TABLE 38.1 Treatment of Hyponatremia

BACKGROUND
Electrolyte disorders are frequently observed in critically ill patients and are associated with increased morbidity and mortality. This chapter reviews the most common electrolyte disturbances and provides a systematic approach to their management.
DISORDERS OF SODIUM
Hyponatremia is a common electrolyte abnormality and may be seen in isolation or as a complication of other medical problems. Its prevalence varies according to the patient population, clinical setting, and serum sodium level used to define it. A normal serum sodium range is generally considered to be 135 to 145 mEq/L; hyponatremia is typically defined as a serum sodium level of <135 mEq/L.
Sodium is the dominant extracellular cation and does not move freely across cell membranes. Therefore, in order for hyponatremia to occur, water intake must exceed water excretion. In healthy individuals, water intake rarely overwhelms the kidneys' ability to excrete sodium, and hyponatremia most commonly results from either impaired renal function or inappropriate antidiuretic hormone (ADH) or vasopressin release.1
Manifestations of hyponatremia include headache, seizures, coma, and, if brain edema results from associated fluid shift, even death. Symptom severity correlates with the rapidity of onset and the magnitude of drop in serum sodium.2
True hyponatremia is always hypoosmolar, but hyperosmolar and iso-osmolar hyponatremia may also occur. Hyperosmolar hyponatremia (>295 mOsm/kg) is due to the presence of another effective osmole, typically excess serum glucose or an osmotic diuretic (e.g., mannitol). Treatment includes stopping the offending infusion, and/or targeting a decrease in glucose concentration of 75 to 100 mg/dL/h. Iso-osmolar hyponatremia (280 to 295 mOsm/kg), also termed pseudohyponatremia, represents artifact due to hyperlipidemia or hyperproteinemia. It is usually asymptomatic and does not require specific treatment. The remainder of this review will focus on hypoosmolar hyponatremia (<280 mOsm/kg).
Hypoosmolar hyponatremia can exist in the setting of elevated (hypervolemic), normal (isovolemic), or low (hypovolemic) plasma volumes. Hypovolemic hyponatremia results from either renal or extrarenal losses of water and salt. Extrarenal hypovolemic hyponatremia typically results from vomiting and diarrhea. Other notable etiologies include burns, trauma, and pancreatitis. In cases of extrarenal losses, the body attempts to retain sodium while simultaneously releasing ADH. Ultimately, however, more water than salt is retained, resulting in low serum sodium levels as well as hypertonic urine (urine sodium <10 mEq/L). Renal causes of sodium and water loss include mineralocorticoid insufficiency, excessive use of diuretics, osmotic diuresis, and cerebral salt wasting syndrome.3 In cases of renal loss, inappropriate elevations in both urine sodium (>20 mEq/L, usually >40 mEq/L) and urine osmolality (>100 mOsm/kg, and frequently >300 mOsm/kg) exist.
Isovolemic hyponatremia results from retention of water without salt. Although a diagnosis of exclusion, the classic example of isovolemic hyponatremia is the syndrome of inappropriate antidiuretic hormone (SIADH) secretion. SIADH is defined as hypotonic hyponatremia that occurs in the face of clinical euvolemia and in the absence of diuretic use, hypothyroidism, or adrenal insufficiency. In SIADH, both urine sodium concentration (>20 mEq/L) and urine osmolality (>100 mOsm/kg and generally >300 mOsm/kg) are elevated.4 SIADH has multiple etiologies, including meningitis, malignancy (e.g., cervical cancer, lymphoma, leukemia, bronchogenic cancers), medications (e.g., cyclophosphamide, vincristine, vinblastine, selective serotonin reuptake inhibitors), and pulmonary or granulomatous diseases.5 Other less common causes of isovolemic hyponatremia include psychogenic polydipsia, hypothyroidism, and adrenal or glucocorticoid insufficiency.
Hypervolemic hyponatremia occurs when the quantity of water retained is greater than that of sodium; it most commonly occurs with congestive heart failure, cirrhosis, and nephrotic syndrome.6 In these disorders, the body attempts to retain sodium, resulting in a low urine sodium level (<20 mEq/L) and a high urine osmolality (>500 mOsm/kg). Of note, acute or chronic renal failure can also lead to hypervolemic hyponatremia, but in these cases the urine sodium level is generally elevated (>20 mEq/L) and the urine isotonic.7
Correction of hypovolemic hyponatremia requires both salt and water supplementation. Factors to consider in management include the severity and duration of symptoms. Chronic hyponatremia (or asymptomatic hyponatremia of unknown duration) should be treated with water restriction and avoidance of extra sodium, with a goal to correct the serum sodium at a rate of ≤0.5 mEq/L/h and avoid neurologic complications associated with overly rapid correction rates. In mild and acute hyponatremia, the sodium correction should not exceed 1 mEq/L/h8 or approximately 8 mEq/L/24 h.9 In acute symptomatic cases (e.g., seizures, altered mental status), hypertonic saline should be used to raise the serum sodium by 2 mEq/L/h, preserving a target increase of ≤12 mEq/L/24 h.10 The most feared consequence of overly rapid correction of chronic hyponatremia is central pontine myelinolysis (CPM), which develops when water abruptly leaves the intracellular space of brain cells to equalize intra- and extracellular osmolalities.11,12 CPM will typically present as paraparesis or quadriparesis with dysarthria and dysphagia. On autopsy, patients with CPM will often demonstrate diffuse demyelinating lesions.
Once the severity and duration of symptoms are clarified, the next steps are to calculate the sodium deficit, total body water (TBW), and the target rate of rise of sodium.
(Y = 0.6 L/kg in children/adult males, 0.5 L/kg in adult females/elderly males, 0.4 L/kg in elderly females)
For example, in a symptomatic 50-kg female with a serum sodium of 112 mEq/L, raise the serum sodium by approximately 10 mEq in the first 24 hours (target serum sodium of 122 mEq/L). The sodium deficit is calculated as follows: (50 kg × 0.5 L/kg) × (122 mEq/L − 112 mEq/L) = 250 mEq. Because the patient is symptomatic, 3% hypertonic saline, containing 500 mEq of sodium per liter, can be used. Therefore, 500 mL (i.e., 250 mEq × [1,000 mL/500 mEq]) of 3% hypertonic saline would be given in the first 24 hours, resulting in an infusion rate of approximately 20 mL/h.
As a general guideline, the increase in serum sodium in mEq/L produced by giving 1 L of any fluid can be estimated as follows:
If the above patient's symptoms are not severe and normal saline (154 mEq/L) was used instead, the expected rise in serum sodium would be (154 − 112)/(25 + 1) = 1.6 mEq/L.
Correction of isovolemic hyponatremia is usually achieved with water restriction and correction of the underlying cause of the imbalance (e.g., SIADH, hypothyroidism, adrenal insufficiency). The use of salt tablets may be considered, and loop diuretics may be needed in cases where urine output is low. In refractory cases, vasopressin antagonists, referred to as vaptans, may be used. ADH has multiple receptors, including V1a, V1b, and V2. The V1a and V1b receptors are largely responsible for vasoconstriction, while the V2 receptors mediate the antidiuretic response.13,14 Vaptans work by selectively causing water diuresis without affecting sodium. The loss of free water corrects the hyponatremia, although the resulting increase in thirst may lead patients to drink more free water, thereby limiting the anticipated rise in sodium. Only two vaptans are currently available in the United States: tolvaptan and conivaptan. Tolvaptan, an oral formulation selective for the V2 receptors, has been shown to increase serum sodium levels significantly when compared to placebo. However, a potential significant adverse effect of tolvaptan is overly rapid correction of hyponatremia.15 In contrast, conivaptan, available either intravenously (IV) or orally (PO), blocks both the V2 and V1a receptors. Trials with the IV16 and oral17 forms have shown statistically significant increases in serum sodium when compared to placebo. Concerns, however, exist about conivaptan's ability to lower blood pressure and potential to increase the risk of variceal bleed in cirrhotic patients via its V1a effect. More research is needed before the regular use of vaptans can be recommended. Additional treatment options include demeclocycline (600 to 1,200 mg/d), a tetracycline antibiotic that renders the collecting ducts unresponsive to ADH, effectively inducing a state of nephrogenic diabetes insipidus (DI), or diphenylhydantoin (40 mg/kg every 6 hours), which prevents the release of ADH and mimics central DI.18,19
Correction of hypervolemic hyponatremia centers on fluid restriction (600 to 1,000 mL/d), treatment of the underlying disorder (e.g., cardiac failure, renal failure, cirrhosis, nephrotic syndrome), and avoidance of extra sodium. Vaptans may also be considered along with loop diuretics.20
Hypernatremia occurs when sodium exceeds water in the body. As previously noted, hyponatremia can be associated with hypo-, iso-, or even hyperosmolality. Hypernatremia, on the other hand, always results in hyperosmolality.21,22 Because hyperosmolality stimulates thirst and water ingestion, hypernatremia only occurs when either a defect in the thirst mechanism or restricted water access exists. Therefore, the elderly or otherwise disabled patients, as well as critically ill hospitalized patients, are at greatest risk. Between 2% and 6% of newly admitted ICU patients are hypernatremic,23 and between 6% and 26% of patients in medical intensive care units (ICUs) and 4% to 10% of patients in surgical ICUs will become hypernatremic during the hospitalization, usually in the first week after admission. This is important because the development of hypernatremia in hospitalized patients has been shown to be an independent risk factor for mortality.24–29
Manifestations of hypernatremia occur as a result of neuronal dehydration as intracellular water shifts to the more hypertonic extracellular space. Lethargy, altered level of consciousness, irritability, hyperreflexia, and spasticity are common. Hypernatremia may be associated with impaired glucose metabolism leading to hyperglycemia,30,31 and, in severe cases, can cause rhabdomyolysis with consequent acute renal failure.32,33 Finally, hypernatremia has been associated with a decrease in cardiac function.34
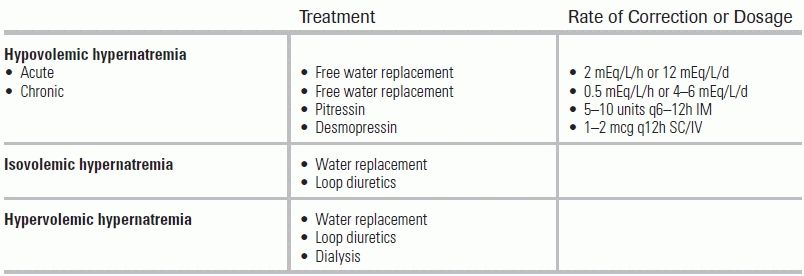
Like hyponatremia, hypernatremia can coexist with decreased, normal, or elevated plasma volumes. Hypovolemic hypernatremia occurs when the body loses hypotonic fluids (water deficit exceeds sodium deficit). This is commonly seen with gastrointestinal losses (e.g., vomiting, diarrhea) and renal losses (e.g., intrinsic renal disease, use of diuretics). Physical exam abnormalities usually are not evident until dehydration reaches ≥10% to 15% (expressed as percentage of body weight) because fluid shifts from the intracellular to the extracellular space to preserve plasma volume.
Isovolemic hypernatremia typically occurs when a patient is unable to sense thirst, usually the result of a congenital or acquired disorder of the hypothalamus (e.g., craniopharyngiomas, primary or metastatic hypothalamic tumors [usually breast or lung], vascular lesions, trauma).35 Other causes of isovolemic hypernatremia include central and nephrogenic DI. Central DI results from either impaired production or release of ADH, and it often follows head trauma or pituitary surgery. Nephrogenic DI results from a defect in the kidneys' response to ADH. In either case, urine output can be as high as 3 mL/kg/h, and the specific gravity will usually be between 1.000 and 1.003.
Hypervolemic hypernatremia is usually iatrogenic in nature and secondary to large infusions of hypertonic fluids, such as 3% saline or sodium bicarbonate (NaHCO3), as well as replacing hypotonic insensible losses (e.g., febrile illness, respiratory distress, gastrointestinal loss) with 0.9% (normal) saline. It can also be seen in accidental salt ingestions and, rarely, with mineralocorticoid excess (e.g., Cushing syndrome).
The first step in the management of hypernatremia is determination of volume status, as hypovolemic hypernatremia is treated differently from isovolemic or hypervolemic hypernatremia. Clinical signs of low volume status include increased thirst, sunken eyes, dry mucous membranes, resting or orthostatic tachycardia, and hypotension, as well as oliguria. Hemodynamic monitoring may reveal a very low central venous pressure, arterial pressure variation in ventilated patients, or increase in arterial pressure with passive leg raise in spontaneously breathing patients. Biochemistries may show rising hematocrit, high serum uric acid, high urine osmolarity, and low urine sodium (extrarenal cases).
Management of hypovolemic hypernatremia begins with fluid resuscitation with a balanced crystalloid solution to correct volume deficit. Fluid resuscitation should be guided by symptom resolution, including improvement in orthostasis, tachycardia, and urine output. Once the volume deficit is corrected, the next step is to calculate the free water deficit, obtained with the following formula:
The free water deficit can then be corrected with 5% dextrose in water (D5W) or a low-sodium crystalloid solution (e.g., half-normal saline).36 As with hyponatremia, a gradual rate of replacement is essential, as overly rapid correction can cause cerebral edema.37,38 In chronic cases, or cases of unknown duration, the rate of correction should not exceed 0.5 mEq/L/h or 8 to 10 mEq/L/24 h. The diagnosis of acute hypernatremia should only be made if the rise in sodium has a documented onset within the last 48 hours prior to presentation. In these cases, rapid correction at a rate of 2 to 3 mEq/L/h or 12 mEq/L/24 h is appropriate.39 For example, in a 50-kg 40-year-old female patient with a serum sodium of 160 mEq/L, the TBW would be 50 kg × 0.5 L/kg = 25 L. Total water deficit would be 25 L × [160/140) −1] = 3.6 L. Thus, a total positive water balance of 3.6 L must be achieved for the sodium to decrease from 160 to 140 mEq/L, or by 20 mEq. However, assuming that the case is not acute, the rate of correction should be ≤0.5 mEq/h, which would require replacement of the water deficit over 40 hours, or approximately 90 mL/h, to which insensible water losses should be added—generally about 30 mL/h—for a total of 120 mL/h.
In the particular case of hypernatremia caused by DI, water loss should be replaced at a rate of 0.5 to 0.75 mL for every 1 mL of urine made. In cases of central DI, vasopressin (5 to 10 units IM q6-12h) and desmopressin acetate or DDAVP (1 to 2 mcg SC/IV q12h) may be considered. These agents are ADH analogs that increase water reabsorption by the renal collecting ducts.
In cases of isovolemic and hypervolemic hyponatremia, treatment requires only replacement of the free water (e.g., D5W) with or without the use of loop diuretics. In renal failure, dialysis may be necessary.
Sodium treatment summary: Tables 38.1 and 38.2
TABLE 38.1 Treatment of Hyponatremia
TABLE 38.2 Treatment of Hypernatremia
POTASSIUM
While sodium is the major extracellular cation, potassium is the dominant intracellular one. The concentration differences of these positively charged particles create a difference in electrical potential between the inside and outside of cells, known as the membrane potential. The membrane potential allows the cells to generate an action potential, an electrical discharge, which is critical for neurotransmission and muscle contraction. For this reason, the serum potassium level is maintained within a very narrow range. In the setting of hypokalemia, where serum levels are low, potassium shifts from the intracellular to the extracellular space. As a result, the cell membranes become hyperpolarized and thus more resistant to depolarization, which makes them less likely to generate an action potential.
Hypokalemia can manifest as generalized muscle weakness, paralytic ileus, and abnormalities in cardiac conduction. Electrocardiogram (ECG) changes that accompany hypokalemia include ST depressions, small amplitude of T waves, and increased height of U waves.40 In severe cases, prolonged PR intervals and wide QRS complexes may also be seen.
Three broad mechanisms lead to hypokalemia: increased intracellular shifts, decreased potassium intake, and increased potassium loss. Insulin, epinephrine, β2 agonists, and α agonists all shift potassium intracellularly41,42; starvation and malnutrition can lead to inadequate potassium intake; and diuretics and gastrointestinal disorders increase potassium loss. Diuretic therapy is the most common cause of potassium wasting. By blocking sodium reabsorption, thiazide and loop diuretic increase sodium delivery to the collecting tubules, creating a favorable electrochemical gradient for potassium secretion in exchange for sodium reabsorption.43 Contrary to popular belief, hypokalemia complicating vomiting or nasogastric suctioning actually results from renal potassium loss, not gastric fluid loss. Intravascular volume depletion from gastric fluid loss stimulates the renin–angiotensin pathway and aldosterone release. Aldosterone, in turn, increases renal sodium absorption at the expense of potassium excretion, similar to other primary or secondary aldosteronism–induced hypokalemia.
Management of asymptomatic hypokalemia is safely achieved with slow enteral correction over several days. For patients with severe hypokalemia, parenteral replacement is preferred with a maximum recommended rate of correction of 10 to 20 mEq/h. Potassium chloride is commonly used, but potassium phosphate is also acceptable. In life-threatening cases, up to 40 mEq/h of potassium chloride can be given through a central line, preferably in an ICU setting.44 Because severe transient hyperkalemia can easily occur during correction of hypokalemia, care must be taken to closely monitor telemetry data as treatment proceeds.45 Low phosphate and magnesium levels often accompany hypokalemia and must also be treated in order for potassium levels to be successfully corrected.46
Hyperkalemia is a potentially lethal electrolyte disturbance. Expeditious recognition and prompt treatment are paramount. Like hypokalemia, hyperkalemia can be caused by increased intake, intracellular-to-extracellular potassium shifts, or defects in renal excretion. Increased intake in hospitalized patients is typically iatrogenic in nature and the result of accidental overdose of IV potassium. Shifts between the intracellular and extracellular fluids occur in the setting of acidosis or cell destruction. Decreased excretion is often the result of renal failure or adrenal insufficiency.
Severe hyperkalemia can present with paresthesias, muscle weakness leading to flaccid paralysis but typically with sparing of the diaphragm, and depressed deep tendon reflexes. Cranial nerves are rarely affected.47 Electrocardiographic changes include peaked and narrow T waves, widened QRS complexes, sine waves, and shortened QT intervals, which, when left untreated, can progress to ventricular fibrillation and asystole.48
Although commonly relied upon for diagnosis, the sensitivity of the ECG to reveal changes related to hyperkalemia has been estimated at around 80%, according to one retrospective review of 90 hyperkalemic patients.49 ECG sensitivity for hyperkalemia increases with the severity of electrolyte derangement, but normal ECGs have been reported even with profound hyperkalemia.50 ECG changes should, therefore, not be considered sine qua non to initiate treatment of severe hyperkalemia.
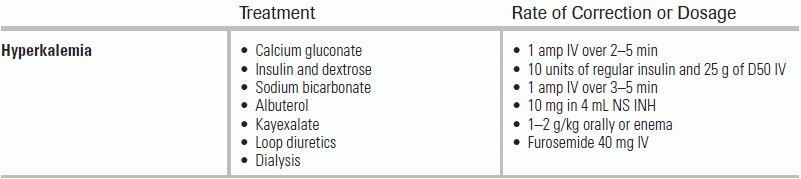
Immediate treatment of hyperkalemia is needed if ECG changes are noted, irrespective of serum potassium level, or if the serum potassium level is >6.5 to 7 mEq/L.51 The goals of therapy are threefold: (1) antagonize the effect of potassium on excitable cell membranes; (2) shift potassium from the extracellular milieu into cells; and (3) enhance elimination of potassium from the body.
Calcium gluconate or calcium chloride should be given first to antagonize the myocardial effects of hyperkalemia and prevent dysrhythmias. Classic teaching recommends an ampule of calcium gluconate, which represents 1 g or 4.6 mEq in 10 mL of a 10% solution, infused over 2 to 5 minutes with expected effect in 2 to 3 minutes.52,53 Calcium gluconate is preferred over calcium chloride—although calcium chloride is more concentrated (13.6 mEq in 10 mL of a 10% solution)—because it is less likely to cause tissue necrosis in the event of extravasation from the peripheral IV.54 A second ampule may be repeated after 5 minutes if there is no improvement in the ECG or if the ECG deteriorates after an initial improvement. The duration of action of 1 ampule is 30 to 60 minutes.55 Of note, reports exist of sudden death in patients taking digitalis glycosides who were given given IV calcium.56,57 Although these cases were anecdotal, prudence warrants either avoidance of IV calcium entirely in this subset of patients or at least very close monitoring during calcium administration.
Insulin lowers potassium levels by shifting potassium into cells. The effect is dose dependent58 and is mediated by the sodium/potassium ATPase pump in the plasma membrane of cells.59 An IV 10-unit dose of regular insulin is standard, and will shift potassium from the extracellular fluid to the intracellular fluid within 15 to 30 minutes, with the effect lasting 4 to 6 hours.60 Studies have shown that this dose will reduce serum potassium level by approximately 0.6 mEq/L. A bolus of 25 g of IV dextrose (50% solution) is generally given with the insulin to prevent hypoglycemia. However, because the effect of insulin on serum potassium levels peaks at 60 minutes, a single bolus of dextrose may be inadequate to prevent later hypoglycemia. For this reason, some advocate starting a dextrose infusion after the initial bolus.61 Insulin should be used without dextrose in hyperglycemic patients (baseline glucose level >250 mg/dL), as the hyperglycemia itself may the cause of hyperkalemia in these patients.62
NaHCO3 use in the emergent treatment of hyperkalemia remains controversial. NaHCO3 is typically formulated as an 8.4% solution (1 mEq/mL) and given in ampules of 50 mL (50 mEq per ampule) infused over 5 minutes. Like insulin, NaHCO3 has been postulated to shift potassium from the extracellular to the intracellular space. In theory, the administration of NaHCO3 should prompt hydrogen ions to move out of the cells via the Na+/H+ exchanger. This, in turn, leads to more sodium entering the cells to maintain electroneutrality. In the setting of hyperkalemia, this increase in intracellular sodium would subsequently activate the Na+/K+ ATPase pump, driving potassium from the extracellular to the intracellular space. Of critical importance, the Na+/H+ exchanger appears to be inactive in a steady state but active in the setting of acidosis.63 Arguments for the benefit of NaHCO3 in hyperkalemia originated with a few small clinical studies conducted in the 1950s and 1970s.64,65 Subsequent research has suggested that short-term infusions or boluses of NaHCO3 are ineffective in the acute setting,66–69 whereas a prolonged (4 to 6 hours) infusion of NaHCO3 decreased potassium levels by about 0.6 mEq/L.70 Given its limited efficacy acutely, while not contraindicated in hyperkalemic patients with acidemia, no significant or rapid change in potassium levels should be expected with NaHCO3 therapy.
The effect of β2-adrenergic stimulation effectively lowers serum potassium.71–78 β2 agonists (e.g., albuterol), like insulin, stimulate the Na+/K+ ATPase pump to shift potassium from the extracellular to the intracellular space. The recommended dose is 10 to 20 mg in 4 to 8 mL of saline, nebulized over 10 to 20 minutes. IV and metered-dose inhaler doses are also sometimes used. The onset of action is typically within 30 minutes, and the effect is maintained for up to 2 hours. Serum potassium will usually decrease by 0.5 to 1.2 mEq/L per 10- to 20-mg dose of albuterol.
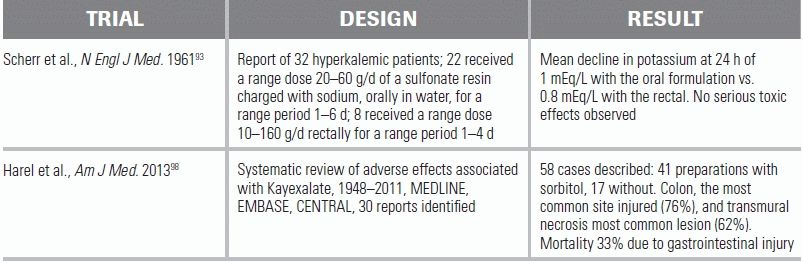
Sodium polystyrene sulfonate (Kayexalate) is a cation-exchange resin that removes potassium from the body by exchanging sodium for secreted potassium in the gastrointestinal tract. Kayexalate is generally given as an oral dose of 1 to 2 g/kg or as a retention enema with sorbitol to prevent constipation. Each gram of sodium polystyrene removes approximately 0.65 mEq/L of potassium, although the effect can be variable.79,80 Two important concerns exist with the use of Kayexalate. The first is its slow onset; when given orally, the onset of action is >2 hours and the maximum effect may not occur for 6 hours. As a retention enema, the effect is more rapid, but the magnitude of effect is less because of a shorter transit time through the gut lumen.81 The second potential problem is the possibility of toxicity. Numerous reports of Kayexalate-induced intestinal necrosis exist, both with the enema82–85 and oral forms.86–90 Although the true incidence of necrosis is unknown, estimates are 0.1% to 0.3% in the general population given the medication, and it occurs almost exclusively in “at-risk” patients (i.e., post–abdominal surgery, bowel injury, other gastrointestinal dysfunction).91 The Food and Drug Administration (FDA) first approved Kayexalate in 1958 after a small case series published in 1953 showed potassium binding in the stool and a hypokalemic effect in four patients with renal failure and a normal volunteer.92 The reported effectiveness of the drug, however, is largely based on the 1961 study, in which Kayexalate suspended in water was used orally or rectally in patients with acute and chronic kidney disease. In 22 of 32 cases, the plasma potassium fell by a mean of 1 mEq/L with the oral formulation versus 0.8 mEq/L with the rectal.93 Soon after, however, it was recognized that Kayexalate could cause life-threatening intestinal impactions, which then led to the practice of concomitantly administering 70% sorbitol, an osmotic laxative. A follow-up study showed a decrease in intestinal impactions with this combination94; however, reports of gastrointestinal necrosis continued to accumulate. With the precise mechanism of injury unclear, it was postulated that the 70% sorbitol rather than Kayexalate itself could be the culprit.95 Since 2007, the FDA has asked all manufacturers of premixed resin to reformulate their products to contain 33%, rather than 70%, sorbitol.
Studies have now called into questions the safety of even the 33% formulation.96 For all these reasons, consensus recommendations are to exhaust alternatives (e.g., diuretics, dialysis) before considering Kayexalate use.97,98 Importantly, Kayexalate continues to play a key role in the treatment of acute hyperkalemia under austere conditions, for example, after a natural or manmade disaster. In situations like these, where dialysis is not available, Kayexalate may be the only option for potassium removal, especially in chronic renal patients in whom diuretics are expected to have no effect. In the recent past, it was used in military facilities in Iraq, in the aftermath of Hurricane Katrina, and after the Haitian earthquake.99–102
If the potassium levels remain elevated despite the aforementioned therapies, a trial of loop diuretics in patients with preserved renal function may be attempted. In patients with end-stage renal disease and refractory cases, dialysis should be considered. Hemodialysis against a potassium-free dialysate can decrease the serum potassium level by as much as 1.5 mEq/h.66 However, a rebound in serum levels will always occur following dialysis, with 35% of the decrease in potassium negated after just 1 hour and nearly 70% after 6 hours as intracellular levels equilibrate with those of the serum. The magnitude of the rebound is thought to be proportional to the predialysis potassium level.103 Due to the risk of ventricular dysrhythmias during dialysis for severe hyperkalemia, which may result from the substantial intravascular volume shifts in the presence of a dysrhythmogenic potassium level, such patients are recommended to undergo continuous ECG monitoring104 during the session.
Potassium treatment summary: Tables 38.3 and 38.4
TABLE 38.3 Treatment of Hypokalemia

Treat concomitant low magnesium and phosphate.
TABLE 38.4 Treatment of Hyperkalemia
CALCIUM
Calcium is the most abundant electrolyte in the body and exists in three forms: (1) a chelated form; (2) an ionized form; and (3) a protein-bound form. The ionized form is the most physiologically active form and is therefore the one needing measurement. Two hormones—parathyroid hormone (PTH) and calcitonin—are responsible for regulating the body's calcium balance. PTH is released in response to hypocalcemia and increases calcium levels by stimulating osteoclasts, enhancing intestinal absorption, and decreasing renal excretion. Calcitonin, conversely, inhibits osteoclast activity and promotes renal excretion of calcium.
Because calcium plays a major role in muscle contraction–excitation, nerve conduction, myocardial function, and coagulation, the effects of hypocalcemia can be varied. Paresthesias in the hands and feet, circumoral numbness, muscle spasms, seizures, anxiety, irritability, psychosis, hypotension, low cardiac output, and QT interval prolongation may all be observed. QT interval prolongation can progress to bradycardia, heart block, or ventricular fibrillation.105
Hypocalcemia is diagnosed by measurement of serum levels. Because serum protein levels affect total serum calcium levels, the ionized calcium level provides a more accurate assessment of the physiologic active calcium available. Ionized calcium of <1.1 mmol/L confirms hypocalcemia (physiologic range is 1.1 to 1.4 mmol/L, or 4.5 to 5.6 mg/dL; 1 mmol/L is roughly equivalent to 4 mg/dL). Common causes of hypocalcemia include hypoparathyroidism; hyperphosphatemia, in which excess phosphate chelates circulating calcium (e.g., rhabdomyolysis, kidney disease); and massive transfusion, in which the preservative citrate binds calcium.106
In severe symptomatic cases, hypocalcemia is treated with 200 mg of elemental calcium given slowly over 10 to 20 minutes. Calcium gluconate can be given through a peripheral IV, but calcium chloride infused through a central line provides three times as many ionized calcium molecules (10 mL of calcium gluconate 10% contains 94 mg of elemental calcium; 10 mL of calcium chloride 10% contains 272 mg of elemental calcium). In less emergent cases, infusions containing 0.5 to 1.5 mg elemental calcium/kg/h may also be used, diluted in dextrose or saline, and given over 4 to 6 hours.107
A magnesium level must be concurrently checked and repleted because hypomagnesemia can impair PTH secretion and induce end-organ resistance to PTH, thus rendering hypocalcemia correction difficult.108 Finally, ionized calcium and H+ ions compete to bind to negatively charged sites on protein molecules, such as albumin. This binding is pH dependent, such that a sudden increase in pH—in the setting of, for example, alkali therapy—would cause proteins to release H+ and then bind calcium instead, potentially precipitously decreasing ionized calcium levels.109 For this reason, if a metabolic acidosis exists concomitantly with hypocalcemia, calcium replacement must take place before attempting to correct the acidosis.
Hypercalcemia is usually encountered in the setting of malignancy or primary hyperparathyroidism. Hyperparathyroidism is the culprit in 90% of ambulatory patients, while cancer causes 65% of hypercalcemia in hospitalized patients.110,111 Other causes of hypercalcemia include hyperthyroidism, Addison disease, and use of thiazide diuretics.
Manifestations of hypercalcemia are varied and frequently nonspecific. Patients will often report nausea, vomiting, and constipation. Weakness and fatigue are common, and altered mental status and coma may also be observed. Dysrhythmias can result from PR interval prolongation and QT interval shortening. Reports of heart block and cardiac arrest exist but are rare.112
As with hypocalcemia, hypercalcemia is generally diagnosed by measuring serum levels. Mild hypercalcemia is defined as total serum level of 12 mg/dL and is usually asymptomatic. Levels between 12 and 16 mg/dL can produce the nonspecific symptoms of weakness, nausea, vomiting, and abdominal pain. Cognitive dysfunction, personality changes, confusion, hallucinations, psychosis, stupor, and coma are expected with concentrations >16 mg/dL.
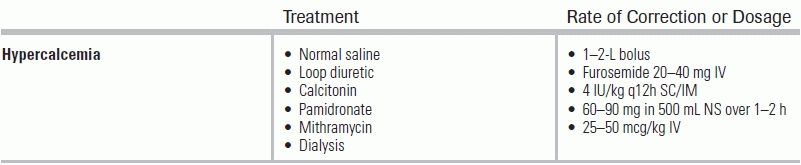
Because hypercalcemic patients are frequently volume-depleted from the associated polyuria (hypercalciuria) and poor oral intake, IV fluids are usually indicated initially. As with hypovolemic hypernatremia, the volume deficit must first be calculated and corrected using isotonic saline (generally 1 to 2 L IV over 1 hour). By increasing the glomerular filtration rate, renal calcium excretion also increases. Once the patient is determined to be euvolemic, a loop diuretic may be added to accelerate calcium excretion by the kidneys.113 Furosemide, 20 to 40 mg IV every 2 hours after correction of dehydration, is commonly used.
Calcitonin may also be used if first-line treatments are ineffective. A standard dose of 4 IU/kg is given either subcutaneously or intramuscularly every 12 hours. Its mechanism of action is inhibition of bone resorption and enhancement of renal excretion of calcium. Its main advantage is its fast onset of action; a response is usually noted within 2 to 4 hours. Unfortunately, its impact is mild (expected lowering in serum calcium level is 1 to 3 mg/dL after 4 to 6 hours, with a nadir within 12 to 24 hours), and tachyphylaxis is known to occur after 2 to 3 days.114,115 Bisphosphonates are good alternatives and inhibit osteoclast activity. The bisphosphonate pamidronate has been used for many years and is generally well tolerated, even in patients with renal disease. Pamidronate is a pyrophosphate analog that binds to hydroxyapatite and inhibits bone crystal dissolution as well as osteoclastic resorption.116 Standard dosing is 60 to 90 mg in 500 mL of isotonic saline given as an infusion over 1 to 2 hours. Unfortunately, it can take up to 48 hours to take effect, and the duration of action is 2 to 4 weeks. For these reasons, it is more appropriate for long-term rather than acute management of hypercalcemia.117
Additional therapies include mithramycin, an antibiotic that works by inhibiting RNA synthesis in osteoclasts. Its calcium-lowering effect is seen after 24 to 48 hours, but its use is limited by its poor side effect profile, including hepatotoxicity, renal failure, and bone marrow suppression.118 In severe or refractory cases of hypercalcemia, dialysis may be considered.119
Calcium treatment summary: Tables 38.5 and 38.6
TABLE 38.5 Treatment of Hypocalcemia

Correct simultaneous hypomagnesemia.
TABLE 38.6 Treatment of Hypercalcemia
MAGNESIUM
Hypomagnesemia is seen in as many as 12% of hospitalized patients, and 60% to 65% of critically ill patients in the ICU.120 Common etiologies include nutritional deficiency, intestinal losses, renal losses, as well as endocrine and metabolic derangements. Like calcium, magnesium exists in three forms: (1) ionized (61%), (2) protein-bound (33%), and (3) complexed (6%). The kidney is primarily responsible for magnesium homeostasis and, because magnesium reabsorption is proportional to urine flow, volume expansion can lead to magnesium wasting. In addition, thiazides and loop diuretics are well known for inhibiting magnesium reabsorption in the kidneys. Finally, many drugs, most notably alcohol, cause renal magnesium loss.121
Magnesium deficiency is often seen in conjunction with hypokalemia, hypocalcemia, and metabolic alkalosis. Thus, signs and symptoms are often varied and nonspecific. Cardiac manifestations include prolonged PR and QT intervals, as well as a widened QRS complex, which can lead to dysrhythmias, notably torsades de pointes.122 Neuromuscular manifestations include generalized weakness, seizures, tetany, lethargy, and coma.
A normal serum magnesium concentration is 1.7–2.1 mg/dL (1.4–1.8 mEq/L) in most cases, a diagnosis of hypomagnesemia can be made from patient history, as magnesium depletion is usually the result of either gastrointestinal or renal losses. In obscure cases, however, calculating the fractional excretion of magnesium or measuring the magnesium excretion over a 24-hour period can help to distinguish between the two causes of wasting. A daily excretion of more than 10 to 30 mg, or a fractional excretion of more than 2%, suggests renal wasting.123,124
If the patient is asymptomatic, oral supplementation is generally sufficient with a daily maintenance requirement of 0.4 mEq/kg/d. Magnesium oxide (49.6 mEq/g) is commonly used for repletion. If the patient is symptomatic, IV magnesium sulfate (8.12 mEq/g) is preferred at a dose of 1 to 2 mEq/kg administered over 8 to 24 hours. In the event of life-threatening dysrhythmias, the patient should be loaded with 25 to 50 mg/kg of magnesium sulfate over 3 to 5 minutes, followed by an infusion of 25 to 50 mg/kg/h for 4 to 6 hours.125
Hypermagnesemia is rare and typically iatrogenic in nature. It tends to occur because of overzealous correction of hypomagnesemia, or in the treatment of preeclampsia and preterm labor.126 However, it may also be seen with parenteral hyperalimentation, use of laxatives and enemas, or use of antacids. Patients with kidney disease are particularly at risk.127
Signs and symptoms of hypermagnesemia are flushing, respiratory depression, pulmonary edema, hypotension, weakness with loss of deep tendon reflexes, or paralysis. ECG manifestations associated with hypermagnesemia include prolonged PR and ST intervals, which may lead to bradycardia, complete heart block, and even cardiac arrest.
Except in severely symptomatic cases, the diagnosis of hypermagnesemia is made on laboratory evaluation. It is usually defined by a serum magnesium concentration >0.95 mmol/L, or 2.2 mg/dL.
Because most cases of hypermagnesemia are iatrogenic, the first-line treatment is to remove the exogenous source of magnesium. Diuretics can also promote renal excretion. In severe cases, calcium gluconate can temporarily antagonize the cardiac and neurologic symptoms. Dialysis may also be considered if initial therapies are unsuccessful.128
Magnesium treatment summary: Tables 38.7 and 38.8.
TABLE 38.7 Treatment of Hypomagnesemia

TABLE 38.8 Treatment of Hypermagnesemia

LITERATURE TABLE

1.Anderson RJ, Chung H-M, Kluge R, et al. Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin. Ann Intern Med. 1985;102:164–168.
2.Arieff AI, Llach F, Massry SG. Neurologic manifestations and morbidity of hyponatremia, correlation of brain water and electrolytes. Medicine (Baltimore). 1976;55:121–129.
3.Wijdicks EF, Ropper AH, Hunnicutt EJ, et al. Atrial natriuretic factor and salt wasting after aneurysmal subarachnoid hemorrhage. Stroke. 1991;22:1519–1524.
4.Fried LF, Palevsky PM. Hyponatremia and hypernatremia. Med Clin North Am. 1997;81:585–609.
5.Kapoor M, Chan GZ. Fluid and electrolyte abnormalities. Crit Care Clin. 2001;17:503–529.
6.Schrier RW. Pathogenesis of sodium and water retention in high output and low output cardiac failure, nephrotic syndrome, cirrhosis, and pregnancy. N Engl J Med. 1988;319:1127–1134.
7.Spalding HK, Goodwin SR. Fluids and electrolyte disorders in the critically ill. Semin Anesth Perioper Med Pain. 1999;18:15–26.
8.Cluitmans FH, Meinders AE. Management of severe hyponatremia: rapid or slow correction? Am J Med. 1990;88:161–166.
9.Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589.
10.Rose BD. Clinical Physiology of Acid–base and Electrolyte Disorders. 4th ed. New York: McGraw-Hill; 1994:651–694.
11.Sterns RH, Thomas DJ, Herndon RH. Brain dehydration and neurologic deterioration after rapid correction of hyponatremia. Kidney Int. 1989;35:69–75.
12.Sterns RH, Riggs JE, Schochet SS Jr. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med. 1986;314:1535–1542.
13.Verbalis JG, Goldsmith SR, Greenberg A, et al. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120:S1.
14.Greenberg A, Verbalis JG. Vasopressin receptor antagonists. Kidney Int. 2006;69:2124–2130.
15.Schrier RW, Gross P, Gheorghiade M, et al.; SALT Investigators. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355:2099–2112.
16.Zeltser D, Rosansky S, van Rensburg H, et al.; Conivaptan Study Group. Assessment of the efficacy and safety of intravenous conivaptan in euvolemic and hypervolemic hyponatremia. Am J Nephrol. 2007;27:447–457.
17.Annane D, Decaux G, Smith N. Efficacy and safety of oral conivaptan, a vasopressin-receptor antagonist, evaluated in a randomized, controlled trial in patients with euvolemic or hypervolemic hyponatremia. Am J Med Sci. 2009;337:28–36.
18.White MG, Fetner CD. Treatment of the syndrome of inappropriate secretion of antidiuretic hormone with lithium carbonate. N Engl J Med. 1975;292:390–392.
19.Forrest JN, Cox M, Hong C, et al. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298:173–177.
20.Chawla R. Hyponatremia. In: Chawla R, Subhash T, eds. ICU Protocols: A Stepwise Approach. India: Springer; 2012:433–440.
21.Rose BD. Clinical Physiology of Acid–Base and Electrolyte Disorders. 5th ed. New York: McGraw-Hill; 2001.
22.Kumar S, Berl T. Sodium. Lancet. 1998;352:220–228.
23.Funk GC, Lindner G, Druml W, et al. Incidence and prognosis of dysnatremias present on ICU admission. Intensive Care Med. 2010;36:304–311.
24.Lindner G, Funk GC, Schwarz C, et al. Hypernatremia in the critically ill is an independent risk factor for mortality. Am J Kidney Dis. 2007;50:952–957.
25.Darmon M, Timsit JF, Francais A, et al. Association between hypernatremia acquired in the ICU and mortality: a cohort study. Nephrol Dial Transplant. 2010;25:2510–2515.
26.Lindner G, Funk GC, Lassnigg A, et al. Intensive care-acquired hypernatremia after major cardiothoracic surgery is associated with increased mortality. Intensive Care Med. 2010;36:1718–1723.
27.Stelfox HT, Ahmed SB, Khandwala F, et al. The epidemiology of intensive care unit-acquired hyponatremia and hypernatremia in medical-surgical intensive care units. Crit Care. 2008;12:R162.
28.Stelfox HT, Ahmed SB, Zygun D, et al. Characterization of intensive care unit acquired hyponatreamia and hypernatremia following cardiac surgery. Can J Anaesth. 2010;57:650–658.
29.O'Donoghue SD, Dulhunty JM, Bandeshe HK, et al. Acquired hypernatremia is an independent predictor of mortality in critically ill patients. Anesthesia. 2009;64:514–520.
30.Bratusch-Marrain PR, DeFronzo RA. Impairment of insulin-mediated glucose metabolism by hyperosmolality in man. Diabetes. 1983;32:1028–1034.
31.Hoorn EJ, de Vogel S, Zietse R. Insulin resistance in an 18-year-old patient with Down syndrome presenting with hyperglycemia coma, hypernatremia, and rhabdomyolysis. J Intern Med. 2005;528:285–288.
32.Alonso PC, Matute SS, Urena SF, et al. Rhabdomyolysis secondary to hypernatremia. An Pediatr (Barc). 2010;73:223–224.
33.Denman JP. Hypernatremia and rhabdomyolysis. Med J Aust. 2007;187:527–528.
34.Kozeny GA, Murdock DK, Euler DE, et al. In vivo effects of acute changes in osmolality and sodium concentration on myocardial contractility. Am Heart J. 1985;109:290–296.
35.Robertson GL, Aycinena P, Zerbe RL. Neurogenic disorders of osmoregulation. Am J Med. 1982;72:339–353.
36.Chawla R. Hypernatremia. In: Chawla R, Subhash T, eds. ICU Protocols: A Stepwise Approach. India: Springer; 2012:441–446.
37.Haddow JE, Cohen DL. Understanding and managing hypernatremic dehydration. Pediatr Clin North Am. 1974;21:435–441.
38.Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342:1493–1499.
39.Lindner G, Funk GC. Hypernatremia in critically ill patients. J Crit Care. 2013;28:216.e11–216.e20.
40.Gennari F. Hypokalemia. N Engl J Med. 1998;339:451–458.
41.Fulop M. Hyperkalemia in diabetic ketoacidosis. Am J Med Sci. 1990;299:164–169.
42.Williams ME, Gervino EV, Rosa RM, et al. Catecholamine modulation of rapid potassium shifts during exercise. N Engl J Med. 1985;312:823–827.
43.Tannen RL. Potassium disorders. In: Kokko JP, Tannen RL, eds. Fluids and Electrolytes. 3rd ed. Philadelphia, PA: WB Saunders; 1996:111–199.
44.Subhash T. Hypokalemia and Hyperkalemia. In: Chawala R, Subhash T, eds. ICU Protocols: A Stepwise Approach. India: Springer; 2012.
45.Kruse JA, Carlson RW. Rapid correction of hypokalemia using concentrated intravenous potassium chloride infusions. Arch Intern Med. 1990;150:613–617.
46.Dyckner T. Relation of cardiovascular disease to potassium and magnesium deficiencies. Am J Cardiol. 1990;65:44–46.
47.Weiner ID, Wingo CS. Hyperkalemia: a potential silent killer. J Am Soc Nephrol. 1998;9:1535–1543.
48.Fisch C. Relation of electrolyte disturbances to cardiac arrhythmias. Circulation. 1973;47:408–419.
49.Montague BT, Ouellette JR, Buller GK. Retrospective review of the frequency of ECG changes in hyperkalemia. Clin J Am Soc Nephrol. 2008;3:324–330.
50.Szerlip HM, Weiss J, Singer I. Profound hyperkalemia without electrocardiographic manifestations. Am J Kidney Dis. 1986;7:461–465.
51.Weisberg LS. Potassium Homeostasis. In: Carlson RW, Geheb MA, eds. Principle and Practice of Medical Intensive Care. Philadelphia, PA: Saunders; 1993.
52.Schwartz AB. Potassium-related cardiac arrhythmias and their treatment. Angiology. 1978;29:194–205.
53.Bisogno JL, Langley A, Von DMM. Effect of calcium to reverse the electrocardiographic effects of hyperkalemia in the isolated rat heart: a prospective, dose–response study. Crit Care Med. 1994;22:697–704.
54.Semple P, Booth C. Calcium chloride: a reminder. Anesthesia. 1996;51:93.
55.Weisberg LS. Management of severe hyperkalemia. Crit Care Med. 2008;36:3246–3251.
56.Bower JO, Mengle HAK. The additive effect of calcium and digitalis. A warning with a report of two deaths. JAMA. 1936;106:1151–1153.
57.Shrager MW. Digitalis intoxication: a review and report of forty cases, with emphasis on etiology. AMA Arch Intern Med. 1957;100:881–893.
58.DeFronzo RA, Felig P, Ferrannini E, et al. Effects of graded doses of insulin on splanchnic and peripheral potassium metabolism in man. Am J Physiol. 1980;238:E421–E427.
59.Clausen T, Everts ME. Regulation of the Na/K-pump in skeletal muscle. Kidney Int. 1989;35:1–13.
60.Allon M, Takeshian A, Shanklin N. Effect of insulin-plus-glucose infusion with or without epinephrine on fasting hyperkalemia. Kidney Int. 1993;43:212–217.
61.Allon M, Copkney C. Albuterol and insulin for treatment of hyperkalemia in hemodialysis patients. Kidney Int. 1990;38:869–872.
62.Goldfarb S, Cox M, Singer I, et al. Acute hyperkalemia induced by hyperglycemia: hormonal mechanisms. Ann Intern Med. 1976;84:426–432.
63.Kamel KS, Wei C. Controversial issues in the treatment of hyperkalemia. Nephrol Dial Transplant. 2003;18:2215–2218.
64.Schwarz KC, Cohen BD, Lubash GD, et al. Severe acidosis and hyperpotassemia treated with sodium bicarbonate infusion. Circulation. 1959;19:215–220.
65.Fraley DS, Adler S. Correction of hyperkalemia by bicarbonate despite constant blood pH. Kidney Int. 1977;12:354–360.
66.Blumberg A, Weidmann P, Shaw S, et al. Effect of various therapeutic approaches on plasma potassium and major regulating factors in terminal renal failure. Am J Med. 1988;85:507–512.
67.Gutierrez R, Schlessinger F, Oster JR, et al. Effect of hypertonic versus isotonic sodium-bicarbonate on plasma potassium concentration in patients with end-stage renal disease. Miner Electrolyte Metab. 1991;17:297–302.
68.Allon M, Shanklin N. Effect of bicarbonate administration on plasma potassium in dialysis patients: interactions with insulin and albuterol. Am J Kidney Dis. 1996;28:508–514.
69.Kim HJ. Combined effect of bicarbonate and insulin with glucose in acute therapy of hyperkalemia in end-stage renal disease patients. Nephron. 1996;72:476–482.
70.Blumberg A, Weidmann P, Ferrari P. Effect of prolonged bicarbonate administration on plasma potassium in terminal renal failure. Kidney Int. 1992;41:369–374.
71.Brown MJ, Brown DC, Murphy MB. Hypokalemia from beta2-receptor stimulation by circulating epinephrine. N Engl J Med. 1983;309:1414–1419.
72.Montoliu J, Lens XM, Revert L. Potassium-lowering effect of albuterol for hyperkalemia in renal failure. Arch Intern Med. 1987;147:713–717.
73.Allon M, Dunlay R, Copkney C. Nebulized albuterol for acute hyperkalemia in patients on hemodialysis. Ann Intern Med. 1989;110:426–429.
74.Montoliu J, Lens XM, Revert L. Treatment of hyperkalemia in renal failure with salbutamol inhalation. J Intern Med. 1990;228:35–37.
75.Liou HH, Chiang SS, Wu SC, et al. Hypokalemic effects of intravenous infusion or nebulization of salbutamol in patients with chronic renal failure. Am J Kidney Dis. 1994;23:266–270.
76.Kemper MJ, Harps E, Müller-Wieffel DE. Hyperkalemia: therapeutic options in acute and chronic renal failure. Clin Nephrol. 1996;46:67–69.
77.Halperin ML. Potassium. Lancet. 1998;352:135–142.
78.Mandelberg A, Krupnik Z, Houri S, et al. Salbutamol metered-dose inhaler with spacer for hyperkalemia: how fast? How safe? Chest. 1999;115:617–622.
79.Frohnert PP, Johnson WJ, Mueiier GJ, et al. Resin treatment of hyperkalemia I. Exchange properties of a cation exchange resin (calcium cycle). J Lab Clin Med. 1968;71:834–839.
80.Frohnert PP, Johnson WJ, Mueiier GJ, et al. Resin treatment of hyperkalemia II. Clinical experience with a cation exchange resin (calcium cycle). J Lab Clin Med. 1968;71:840–846.
81.Emmett M, Hootkins RE, Fine KD, et al. Effect of three laxatives and a cation exchange resin on fecal sodium and potassium excretion. Gastroenterology. 1995;108:752–760.
82.Rogers RB, Li SC. Acute colonic necrosis associated with sodium polystyrene sulfonate (Kayexalate) enemas in critically-ill patients: case report and review of the literature. J Trauma. 2001;51:395–397.
83.Rashid A, Hamilton SR. Necrosis of the gastrointestinal tract in uremic patients as a result of sodium polystyrene sulfonate (Kayexalate) in sorbitol: an under-recognized condition. Am J Surg Pathol. 1997;21:60–69.
84.Scott TR, Graham SM, Schweitzer EJ, et al. Colonic necrosis following sodium polystyrene sulfonate (Kayexalate)–sorbitol enema in a renal transplant patient. Report of a case and review of the literature. Dis Colon Rectum. 1993;36:607–609.
85.Wootton FT, Rhodes DF, Lee WM, et al. Colonic necrosis with kayexalate-sorbitol enemas after renal transplantation. Ann Intern Med. 1989;111:947–949.
86.Abraham SC, Bhagavan BS, Lee LA, et al. Upper gastrointestinal tract injury in patients receiving kayexalate (sodium polystyrene sulfonate) in sorbitol: clinical, endoscopic, and histopathologic findings. Am J Surg Pathol. 2001;25:637–644.
87.Cheng ES, Stringer KM, Pegg SP. Colonic necrosis and perforation following oral sodium polystyrene sulfonate (resonium A/kayexalate) in a burn patient. Burns. 2002;28:189–190.
88.Dardik A, Moesinger RC, Efron G, et al. Acute abdomen with colonic necrosis induced by kayexalate-sorbitol. South Med J. 2000;93:511–513.
89.Gardiner GW. Kayexalate (sodium polystyrene sulfonate) in sorbitol associated with intestinal necrosis in uremic patients. Can J Gastroenterol. 1997;11:573–577.
90.Roy-Chaudhury P, Meisels IS, Freedman S, et al. Combined gastric and ileocecal toxicity (serpiginous ulcers) after oral kayexalate in sorbitol therapy. Am J Kidney Dis. 1997;30:120–122.
91.Watson M, Abbott KC, Yuan CM. Damned if you do, damned if you don't: potassium binding resins in hyperkalemia. Clin J Am Soc Nephrol. 2010;5:1723–1726.
92.Evans BM, Evans BM. Ion-exchange resins in the treatment of anuria. Lancet. 1953;262:791–795.
93.Scherr L, Ogden DA, Mead AW, et al. Management of hyperkalemia with a cation-exchange resin. N Engl J Med. 1961;264:115–119.
94.Flinn RB, Merrill JP, Welzan WR. Treatment of the oliguric patient with a new sodium ion-exchange resin and sorbitol: a preliminary report. N Engl J Med. 1961;264:111–115.
95.Lillemoe KD, Romolo JL, Hamilton SR, et al. Intestinal necrosis due to sodium polystyrene (Kayexalate) in sorbitol enemas: clinical and experimental support for the hypothesis. Surgery. 1987;101:267–272.
96.McGowan CE, Saha S, Chu G, et al. Intestinal necrosis due to sodium polystyrene sulfonate (Kayexalate) in sorbitol. South Med J. 2009;102:493–497.
97.Sterns RH, Rojas M, Bernstein P, et al. Ion-exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol. 2010;21:733–735.
98.Harel Z, Harel S, Shah PS, et al. Gastrointestinal adverse events with sodium polystyrene sulfonate (Kayexalate) use: a systematic review. Am J Med. 2013;126:264e.
99.Kopp JB, Ball LK, Cohen A. Kidney patient care in disasters: lessons from hurricanes and earthquakes of 2005. Clin J Am Soc Nephrol. 2007;2:814–824.
100.Centers for Medicare and Medicaid Services. Preparing For Emergencies: A Guide for People on Dialysis. CMS publication No. 10150. Baltimore, MD: Department of Health and Human Services, Centers for Medicare and Medicaid Services; 2007.
101.Miller AC, Arquilla B. Chronic diseases and natural hazards: impact of disasters on diabetic, renal and cardiac patients. Prehosp Disaster Med. 2008;23:185–194.
102.Amundson D, Dadekian G, Etienne M, et al. Practicing internal medicine onboard the USNS COMFORT in the aftermath of the Haitian earthquake. Ann Intern Med. 2010;152:733–737.
103.Zehnder C, Gutzwiller JP, Huber A, et al. Low-potassium and glucose-free dialysis maintains urea but enhances potassium removal. Nephrol Dial Transplant. 2001;16:78–84.
104.Ahmed J, Weisberg LS. Hyperkalemia in dialysis patients. Semin Dial. 2001;14:348–356.
105.Shane E, Irani D. Hypercalcemia: pathogenesis, differential diagnosis, and management. In: Favus MJ, ed. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Philadelphia, PA: Lippincott-Raven; 1996:217–219.
106.Wilson RF, Binkley LE, Sabo FM Jr, et al. Electrolyte and acid–base changes with massive blood transfusions. Am Surg. 1992;9:535–544.
107.Weiss-Guillet E-M, Takala J, Jakob SM. Diagnosis and management of electrolyte emergencies. Best Pract Res Clin Endocrinol Metab. 2003;17(4):623–651.
108.Cholst IN, Steinberg SF, Tropper PJ, et al. The influence of hypermagnesemia on serum calcium and parathyroid hormone levels in human subjects. N Engl J Med. 1984;310:1221–1225.
109.Suki WN, Massry MG, eds. Therapy of Renal Diseases and Related Disorders. 2nd ed. Norwell, MA: Kluwer Academic Publishers; 1991.
110.Fisken RA, Heath DA, Somers S, et al. Hypercalcemia in hospital patients: clinical and diagnostic aspects. Lancet. 1981;1:202–207.
111.Klee GG, Kao, PC, Heath, H III. Hypercalcemia. Endocrinol Metab Clin North Am. 1988;17:573–600.
112.Bajorunas DR. Clinical manifestations of cancer-related hypercalcemia. Semin Oncol 1990;17(2)(suppl 5):16–25.
113.Suki WN, Yium JJ, Von Minden M, et al. Acute treatment of hypercalcemia with furosemide. N Engl J Med. 1970;283:836–840.
114.Kammerman S, Canfield RE. Effect of porcine calcitonin on hypercalcemia in man. J Clin Endocrinol Metabol. 1970;31:70–75.
115.Wisneski LA. Salmon calcitonin in the acute management of hypercalcemia. Calcif Tissue Int. 1990;46(suppl):S26–S30.
116.McCurdy MT, Shanholtz CB. Oncologic Emergencies. Crit Care Med. 2012;40:2212–2222.
117.Wimalawansa SJ. Optimal frequency of administration of pamidronate in patients with hypercalcemia of malignancy. Clin Endocrinol (Oxf). 1994;41:591–595.
118.Smith IE, Powles TJ. Mithramycin for hypercalcemia associated with myeloma and other malignancies. BMJ. 1975;1:268–269.
119.Bilezikian JP. Management of acute hypercalcemia. N Engl J Med. 1992;326:1196–1203.
120.Wong ET, Rude RK, Singer FR, et al. A high prevalence of hypomagnesemia and hypermagnesemia in hospitalized patients. Am J Clin Pathol. 1983;79:348–352.
121.Elisaf M, Merkouropoulos M, Tsianos EV, et al. Pathogenetic mechanisms of hypomagnesemia in alcoholic patients. J Trace Elem Med Biol. 1995;9:210–214.
122.Dyckner T. Serum magnesium in acute myocardial infarction. Relation to arrhythmias. Acta Med Scand. 1980;207:59–66.
123.Al Ghamdi SM, Cameron EC, Sutton RA. Magnesium deficiency: pathophysiologic and clinical overview. Am J Kidney Dis. 1994;24:737–752.
124.Elisaf M, Panteli K, Theodorou J, et al. Fractional excretion of magnesium in normal subjects and in patients with hypomagnesemia. Magnes Res. 1997;10:315–320.
125.Agus ZA, Wasserstein A, Gold Farb S. Disorders of calcium and magnesium homeostasis. Am J Med. 1982;72:473–488.
126.Morisaki H, Yamamoto S, Morita Y, et al. Hypermagnesemia-induced cardiopulmonary arrest before induction of anesthesia for emergency cesarean section. J Clin Anesth. 2000;12:224–226.
127.Schelling JR. Fatal hypermagnesemia. Clin Nephrol. 2000;53:61–65.
128.Mordes JP, Wacker WE. Excess magnesium. Pharmacol Rev. 1977;29:273–300.