LITERATURE TABLE
RCT, randomized controlled trial.
BACKGROUND
Sickle cell disease (SCD) is one of the most common genetic disorders in North America, affecting an estimated 70,000 individuals. It results from a point mutation (valine for glutamate) at codon 6 of the beta-globin gene on chromosome 11. The most common genotype, HbSS (SCD-SS, sickle cell anemia), is due to mutation of both genes leading to the production of sickle hemoglobin (HbS) and no normal adult hemoglobin (HbA). HbSC and HbS/beta-thalassemia are two other commonly encountered genotypes of SCD.
Under physiologic stress, the HbS undergoes conformational change, creates polymers in the red blood cell (RBC), and deforms the cell to a sickle shape. This change has two consequences. First, it results in hemolysis of the RBC, which causes anemia and the release of free Hb into the circulation; this, in turn, produces a vasculopathy by triggering an abnormal increase in nitric oxide consumption and, by modulating arginine pathways, nitric oxide underproduction. Second, it results in vasoocclusion of microcirculatory organ beds and an ischemic–reperfusion pattern of injury.1 It is this second consequence that produces bony pain, the most common clinical presentation of SCD.2
SCD is a lifelong, multisystem disorder with variable and intermittent severity. Many adult patients with SCD are not registered in a comprehensive care center and are therefore at risk of developing significant end-organ dysfunction over time. This inadequate access to care is made worse by the fact that hydroxyurea, the only U.S. FDA-approved disease-modifying treatment, is underutilized.3
DIAGNOSTIC EVALUATION
An acute sickle cell vasoocclusive episode or “crisis” (VOC) is a frequent complication of SCD and the most common reason these patients present to the emergency department (ED).4 Patients typically present with limb, back, or chest pain caused by vasoocclusion in the bone marrow and resultant severe generalized bony pain.
Prompt clinical assessment and provision of rapid, adequate, and sustained analgesia are key to successful treatment. Clinical assessment should focus on the location of pain, severity of pain (using an objective pain scale such as the visual analogue scale), and duration of pain; precipitating factors (extremes of temperature, dehydration, infection, psychological distress, menstruation in females, excessive exercise); and home analgesic use. A systems-based patient history and physical exam should work to identify additional complications—related to SCD or to other general medical/surgical conditions—that may require specific treatment. Complications commonly associated with VOCs include infection (particularly respiratory tract), stroke, cholecystitis, sequestration syndrome (presenting with organomegaly), and, in males, priapism.
A detailed medical history can help identify SCD patients with severe sickle cell phenotype and should include the use of hydroxyurea, a history of multiple transfusions, previous exchange transfusion, prior acute chest syndrome (ACS), or intensive care unit admission. A thorough transfusion history will also assist the blood bank in sourcing safe units of blood, when required.
Laboratory and imaging testing rarely provide much assistance in the management algorithm of an uncomplicated VOC, with the exception of a complete blood count and reticulocyte count. The reticulocyte count is helpful in determining whether an unexpectedly severe anemia is due to marrow aplasia (usually viral in etiology) or simply brisk hemolysis. There is no indication for chest radiograph (CXR) in the absence of hypoxia or respiratory symptoms and signs. Routine biochemistry will usually confirm the hemolytic process and normal renal function. All SCD patients presenting with a fever, even if they otherwise appear well, should undergo a full septic screen, as they are predisposed to infection as a result of functional asplenia. In patients with new and unexplained hypoxia and an unremarkable CXR, a diagnosis of pulmonary embolism should be considered.5
MANAGEMENT OF AN UNCOMPLICATED PAIN EPISODE
The mainstay of the management of an acute, uncomplicated VOC is supportive care, including analgesia, hydration, and oxygenation.6,7
Rapid administration of adequate analgesia optimizes chances for patient discharge. Analgesia should be initiated within 30 minutes of presentation, with adequate pain control achieved ideally within 60 minutes.8 Pain should be reassessed, and vital signs checked, on a frequent basis until pain is controlled. Patients with SCD usually have had previous exposure to opiate analgesia and often require higher doses of opiates to achieve analgesia when compared to opiate-naive patients. Despite a paucity of supporting clinical trials, multimodal analgesia is recommended, including combination of a nonsteroidal anti-inflammatory drug (NSAID) and acetaminophen with an opiate.1,2,9 Consultation with a pain service may be helpful for patients with difficult-to-control pain (i.e., pain that cannot be controlled without inducing significant side effects or excessive sedation).
The route and formulation of analgesia depend on local institutional policies; there is little high-quality clinical trial evidence to guide specific recommendations. Analgesics are often administered intravenously, but this can be challenging in patients with poor venous access, and subcutaneous or oral administration can be equally effective.10 Intramuscular administration is not recommended due to pain at the injection site and unpredictable absorption. Both morphine sulfate11 and hydromorphone are available for intravenous and subcutaneous administration, as well as in both immediate-acting and slow-release oral liquid and tablet formulations. Using these agents in a combination of intravenous and oral administration allows for background analgesia with breakthrough dosing, permits patients to be discharged on a weaning dose of opiates, and removes the need to switch class of agent. If switching from one opiate to another is unavoidable, care should be taken to ensure bioequivalent dosing.
Certain analgesics have specific risks. Morphine sulfate has been weakly associated with an increased risk of developing ACS,12 and oxycodone has been associated with an increased risk of opiate dependency. Meperidine is contraindicated due to cerebral agitation and risk of seizure. Adjunct laxatives and an antihistamine should be prescribed as required. Acute painful episodes in pregnancy should be managed as at other times but with close monitoring of fetal movements and avoidance of NSAIDs, especially in the first trimester and after 32 weeks' gestation.13
Reduced renal tubular concentrating ability is common in patients with SCD and predisposes to dehydration. Continued fluid loss without adequate replacement causes a reduction in plasma volume with an increased blood viscosity and aggravation of the sickling process. However, the concurrent use of opiates for pain control can increase vascular leak and predisposes SCD patients to pulmonary edema. The goal of hydration should, therefore, be to replace estimated deficits and provide adequate maintenance while avoiding excessive hydration. Oral hydration, if tolerated, is preferred.
Patients often feel symptomatic benefit from supplemental oxygen, even when pulse oximetry is normal. Since SCD is a prothrombotic disorder, once a patient is admitted to the hospital, or in cases of extended ED stay, pharmacologic venous thromboembolism prophylaxis should be instituted.14 In the absence of infection, current hydroxyurea treatment should not be withheld during a pain episode. Its efficacy in the treatment of VOC is thought to be due to its ability to increase fetal hemoglobin and reduce neutrophil and platelet activation.15
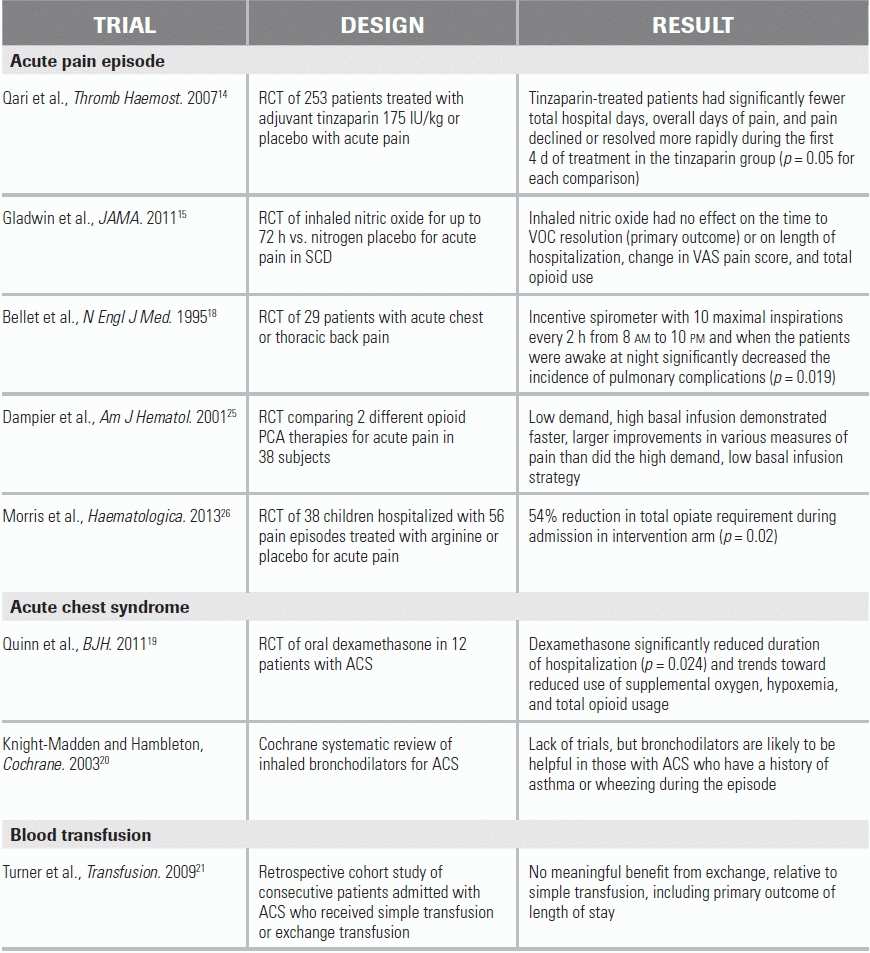
Limited data have shown dexamethasone therapy to be associated with reduced hospital length of stay and trends toward improvement in oxygen and opiate use; however, because of a potential to cause rebound pain, its use is currently not recommended. Various investigational and novel therapies—such tinzaparin, arginine, and inhaled nitric oxide—have been trialed in the setting of VOC, but there are insufficient supporting data at this time to formally recommend their use or their place in a treatment algorithm (see literature summary table for details of recent trials). These therapies remain an area of active research in SCD.
Common infectious organisms in SCD include Streptococcus pneumoniae, Haemophilus influenzae, and Salmonella spp., while atypical organisms such as Mycoplasma, Chlamydia, and Legionella spp. are common in patients with ACS. If there are signs of a lower respiratory tract infection, a macrolide antibiotic should be added to a broad-spectrum, third-generation cephalosporin. In suspected sepsis, hydroxyurea and iron chelation therapy should be avoided due to the risk of cytopenia and promoting growth of siderophore organisms, respectively. Transfusion therapy carries specific risks in patients with SCD, and in the great majority of cases, there is no role for transfusion therapy in management of uncomplicated acute pain.
Patients may be discharged from the ED if their pain can be adequately controlled with oral analgesia.16 Due to the complex interrelationship between pain and psychosocial stressors in patients with SCD, a social work consultation can be very useful in coordinating patient disposition.
DIFFERENTIAL DIAGNOSIS OF AN UNCOMPLICATED PAIN EPISODE
Patients presenting with features other than simple bone pain should be referred to internal medicine and/or hematology.
ACS is an acute illness characterized by fever (>38.50°C), respiratory symptoms, and new pulmonary infiltrates on CXR. Precipitants are commonly infection, postoperative atelectasis, and pulmonary fat emboli (a known complication of marrow infarction in patients with SCD). ACS is the second most common cause of hospitalization in SCD and carries a mortality of approximately 5%.17 Previous pulmonary events, including prior ACS, are risk factors. The pain of ACS is characterized by a “T-shirt” distribution; its severity will usually cause splinting of the diaphragm, further impairing oxygenation and resulting in progressive hypoxia. Signs of lung consolidation may varyingly accompany tachycardia and tachypnea; cough is a late symptom.
If ACS is suspected, the following laboratory tests should be ordered in addition to routine investigations and chest radiography: RBC transfusion crossmatch, hemoglobin electrophoresis, arterial blood gas, pan-culture, and an atypical organism infectious disease serology screen. Specific management of ACS should incorporate inspired O2 to maintain oxygen saturations >96%, bronchodilators if there is history of obstructive/reversible airways disease or in the presence of bronchospasm or wheeze, antimicrobials, incentive spirometry, and blood transfusion.18–20 Early transfusion is appropriate and can prevent the need for ventilator support. The purpose of transfusion is to enhance oxygen-carrying capacity, improve tissue oxygen delivery, and reduce HbS concentration and RBC sickling, all of which can together help prevent progression to acute respiratory failure. Transfusion commonly results in impressive improvement within hours. Patients presenting with mild or moderate ACS, or severe anemia, can be managed with a simple transfusion, aiming for a maximum Hb level of no more than 10 g/dL.21
For severe ACS, or in the presence of rapid or significant clinical deterioration, worsening chest radiography, a pO2 < 70 mm Hg, or baseline Hb > 9 g/dL that preludes use of simple transfusion due to risk of hyperviscosity, consensus opinion is that an exchange transfusion is indicated. Under these circumstances, critical care support is advised. A randomized control trial on the use of exchange transfusion in this setting is still needed to provide more definitive evidence-based guidelines. To ensure that the most appropriate units of blood are selected (given the prevalence of alloimmunization in patients with SCD who have received multiple transfusions), it is imperative that the patient's diagnosis of SCD be communicated to the blood bank with any transfusion request. Additional information may be available if the patient is carrying an antibody warning card documenting clinically significant antibodies whose titers have fallen below currently detectable levels.
Ischemic or hemorrhagic stroke is a common complication of SCD, and the diagnosis should be confirmed with a CT or MRI of the brain. The management of acute stroke in patients with SCD should include most aspects of care provided for non-SCD patients, including aggressive control of blood pressure, administration of antiplatelet therapy, and deep venous thrombosis (DVT) prophylaxis. Thrombolysis, however, is generally not used due to the increased risk of intracerebral hemorrhage. SCD patients with stroke should also undergo immediate exchange transfusion.22 The goal of exchange transfusion support is to increase the HbA to 70% while keeping total Hb < 11 g/dL.
Chronic hemolysis with accelerated bilirubin turnover leads to a high incidence of pigment gallstones. Certain antimicrobials, such as ceftriaxone, are also known to promote biliary sludge formation and should be used with caution in patients with SCD. Management of acute cholecystitis in SCD patients is the same as for the general population.
Hepatic sequestration presents as a rapidly enlarging liver with a significant drop in hemoglobin, accompanied by reticulocytosis. Exchange transfusion may be required, and simple transfusion should be performed judiciously as it carries a risk of hyperviscosity due to desequestering of the RBCs when the episode resolves. Splenic sequestration is less common in adults.
Priapism—a sustained, painful, and unwanted erection of the penis—may go unrecognized by patients as a complication of SCD; they may be reluctant to discuss it and/or may present late to ED. Priapism is caused by vasoocclusion obstructing venous drainage of the penis and typically affects the corpora cavernosa. Penile ischemia and acidosis begin to occur approximately 6 hours into a sustained priapic episode, and recurrent episodes can result in fibrosis and impotence. Treatment centers on management of the underlying sickling process should include urologic consultation for penile aspiration and epinephrine irrigation if the condition persists beyond 6 hours.23 There is little evidence to support transfusion for priapism, and there has been a reported association between priapism, exchange transfusion, and adverse neurologic events.24
CONCLUSION
SCD is a multisystem, inherited blood disorder characterized by ischemia–reperfusion injury and vasculopathy. The most common ED presentation is simple VOC causing generalized bone pain. Assessment of the patient is targeted to detection of complications that may warrant blood transfusion. Management of a VOC should be focused on rapid, adequate, and sustained multimodal analgesia.
LITERATURE TABLE
RCT, randomized controlled trial.
1.Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647– 3656.
2.Ballas SK. Current issues in sickle cell pain and its management. Hematology. 2007;2007(1):97–105.
3.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317–1322.
4.Brousseau DC, Owens PL, Mosso AL, et al. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303(13):1288–1294.
5.Stein PD, Beemath A, Meyers FA, et al. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am J Med. 2006;119(10):897 e7–e11.
6.Mousa SA, Al Momen A, Al Sayegh F, et al. Management of painful vaso-occlusive crisis of sickle-cell anemia: consensus opinion. Clin Appl Thromb Hemost. 2010;16(4):365–376.
7.NIH, The Management of Sickle Cell Disease. US Department of Health and Human Services, Editor; 2002.
8.Rees DC, Olujohungbe AD, Parker NE, et al. Guidelines for the management of the acute painful crisis in sickle cell disease. Br J Haematol. 2003;120(5):744–752.
9.Bartolucci P, et al. A randomized, controlled clinical trial of ketoprofen for sickle-cell disease vaso-occlusive crises in adults. Blood. 2009;114(18):3742–3747.
10.Dunlop RJ, Bennett KC. Pain management for sickle cell disease. Cochrane Database Syst Rev. 2006(2):CD003350.
11.Darbari DS,Neely M, et al. Morphine pharmacokinetics in sickle cell disease: implications for pain management. ASH Annual Meeting Abstracts. 2009;114(22):2574.
12.Kopecky EA, Jacobson S, Joshi P, et al. Systemic exposure to morphine and the risk of acute chest syndrome in sickle cell disease. Clin Pharmacol Ther. 2004;75(3):140–146.
13.Marti-Carvajal AJ, Peña-Martí GE, Comunián-Carrasco G, et al. Interventions for treating painful sickle cell crisis during pregnancy. Cochrane Database Syst Rev. 2009(1):CD006786.
14.Qari MH, Aljaouni SK, Alardawi MS, et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost. 2007;98(2):392–396.
15.Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305(9):893–902.
16.Tanabe P, Artz N, Mark Courtney D, et al. Adult emergency department patients with sickle cell pain crisis: a learning collaborative model to improve analgesic management. Acad Emerg Med. 2010;17(4):399–407.
17.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342(25):1855–1865.
18.Bellet PS, Kalinyak KA, Shukla R, et al. Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases. N Engl J Med. 1995;333(11):699–703.
19.Quinn CT, Stuart MJ, Kesler K, et al. Tapered oral dexamethasone for the acute chest syndrome of sickle cell disease. Br J Haematol. 2011;155(2):263–267.
20.Knight-Madden JM, Hambleton IR. Inhaled bronchodilators for acute chest syndrome in people with sickle cell disease. Cochrane Database Syst Rev. 2003; doi: 10.1002/14651858.CD003733
21.Turner JM, Kaplan JB, Cohen HW, et al. Exchange versus simple transfusion for acute chest syndrome in sickle cell anemia adults. Transfusion. 2009;49(5):863–868.
22.Adams RJ. Big strokes in small persons. Arch Neurol. 2007;64(11):1567–1574.
23.Mantadakis E, Ewalt DH, Cavender JD, et al. Outpatient penile aspiration and epinephrine irrigation for young patients with sickle cell anemia and prolonged priapism. Blood. 2000;95(1):78–82.
24.Merritt AL, Haiman C, Henderson SO. Myth: blood transfusion is effective for sickle cell anemia-associated priapism. CJEM. 2006;8(2):119–122.
25.Dampier CD, Smith WR, Kim HY, et al. Opioid patient controlled analgesia use during the initial experience with the IMPROVE PCA trial: a phase III analgesic trial for hospitalized sickle cell patients with painful episodes. Am J Hematol. 2011;86(12):E70–E73.
26.Morris CR, Kuypers FA, Lavrisha L, et al. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica. 2013;98(9):1375–1382.