TABLE 43.1 Causes of Adrenal Insufficiency

BACKGROUND
The adrenal glands are two small, irregularly shaped bodies located superior to each renal pole. Each gland contains two distinct structures, the outer adrenal medulla and the inner adrenal cortex. The adrenal cortex can be divided into three zones: the zona glomerulosa, which produces the mineralocorticoid aldosterone, and the zona fasciculata and zona reticularis, which produce the glucocorticoid cortisol as well as androgens. The outer medulla is responsible for catecholamine production, including epinephrine and norepinephrine.
Cortisol has multiple effects, including increasing blood glucose and gluconeogenesis, suppressing the immune system, decreasing bone formation, and aiding in fat, protein, and carbohydrate metabolism. Aldosterone works on the kidneys to promote reabsorption of sodium and of water and secretion of potassium. Adrenal insufficiency is defined as a condition in which the adrenal glands fail to produce an adequate amount of steroid hormones to meet the needs of the body. Adrenal insufficiency can be a devastating complication of critical illness and, in high-risk patients (e.g., those with hypotension, shock, sepsis), has an incidence of approximately 30% to 40%.1
Adrenal gland production of cortisol is regulated through the hypothalamus–pituitary–adrenal gland axis. The hypothalamus responds to external stimuli, including low cortisol levels, by secreting corticotropin-releasing hormone (CRH). This causes the pituitary gland to release corticotropin (adrenocorticotropic hormone [ACTH]), which is the primary regulator of cortisol production and release. Approximately 95% of body cortisol is protein bound; it is the remaining 5% of free cortisol that produces effects in the body. In normal individuals, daily cortisol secretion ranges from 40 to 80 μmol and has a pronounced circadian rhythm. Severe physical or emotional stresses stimulate the secretion of CRH and ACTH, which results in large increases (two- or threefold) in serum cortisol concentrations.
Aldosterone production is mediated by the renin–angiotensin–aldosterone system (RAAS). The RAAS is involved with the regulation of vasoconstriction and extracellular blood volume. Renin is an enzyme secreted by specialized cells that encircle the arterioles at the entrance to the glomeruli of the kidneys. These specialized cells modulate renin production in response to changes in blood flow and blood pressure. Low blood flow to the kidneys from any reason, including low blood pressure, will result in increased renin production and release. Renin promotes conversion of the plasma protein angiotensinogen into angiotensin I, which is subsequently converted into angiotensin II—a potent vasoconstrictor—by the angiotensin-converting enzyme. Angiotensin II acts on receptors in the adrenal glands to stimulate the secretion of aldosterone, which in turn promotes renal resorption of sodium and water.
The adrenal medulla makes up about 10% of the adrenal gland and is an integral part of the sympathetic nervous system. The cells of the medulla, known as chromaffin cells, house chromaffin granules that contain epinephrine, norepinephrine, and dopamine, which are released in response to sympathetic nerve stimulation. In patients with adrenal insufficiency, the adrenal medulla is typically not dysfunctional.
ETIOLOGY
Recognizing adrenal insufficiency is the most challenging aspect of its treatment. The hallmark presentation of this condition is hypotension unresponsive to fluids in a patient with sepsis or another acute stressor; however, its presentation can be much subtler. The provider's goal should be to recognize the disease and start treatment before the patient is in shock; once initiated, treatment is generally straightforward.
In subacute or chronic presentations, the symptoms of adrenal insufficiency may include fatigue, anorexia, nausea, vomiting, muscle aches, weight loss, and a low blood pressure (<110 mm Hg) that may be orthostatic. More than 90% of cases will demonstrate skin hyperpigmentation (due to increased ACTH release) in areas exposed to light, chronic friction, or pressure, as well as the palmar creases.2
There are three major types of adrenal insufficiency. Primary adrenal insufficiency is caused by destruction or dysfunction of the adrenal gland itself; this will not manifest until 90% of the gland is destroyed.3 Because there is direct damage to the gland, glucocorticoid and mineralocorticoid secretion are affected; destruction of the entire gland can affect sympathetic nervous system function as well. In the United States, the most common cause (80%) of primary adrenal insufficiency is autoimmune adrenalitis (Addison disease); half of these cases exist as part of the polyglandular autoimmune syndrome type I or II, which can include diabetes and hypothyroidism in addition to adrenal insufficiency. Worldwide, the most common cause of primary adrenal insufficiency is destruction of the adrenal gland by Mycobacterium tuberculosis.
Secondary (and tertiary) adrenal insufficiency results from any process involving the pituitary gland or hypothalamus that interferes with ACTH secretion. Such processes include tumors of the pituitary or hypothalamus, infiltrative processes such as sarcoid or TB, surgery, radiation, trauma, and postpartum pituitary necrosis (Sheehan syndrome). Secondary adrenal insufficiency affects only glucocorticoid production (ACTH only affects cortisol production and release). Mineralocorticoid production and sympathetic function are generally not affected.
By far, the most common etiology of secondary adrenal insufficiency is hypoaldosteronism caused by prolonged glucocorticoid therapy. When exogenous glucocorticoids are administered, the body's production of cortisol can be suppressed, which can lead to decreased adrenal gland responsiveness and atrophy. Inhibition of ACTH secretion depends on the dose, duration, and frequency of glucocorticoid therapy. While there is variation among individual patients, adrenal insufficiency should be considered in symptomatic patients who have received doses of prednisone >7.5 mg per day or another steroid equivalent for longer than 3 weeks.4,5 Inhaled and topical steroids are also demonstrated culprits in adrenal suppression.6,7 Patients taking medications that inhibit the cytochrome P450 enzyme CYP3A4 (diltiazem, protease inhibitors, azole antifungals, grapefruit juice) will have a prolonged biologic half-life of glucocorticoids and thus an enhanced suppression of adrenal function.8 The adrenal glands may require 6 to 12 months for full recovery of function following prolonged use of exogenous glucocorticoids.9 Tertiary adrenal insufficiency results when an insufficient amount of CRH is produced by the hypothalamus (Table 43.1).
TABLE 43.1 Causes of Adrenal Insufficiency
SPECIAL CONSIDERATIONS
Adrenal insufficiency has been demonstrated in 5% to 20% of tested patients with HIV.10 The prevalence increases with the progression of HIV. HIV may result in primary adrenal insufficiency by itself or through associated infection (TB, cytomegalovirus [CMV], Mycobacterium avium-intracellulare (MAI) or malignancy (Kaposi sarcoma, lymphoma). HIV is also associated with secondary adrenal insufficiency through infectious agents that affect the pituitary gland (toxoplasmosis, CMV), drugs that interfere with adrenal function (ketoconazole, megestrol acetate), or drugs that increase degradation of cortisol (rifampin, phenytoin, opiates).11 Autopsies of patients with AIDS show adrenal injury in over 50% of cases and pituitary involvement injury in 30% of cases.12
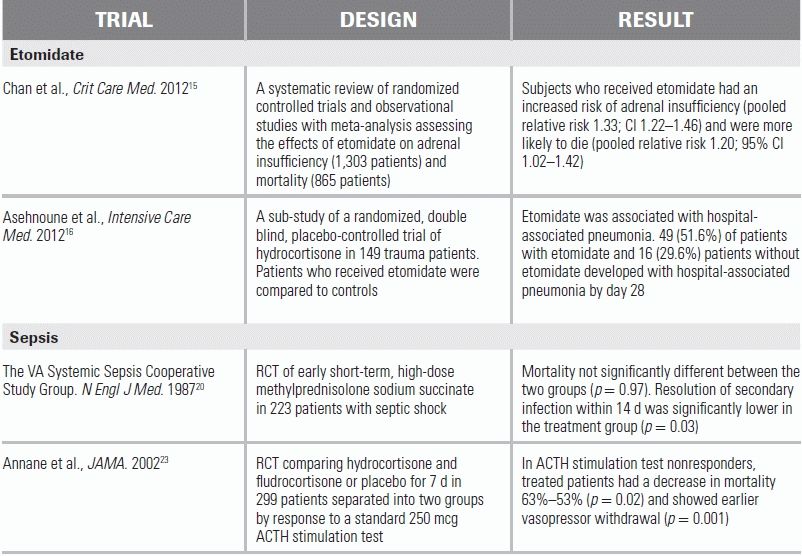
Etomidate is a first-line anesthetic used in rapid sequence intubation; it is particularly useful in critically ill patients because it is hemodynamically well tolerated. Etomidate, however, decreases available cortisol by inhibiting the 11β-hydroxylase enzyme that converts 11β-deoxycortisol into cortisol in the adrenal gland. Use of continuous etomidate infusion in the critically ill has been associated with an increase in mortality and thus has been curtailed.13 A single dose of etomidate has been shown to cause laboratory evidence of adrenal suppression for 4 to 24 hours. In one study, 80% of patients showed adrenal suppression when tested at 12 hours after a single dose of etomidate.14 However, the clinical effects of this effect are still in debate. A multitude of smaller and retrospective studies showed no difference in outcome with etomidate. A large 2012 meta-analysis/systemic review demonstrated that in critically ill patients, the relative risk of death with etomidate use was 1.20 (95% CI 1.04–1.42).15 Smaller studies have also linked etomidate to a higher risk of pneumonia (56% vs. 26%) in trauma patients.16 Although evidence is inconclusive, it may be prudent to consider other induction agents if they are available. While some practitioners have opted to give steroids for 24 to 48 hours after an etomidate-assisted intubation in order to compensate for the decreased adrenal function, small studies have failed to demonstrate any changes in patient outcomes with the use of adjunctive steroids.
Infection stimulates an inflammatory, coagulation, and immunologic response that works synergistically to either eliminate or control the infection and to repair associated tissue damage. If left unchecked or unregulated, however, these host defenses may themselves become counterproductive and lead to deterioration in organ function rather than to restoration of homeostasis. It is believed that in some cases of sepsis, it is overaggressive and protracted host defense, rather than the precipitating insult (e.g., pneumonia), that primarily determines outcome.
Infection also stimulates the hypothalamic–pituitary–adrenal axis, causing an increase in glucocorticoid release. Glucocorticoids exert a protective effect by restraining the host defense response at many levels, including suppression of cytokine production. Studies have shown that sepsis nonsurvivors exhibit a persistent and exaggerated increase in circulating inflammatory cytokine concentrations when compared with survivors.17,18 Research also suggests that cytokines can produce a concentration-dependent resistance to glucocorticoids in target tissues by reducing glucocorticoid receptor binding affinity for cortisol.19 Finally, inflammatory mediators (including cytokines) are also known to alter the hypothalamic–pituitary–adrenal axis and contribute directly to adrenal insufficiency.
Because of these associations, it was once thought that all patients with severe septic shock should be treated with exogenous steroids. Early treatment and studies used “high-dose” steroids (30 mg/kg methylprednisolone), but showed limited success; subsequent studies of “high-dose” steroids showed at first no improvement, and later, harm.20,21 The negative effects of glucocorticoid therapy included hyperglycemia, superinfection, muscular weakness, hypernatremia, upper gastrointestinal bleeding, psychosis, and poor wound healing.
More recently, studies have focused on the effect of “low-dose” steroids (e.g., 200 to 300 mg of hydrocortisone or equivalent). Patients considered for exogenous glucocorticoid therapy are those in septic shock who remain unstable after fluid and vasopressor therapy. The endpoints examined were earlier reversal of shock and effect on mortality. A representative study with “low-dose” steroids showed effect on the timing of shock reversal, but no effect at 28-day mortality.22 A 2002 landmark study separated patients with a normal corticotrophin stimulation (responders) from patients with an abnormally low response (nonresponders). This study showed the 28-day mortality was decreased by corticosteroid therapy in the overall patient population (61% vs. 55%) and in the ACTH nonresponder group of patients (63% vs. 53%), with no increase in adverse events.23 This study led to the inclusion of steroid therapy in the Surviving Sepsis Campaign in 2004.
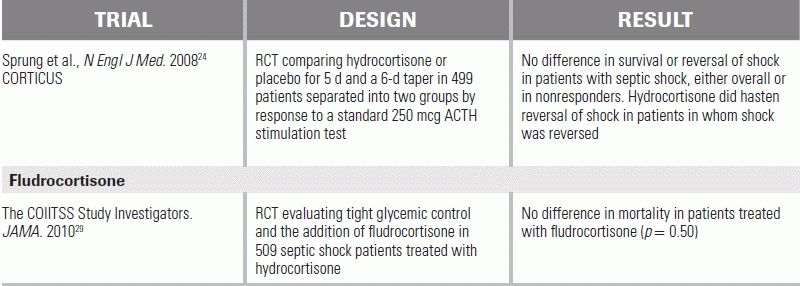
An important follow-up trial in 2008 reopened the question of steroids in sepsis.24 This large study also separated septic shock patients into responders and nonresponders, but found no difference in nonresponders who received glucocorticoids from placebo (39% vs. 36%). Shock reversal did occur more quickly in the treated group (3.3 days vs. 5.8 days); however, no difference in mortality was proven, and the study showed an increase in hyperglycemia, hypernatremia, and superinfections in steroid-treated patients.
Presently, the Surviving Sepsis Campaign 2012 guidelines recommend hydrocortisone at a dose of 200 mg per day for patients who remain hemodynamically unstable after adequate fluid and vasopressor therapy. An ACTH stimulation test is not a prerequisite for initiating steroid therapy (see below).25
DIAGNOSTIC EVALUATION
The diagnosis of adrenal insufficiency involves testing cortisol levels. In an unstressed patient, a single morning cortisol level can be sufficient. A level of <3 mcg/dL is diagnostic for adrenal insufficiency, a level of 4 to 10 mcg/dL is suggestive, and a level of >20 mcg/dL excludes the condition. However, as any form of stress increases cortisol levels, this metric is rarely helpful in the emergency or ICU setting. In septic patients, a random level of <10 mcg/dL has been used as an indicator of adrenal insufficiency, while a level of >33 mcg/dL makes the diagnosis unlikely.26 One study evaluated multiple approaches to diagnosing adrenal insufficiency in critically ill patients with septic shock and concluded that the standard 250 mcg cosyntropin (ACTH) stimulation test—with a result of ≤9 mcg/dL increase from baseline in total cortisol 60 minutes after administration—was the best predictor of decreased adrenal function.27 A “low-dose” 1 mcg cosyntropin (ACTH) stimulation test has also been in limited use; this more sensitive assay has been shown to be a better predictor of survival, but is not readily available and is not as widely accepted.28 The American College of Critical Care Medicine currently recommends that adrenal insufficiency in critically ill patients is best identified by an increase in serum cortisol level of <9 mcg/dL after a 250 mg ACTH stimulation test, or a random total cortisol level <10 mcg/dL.26 It also recommends that these tests be performed only in patients with suspected adrenal insufficiency. This means that not all septic shock patients who are put on steroids require testing.
MANAGEMENT GUIDELINES
In the critical care setting, management of adrenal insufficiency focuses on treating concomitant stresses, resuscitating the patient, and giving glucocorticoids. Hydrocortisone is the glucocorticoid of choice, as it has both glucocorticoid and mineralocorticoid activities; it also adequately treats associated electrolyte disturbances caused by mineralocorticoid deficiency that are seen in primary adrenal insufficiency. Fludrocortisone, a mineralocorticoid, has been studied and is not considered necessary to supplement in the acute setting. Dexamethasone has been suggested for patients who may eventually need an ACTH stimulation test, as it does not interfere with cortisol levels; however, it is not indicated in critically ill patients, especially those with electrolyte disturbances, as it has no mineralocorticoid activity. In all patients receiving hydrocortisone, glucose should be monitored, as hyper- and hypoglycemic episodes are possible. Patients should also be monitored for hypernatremia, and hypertonic saline should generally be avoided.
In patients with acute adrenal crisis, treatment dose hydrocortisone is 50 to 100 mg IV q6h. Alternatively, a dose of hydrocortisone 50 to 100 mg followed by an infusion of 20 mg/hour has been used. Improvement is typically observed within 4 to 6 hours. In the setting of suspected adrenal insufficiency with concomitant septic shock, no standard dosing has been established; a common approach is to give hydrocortisone at a dose of either 200 to 300 mg per day or 50 mg q6h. This is often continued for 5 to 7 days with or without a taper.25,29 In patients who have a history of adrenal insufficiency who are undergoing an acute stress/procedure, a single dose of 100 mg IV of hydrocortisone has been recommended.
CONCLUSION
Adrenal insufficiency can be a devastating complication of critically illness and is an important entity to consider in the differential diagnosis of patients with persistent hypotension, shock, or sepsis. The use of steroids in sepsis should be strongly considered in patients with vasopressor refractory shock.
LITERATURE TABLE
CI, confidence interval.
1.Gary P, Zaloga MD, Paul Marik MD. Hypothalamic-Pituitary-adrenal insufficiency. Crit Care Clin. 2001;17:25–41.
2.Dunlop D. Eighty-six cases of addison's disease. Br Med J. 1963;2(5362):887.
3.Munver R, Volfson IA. Adrenal insufficiency: diagnosis and management. Curr Urol Rep. 2006;7:80–85.
4.Bouillon R. Acute adrenal insufficiency. Endocrinol Metab Clin North Am. 2006;35:76797.
5.Marik PE. Critical illness-related corticosteroid insufficiency. Chest. 2009;135:181–193.
6.Hengge UR, Ruzicka T, Schwartz RA, et al. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54(1):1–15.
7.Zollner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (Part 2). Pediatr Allergy Immunol. 2007;18:469.
8.Varis T, Kivisto KT, Backman JT, et al. The cytochrome P450 3A4 inhibitor itraconazole markedly increases the plasma concentrations of dexamethasone and enhances its adrenal-suppressant effect. Clin Pharmacol Ther. 2000;68:487–494.
9.Axelrod L. Perioperative management of patients treated with glucocorticoids. Endocrinol Metab Clin North Am. 2003;32:367–383.
10.Masharani U, Schambelan M. The endocrine complications of acquired immunodeficiency syndrome. Adv Intern Med. 1993;38:323–336.
11.Prasnthai V, Sunthornyothin S, Phowthongkum P, et al. Prevalence of adrenal insufficiency in critically ill patients with AIDS. J Med Assoc Thai. 2007;90:1768.
12.Eledrisi MS, Verghese AC. Adrenal Insufficiency in HIV Infection: a review and recommendations. Am J Med Sci. 2001;321(2):137–144.
13.Wagner RL, White PF, Kan PB, et al. Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med. 1984;310:1415–1421.
14.Vinclair M, Broux C, Faure P, et al. Duration of adrenal inhibition following a single dose of etomidate in critically ill patients. Intensive Care Med. 2008;34:714–719.
15.Chan CM, Mitchell AL, Shorr AF. Etomidate is associated with mortality and adrenal insufficiency in sepsis: a meta-analysis*. Crit Care Med. 2012;40(11):2945–2953.
16.Asehnoune K, Mahe PJ, Seguin P. Etomidate increases susceptibility to pneumonia in trauma patients. Intensive Care Med. 2012;38(10):1673–1678.
17.Hermus ARMM, Sweep CGJ. Cytokines and the hypothalamic-pituitary-adrenal axis. J Steroid Biochem Mol Biol. 1990;37:867–871.
18.Damas P, Ledoux D, Nys M, et al. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann Surg. 1992;215:356–362.
19.Kam JC, Szefler SJ, Surs W, et al. Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. J Immunol. 1993;151:3460–3466.
20.The Veterans Administration Systemic Sepsis Cooperative Study Group. Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N Engl J Med. 1987;317:659–665.
21.Cronin L, Cook DJ, Carlet J, et al. Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;24:1430–1439.
22.Cicarelli DD, Viera JE, Martin Besenor FE. Early dexamethasone treatment for septic shock patients: a prospective randomized clinical trial. Sao Paulo Med J. 2007;125:237–241.
23.Annane D, Sebille V, Charpentier C, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–870.
24.Sprung CL, Annane D, et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med. 2008;10;358:111–124.
25.Dellinger P, Levy M, Rhodes A. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41:580–637.
26.Marik PE, Pastores SM, Annane D, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med. 2008;36:1937–1949.
27.Salgado DR, Verdeal JCR, Rocco JR. Adrenal function testing in patients with septic shock. Crit Care. 2006;10:R149.
28.Siraux V, De Backer D, Yalavatti G. Relative adrenal insufficiency in patients with septic shock: comparison of low-dose and conventional corticotropin tests. Crit Care Med. 2005;33:2479–2486.
29.The COIITSS Study Investigators. Corticosteroid treatment and intensive insulin therapy for septic shock in adults: randomized controlled trial. JAMA. 2010;303:341–348.