure. J Am Coll Cardiol. 2010;56(18):1435–1446.
57.Khoo CW, Lip GY. Acute management of atrial fibrillation. Chest. 2009;135(3):849–859.
58.Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745.
59.Rich S, Seidlitz M, Dodin E, et al. The short-term effects of digoxin in patients with right ventricular dysfunction from pulmonary hypertension. Chest. 1998;114(3):787–792.
60.Romero-Bermejo FJ, Ruiz-Bailen M, Gil-Cebrian J, et al. Sepsis-induced cardiomyopathy. Curr Cardiol Rev. 2011;7(3):163–183.
61.Kucher N, Goldhaber SZ. Management of massive pulmonary embolism. Circulation. 2005;112(2):e28.
62.Pinsky MR. Cardiovascular issues in respiratory care. Chest. 2005;128(5 suppl 2):592S–597S.
63.Franklin WJ, Benton MK, Parekh DR. Cardiac disease in pregnancy. Tex Heart Inst J. 2011;38(2):151–153.
64.Chan CY, Chen YS, Ko WJ, et al. Extracorporeal membrane oxygenation support for single lung transplantation in a patient with primary pulmonary hypertension. J Heart Lung Transplant. 1998;17:325–327.
65.Gregoric ID, Chandra D, Myers TJ, et al. Extracorporeal membrane oxygenation as a bridge to emergency heart-lung transplantation in a patient with idiopathic pulmonary arterial hypertension. J Heart Lung Transplant. 2008;27:466–468.
66.Deehring R, Kiss AB, Garrett A, et al. Extracorporeal membrane oxygenation as a bridge to surgical embolectomy in acute fulminant pulmonary embolism. Am J Emerg Med. 2006;24:879–880.