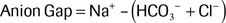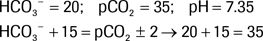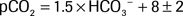37
Acid–Base Disorders
Tara Scherer and Corey Slovis
BACKGROUND
Acid–base homeostasis influences protein function, which in turn affects tissue and organ performance. Disturbances of the acid–base system are common in the critically ill patient and must be promptly identified and corrected to prevent harm. Optimal cellular function occurs with a pH of 7.35 to 7.45, and the body employs several compensatory mechanisms to tightly regulate its pH. It is helpful to use accurate terminology when describing acid–base disturbances. Acidemia refers to a pH ≤ 7.35, while alkalemia refers to a pH ≥ 7.45. Acidosis denotes a process that increases hydrogen ion concentration, while alkalosis denotes a process that decreases hydrogen ion concentration. Patients with an acid–base disorder will either be acidemic or alkalemic or have a normal pH.1,2
Acid–base disturbances are classified as either primarily respiratory or metabolic in origin. Respiratory disturbances are caused by changes in the partial pressure of carbon dioxide (pCO2). The pCO2 is elevated in a respiratory acidosis and decreased in a respiratory alkalosis. Metabolic disturbances are caused by primary changes in the bicarbonate concentration (HCO3−). The HCO3− is elevated in a metabolic alkalosis and decreased in a metabolic acidosis. Each primary disturbance has a compensatory mechanism that leads to a change in pH opposite of the primary problem. For example, a metabolic acidosis is compensated for by hyperventilation, leading to a decrease in pCO2 and a compensatory respiratory alkalosis, resulting in a corrective increase in pH. The approaches described in this chapter allow rapid detection of acid–base disturbances and identification of their underlying etiology.
AN APPROACH TO ACID–BASE PROBLEMS
Blood Gas Analysis
Analyzing blood gas results is a rapid way to determine a patient's acid–base status. Blood gas values include pH, pCO2, and partial pressure of oxygen (pO2). Traditionally, blood gases have been obtained via arterial puncture. Normal arterial blood gas (ABG) values are a pH of 7.36 to 7.44, HCO3− of 21 to 27 mEq/L, pCO2 of 35 to 45 mm Hg, and pO2 of 80 to 100 mm Hg. In an ABG, the pH, pCO2, and pO2 are measured directly, while the HCO3− is calculated using the Henderson-Hasselbalch Equation. Recently, venous blood gas (VBG) measurements have been suggested as a less invasive alternative to arterial blood sampling. Studies have shown that both venous pH and bicarbonate levels can serve as substitutes for arterial pH in normotensive patients.3–8 Values from arterial and venous samples are not identical, but their differences are thought to be minimal. In a large prospective study of 246 emergency department (ED) patients, simultaneous arterial and venous samples demonstrated high correlation between pH and bicarbonate (r = 0.97 and r = 0.95, respectively).7 In another study, arterial and central venous samples were obtained from 26 patients with normal cardiac output, 36 patients with moderate cardiac output, 5 patients with severe circulatory failure, and 38 patients in cardiac arrest. In patients with normal cardiac output, the venous pH was 0.03 less than the arterial pH, and venous pCO2 was higher than arterial values by 5.7 mm Hg. In severe circulatory failure and cardiac arrest, there were substantial differences between pH and pCO2.5 Observed differences were thought to be due to the divergence of the arterial and venous systems that occur as a patient becomes more hypotensive. Specifically, hypotension leads to hypoperfusion at the tissue level, which causes an increased proportion of CO2 to enter the blood at the capillary level. In a separate study that compared arterial and venous blood gas results in 16 patients in cardiac arrest, venous pH was shown to be 0.3 less than a simultaneously drawn arterial sample.9 As a rule of thumb, arterial samples should be obtained in any patient with shock, respiratory distress leading to cardiovascular collapse, or cardiac arrest, while venous measurements can be used in all other patients, including those with diabetic or alcoholic ketoacidosis (AKA).
The pO2 has not been shown to correlate accurately between arterial and venous samples. A prospective study of 95 pathologically diverse ED patients demonstrated venous pH, pCO2, and HCO3− to be reliable substitutes for ABG analysis (pH lower by 0.02 to 0.04, pCO2 higher by 3 to 8 mm Hg, and HCO3− higher by 1 to 2 mEq/L) but reported poor agreement in pO2.6
The following is a simple, three-step approach to the interpretation of blood gas values.10
1.Does the patient have an acidosis or alkalosis?
A pH of 7.35 or less indicates the presence of an acidosis. A pH > 7.45 indicates the presence of an alkalosis.
2.Is the acidosis/alkalosis a respiratory or metabolic process?
If the pCO2 and pH move in opposite directions, then there is a primary respiratory process. If the pCO2 and pH move in the same direction, then there is a primary metabolic process.
3.If a respiratory acidosis or alkalosis is present, is it a pure respiratory process or is there a concurrent metabolic component?
In a pure acute respiratory process, for every 10 mm Hg change in pCO2, the pH should move in the opposite direction by 0.08 ± 0.02. For example, if the pCO2 is 50 mm Hg (a 10 mm Hg increase), the pH should be 7.32 (a decrease of 0.08). If this rule is not followed, a simultaneous metabolic process is present: If the pH is higher than expected, there is a simultaneous metabolic alkalosis; if the pH is lower than expected, there is a simultaneous metabolic acidosis.
METABOLIC ACIDOSIS
The rapid identification and interpretation of acid–base disorders permits optimal patient management and disposition. Historically, physicians have been poor at acid–base analysis11–13 despite multiple approaches to interpreting acid–base disorders being available.14–16 Metabolic acidosis is the most common acid–base abnormality encountered in the ED. The following is a simplified five-step approach to interpretation and management of metabolic acidoses using the basic metabolic panel (BMP) and blood gas values as described below.
1.Identify abnormal values on the BMP: Prior to calculating the anion gap, be sure to identify other abnormalities (e.g., hyperkalemia) that commonly accompany acid–base disorders.
2.Calculate the anion gap: The anion gap is the difference in the measured serum cations and anions.17–19 Using the values from the BMP, the anion gap is calculated using the following formula:
A normal anion gap ranges from 8 to 12 ± 2. Values above this indicate the presence of unmeasured anions. An elevated, or wide, anion gap indicates the presence of a metabolic acidosis regardless of the serum bicarbonate or pH value.
3.If a wide or normal gap acidosis is present, apply the Rule of 15 to evaluate for a “hidden” respiratory process.20
When an acidosis is identified, further evaluation must be performed to determine if there is an appropriate respiratory compensatory process or a concurrent primary respiratory process. If an appropriate respiratory compensation for a metabolic acidosis is present, the respiratory rate will be increased in order to lower the pCO2 and correct the low serum pH.
The Rule of 15 is used to predict a patient's expected compensatory pCO2 and pH based on the bicarbonate concentration. The rule states that HCO3− + 15 should equal the pCO2 and the last two digits of the pH as described below:
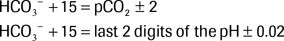
If the pCO2 and pH equal to the predicted values, there is a pure metabolic acidosis with appropriate secondary respiratory alkalosis. If the Rule of 15 is not followed, a simultaneous primary respiratory process must be present. If the pCO2 is lower than predicted, a primary respiratory alkalosis exists in addition to a metabolic acidosis. If the pCO2 is higher than predicted, a primary respiratory acidosis exists in addition to the metabolic acidosis.
The following is an example in which the Rule of 15 is satisfied:
Because the actual pCO2 is within ± 2 of predicted pCO2 using the Rule of 15, this is a pure metabolic acidosis with appropriate respiratory compensation. The last two digits of the pH are also within 0.02 of the predicted pH.
The following is an example in which the Rule of 15 is not satisfied:
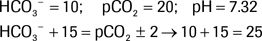
The expected pCO2 is 25 (± 2), but the actual pCO2 is 20. Therefore, the Rule of 15 is not followed, and a simultaneous respiratory process is also present. Because the actual pCO2 is lower than the expected pCO2, there is a concurrent primary respiratory alkalosis in addition to the metabolic acidosis.
A corollary to the Rule of 15 is that as HCO3− falls below 10 and approaches 5, then the pCO2 should equal 15. Recall that on an ABG, the HCO3− is estimated using the Henderson-Hasselbalch Equation, while on a BMP, it is the directly measured total serum CO2 that is used in lieu of HCO3− (total serum CO2 represents both serum bicarbonate and other forms of CO2 such as dissolved CO2 and carbonic acid (H2CO3)). A bicarbonate buffering system exits to maintain the balance between CO2 and HCO3, as described by the following Equation:
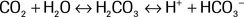
In a patient with metabolic acidosis (i.e., increased H+), this Equation is driven to the left as compensatory hyperventilation increases the loss of CO2. In a patient with a respiratory acidosis (i.e., increased CO2), this process is driven to the right with a concomitant increase in bicarbonate. Winter's formula (below) is used to evaluate respiratory compensation—the change in pCO2 for a given HCO3−—in the setting of a metabolic acidosis20:
The Rule of 15 is extrapolated from this formula. The maximal fall in pCO2 in adults, however, is approximately 15; thus, once HCO3− drops below 10, the Rule of 15 can no longer be applied, and Winter's formula should be used instead. For example, in a patient with a HCO3− of 8, the pCO2 should be 20.
4.If an acidosis is present, check the delta gap to evaluate for a “hidden” metabolic process.
The next step is to evaluate for the presence of an additional primary metabolic process by calculating the delta gap. In an uncomplicated anion gap acidosis, for every 1 mmol/L rise in the anion gap, there should be a concomitant fall of 1 mmol/L in the HCO3− ± 4.21–23 The delta gap (Δ gap) is defined as the difference between the rise in the anion gap and the fall in the bicarbonate concentration24:
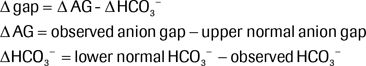
For this approach, the upper normal anion gap is defined as 15 mmol/L, and the lower normal bicarbonate concentration is 25 mmol/L. If the Δ HCO3− equals the Δ AG and the delta gap is zero, then there is no hidden metabolic process. If the bicarbonate is higher than expected, leading to a positive delta gap, then there is an additional metabolic alkalosis. If the bicarbonate is lower than expected, leading to a negative delta gap, then there is a concomitant primary non–anion gap metabolic acidosis.
5.For an unexplained wide gap metabolic acidosis, check the osmolar gap.
In an unexplained anion gap metabolic acidosis, or in a patient with a history of toxic alcohol ingestion, the osmol gap should be calculated to determine the presence of substances with osmotic activity omitted from the calculated osmolarity, such as ethylene glycol or methanol:
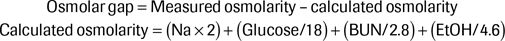
Traditional teaching is that a normal osmolar gap is 10 or less and that in the setting of an elevated anion gap metabolic acidosis, an osmol gap of >10 indicates the presence of a toxic alcohol. While this is a good general guide, a more accurate calculation of a normal osmol gap is approximately −2 ± 6, the range that accounts for 95% of the population, which has a baseline osmol gap of −10 to +14. As such, a normal gap measurement can be misleading. For example, in a patient with a baseline osmol gap of −5 and a calculated osmol gap of 12, the true osmol gap would be 17.25 Since a patient's baseline osmol gap is not known, it can be difficult to be certain whether the calculated gap is in fact elevated.
Management Guidelines
Metabolic acidosis is frequently seen in the ED and results from either a loss of bicarbonate or an accumulation of a nonvolatile acid. Severe acidemia can be devastating to the cardiovascular system (producing arrhythmias, decreased cardiac contractility, and arteriolar vasodilation) and the neurologic system (producing coma and seizures). Severe acidemia is often accompanied by profound hypotension and shock, which only further exacerbate acid production.
Anion Gap Metabolic Acidosis
Anion gap acidosis results from the presence of unaccounted-for anions such as sulfate, phosphate, and organic anions or weak acid proteins not measured on a basic metabolic profile.26 Common etiologies of an elevated anion gap acidosis can be recalled using the mnemonics KULT (ketones, uremia, lactate, and toxins) or the more comprehensive MUDPILES (methanol, uremia, DKA (and AKA along with starvation ketoacidosis), phenformin (and metformin), paracetamol (acetaminophen), Isoniazid (INH) and iron, lactic acidosis, ethylene glycol, salicylates, and solvents.
Causes of Anion Gap Acidosis
1.Lactic acidosis:
a.Type A lactic acidosis: Impaired systemic perfusion due to shock, severe hypoxemia, or severe anemia
b.Type B lactic acidosis:
i.Type B1 (underlying disease): Impaired clearance of lactate due to liver or renal dysfunction or increased production of lactate due to seizures, hypothermic shivering, strenuous exercise, and ischemic colitis
ii.Type B2 (medication/intoxication): Metformin, linezolid, isoniazid (INH), HIV medications
iii.Type B3 (inborn errors of metabolism)
2.Ketoacidosis: Diabetic ketoacidosis (DKA), AKA, starvation ketoacidosis
3.Renal failure: Decreased excretion of organic anions (urea, phosphates, sulfates)
4.Toxic ingestions: Methanol, ethylene glycol, toluene, salicylates
Lactic Acidosis
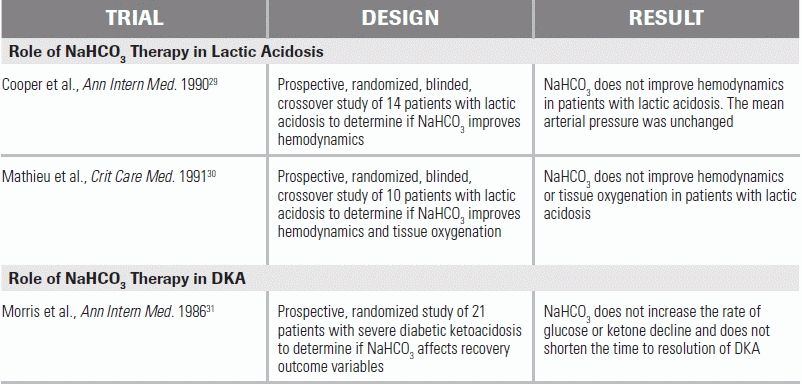
Lactic acidosis is the most common cause of an anion gap metabolic acidosis and is defined as a pH of <7.35 with a lactate concentration of >5 mmol/L.27 Lactate is most commonly a product of anaerobic metabolism (i.e., a type A lactic acidosis), and elevated levels can be observed in a variety of conditions, including severe hypoxia, seizures, sepsis, shock, and cyanide poisoning. Patients with severe lactic acidosis have mortality rates as high as 80% at 10 days.28 The mainstay of lactic acidosis treatment is correction of the underlying or precipitating illness and aggressive patient resuscitation. The role of supplemental therapeutic buffers, such as sodium bicarbonate (NaHCO3), is controversial. In a prospective randomized study, 14 hemodynamically unstable patients with lactic acidosis were given NaHCO3- and sodium chloride–containing infusions. While the NaHCO3 infusions helped correct the patient's acidemia, the hemodynamic response, including response to catecholamines, was the same to both solutions.29 A second similarly designed study in 10 patients yielded comparable results.30 As a consequence of these and other studies, current guidelines recommend avoiding NaHCO3 treatment in patients with lactic acidosis unless the pH falls below 7.15 or when bicarbonate levels fall below 5 mEq/L, at point at which small changes in bicarbonate concentration can lead to profound and potentially fatal decreases in serum pH.26
Diabetic Ketoacidosis and Alcoholic Ketoacidosis
In DKA and AKA, an anion gap metabolic acidosis occurs as the result of decreased availability of cellular glucose, leading to fatty acid metabolism and associated ketoacid production. DKA occurs because of a relative insulin deficiency; AKA is the result of a starvation state. In DKA, treatment centers on the provision of fluid resuscitation and insulin. The role of NaHCO3 in DKA management is controversial. In a prospective study, 21 patients with severe DKA (defined as pH of 6.9 to 7.14) were randomized to either receive or not receive supplemental NaHCO3.31 The group receiving NaHCO3 showed no benefit in terms of clinical recovery.31 No randomized prospective studies have examined the effect of NaHCO3 on DKA patients with a pH of <6.9. In these cases, careful, judicious NaHCO3 administration is recommended to prevent possible cardiovascular collapse.32
In AKA, treatment centers on volume resuscitation; repletion of glucose, potassium, and magnesium; and provision of intravenous vitamins, most importantly thiamine. It should be noted that a similar starvation ketoacidosis can be seen early in pregnancy in women with hyperemesis gravidarum.
Uremia
As kidneys fail, they lose their ability to excrete ammonium and hydrogen ions, leading to a non–anion gap metabolic acidosis. Ammonia is converted to urea in the liver, and the urea is subsequently excreted in the urine. As the renal dysfunction progresses, the kidneys lose the ability to effectively excrete urea, phosphates, sulfates, and other organic acids, which results in an anion gap metabolic acidosis.33 Treatment is hemodialysis, which corrects the acidosis by removing nitrogenous waste products.
Toxic Alcohols
Ingestion of toxic alcohols such as methanol or ethylene glycol can result in the accumulation of toxic metabolites and an associated anion gap metabolic acidosis. The metabolism of methanol, a substance found in products such as windshield wiper fluid and “moonshine,” leads to the formation of formate, an organic acid that can cause acidosis, blindness, and pancreatic injury. The formation of formate is catalyzed by the enzyme alcohol dehydrogenase. The acidosis in methanol toxicity leads to the protonation of formate to formic acid, an uncharged molecule that is more likely to penetrate tissues. Treatment begins with administration of NaHCO3 to reverse the acidosis, which decreases formic acid production and results in less tissue penetration and damage. Another treatment modality is 4-methylpyrazole (trade name Fomepizole). Fomepizole is a competitive inhibitor of alcohol dehydrogenase, and thus serves to block the formation of formate. Hemodialysis to remove the toxic metabolite is indicated in severe cases.34
Ethylene glycol, the primary ingredient of antifreeze, is another important toxic alcohol capable of producing an anion gap metabolic acidosis. Following ingestion, alcohol dehydrogenase metabolizes ethylene glycol to glycolic and oxalic acids, which result in metabolic acidosis and renal injury, respectively. The treatment for ethylene glycol ingestion is the same as for methanol (bicarbonate, 4-methylpyrazole, and hemodialysis). A recent study evaluated available treatment algorithms for toxic alcohol ingestion by combining therapeutic interventions with a physiologically based pharmacokinetic model. The study found that if administered early enough, fomepizole was more effective than hemodialysis. However, if renal injury had already occurred or toxic metabolites had already formed, then hemodialysis was the appropriate treatment.35
Other Toxins
INH, a drug used to treat tuberculosis, inhibits GABA synthesis and lowers the seizure threshold. Frequently, patients with INH overdoses will present with refractory seizures. The anion gap metabolic acidosis is a result of both the seizure activity and INH's interference with nicotine adenine dinucleotide, an essential cofactor in the conversion of lactate to pyruvate. INH also binds to pyridoxine, making it inactive. Pyridoxine is a necessary cofactor for the production of GABA, and in the setting of an INH overdose, GABA stores are depleted, which leads to seizure activity. Treatment of INH overdoses requires pyridoxine therapy to replete the GABA stores.36
Acute iron poisoning can also lead to an anion gap metabolic acidosis. This is due in part to the hydration of ferric ions, a process that results in the release of three protons. Iron also causes mitochondrial dysfunction, which leads to anaerobic metabolism and subsequent lactic acid formation. Treatment of iron overdose is chelation with deferoxamine.37
An anion gap metabolic acidosis may also be seen with salicylate overdose. Salicylates uncouple oxidative phosphorylation, which results in an increase in anaerobic metabolism and an associated lactic acidosis and ketoacidosis. Treatment focuses on administration of NaHCO3 to alkalinize the urine and on hemodialysis when indicated. Urine alkalinization enhances the renal elimination of salicylates; in alkaline urine, salicylates will ionize and become “ion trapped,” limiting reabsorption.38
Finally, inhalation of solvents such as toluene can lead to an anion gap metabolic acidosis when they are metabolized to hippuric acid. Treatment is supportive.39
Non–Anion Gap Metabolic Acidosis
Non–anion gap metabolic acidoses are rarely life threatening and typically resolve with correction of the underlying etiology. The most common causes of a non–anion gap acidosis are loss of base from either the kidneys or the gastrointestinal system. Etiologies of an elevated anion gap acidosis may be recalled using the mnemonic HARDUP: hyperalimentation or hyperventilation, acetazolamide, renal tubular acidosis (RTA), diarrhea, ureteral diversions, and pancreatic fistula.
Gastrointestinal Etiologies
Gastrointestinal loss of bicarbonate-rich fluid occurs in diarrhea, ureteral diversions, and pancreatic fistulas. In severe diarrhea, excessive loss of this fluid can result in a non–anion gap metabolic acidosis. Therapy consists of fluid replacement and prevention of further loss. Ureteral diversions (e.g., ileal conduits) lead to a non–anion gap metabolic acidosis when chloride from the urine enters the colon. The colonic mucosa has an anion exchanger to reabsorb chloride in exchange for bicarbonate. This leads to increased gastrointestinal loss of bicarbonate.40 Pancreatic fluids are also high in bicarbonate, and when a pancreatic fistula is present, this fluid is lost. Treatment consists of repairing the fistula.41
Renal Etiologies
An RTA results when the kidneys are unable to adequately manage the body's acid. A distal, or type 1 RTA, occurs in the setting of impaired H+ secretion. There are many causes of a distal RTA. The most common etiologies in adults are autoimmune disorders such as lupus. In children, distal RTAs are frequently hereditary. A proximal, or type 2 RTA, occurs when there is a defect in bicarbonate reabsorption leading to excessive bicarbonate loss. Type 2 RTAs can be caused by multiple myeloma, familial disorders, amyloidosis, heavy metal toxicity, and renal transplantation. Medications, notably carbonic anhydrase inhibitors such as acetazolamide, can mimic a proximal RTA by inhibiting the renal absorption of bicarbonate.42 A type 4 RTA occurs in the setting of hypoaldosteronism and decreased ammonium secretion and is associated with electrolyte disturbances including hyperkalemia (type 3 RTA is now excluded from modern classifications). Renal losses of bicarbonate can also occur in the setting of prolonged hyperventilation—for example, in patients with severe asthma or COPD—leading to a compensatory metabolic acidosis. If the respiratory condition is corrected quickly (e.g., with sedation and mechanical ventilation), the underlying metabolic acidosis will be unmasked.43
Iatrogenic Etiologies
Rapid administration of chloride-rich and bicarbonate-poor solutions, such as normal saline, can also produce a non–anion gap metabolic acidosis. Normal saline has a chloride concentration of 154 to 155 mmol/L and a pH of 5.5. Normal plasma has a chloride concentration of 100 mmol/L and a pH of 7.4. Administration of a large amount of normal saline during volume resuscitation can result in a hyperchloremic non–anion gap metabolic acidosis. No anion gap is seen because chloride is accounted for in the anion gap formula. Resolution occurs after stopping administration of high–chloride content fluids and/or switching to a more pH neutral alternative such as lactated Ringer's.44,45 Iatrogenic addition of acids, such as hydrochloric acid and ammonium chloride, can also lead to a non–anion gap metabolic acidosis.
METABOLIC ALKALOSIS
Metabolic alkalosis is defined by a primary elevation in the serum bicarbonate concentration. While not as common as metabolic acidosis, severe alkalemia can be equally dangerous. Neurologic complications include altered mental status, coma, and seizures. Cardiovascular complications include increased risk of arrhythmias and arteriolar vasoconstriction, which can cause decreased coronary blood flow. Alkalemia is also associated with hypokalemia, hypocalcemia, and hypophosphatemia.
Metabolic alkalosis occurs in the setting of acid loss by gastrointestinal or renal routes or exogenous base administration. Metabolic alkalosis may be categorized as either chloride responsive or chloride unresponsive:
1.Chloride responsive
a.GI losses: vomiting, gastric drainage
b.Contraction alkalosis
c.Diuretics
2.Chloride unresponsive
a.Hyperaldosteronism
b.Hypokalemia
c.Exogenous alkali load
Chloride-/Saline-Responsive Conditions
Gastric fluid contains a high concentration of hydrochloric acid. Loss of this fluid through vomiting and nasogastric suctioning can lead to a metabolic alkalosis. Therapy is directed at fluid replacement and preventing future loss of gastric fluid. Potassium repletion may also be required. A rare congenital chloride-losing diarrhea results from a defect in the chloride/bicarbonate transporter; in this case, large amounts of chloride are lost in the stool, leading to a metabolic alkalosis that is refractory to antidiarrheal agents.
Contraction alkalosis can occur with the setting of thiazides or loop diuretics use. These diuretics result in enhanced sodium and chloride excretion without a proportional loss of bicarbonate. Treatment involves administration of IV fluids.
Saline-/Chloride-Unresponsive Conditions
Hyperaldosteronism results in renal acid loss. Aldosterone directly enhances sodium and chloride resorption in the cortical collecting tubule. This creates a more electronegative environment promoting hydrogen and potassium secretion. It also stimulates the apical H-ATPase in the collecting tubule. Primary hyperaldosteronism is seen with adrenal hyperplasia and adrenal adenomas. Secondary hyperaldosteronism occurs in the setting of congestive heart failure, chronic renal insufficiency, and hepatic failure. Aldosterone excess is also seen in Bartter syndrome. In patients with concurrent hypokalemia, potassium repletion will improve alkalosis due to transcellular hydrogen/potassium ion exchange.46
Acetazolamide decreases the proximal tubule's reabsorption of bicarbonate and is commonly used to correct a metabolic alkalosis in critically ill patients.47 Case reports exist of large bicarbonate ingestions causing severe metabolic alkalosis. If a patient has a severe alkalosis with a pH > 7.7 or experiences arrhythmias, dilute hydrochloric acid is indicated. When administering hydrochloric acid, it should be given through a central line at 100 mL/h with hourly pH checks.48
RESPIRATORY ACIDOSIS
Respiratory acidosis is defined as a primary increase in pCO2. Etiologies stem from disturbances in the airway, pulmonary system, central nervous system, and neuromuscular system. Airway causes include obstruction and spasm. Pulmonary etiologies include COPD, asthma, pulmonary edema, pneumothorax, mass, and infection. Narcotics, sedative hypnotics, and brain tumors can suppress the central respiratory center. Neuromuscular disorders, including myopathies and neuropathies, can also lead to respiratory acidosis. Treatment aims to remove or correct the underlying cause while ensuring adequate oxygenation and ventilation using either noninvasive positive pressure ventilation or orotracheal intubation.1
RESPIRATORY ALKALOSIS
Respiratory alkalosis occurs when the primary disturbance is a decrease in pCO2. The differential diagnosis is broad and includes a variety of benign and pathologic causes. Normal pregnancy, high-altitude residence, anxiety, pain, and withdrawal can all lead to a respiratory alkalosis. Pathologic causes of respiratory alkalosis include sepsis, pulmonary embolus, hypoxia, and salicylate overdose. The respiratory alkalosis from salicylate toxicity occurs due to the stimulation of the respiratory center. Management of respiratory alkalosis is directed toward correction of its underlying cause.2
MIXED ACID–BASE DISORDERS
There are myriad potential mixed acid–base disturbances. The most important are (1) an anion gap metabolic acidosis and primary respiratory alkalosis, (2) an anion gap metabolic acidosis and respiratory acidosis, and (3) an anion gap metabolic acidosis and metabolic alkalosis.
An anion gap metabolic acidosis accompanied by a primary respiratory alkalosis is most commonly seen in patients with hypotension from traumatic blood loss and hyperventilation due to pain. This mixed disorder can also be seen in patients with AKA and withdrawal leading to hyperventilation. Aspirin toxicity (salicylic acid) and sepsis (lactic acidosis) should also be considered with this acid–base abnormality.
An anion gap metabolic acidosis accompanied by a primary respiratory acidosis is seen in patients unable to appropriately compensate for their acidosis. This may be seen in patients with severe acidosis or cerebral edema and/or elevated intracranial pressure or in the presence of CNS depressants (e.g., opiates). Treatment focuses on correction of the underlying disease process with accompanying supportive care and, frequently, ventilatory assistance.
Finally, an anion gap metabolic acidosis accompanied by a primary metabolic alkalosis is seen in patients with renal failure (RTA) or DKA and emesis (contraction alkalosis) or in similarly acidemic patients receiving intravenous NaHCO3 therapy.
CONCLUSION
Acid–base disturbances are common in critically ill patients. The systematic approach outlined in this chapter is designed to enable prompt recognition and response to these disorders in order to optimize cellular function and improve patient outcomes.
LITERATURE TABLE

REFERENCES
1.Adrogue HJ, Madias NE. Management of life-threatening acid–base disorders. First of two parts. N Engl J Med. 1998;338:26–34.
2.Adrogue HJ, Madias NE. Management of life-threatening acid–base disorders. Second of two parts. N Engl J Med. 1998;338:107–111.
3.Gennis PR, Skovron ML, Aronson ST, et al. The usefulness of peripheral venous blood in estimating acid–base status in acutely ill patients. Ann Emerg Med. 1985;14:845–849.
4.Kelly AM, McAlpine R, Kyle E. Venous pH can safely replace arterial pH in the initial evaluation of patients in the emergency department. Emerg Med J. 2001;18:340–342.
5.Adrogue H, et al. Assessing acid–base status in circulatory failure: differences between arterial and central venous blood. N Engl J Med. 1989;320:1312–1316.
6.Malatesha G, et al. Comparison of arterial and venous pH, bicarbonate, PCO2 and PO2 in initial emergency department assessment. Emerg Med J. 2007;24(8):569–571.
7.Brandenburg MA, Dire DJ. Comparison of arterial and venous blood gas values in the initial emergency department evaluation of patients with diabetic ketoacidosis. Ann Emerg Med. 1998;31:459–465.
8.McCanny P, Bennett K, et al. Venous vs arterial blood gases in the assessment of patients presenting with an exacerbation of chronic obstructive pulmonary disease. Am J Emerg Med. 2012;30:896–900.
9.Weil M, Rachow E, et al. Difference in acid–base state between venous and arterial blood during cardiopulmonary resuscitation. N Engl J Med. 1986;315:153–156.
10.Isenhour JL, Slovis CM. Arterial blood gas analysis: a simple, 3-step approach: When should you suspect a mixed acid–base disturbance? J Respir Dis. 2008;29:74–82.
11.O'Sullivan I, Jeavons R. Survey of blood gas interpretation. Emerg Med J. 2005;22:391–392.
12.Schreck D, et al. Diagnosis of complex acid–base disorders: physician performance versus the microcomputer. Ann Emerg Med. 1986;15:164–170.
13.Austin K, Jones P. Accuracy of interpretation of arterial blood gases by emergency medicine doctors. Emerg Med Australas. 2010;22:159–165.
14.Haber R. A practical approach to acid–base disorders. West J Med. 1991;155(2):146–151.
15.Palmer B. Approach to fluid and electrolyte disorders and acid–base problems. Prim Care. 2008;35(2):195–213.
16.Carmody JB, Norwood VF. A clinical approach to paediatric acid–base disorders. Postgrad Med J. 2012:88:143–151.
17.Emmett M, Narins R. Clinical use of the anion gap. Medicine. 1977;56:38–54.
18.Gabow P, et al. Diagnostic importance of an increased serum anion gap. N Engl J Med. 1980;303:854–858.
19.Oh M, Carroll H. The anion gap. N Engl J Med. 1977;297:814–817.
20.Albert MS, Dell RB, Winters RW. Quantitative displacement of acid base equilibrium in metabolic acidosis. Ann Intern Med. 1967;66:312–322.
21.Narins R, Emmett M. Simple and mixed acid–base disorders: a practical approach. Medicine. 1980;59:161–187.
22.Dubose T. Clinical approach to patients with acid–base disorders. Med Clin North Am. 1983;67:799–813.
23.Goodkin D, et al. The role of the anion gap in detecting and managing mixed metabolic acid–base disorders. Clin Endocrinol Metab. 1984;13:333–349.
24.Wrenn K. The delta gap: an approach to mixed acid–base disorders. Ann Emerg Med. 1990;19:1310–1313.
25.Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: normal values and limitations. J toxicol Clin toxicol. 1993;31(1):81–93.
26.Gauthier P, Szerlip H. Metabolic acidosis in the intensive care unit. Crit Care Clin. 2002;18:289–308.
27.Mizock B, Falk J. Lactic acidosis in critical illness. Crit Care Med. 1992;20:80–93.
28.Stacpoole P, Wright E, et al. Natural history and course of acquired lactic acidosis in humans: the DCA-lactic acidosis study group. Am J Med. 1994;97:47–54.
29.Cooper DJ, et al. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. Ann Intern Med. 1990;112:492–498.
30.Mathieu D, et al. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med. 1991;19(11):1352–1356.
31.Morris LR, et al. Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986;105:836–840.
32.Kitabchi A, et al. Hyperglycemic crises in adult patients with diabetes: a consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(12):2739–2748.
33.Walls J. Metabolic acidosis and uremia. Perit Dial Int. 1995;15(5):S36–S38.
34.Burns M, et al. Treatment of methanol poisoning with intravenous 4-methylpyrazole. Ann Emerg Med. 1997;30(6):829–832.
35.Corley R, McMartin K. Incorporation of therapeutic interventions in physiologically based pharmacokinetic modeling of human clinical case reports of accidental or intentional overdosing with ethylene glycol. Toxicol Sci. 2005;85(1):491–501.
36.Morrow L, et al. Acute isoniazid toxicity and the need for adequate pyridoxine supplies. Pharmacotherapy. 2006;26(10):1529–1532.
37.Britton R, et al. Iron toxicity and chelation therapy. Int J Hematol. 2002;76(3):219–228.
38.O'Malley G. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007;25(2):333–346.
39.Dickson R, Luks A. Toluene toxicity as a cause of elevated anion gap metabolic acidosis. Respir Care. 2009;54(8):1115–1117.
40.Davidsson T, et al. Long-term metabolic and nutritional effects of urinary diversion. Urology. 1995;46:804–809.
41.Callery M, et al. Prevention and management of pancreatic fistula. J Gastrointest Surg. 2009;13(1):163–173.
42.Heller I, et al. Significant metabolic acidosis induced by acetazolamide. Not a rare complication. Arch Intern Med. 1985;145(10):1815–1817.
43.Morris C, Low J. Metabolic acidosis in the critically ill: Part 2. Causes and treatment. Anaesthesia. 2008;63:396–411.
44.Kellum J. Saline induced hyperchloremic metabolic acidosis. Crit Care Med. 2002;30:259–261.
45.Prough D, Bidani A. Hyperchloremic metabolic acidosis is a predictable consequence of intraoperative infusion of 0.9% saline. Anesthesiology. 1999;90:1247–1249.
46.Khanna A, Kurtzman N. Metabolic alkalosis. J Nephrol. 2006;19(suppl 9):S86–S96.
47.Mazur J, et al. Single versus multiple doses of acetazolamide for metabolic alkalosis in critically ill medical patients: a randomized, double-blind trial. Crit Care Med. 1999;27:1257–1261.
48.Mennen M, Slovis C. Severe metabolic alkalosis in the emergency department. Ann Emerg Med. 1988;17(4):354–357.