29
Platelet Disorders and Hemostatic Emergencies
Shawn K. Kaku and Catherine T. Jamin
BACKGROUND
Hemostasis is the process by which a blood clot is formed at a site of vessel injury. For simplicity, this process may be thought of as occurring in two steps. The first step, primary hemostasis, is the formation of a platelet aggregate at the site of injury. The second step, termed secondary hemostasis, is the activation of the coagulation cascade, which results in the formation of crossed-linked fibrin that strengthens the platelet aggregate. The fibrinolysis system limits the coagulation cascade, thus preventing excess clot formation. A properly functioning hemostatic system requires a functioning liver to synthesize coagulation factors, a sufficient number of platelets and cofactors, and appropriate coordination between the coagulation and fibrinolysis systems. This chapter provides an overview of the etiology and management of hemostatic dysfunction.
HISTORY AND PHYSICAL EXAM
A thorough history and physical will help to identify the etiology of the hemostatic or platelet disorder. In addition to a standard patient history, the provider should review the details of any bleeding events, including triggers, location, frequency, duration, and severity.1 The physical exam should assess for bruising and petechiae, liver size and stigmata of cirrhosis, joint hemarthrosis, signs of anemia, and evidence of an infection.
Details of the history and physical can indicate a primary or secondary hemostatic disorder. Petechiae, bruising, mucosal bleeding, epistaxis, menorrhagia, and persistent bleeding are suggestive of disorders of platelets or primary hemostasis. Bleeding into soft tissues, muscles, and joints, or delayed bleeding, implies the presence of a coagulation factor deficiency or a disorder of secondary hemostasis.2
PLATELET DISORDERS
Idiopathic Thrombocytopenic Purpura
Idiopathic thrombocytopenic purpura (ITP) is an autoimmune disorder that results in the destruction of platelets through an IgG-mediated antibody. Platelets coated with the antibody are rapidly cleared by macrophages in the liver and spleen.3 ITP is characterized by an isolated thrombocytopenia, defined as a platelet count <100 × 109/L, in the absence of an obvious initiating or underlying cause.4,5 As there is no gold standard for the diagnosis of ITP, it is considered a diagnosis of exclusion.4,6 Therefore, a myriad of potential causes of thrombocytopenia must be evaluated prior to diagnosis, including systemic diseases, thrombotic thrombocytopenic purpura (TTP), drug reactions, primary hematologic disorders, liver dysfunction, infections, and recent transfusions.6 A secondary form of ITP may also occur in association with underlying conditions such as human immunodeficiency virus, systemic lupus erythematosus, lymphoproliferative disorder, and antiphospholipid syndrome.3
In contrast to the self-limited presentation of ITP that is typical in children, ITP in adults is generally chronic, with a gradual onset.3 Mucocutaneous bleeding, purpura, petechiae, epistaxis, and gum bleeding are the most common initial manifestations.4
No definitive evidence exists to guide an exact threshold at which to initiate medical therapy, such as glucocorticoids, in adults with ITP. Most patients will not require therapy; however, it is generally accepted that a platelet count <30 × 109/L should be treated, regardless of the presence of bleeding.5 Therapy must be individually tailored, and the decision to treat should weigh the patient's risk of bleeding—that is, previous bleeding episodes, age, presence of other comorbidities, level of activity, etc.6
In the critically ill patient with ITP and hemorrhage, initial therapy consists of high-dose intravenous steroids, such as methylprednisolone (30 mg/kg/d × 3 days for children and 1 g/day × 3 days for adults). Intravenous immunoglobulin (IVIG) 1 g/kg and transfusions may also be used.5,6 Although the exact mechanism of IVIG in treating ITP remains to be elucidated, it is thought to play a role in preventing the uptake of antibody-coated platelets through the blockage of the Fc receptor on macrophages.7 Platelet transfusion is not typically advised in the treatment of ITP, since any transfused platelets will eventually also be destroyed by circulating autoantibodies. However, platelet transfusions have been shown to help sustain the treatment response and may temporarily aid hemostasis in the bleeding patient.7,8 The use of anti-Rho(D) immune globulin (anti-D) has been shown to be effective, though only in Rh-positive patients who have not had a splenectomy.9 The anti-D binds Rh-positive erythrocytes, occupying the receptor in the splenic macrophages that would otherwise be used for removal of platelets. Emergency splenectomy may also be considered. As a nonemergent second-line therapy, splenectomy is associated with an 80% response rate.6 Its use in an emergency situation must be individualized, as the bleeding thrombocytopenic patient makes a challenging ideal surgical candidate.
Heparin-Induced Thrombocytopenia
Heparin-induced thrombocytopenia (HIT) is a life-threatening disorder caused by antibodies against complexes of heparin bound to platelet factor IV. It should be suspected when a patient has a low platelet count, or at least a 50% drop in the platelet count, approximately 5 to 10 days after heparin exposure.10,11 The frequency of HIT is 1% to 5% when unfractionated heparin is used and <1% with low molecular weight heparin (LMWH).10 Although HIT causes thrombocytopenia, thrombosis—not bleeding—is the major clinical concern.10 This is due to platelet activation and the generation of platelet microparticles that leads to thrombin generation and thrombosis.11,12 Thrombotic complications develop in approximately 20% to 50% of patients and can persist for days to weeks after heparin therapy is stopped.11 Complications include arterial and venous thrombosis, limb ischemia, and cerebral venous sinuses thrombosis.11
The laboratory testing for HIT includes a heparin–platelet factor 4 (H-PF4) ELISA antibody test and functional assay tests. The H-PF4 test is widely available and often the first diagnostic test sent. The functional assay tests, while becoming more common, are not always available and are often send-out tests that require up to a week to result. The H-PF4 test has a high sensitivity (>97%) but a poor specificity (74% to 86%), as only a subset of these detected antibodies can cause HIT.11 This is especially true in surgical patients; up to 20% to 50% of postoperative cardiac patients and 81% of surgical ICU patients can have a false positive H-PF4.10,12 Given the high negative predictive value of the H-PF4 test, patients deemed to have a high to intermediate risk of HIT with a negative H-PF4 should be evaluated for alternative diagnoses of their thrombocytopenia.11
The heparin-induced platelet aggregation test is a functional assay test, with a sensitivity of >90% and a specificity ranging from 77% to 100%.11 The c-serotonin functional assay test measures serotonin release from activated platelets and is considered the “gold standard” for the diagnosis of HIT, with a sensitivity and specificity of >95%.11,13–15 Unfortunately, this test is often not available in the emergency department (ED).
Waiting for the send-out test to confirm a diagnosis of HIT can be problematic, given both the dangers of treatment delay and the potentially serious side effects of the treatment itself. The “4Ts” clinical scoring system shown in Table 29.1 provides a real-time evaluation of HIT.16 A recent meta-analysis confirmed its utility in a wide range of patient population, demonstrating a negative predictive value of 99.8% for those with a low score.17
TABLE 29.1 4T's Pretest Scoring System for HIT
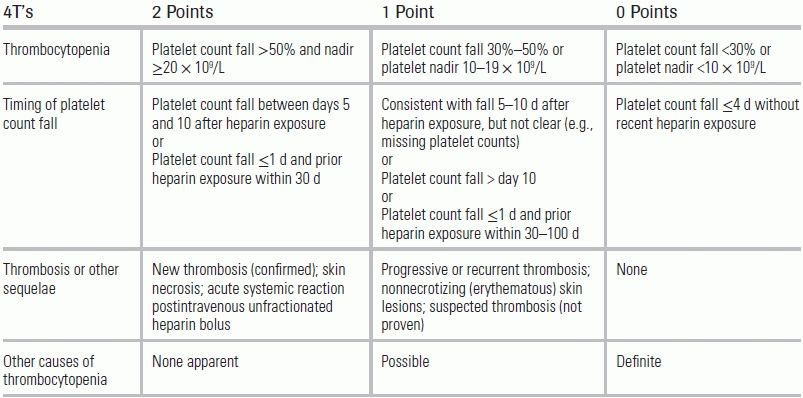
Source: Lo GK, Juhl D, Warkentin TE, et al. Evaluation of pretest clinical score (4T's) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759–765.
Test interpretation: 0 to 3, low probability; 4 to 5, intermediate probability; 6 to 8, high probability.
Treatment of HIT—to suppress thrombotic events—consists of stopping all sources of heparin, including LMWH, and initiating an alternate form of systemic anticoagulation. There are currently three FDA–approved medications for the treatment of HIT: argatroban, bivalirudin, and lepirudin; however, the manufacture of lepirudin has recently been discontinued. While there are no prospective randomized studies examining their efficacy, argatroban has shown superior efficacy in two prospective trials compared to historical controls in reducing thrombotic events and death from thrombosis without an increase in bleeding rates.18,19 Argatroban should be dose adjusted in patients with hepatic dysfunction. Bivalirudin is approved only for patients with HIT undergoing percutaneous coronary intervention. The American College of Chest Physician (ACCP) guidelines notes that fondaparinux may have a theoretical role in treating HIT; at this time, however, it is not approved for this use.20
Patients with HIT are in a prothrombotic state and should remain on anticoagulation for 4 to 12 weeks after diagnosis; this may be accomplished via transition to warfarin therapy.20 The initiation of warfarin must be done cautiously, as warfarin rapidly decreases protein C levels, which can exacerbate the prothrombotic state and lead to skin necrosis and limb gangrene.20 The 2012 ACCP guidelines recommend starting warfarin only after the patient shows platelet recovery of at least 150 × 109/L and stable anticoagulation on thrombin inhibitors; if warfarin has already been started when a patient is diagnosed with HIT, then vitamin K should be administered until the above criteria are met.20 Finally, since spontaneous bleeding is uncommon with HIT, platelets should be transfused only in patients who are bleeding or during the performance of an invasive procedure with a high risk of bleeding.20
HELLP Syndrome
HELLP syndrome is a serious complication of pregnancy, characterized by hemolysis (H), elevated liver enzymes (EL), and low platelets (LP). Controversy exists as to whether HELLP is a severe manifestation of preeclampsia or a separate disease process. Although it can occur earlier, HELLP syndrome usually presents after 28 weeks' gestation.10 Classic symptoms include epigastric or right upper quadrant abdominal pain, nausea, and vomiting.21,22 Patients also may have nonspecific symptoms, such as malaise or headache, which can be mistaken for a viral syndrome.21,22 Although there is no consensus on laboratory values for the diagnosis of HELLP syndrome, patients ideally should demonstrate all components of its acronym, namely, microangiopathic hemolytic anemia (MAHA), EL, and decreased platelets.10,21,22
HELLP syndrome increases the chance of maternal death and is associated with disseminated intravascular coagulopathy, abruptio placentae, severe postpartum bleeding, pulmonary and cerebral edema, liver infarct and rupture, and cerebral infarcts and hemorrhages.21,22 While delivery of the fetus is the cornerstone of treatment, the exact timing of delivery is unclear and is dependent on the gestational age as well as the stability and condition of the mother and fetus.21,22 Laboratory abnormalities may reverse in a subgroup of patients who are managed expectantly; however, this approach needs to be more rigorously investigated.23,24 All patients with HELLP syndrome should be admitted to the hospital and treated for severe preeclampsia, with intravenous magnesium as prophylaxis against convulsions and antihypertensive medications to keep systolic blood pressure below 160 mm Hg, diastolic blood pressure below 105 mmHg, or both.22 Corticosteroid administration to aid fetal lung maturation is often recommended if the fetus is between 24 and 34 weeks' gestational age.22 Platelets should be transfused for significant bleeding or platelet counts <20 × 109/L.22 The use of corticosteroids to improve maternal outcome remains controversial and experimental, as the benefits seen by early small randomized and observational studies could not be reproduced in two larger, randomized, double-blind, placebo-controlled trials.25,26
Thrombotic Thrombocytopenic Purpura and Hemolytic Uremic Syndrome
TTP and hemolytic uremic syndrome (HUS) describe two diseases of a broader category called thrombotic microangiopathies. The thrombotic microangiopathies are microvascular occlusive disorders characterized by aggregation of platelets, thrombocytopenia, and mechanical injury to erythrocytes.27 While sharing similar characteristics, the adult form of TTP and the childhood form of diarrheal HUS are two separate disorders.
TTP is thought to be due to a deficiency of the ADAMTS13 enzyme, with an inhibitory antibody being the cause in the majority of the classic cases.10 The ADAMTS13 enzyme is responsible for cleaving the newly synthesized von Willebrand factor (vWF) multimer. When not cleaved, these unusually large vWF multimers lead to spontaneous platelet aggregation and the clinical syndrome of TTP.10 TTP can be both congenital and acquired, with the congenital form being extremely rare. While the majority of acquired causes are idiopathic, examples of secondary causes of TTP include medications, infections, pregnancy, lupus, malignancy, and transplantation.28
The classic pentad of TTP is thrombocytopenia, MAHA, fluctuating neurologic signs, renal impairment, and fever27 MAHA is caused by erythrocytes passing through areas of the microcirculation that are partially occluded by aggregated platelets.27 This causes fragmented erythrocytes, termed schistocytes or helmet cells, as well as elevated lactate dehydrogenate and indirect bilirubin.27,28 Some patients, however, may not present with neurologic symptoms, renal failure, or fever. Therefore, the diagnosis of TTP may be made in the presence of an MAHA and thrombocytopenia in the absence of any other identifiable cause.28
Treatment for TTP should be initiated even if diagnostic uncertainty exists, as the untreated mortality rate can be as high as 95% to 100%.29–31 Plasma exchange, or the removal of a patient's plasma and replacement with another fluid (donor plasma, colloid, etc.), is the mainstay of treatment, as it removes the inhibitory antibody and supplies new ADAMTS13. Plasma exchange has decreased the TTP mortality rate to <20%.10,27–31 While not as effective as plasma exchange, plasma infusion alone has been shown to decrease the mortality rate to 37%.28,32 Therefore, plasma infusion (30 mL⁄kg⁄d) may be indicated as the initial treatment if there is to be an unavoidable delay in plasma exchange.28 Although steroids have been widely used for TTP as an adjunctive immunosuppressive treatment, there is minimal evidence for their efficacy and no consensus on dosing or route.28 Since patients may benefit from their use, steroids can be given as adjuvant therapy. A reasonable approach is methylprednisolone 2 mg⁄kg⁄d, although pulse doses of 1 g/day × 3 days may also be used.28 Rituximab, an anti-CD20 antibody, should be considered for patients refractory to plasma exchange.33,34 Platelet transfusions are contraindicated as they can worsen the platelet aggregation and effects of TTP. They should be reserved for life-threatening hemorrhage or for invasive procedure preparation.28
HUS is characterized by MAHA, thrombocytopenia, and acute renal failure. HUS is commonly associated with a prodrome of bloody diarrhea caused by the Shiga toxin–producing Escherichia coli 0157:H7.27,28 The toxin damages endothelial cells, causing platelet aggregation and intravascular thrombogenesis. Much of the treatment for HUS is supportive in nature. The optimal care requires careful management of fluid and electrolyte balance and blood pressure. The use of hemodialysis may be required if renal failure is severe.28 Antibiotics and antimotility agents should be avoided as they can worsen the outcome.27,28 Two prospective studies on plasma infusion in HUS failed to show any outcome benefit.35,36 There are no randomized controlled prospective studies evaluating the use of therapeutic plasma exchange for HUS caused by Shiga toxin–producing Escherichia coli, and it is currently reserved only for severe cases, often those involving the nervous system. As in the case of TTP, blood transfusion for HUS should be given based on clinical evaluation and need, rather than on a strict hemoglobin threshold; platelet transfusions should be avoided if possible.
DISORDERS OF COAGULATION
Disseminated Intravascular Coagulation
Disseminated intravascular coagulation (DIC) is characterized by the widespread activation of the coagulation cascade, which results in fibrin formation, thrombotic occlusion of small and midsize vessels, and subsequent organ failure. Simultaneously, the consumption of platelets and coagulation proteins can induce severe bleeding.37 DIC is not a disease in itself but is instead a complication of an underlying disorder. These disorders include sepsis, trauma, organ dysfunction (pancreatitis), obstetric emergencies, malignancy, and toxic and transfusion reactions.38
No single laboratory test can diagnose or rule out DIC. Instead, in a patient at risk for DIC, a combination of test results can be used to diagnose the disorder with reasonable certainty.37 Common laboratory abnormalities include thrombocytopenia, elevated fibrin degradation products, prolongation of clotting times including the prothrombin time (PT) and the activated partial thromboplastin time (PTT), and a low fibrinogen.37,39 Schistocytes may also be present on the blood smear.37 Caution must be exercised when interpreting fibrinogen levels, as it is an acute-phase reactant that can remain within the normal range or elevated for a long period of time.37,39
The cornerstone of treatment of DIC is treatment of the precipitating condition.37–39 Transfusion of platelets or plasma should be reserved for patients with active or high risk of bleeding. Platelet transfusion is indicated in bleeding patients with platelet counts <50 × 109/L and in nonbleeding patients with platelet counts <10 to 20 × 109/L.39 Fresh frozen plasma (FFP) and/or cryoprecipitate are recommended for patients who are bleeding with an INR > 2 or a fibrinogen level <100 mg/dL.40 In cases of DIC where a thrombotic picture predominates (e.g., arterial or venous thromboembolism, severe purpura fulminans, or vascular skin infarctions), therapeutic doses of heparin should be considered.39 In critically ill, nonbleeding patients with DIC, prophylaxis for venous thromboembolism with heparin or LMWH is recommended.39 The use of antithrombin (formerly antithrombin III) has not improved outcomes in DIC, and further investigation is warranted in the use of recombinant human factor VIIa.41,42 For patients with inherited or acquired protein C deficiencies, protein C concentrate has demonstrated some benefit.43
HEMOPHILIA
Hemophilia is an X-linked heritable coagulopathy, most often referring to a deficiency of factor VIII (hemophilia A) or factor IX (hemophilia B, Christmas disease). While these deficiencies are difficult to distinguish clinically, factor VIII deficiency comprises approximately 80% of cases and factor IX deficiency the remaining 20%.44,45 The severity of hemophilia is defined by the level of serum clotting factors as compared to the general population: <1% of normal is defined as severe, 1% to 5% of normal as moderate, and >5% of normal as mild.44,46 Patients with hemophilia are at risk for hemarthrosis (especially knee, ankle, and elbow joints), soft tissue hematomas, bruising, retroperitoneal bleed, intracranial hemorrhage, and postsurgical bleeding.44,47
Due to the heritability of the disease, a family history of hemophilia or abnormal bleeding is extremely helpful in making the diagnosis. Approximately 30% of cases, however, have no known family history and are caused by spontaneous mutations. Laboratory values for patients with hemophilia A and B will demonstrate normal platelets and PT, with a prolonged PTT. Specific assays for each factor can be used to identify the type of hemophilia.
Administration of the deficient factor is needed to limit or stop an episode of bleeding. The amount of factor replaced is dependent on the location of the bleeding. According to the guideline from the World Federation of Hemophilia, a factor level of 40% to 60% is recommended for deep lacerations, joint, and most muscle bleeding, and a factor level of 80% to 100% is recommended for CNS, throat and neck, GI, and iliopsoas muscle bleeding.48 The formulas for estimating the dose of factor required to be administered are shown in Table 29.2. If unknown, the baseline factor should be presumed to be 0%. The patient's replaced factor level should be measured approximately 15 minutes after infusion to verify calculated doses.48 The half-life of factors VIII and IX is 8 to 12 hours and 18 to 24 hours, respectively. Redosing will be needed at that time.48
TABLE 29.2 Formulas for Calculating Dosage of Required Factor
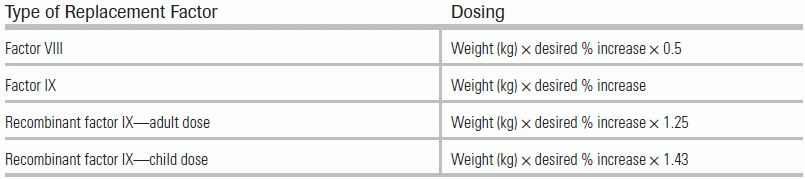
Source: Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Treatment Guidelines Working Group on behalf of The World Federation of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47.
When specific factor replacement is unavailable, other options do exist. FFP contains all coagulation factors, in a concentration of 1 unit of factor in 1 mL, and can be used for both hemophilia A and B.48 Concerns with use of FFP include the large volume required and the inherent risks of transfusion (e.g., transfusion reaction, volume overload, and TRALI). Cryoprecipitate contains about 70 to 80 units of factor VIII and can be used as an alternative for treatment of hemophilia A.48 However, similar safety concerns make it second-line therapy. Other treatment options include prothrombin complex concentrates, recombinant factor VII, and antifibrinolytic agents. These treatments should be used in consultation with a hematologist.
LIVER DISEASE
The liver's essential contribution to maintenance of normal hemostasis is severely disrupted in advanced liver disease, which can lead to coagulopathy and severe bleeding. The loss of hepatic parenchymal cells leads to decreased production of the hemostatic factors II, V, VII, IX, X, XI, XII, and fibrinogen (both the liver and endothelium synthesize factor VIII, allowing for a normal to elevated level in liver disease).49 Impaired bile production decreases the absorption of vitamin K, an essential cofactor for factors II, VII, IX, and X. Although its clinical significance is unclear, vitamin K deficiency may also contribute to the coagulopathy.49,50 Finally, decreased clearance of tissue plasminogen activator and diminished production of fibrinolytic inhibitors are thought to be responsible for the low-grade fibrinolysis found in 30% to 46% of patients with end-stage liver disease.49,51
Platelet number and function are also affected in advanced liver disease. Thrombocytopenia (in the setting of liver disease) is thought to be multifactorial, with factors including excessive trapping and clearing of platelets from portal hypertension–induced hypersplenism; impaired platelet production from decreased synthesis of thrombopoietin; and immune and nonimmune platelet destruction.49–51 Immune-mediated platelet destruction is often seen with chronic liver disease, and particularly in hepatitis C, a disease state associated with antiplatelet antibodies, including the glycoprotein autoantibody associated with ITP.49,50
In addition to decreasing the quantity of platelets, advanced liver disease impairs platelet function through a variety of mechanisms. Increased circulating platelet inhibitors, excess nitric oxide synthesis, a deficiency of platelet receptors, defective signal transduction, and impaired thromboxane A2 synthesis all contribute to impaired platelet aggregation, defective platelet–vessel wall interaction, and enhanced platelet inhibition associated with liver disease.49,51
While liver disease is most often associated with bleeding disorders, hypercoagulability can also be present. Liver disease results in the decreased production of anticoagulation factors such as antithrombin and proteins C and S; in turn, this may either balance the disruption of the pro- and anticoagulation systems or may result in a hypercoagulable state.50–52 In fact, the notion that elevated PT/INR levels in patients with liver disease reflect “autoanticoagulation” may be unfounded.51,53,54 An elevated PT does not necessarily reflect a uniform decrease in vitamin K clotting factors, as in warfarin therapy; it can also reflect an unbalanced decrease of the short-lived factor VII without the concomitant protection from thrombosis.51 Moreover, INR and PT results vary widely in patients with liver disease based on the reagent and device used.50 Thus, clinical context—such as sepsis, recent surgery, or bleeding—is more important than any single laboratory value in assessing coagulation balance in these patients.
In the acutely bleeding patient with liver disease, aggressive treatment of hemostatic deficits is essential. Therapy should be aimed not at complete correction of abnormal laboratory values but at achieving hemostatic competence.51 To guide therapy, laboratory testing should include PT/INR, PTT, platelet count, and fibrinogen level. FFP should be administered to correct an elevated PT/INR and PTT levels as it contains all coagulation factors; however, this correction can be difficult to achieve and may have only transient effect.50,51 Also, note again that an elevated PT/INR may not reflect an increased bleeding risk; and transfusion of FFP may lead to volume overload and other transfusion-related complications. Cryoprecipitate should be used to keep the fibrinogen level >100 mg/dL, and platelets transfused to keep a level >50 × 109/L.51 A trial of desmopressin may be used to aid platelet function in cases of refractory bleeding.51 The mechanism of action of desmopressin is unclear, but it is thought to improve platelet adhesiveness through its release of vWF.51 As previously noted, vitamin K deficiency may also contribute to coagulopathy, and a 3-day trial of vitamin K (5 to 10 mg/day) may be given.51 The efficacy and safety of recombinant factor VIIa is currently under study, and it should be reserved as a rescue therapy.50,51
CONCLUSION
Emergency departments and intensive care units frequently admit patient with platelet disorders and hemostatic emergencies. While many of these conditions share common laboratory values, they vary widely in pathology and appropriate course of treatment. Proper identification of, and tailored therapy for, the platelet disorders ITP, HIT, HELLP, HUS, and TTP, as well as the coagulopathies of hemophilia, DIC, and liver disease, is an essential skill for the emergency physician.
LITERATURE TABLE
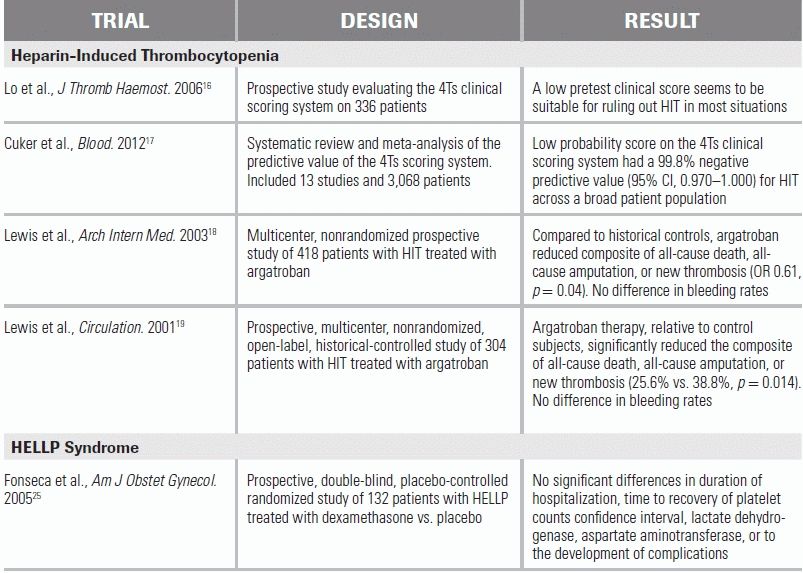
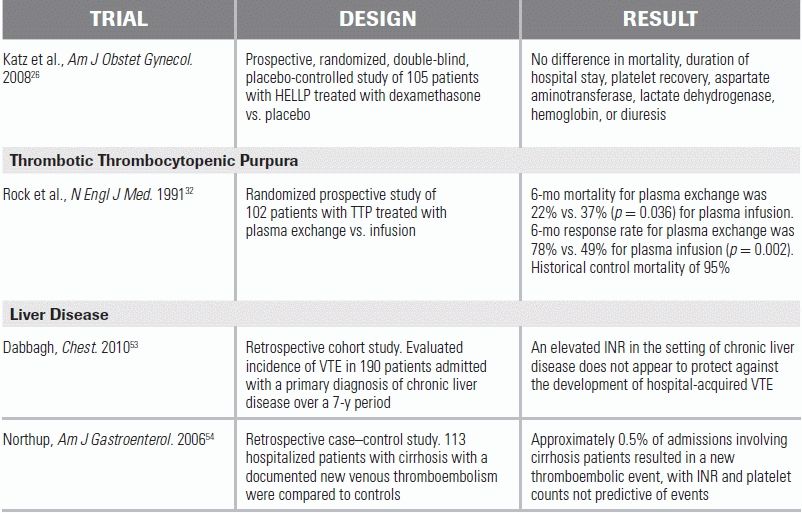
CI, confidence interval; OR, odds ratio.
REFERENCES
1.Rydz N, James PD. Why is my patient bleeding or bruising? Hematol Oncol Clin North Am. 2012;26(2): 321–344.
2.van Ommen CH, Peters M. The bleeding child. Part I: primary hemostatic disorders. Eur J Pediatr. 2012;171(1):1–10.
3.Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346(13):995–1008.
4.Liebman HA, Pullarkat V. Diagnosis and management of immune thrombocytopenia in the era of thrombopoietin mimetics. Hematology Am Soc Hematol Educ Program. 2011;2011:384–390.
5.Bussel JB, Cines DB, Kelton JG, et al. Idiopathic thrombocytopenic purpura: a practice guideline developed by explicit methods for the American Society of Hematology. Blood. 1996;88(1):3–40.
6.Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115(2):168–186.
7.Baumann MA, Menitove JE, Aster RH, et al. Urgent treatment of idiopathic thrombocytopenic purpura with single-dose gammaglobulin infusion followed by platelet transfusion. Ann Intern Med. 1986;104:808.
8.Carr JM, Kruskall MS, Kaye JA, et al. Efficacy of platelet transfusion in immune thrombocytopenia. Am J Med. 1986;80:1051.
9.Ramadan KM, El-Agnaf M. Efficacy and response to intravenous anti-D immunoglobulin in chronic idiopathic thrombocytopenic purpura. Clin Lab Haematol. 2005;27:267.
10.DeLoughery TG. Critical care clotting catastrophies. Crit Care Clin. 2005;21(3):531–562.
11.Arepally GM, Ortel TL. Clinical practice. Heparin-induced thrombocytopenia. N Engl J Med. 2006;355(8):809–817.
12.Berry C, Tcherniantchouk O, Ley EJ, et al. Overdiagnosis of heparin-induced thrombocytopenia in surgical ICU patients. J Am Coll Surg. 2011;213(1):10–17.
13.Sheridan D, Carter C, Kelton JG. A diagnostic test for heparin-induced thrombocytopenia. Blood. 1986;67(1):27–30.
14.Napolitano LM, Warkentin TE, Almahameed A, et al. Heparin-induced thrombocytopenia in the critical care setting: diagnosis and management. Crit Care Med. 2006;34(12):2898–2911.
15.Warkentin TE. Platelet count monitoring and laboratory testing for heparin-induced thrombocytopenia. Arch Pathol Lab Med. 2002;126(11):1415–1423.
16.Lo GK, Juhl D, Warkentin TE, et al. Evaluation of pretest clinical score (4 T's) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759–765.
17.Cuker A, Gimotty PA, Crowther MA, et al. Predictive value of the 4Ts scoring system for heparin-induced thrombocytopenia: a systematic review and meta-analysis. Blood. 2012;120(20):4160–4167.
18.Lewis BE, Wallis DE, Leya F, et al. Argatroban-915 Investigators. Argatroban anticoagulation in patients with heparin-induced thrombocytopenia. Arch Intern Med. 2003;163(15):1849–1856.
19.Lewis BE, Wallis DE, Berkowitz SD, et al. ARG-911 Study Investigators. Argatroban anticoagulant therapy in patients with heparin-induced thrombocytopenia. Circulation. 2001;103(14):1838–1843.
20.Linkins LA, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed.: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e495S–530S.
21.Haram K, Svendsen E, Abildgaard U. The HELLP syndrome: clinical issues and management. A Review. BMC Pregnancy Childbirth. 2009;9:8.
22.Sibai BM. Diagnosis, controversies, and management of the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Obstet Gynecol. 2004;103(5 Pt 1):981–991.
23.Visser W, Wallenburg HC. Temporising management of severe pre-eclampsia with and without the HELLP syndrome. Br J Obstet Gynaecol. 1995;102(2):111–117.
24.van Pampus MG, Wolf H, Westenberg SM, et al. Maternal and perinatal outcome after expectant management of the HELLP syndrome compared with pre-eclampsia without HELLP syndrome. Eur J Obstet Gynecol Reprod Biol. 1998;76(1):31–36.
25.Fonseca JE, Méndez F, Cataño C, et al. Dexamethasone treatment does not improve the outcome of women with HELLP syndrome: a double-blind, placebo-controlled, randomized clinical trial. Am J Obstet Gynecol. 2005;193(5):1591–1598.
26.Katz L, de Amorim MM, Figueiroa JN, et al. Postpartum dexamethasone for women with hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome: a double-blind, placebo-controlled, randomized clinical trial. Am J Obstet Gynecol. 2008;198(3):283.e1–e8.
27.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589–600.
28.Allford SL, Hunt BJ, Rose P, et al. Haemostasis and Thrombosis Task Force, British Committee for Standards in Haematology. Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol. 2003;120(4):556–573.
29.von Baeyer H. Plasmapheresis in thrombotic microangiopathy-associated syndromes: review of outcome data derived from clinical trials and open studies. Ther Apher. 2002;6(4):320–328.
30.Lara PN Jr, Coe TL, Zhou H, et al. Improved survival with plasma exchange in patients with thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Am J Med. 1999;107(6):573–579.
31.Bell WR, Braine HG, Ness PM, et al. Improved survival in thrombotic thrombocytopenic purpura- hemolytic uremic syndrome–clinical experience in 108 patients. N Engl J Med. 1991;325:398–403.
32.Rock GA, Shumak KH, Buskard NA, et al. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. Canadian Apheresis Study Group. N Engl J Med. 1991;325(6):393–397.
33.Fakhouri F, Vernant JP, Veyradier A, et al. Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13-deficient thrombotic thrombocytopenic purpura: a study of 11 cases. Blood. 2005;106(6):1932.
34.Scully M, Cohen H, Cavenagh J, et al. Remission in acute refractory and relapsing thrombotic thrombocytopenic purpura following rituximab is associated with a reduction in IgG antibodies to ADAMTS-13. Br J Haematol. 2007;136(3):451.
35.Rizzoni G, Claris-Appiani A, Edefonti A, et al. Plasma infusion for hemolytic uremic syndrome in children: results of a multicenter controlled trial. J Pediatr. 1988;12:284–290.
36.Loirat C, Sonsino E, Hinglais N, et al. Treatment of childhood haemolytic uraemic syndrome with plasma. A multicentre randomised controlled trial. Pediatr Nephrol. 1988;2:279–285.
37.Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341(8):586–592.
38.Levi M, de Jonge E, van der Poll T. New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann Med. 2004;36(1):41–49.
39.Levi M, Toh CH, Thachil J, et al. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol. 2009;145(1): 24–33.
40.Wada H, Asakura H, Okamoto K, et al. Japanese Society of Thrombosis Hemostasis/DIC subcommittee. Expert consensus for the treatment of disseminated intravascular coagulation in Japan. Thromb Res. 2010;125(1):6–11.
41.Warren BL, Eid A, Singer P, et al. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286(15):1869.
42.Franchini M, Manzato F, Salvagno GL, et al. Potential role of recombinant activated factor VII for the treatment of severe bleeding associated with disseminated intravascular coagulation: a systematic review. Blood Coagul Fibrinolysis. 2007;18(7):589.
43.Smith OP, White B, Vaughan D, et al. Use of protein-C concentrate, heparin, and haemodiafiltration in meningococcus-induced purpura fulminans. Lancet. 1997;350(9091):1590.
44.Knobe K, Berntorp E. Haemophilia and joint disease: pathophysiology, evaluation, and management. J Comorbidity. 2011;1:51–59.
45.Coppola A, Di Capua M, Di Minno MN, et al. Treatment of hemophilia: a review of current advances and ongoing issues. J Blood Med. 2010;1:183–195.
46.White GC II, Rosendaal F, Aledort LM, et al. Factor VIII and Factor IX Subcommittee. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(3):560.
47.Ljung R, Petrini P, Nilsson IM. Diagnostic symptoms of severe and moderate haemophilia A and B. A survey of 140 cases. Acta Paediatr Scand. 1990;79(2):196–200.
48.Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Treatment Guidelines Working Group on behalf of The World Federation of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47.
49.Senzolo M, Burra P, Cholongitas E, et al. New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol. 2006;12(48):7725–7736.
50.Trotter JF. Coagulation abnormalities in patients who have liver disease. Clin Liver Dis. 2006;10(3): 665–678.
51.Kujovich JL. Hemostatic defects in end stage liver disease. Crit Care Clin. 2005;21(3):563–587.
52.Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011;365(2):147–156.
53.Dabbagh O, Oza A, Prakash S, et al. Coagulopathy does not protect against venous thromboembolism in hospitalized patients with chronic liver disease. Chest. 2010;137(5):1145–1149.
54.Northup PG, McMahon MM, Ruhl AP, et al. Coagulopathy does not fully protect hospitalized cirrhosis patients from peripheral venous thromboembolism. Am J Gastroenterol. 2006;101(7):1524–1528.