44
Thyroid Storm and Myxedema Coma
James Lantry III, John E. Arbo, and Geoffrey K. Lighthall
BACKGROUND
Thyrotoxicosis and myxedema coma are life-threatening syndromes representing the extremes of thyroid dysfunction. The rapid deterioration seen in these two conditions can result in significant morbidity and mortality if not promptly recognized.1 Delays in diagnosis and treatment are attributable in part to the nonspecific symptoms found in each condition.2 Success in management depends on developing a high level of suspicion for these disease processes, initiating early patient transfer to an intensive care setting, and delivering prompt targeted treatment.3,4
THYROTOXICOSIS
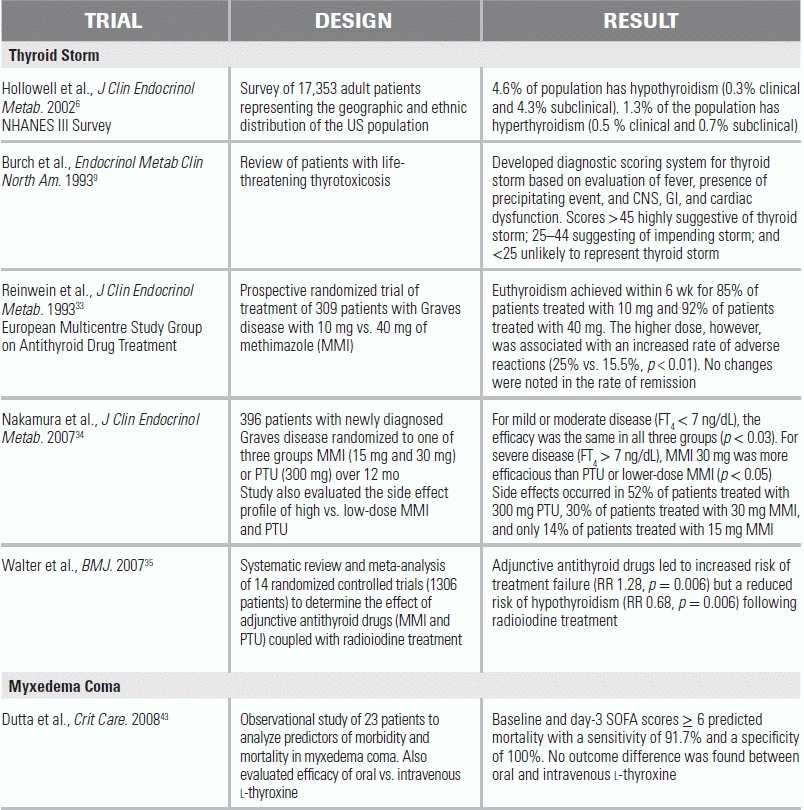
Hyperthyroidism refers to any state of elevated production of thyroid hormone; thyrotoxicosis is defined as a pathologic process that results from excess hormone secretion.3 The overall prevalence of hyperthyroidism in the United States is approximately 1.3%.2,5,6 Only 0.5% of this population will demonstrate the symptoms of thyrotoxicosis.5 In the thyrotoxic population, 1% to 2% will progress to a severe, exaggerated, and life-threatening manifestation of thyrotoxicosis called thyrotoxic crisis or thyroid storm.7,8 The point at which thyrotoxicosis becomes thyroid storm is controversial and somewhat subjective. Efforts to standardize a definition of thyroid storm include a scoring system that evaluates degrees of dysfunction in affected systems (thermoregulatory, cardiac, gastrointestinal, and neurologic).9 In clinical practice, patients presenting with symptoms of thyrotoxicosis should always be evaluated for impending thyroid storm (Table 44.1).
TABLE 44.1 Diagnostic Criteria for Thyroid Storm
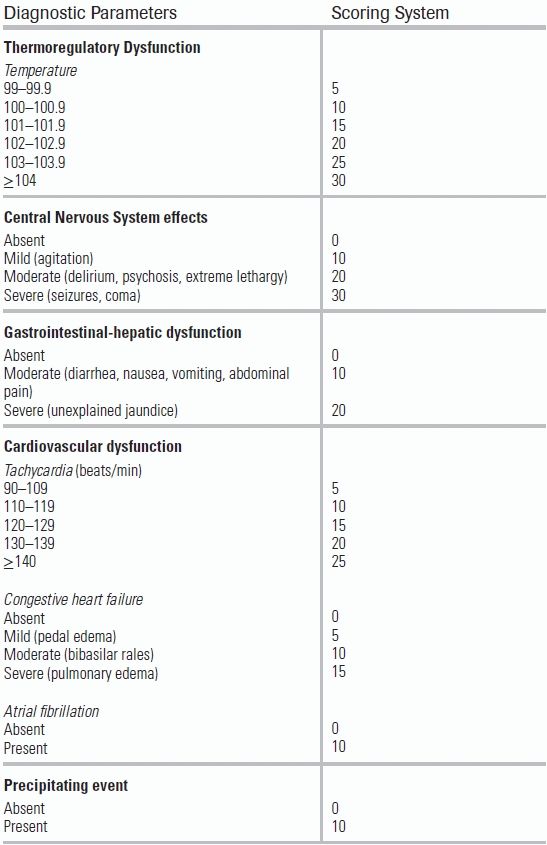
Scoring system: A score of 45 or greater is highly suggestive of thyroid storm; a score of 25–44 is suggestive of impending storm, and a score below 25 is unlikely to represent thyroid storm.
Adapted from Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am. 1993;22(2):263–277.
The most common cause of thyrotoxicosis is Graves disease, a condition in which autoantibodies bind to and stimulate thyroid-stimulating hormone (TSH) receptors on the surface of thyroid follicular cells, leading to unregulated release of the thyroid hormones triiodothyronine (T3) and thyroxine (T4).3 Graves disease occurs most commonly in adults 30 to 40 years of age and is associated with other autoimmune diseases, such as rheumatoid arthritis, as well as with tobacco use, emotional stress, and infection with Yersinia enterocolitica.7 Studies in twins suggest that approximately 80% of susceptibility to Graves disease is driven by genetics.3 The second most common cause of thyrotoxicosis is excess hormone production by a thyroid nodule, either a solitary toxic adenoma or a toxic multinodular goiter (TMNG). The prevalence of TMNG is higher in women and increases with age.3
The prevalence of both Graves disease and TMNG in a population is determined by dietary iodine content.10 In populations with adequate iodine intake, Graves disease represents nearly 80% of cases of thyrotoxicosis; in populations with inadequate iodine intake, the incidence of TMNG increases and can be responsible for half of all clinical cases.10,11 Approximately 10% of cases of thyrotoxicosis are linked to thyroid cell inflammation, or thyroiditis, triggers for which include radiation, drug side effects, and autoantibodies as seen with Hashimoto thyroiditis.1 Subacute thyroiditis, or de Quervain thyroiditis, is a transient hyperthyroid state associated with upper respiratory infections; it presents with neck swelling, malaise, and fatigue. Thyrotoxicosis can be seen in up to 50% of patients with de Quervain thyroiditis, and typically resolves within 8 months of the inciting illness.10 Postpartum thyroiditis—another cause of transient hyperthyroidism—affects 5% to 10% of women in the first 3 to 6 months after delivery.12 Finally, thyrotoxicosis can be attributed to exogenous thyroid hormone use in the treatment of hypothyroid disease.13,14
Of the numerous causes of thyrotoxicosis, Graves disease is the most common condition associated with thyroid storm.15 However, thyroid storm can result in any patient from excessive thyroid hormone release of any cause, including excessive iodine exposure from radiocontrast dye or iodine-containing drugs such as amiodarone.1 The only impetus required to induce thyroid storm from an otherwise stable thyrotoxic state is a stressful event, most commonly from infection or surgery.10 A recent study of Japanese hospitalized patients estimates the overall incidence of thyroid storm to be 0.2 per 100,000 patients per year; however, the true incidence remains unknown due to significant underdiagnosis.16
Physiology and Organ-Specific Effects
Pituitary-derived TSH induces the release of both T3 and T4.2 Both hormones regulate basal metabolism, but T3 is three to four times more potent than T4.3 TSH-induced release accounts for only 20% of circulating T3; the remainder occurs through peripheral conversion of T4 to T3 by the liver and kidney.5,17 The excess circulating thyroid hormone in a thyrotoxic state can produce a range of detrimental systemic effects, the degree and extent of which determine the presence of thyroid storm (Table 44.1).
Excess thyroid hormone activity produces an adrenergic state characterized by tachycardia, nervousness, and anxiety.15 This increase in metabolic activity can manifest as heat intolerance, increased perspiration, and lipolysis, eventually leading to as much as a 15% loss of basal body weight.7
Cardiovascular manifestations can include tachycardia and other atrial and ventricular arrhythmias, as well as systolic hypertension and widening of pulse pressure.18 New-onset dilated cardiomyopathy and congestive heart failure have also been reported in previously healthy patients with thyrotoxicosis.1 These hyperadrenergic effects can lead to increased myocardial oxygen demand and/or coronary artery vasospasm, producing angina and even myocardial infarctions. The thyrotoxic state may also have metabolic consequences, such as acidemia resulting from lipolysis and ketogenesis and tissue acidosis from mismatch between oxygen demand and supply.1
Respiratory signs include dyspnea and orthopnea from respiratory muscle weakness, high output cardiac failure, and engorgement of the pulmonary vasculature.15 A more common finding, however, is tachypnea at rest, which can herald impending respiratory fatigue and collapse.1,7 Higher respiratory demand may be the result of either adrenergic stimulation or compensation for acidemia.1
Gastrointestinal symptoms include hypermotility, which can lead to diarrhea, nausea, and vomiting.8 Concomitant loss of fluid can exacerbate postural hypotension and vascular collapse and can precipitate a state of shock.1 Impairment of neurologic regulation of gastric and intestinal activity can lead to gastroparesis and/or pseudo-obstruction.7
Hematologic changes include hypercoagulability, leukocytosis, and anemia. Hypercoagulability results from higher concentrations of fibrinogen, factors VIII and IX, plasminogen activator inhibitor 1, and von Willebrand factor.1 Moderate leukocytosis with a left shift is common, and approximately 22% of patients will suffer from symptomatic anemia.3 There is also an increase in red blood cell mass secondary to increased erythropoietin levels and an augmentation of platelet plug formation.19 Thromboembolic complications are responsible for 18% of thyrotoxicosis-related deaths.18
Thyrotoxic periodic paralysis (TPP) is an unusual complication of thyrotoxicosis seen in only 0.1% to 0.2% of thyrotoxic patients, with increased incidence in Asians (1.8% to 2.0%) and males (20:1).1,20 TPP is characterized by transient but recurrent flaccid paralysis of the proximal extremities, decreased deep tendon reflexes, and cardiac conduction abnormalities including atrioventricular blocks and asystole.20 The specific distribution of muscular findings in TPP contrasts with the generalized myopathy affecting approximately 50% of thyrotoxic patients, where fatigability and global weakness are the main findings.7 Proximal muscle weakness can also be characteristic of a thyrotoxic state, but to a lesser degree than with TPP.21
History and Physical Exam
Thyroid storm occurs most frequently in patients with either undiagnosed or poorly controlled thyrotoxicosis who are exposed to a systemic insult or stress.14,15 Infection is the most common inciting event; however, case reports have implicated nearly all known forms of physiologic stress.1 Regardless of the inciting event, untreated thyroid storm is uniformly fatal; even with appropriate treatment, the mortality rate is nearly 50%.8 The significant morbidity and mortality result from serial decompensation of multiple organ systems.13
The four principle findings of thyroid storm are (1) fever out of proportion to infection accompanied by significant diaphoresis, (2) sinus tachycardia or supraventricular arrhythmia (paroxysmal atrial tachycardia or atrial flutter or atrial fibrillation) leading to congestive heart failure, (3) gastrointestinal symptoms (vomiting, diarrhea, or bowel obstruction), and (4) central nervous system symptoms (agitation, restlessness, confusion, delirium or coma).1,7,22 The diagnosis is made clinically, as laboratory tests and imaging are not specific.
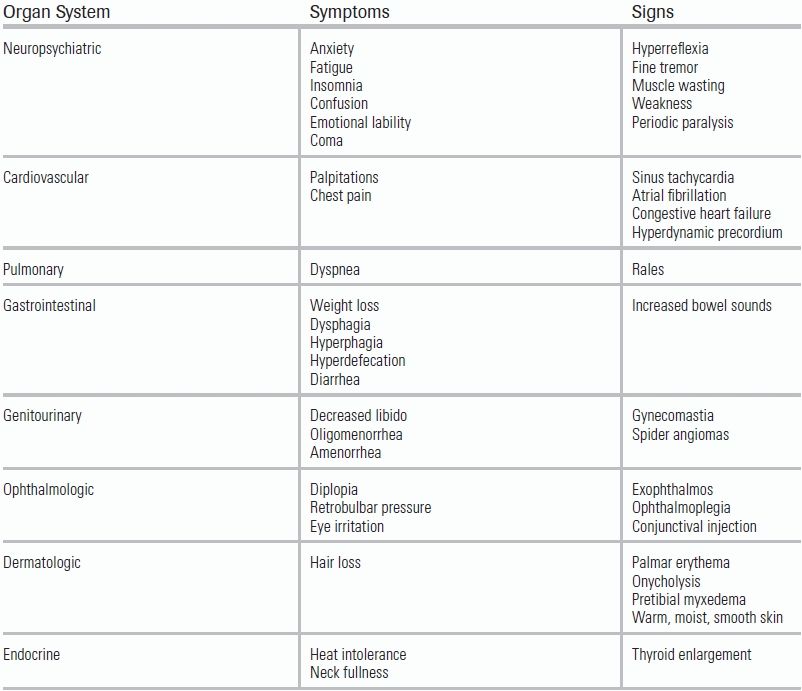
On examination, the thyroid gland will be enlarged and, depending on the etiology of thyrotoxicosis, may contain nodules; additionally, a bruit may be present because of increased thyroid vascularity.15 The skin will be warm, moist, and velvety, with softening of the hair and nails often leading to nail bed separation (onycholysis) and alopecia.2,7 Significant hyperpigmentation and raised, asymmetric lesions of the skin may be present.23 These plaques are often located on the lower extremities and accompanied by significant pretibial myxedema.7 Hands and feet are frequently swollen and may be accompanied by clubbing.18 In Graves disease, ophthalmopathy may be observed (lid lag, lid retraction, and proptosis) leading to burning and irritation with blurring of the vision and diplopia.15 Difficulty with eye closure can lead to corneal ulceration or vision loss unless properly treated.24 Impairment of mental status, including psychosis and delirium, may also be observed.11 Patients may appear restless and complain of heat intolerance, palpitations, anxiety, and fatigue and may demonstrate a fine, rapid tremor at rest.10 Finally, patients may report weight loss despite a significant appetite (Table 44.2).15
TABLE 44.2 Clinical Manifestations of Thyrotoxicosis
Laboratory Testing and Imaging
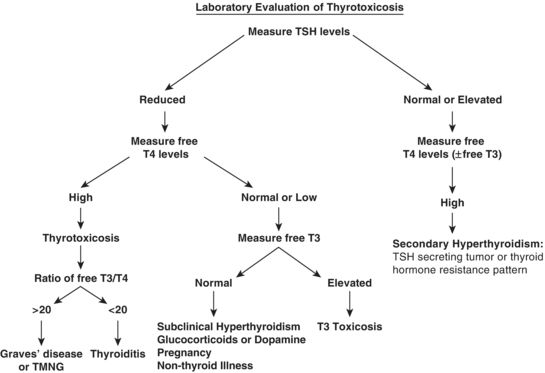
Emergency department (ED) laboratory testing for suspected thyrotoxicosis should include a TSH level and a free T4. TSH levels will often be undetectable (<0.01 microIU/L) due to negative feedback from excess thyroid hormone.3 The TSH assay is reported in a logarithmic scale, such that a small change in T4 levels can lead to a larger change in measured TSH; thus, the assay has a high sensitivity to thyroid hormone excess.25 If thyrotoxicosis is suspected, testing both TSH and free serum T4 improves diagnostic accuracy by allowing confirmation that the drop in TSH is due to thyroid dysfunction and not due to indirect causes (e.g., glucocorticoid or dopamine use).3,10 A rise in T4 is seen in 95% of patients suffering from thyrotoxicosis, with the remaining 5% experiencing an increased free T3 and normal T4 levels.7 This latter pattern can develop in early Graves disease or TMNG; thus, experts recommend checking free T3 levels to increase sensitivity if thyrotoxicosis figures prominently on the differential diagnosis.10 The ratio of free T3 to T4 helps distinguish increased and decreased thyroid gland metabolism.18 Patients with Graves disease or TMNG will have an increased ratio of free T3/T4 (>20); by contrast, patients with thyroiditis will have a decreased ratio (<20).15 Testing for total T3 or T4 is no longer recommended as liver disease, exogenous hormone use, or pregnancy can cause unreliable protein binding of free thyroid hormones (Fig. 44.1).7
Other laboratory abnormalities may include an initial hyperglycemia, mediated by increased glycogenolysis and an increase in insulin clearance; profound hypoglycemia can subsequently result from this depletion of glycogen stores.1 Hepatic dysfunction can lead to accumulation of lactate dehydrogenase, aspartate aminotransferase, and bilirubin.7 Acidemia may occur due to lipolysis and dehydration ketosis.26 Hypercalcemia, the result of hemoconcentration from fluid shifts and an increase in bone resorption, is also common.15 This increase in osteoblastic bone activity can also lead to elevations in alkaline phosphatase.15
Prevention of thyroid storm is dependent on early detection of thyrotoxicosis. While thyroid storm is a clinical diagnosis, thyrotoxicosis is confirmed when laboratory findings corroborate suspicion developed from the history and physical exam.1 While the trigger of a patient's thyroid storm (e.g., infection) may not be evident initially, clinical stabilization should proceed at the same time as the diagnostic evaluation.27 Testing for thyroid receptor antibody levels is rarely needed for evaluation of thyrotoxicosis; however, in the pregnant patient, who cannot undergo radionucleotide scanning, the level can help distinguish between Graves disease and gestational thyrotoxicosis.11,28 Additionally, maternal TSH can cross the placental barrier and have a direct effect on the fetus; it is therefore recommended that TSH levels be drawn between 22 and 26 weeks of gestation in order to determine need for aggressive neonatal monitoring.3 Finally, 10% of patients with proptosis will be diagnosed with Graves disease by antibody levels alone, as TSH and T4 may not be abnormal.15
Radionucleotide testing and ultrasound can help differentiate between thyrotoxicosis caused by hyperthyroidism (i.e., Graves disease or TMNG) and thyrotoxicosis not caused by hyperthyroidism (i.e., thyroiditis or ingestion of exogenous thyroid hormone).3,15 Radioactive iodine uptake (RAIU) uses either technetium-99m (Tc 99m) or radioiodine29 to assess the activity of the sodium/iodide symporter on the thyroid gland.17 Each molecule has advantages in testing: radioiodine is incorporated into hormone production and is thought to be more reflective of true physiology, while Tc 99m requires less acquisition time for similar results—but at the cost of an increased exposure to radiation.10 Uptake measurements are taken at 4 hours and 24 hours after administration.29 The pattern of uptake reflects the cause of thyrotoxicosis and helps narrow the differential diagnosis.3 With ultrasonography, an increase in thyroid total area blood flow (calculated as thyroid artery blood flow/glandular area) of 4% to 8% can differentiate Graves disease from destructive thyroiditis with sensitivity and specificity of 84% and 90%, respectively.10 This diagnostic approach is operator dependent but is a viable option for patients who cannot tolerate radioactive screening, such as those who are pregnant or breast-feeding.22
Differential Diagnosis
Thyroid storm should be suspected in patients with mental status changes, hyperadrenergic state, and any of the systemic manifestations noted above.8 The differential diagnosis includes generalized infections and sepsis, anxiety, depression, pheochromocytoma, atrial fibrillation/flutter, chronic fatigue syndrome, Plummer-Vinson syndrome, as well as various malignancies.2,8,11,18 Use of methamphetamine, cocaine, or other nutritional supplements can confuse the clinical picture and should be excluded.30 In most cases, thyroid testing will help narrow the differential; however, certain medical conditions, such as euthyroid sick syndrome, pregnancy, and hyperemesis gravidarum, will lower TSH levels and affect T4 assays.2,7,22 Additionally, the use of glucocorticoids, dopamine, and heparin will lower the TSH level and can confound the diagnosis.22
Management Guidelines
Treatment of thyrotoxicosis in the acute care setting focuses on attenuating the hyperadrenergic state, controlling the production and release of thyroid hormone, inhibiting peripheral conversion of T4 to T3, and treating the precipitating cause.12,27 Definitive therapy is achieved with radioactive iodine or surgery (e.g., subtotal thyroidectomy).
Beta-blockers are the primary agents used to attenuate the cardiovascular complications of thyrotoxicosis; propranolol is a first-line agent because of its additional ability to reduce peripheral conversion of T4 to T3.8 The application of adrenergic blockage leads to an improvement in heart rate and cardiac output and a decrease in cardiac oxygen consumption.27 In patients with preexisting heart failure, because of the concern for abrupt clinical deterioration, continuous cardiac monitoring and, in some cases, a screening echocardiography are required prior to initiation of beta-blocker therapy.31 When beta-blockers are used in patients with a history of obstructive lung disease, including asthma and COPD, there exists an additional risk of reactive airway disease exacerbation.15 In these patients, a beta-1–selective agent, such as metoprolol or esmolol, is a reasonable alternative to propranolol. Amiodarone should always be avoided as an antidysrhythmic because of its iodine content.
Thionamides have been successfully used for over 70 years to decrease circulating hormone levels.15 Propylthiouracil (PTU) and methimazole (MMI) are available in the United States, while Carbimazole (CBZ, metabolized peripherally to MMI) is available in Europe and Asia.3,7,27 All three agents inhibit the intrathyroid hormone synthesis and have high oral bioavailability, leading to effects within 1 to 2 hours of ingestion.32 PTU has the added effect of inhibiting peripheral conversion of T4 to T3, reducing the concentration of the active hormone.12 MMI has the advantage of allowing once-daily dosing, which improves compliance, and is 10 to 12 times more potent than PTU, leading to more rapid normalization of thyroid function.27 All three agents also have an immunomodulatory effect and decrease both natural killer and T-cell substrates and autoantibodies, which may be relevant in patients with Graves disease.32
Several randomized trials have examined the efficacy of differing doses of and combinations of these treatments. In one RCT that assessed the treatment efficacy of 10 versus 40 mg of MMI in patients with Graves disease, both groups achieved acceptable levels of euthyroidism within 6 weeks (85% and 92%, respectively), but the higher dose was associated with an increased rate of complications (25% vs. 15.5%).33 In another RCT of patients with newly diagnosed Graves disease, the clinical efficacy of 15 mg and 30 mg MMI versus 300 mg PTU was assessed over a period of 12 months. For mild or moderate disease (Free T4 ( FT4) < 7 ng/dL), efficacy was the same in all three groups. For severe disease (FT4 > 7 ng/dL), MMI 30 mg was more efficacious than PTU or lower-dose MMI.34
Side effects of PTU and MMI occur in 14% to 52% of patients; they are dose dependent, usually limited to fever, rash, urticaria, and arthralgias, and are typically resolved by switching from one agent to the other.33,34 One severe side effect, agranulocytosis, affects 0.5% of patients treated with any of the three medications and requires immediate cessation of all thionamides.27 This complication usually occurs within the first 3 months of treatment, and patients are advised to monitor for oropharyngeal infections commonly associated with this development.18 Other, less common side effects, including hepatotoxicity and anti-neutrophil cytoplasmic antibody (ANCA)-mediated vasculitis, also require the cessation of thionamides.32 For these reasons, thionamides are used as primary treatment of thyrotoxicosis only in select populations in whom surgical resection or radiation therapy is undesirable: young patients with mild to moderate illness; patients with only a slight increase in glandular volume; pediatric or adolescent patients; and pregnant or breast-feeding patients.3 Otherwise, these agents are primarily used as the initial medical therapy for patients awaiting definitive treatment with radioactive iodine or surgical resection.32 Patients with TMNG benefit greatly from premedication (prior to RAIU or surgery) with thionamides because any delay in treatment leads to high rates of relapse.32 Caution should be taken with continued use of MMI or PTU in the week prior to radioiodine therapy as either medication can decrease the iodine uptake into the thyroid gland and lead to treatment failure.35 This decreased in iodine uptake, however, exerts a small protective effect against long-term hypothyroidism by minimizing damage to healthy thyroid tissue.35
For those patients unable to tolerate the thionamides, lithium is an alternative agent that blocks thyroid hormone release.8 Lithium is taken up by the thyroid gland in a manner similar to iodine32; however, its effects are transient, and the value of long-term use is undefined.18
Other medications are available for short-term symptomatic relief.7 Glucocorticoids may be used to inhibit the peripheral conversion of T4 to T3 and are useful in cases associated with secondary adrenal insufficiency.32 Graves ophthalmopathy is also improved with long-term (6 to 8 weeks) treatment with glucocorticoids.3 Cholestyramine is an anion-exchange resin that binds thyroid hormones in the enterohepatic circulation and can increase their fecal excretion.8 Potassium perchlorate, a competitive inhibitor of iodine transport into the thyroid, is used in patients with iodine-induced thyrotoxicosis.31 In extreme cases, peritoneal dialysis, plasma exchange, or hemodialysis can be utilized to abruptly lower thyroid hormone concentrations.18
Essential supportive care includes antipyretics, cooling, and correction of intravascular fluid deficits.8 For fever, acetaminophen is the medication of choice, as salicylates decrease thyroid-binding protein and increase free thyroid levels.7,15 To avoid Wernicke encephalopathy, a condition associated with thyrotoxicosis, thiamine should be given along with a general multivitamin.26 Treatment of infection, myocardial injury, and other stressors should proceed according to best practices.27
Special Populations
The Pregnant Patient
One in 500 pregnancies is complicated by Graves disease, which can lead to significant morbidity including miscarriage, premature labor, low birth weight, and eclampsia.3,11 Pregnant patients presenting with >5% weight loss, goiter, ophthalmopathy, or onycholysis will require a thorough evaluation for Graves disease.7,11 Normal hormonal changes in pregnancy can make diagnosis challenging: increased thyroid-binding globulin production will lower free T4 levels; and human chorionic gonadotropin will lower TSH production in the first trimester. In addition, several classic signs of thyrotoxicosis may be present in normal pregnancy, including a widened pulse pressure and heat intolerance.3,7,28 Treatment of pregnant patients is difficult, as both PTU and MMI cross the placenta and can cause fetal hypothyroidism and goiter.27 Of the two, PTU is a better choice; it is more protein bound, slightly reducing its ability enter the fetal circulation18 and does not carry the same increased risk that MMI does of causing fetal cutis aplasia and gastrointestinal atresia.31 Treatment aims to use the lowest effective dose of PTU to keep T4 levels in a high-normal to slightly thyrotoxic range.27 The level of thyrotoxicosis can wane during pregnancy, and up to 30% of women discontinue use of thionamides in the third trimester.11 Thyroidectomy is reserved for the second-trimester or for severely decompensated patients, as there is an increased risk of miscarriage.27 In women with previous thyroid dysfunction, 10% will experience thyrotoxicosis in the postpartum period.3 Additionally, 80% of women whose pregnancies are complicated by Graves disease will have a relapse in future pregnancies, with 50% developing permanent thyrotoxicosis.3
The Elderly
Thyrotoxicosis in the elderly is difficult to diagnose because suggestive symptoms, such as hyperkinesis and ophthalmopathy, are often lacking, and because clinical manifestation is often limited to a single organ system (e.g., heart failure or atrial fibrillation).7 Up to 70% of elderly patients with thyrotoxicosis demonstrate no clinical signs or symptoms of a goiter and may even have depressive signs, such as apathy and fatigue.7 This genre of clinical symptoms is termed “apathetic hyperthyroidism” and is often diagnosed after a lengthy workup for cardiotonic-resistant cardiovascular disease.36
Amiodarone
Amiodarone, an iodine-containing antiarrhythmic agent, will induce thyrotoxicosis in approximately 6% to 10% of patients.3 This condition doubles the adverse cardiac effects of the drug and can lead to even worse outcomes in patients with preexisting thyroid disease.37 Diagnosis is similar to other forms of thyrotoxicosis; however, conditions such as atrial and ventricular arrhythmias, for which the patient would normally take amiodarone, need to be carefully differentiated from thyroid hormone excess.3 There are two types of amiodarone-related thyroid disease.38 Type 1 is an iodine-induced thyrotoxicosis that occurs in individuals with preexisting nodules or autoimmune thyroid disease. Type 2 is an amiodarone-induced destruction of the thyroid gland itself.3,38 Color-flow Doppler will show increased uptake in type 1, which is treated with thionamides and potassium perchlorate.11,37 Glucocorticoids are the preferred medication for type 2 and often lead to complete resolution.10 Since the subtype is not always apparent, experts recommend a combination of the three drugs for 6 to 12 months; ongoing consultation by cardiology and endocrinology is recommended because of the long half-life of amiodarone and the complex disease states that it treats.37
MYXEDEMA COMA
Hypothyroidism affects approximately 4.6% of the US population and is characterized by a generalized slowing of the body's metabolic processes leading to an overall depression of both physical and mental activity.39 Myxedema coma is a rare complication of untreated hypothyroidism and describes a state of severely decompensated hypothyroidism in which the body cannot maintain thermal energetic homeostasis.40 Hypothyroidism is four times more common in women than in men, and 80% of all reported cases of myxedema coma occur in females, a majority over the age of 60.40 Hallmarks of laboratory diagnosis are severely depressed levels of T4 and T3 and an elevation in TSH41; however, lab values correlate poorly with the severity of the clinical disease.42 In the past, their poor predictive value contributed to delays in diagnosis and a mortality rate of 60% to 70%.41 More recently, advances in physician education have resulted in earlier recognition of this disease and an improved patient mortality rate of 20% to 25%.31,41 A recent review of risk factors showed higher mortality rates associated with advanced age, hemodynamic instability, severe bradycardia, respiratory failure (requiring intubation), hypothermia, sepsis, depressed Glasgow Coma Scale (GCS), and a higher APACHE II score.43 This same study also showed that the sequential organ failure assessment (SOFA) score offered the most effective prediction model, with baseline and 3-day SOFA scores of 6 or greater predicting mortality with a sensitivity and specificity of 91.7% and 100%, respectively.
Myxedema coma is most commonly precipitated by a significant systemic stressor in the undiagnosed or poorly managed hypothyroid patient. Infection—notably pneumonia, urinary tract infection, and cellulitis—is the most common trigger.1 Hypothermia during the winter months is also thought to be responsible for a large number of cases.44 The seasonal pattern is explained by an age-related loss of temperature regulation coupled with the depressed heat production common to hypothyroidism.1 Other triggers of myxedema coma include cerebrovascular accidents, congestive heart failure, gastrointestinal bleeds, the use of centrally acting depressants such as sedatives and lithium, or the abrupt discontinuation of thyroid supplements in critically ill patients.14,26,45 A recent report detailed the development of myxedema crisis following the consumption of raw bok choy, which contains cyanates, nitriles, and oxazolidines that inhibit iodine uptake in the thyroid.46
Physiology and Organ-Specific Effects
Thyroid hormones affect the metabolism and development of nearly every cell in the body.11 In hypothyroidism, there is both an inadequate production of thyroid hormones and decreased peripheral conversion of T4 to the active hormone T3.47 Patients suffering from hypothyroidism rely upon both arms of the autonomic nervous system to maintain circulatory homeostasis and a normal core temperature.39,48 Any further reduction in intravascular volume (dehydration, blood loss), compromise to ventilation (infection), or insult to the central nervous system (drugs) can overwhelm these mechanisms and lead to myxedema coma.40
Clinically, patients will demonstrate global depressed physiologic function manifested as hypothermia, bradycardia, hypertension, respiratory acidosis, and depressed mental status leading to a comatose state.44 Respiratory failure results from decreased central nervous system sensitivity to hypoxia and hypercarbia, as well as airway problems from macroglossia, respiratory muscle weakness, and nonpitting edema (myxedema) of the nasopharynx.41 Altered vascular permeability leads to effusions and ascites; renal injury leads to water retention and hyponatremia; and depressed inotropy and chronotropy lead to intractable cardiogenic collapse.41
History and Physical Exam
Myxedema coma can be difficult to distinguish from other life-threatening conditions, such as heart failure, hypothermia, or respiratory dysfunction, that present with similar manifestations.42 Even more difficult is recognizing this disease in an unstable, altered, or septic patient.41 Providers must maintain a high clinical suspicion, guided by a detailed patient history noting any previous use of thyroid hormones or recent discontinuation of thyroid supplements, to make the diagnosis.1
The physical exam should focus on identifying the classic features of hypothyroidism: dry skin, brittle nails, hair loss, delayed tendon reflexes, and goiter.49 Additionally, mucin deposits can lead to swelling of the hands, ptosis, periorbital edema, macroglossia, laryngeal edema, and other nondependent sites of nonpitting edema.44,48 In patients with hypothyroidism due to prior treatment for Graves disease, a subtle clue is the presence of Graves orbitopathy, which does not resolve with treatment of the thyrotoxic state and can signal that the patient has previously received thyroid depressant treatments.41 Providers should also look for signs of prior thyroid surgery, such as a midline incision in the anterior neck or documentation of a previous radioactive iodine ablation.44
A comatose state is not required for the diagnosis of myxedema coma, but all patients will demonstrate some degree of central nervous system depression, including diminished cognition, lethargy, and somnolence.1 This alteration of mental status may be worsened by the presence of concomitant hyponatremia, an electrolyte abnormality common in severe hypothyroidism.41 The pathogenesis of the depressed neurologic function is thought to be related to a depressed respiratory drive leading to hypercarbia; a decrease in cerebral blood flow; and decreased brain glucose utilization coupled with an overall hypoglycemic state.48 The depressed cerebral function, hyponatremia, hypoglycemia, hypoxemia, and reduced cerebral blood flow can combine to result in generalized seizures that, without early intervention and treatment, can progress to status epilepticus, further clouding the clinical picture.41,44
A decreased physiologic response to hypoxia and hypercarbia results in alveolar hypoventilation in patients with previously healthy lung tissue.4 Studies have shown that both myxedema coma and brief hypothyroid states produce a depressed hypoxic ventilatory drive that reverses with thyroid hormone replacement; however, the hypercapnic respiratory depression is not affected.41 Associated hypothermia, obesity hypoventilation syndrome, and macroglossia often contribute to the need for ventilator support.44 Due to the severity of respiratory failure in myxedema coma—even following the initiation of appropriate treatment with levothyroxine—a 3- to 6-month period of mechanical ventilation may be required.41
Effects of a decompensated hypothyroid state on the cardiovascular system include bradycardia, hypertension, and narrowing of the pulse pressure.41 Common electrocardiogram findings include sinus bradycardia, complete heart blocks, QT prolongation, and nonspecific ST-segment changes.4 An early echocardiogram is recommended to evaluate for pericardial effusions.44 Despite an overall reduction in cardiac function, overt heart failure in myxedema coma is uncommon.41 In extreme cases of decompensated hypothyroidism, dilated cardiomyopathy and associated left ventricular failure may develop.43 Fortunately, improvement in both cardiac output and ejection fraction is seen with prompt initiation of thyroxine therapy.48
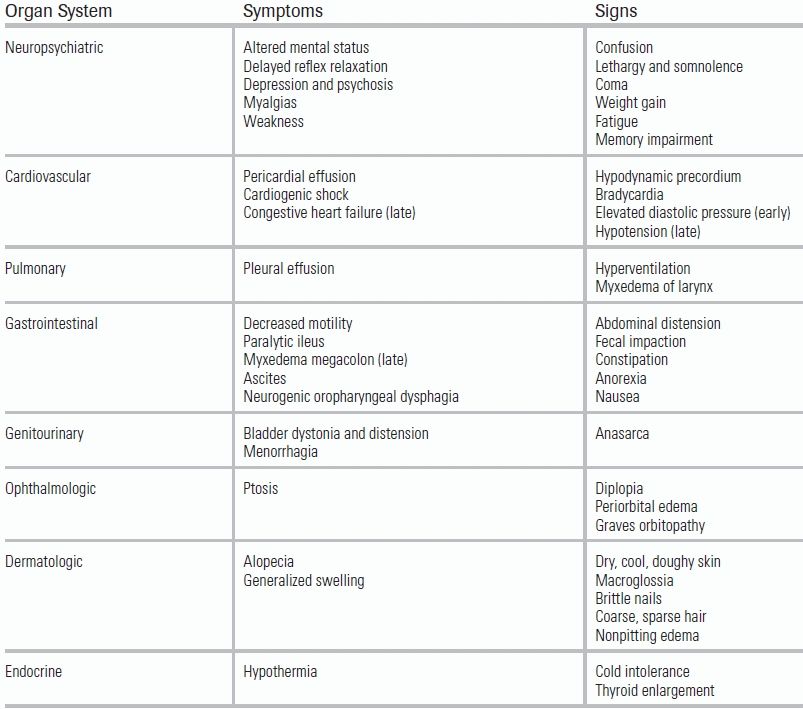
Gastrointestinal dysmotility can lead to constipation and, without treatment, can progress to paralytic ileus.42 The associated abdominal pain, nausea, and anorexia can mimic the appearance of a surgical abdomen.4 This devastating complication can lead to unnecessary exploratory surgery, worsening physiologic stress and precipitating further decompensation of the myxedema coma state (Table 44.3).41
TABLE 44.3 Clinical Manifestations of Myxedema Coma
Laboratory Testing and Imaging
ED laboratory testing in patients with suspected decompensated hypothyroidism should include a TSH level and a free T4. In early hypothyroid disease, when production of T3 is decreased, the peripheral conversion of T4 to T3 increases in an attempt to maintain physiologic levels.48 T3 and T4 are bound in the peripheral circulation by proteins, including the high-affinity thyroxine-binding globulin (TBG) and lower-affinity but more abundant albumin.2 Only the free hormone is able to bind to receptors and create biologic activity.39 In a nondiseased state, approximately 0.03% of T4 and 0.5% of T3 are unbound.48 A clinical picture consistent with hypothyroid disease coupled with low T4 levels (T3 is rarely measured directly) typically confirms the diagnosis.26 However, changes either in the quantity or in the affinity of available binding proteins can effect a pseudonormalization of thyroid hormone levels, clouding the diagnosis.48 Notably, certain infections, such as hepatitis and HIV, or any increase in estrogen (e.g., pregnancy), will result in an increase in TBG and can mimic the diagnostic criteria for hypothyroidism.2
Elevated TSH is a very specific marker of a hypothyroid state.14 However, TSH levels can be poorly sensitive for hypothyroid disease resulting from secondary or tertiary causes, also known as central hypothyroidism.49 In this state, hypothalamic–pituitary–thyroid (HPT) axis dysfunction leads to a decrease in the production of thyrotropin-releasing hormone and thus in serum TSH.48 The most common culprit is direct damage to the pituitary gland (e.g., pituitary adenoma), although systemic diseases, including sarcoidosis and hemochromatosis may also damage the HPT axis.48 Central hypothyroidism accounts for approximately 5% of myxedema coma cases and may present with normal or even low levels of TSH.4 The use of corticosteroids and vasopressors such as dopamine can also result in decreased TSH secretion.41
Other laboratory abnormalities commonly seen in hypothyroid disease include a marked reduction in the glomerular filtration rate (GFR), which occurs because of decreased renal plasma flow and increased vascular resistance in both the afferent and efferent arterioles.41 Reduced GFR results in creatinine elevation and an increased risk of hyponatremia.42 This is thought to be due to the loss of the aldosterone-like effect that T3 and T4 have on the Na–K channels of proximal tubular cells, leading to increase in sodium excretion.4 Renal dysfunction also results in impaired free water clearance and the development of myxedema.44 Thyroxine replacement has been shown to successfully reverse changes in renal function.41
Decreased lipid clearance may also be present, leading to hypercholesterolemia and hypertriglyceridemia.48 Normocytic anemia may develop due to decreased oxygen requirements and decreased levels of erythropoietin, while alteration to von Willebrand factor synthesis, caused by low thyroxine levels, can result in coagulopathies, including prolonged bleeding and clotting times, decreased platelet adhesiveness, and prolongation of aPTT.1,41 Elevations of creatinine phosphokinase, lactate dehydrogenase, and aspartate transaminase may also be seen, as can hypoglycemia due to decreased gluconeogenesis (Fig. 44.2).40,45,49
Differential Diagnosis
Myxedema coma has no classic presentation and will often present simply as a patient with depressed mental status and hemodynamic instability.39 A broad differential is required on initial presentation, as adrenal insufficiency, congestive heart failure, hepatic encephalopathy, hypothermia, and septic shock can present in similar fashion.4 The neurologic manifestations of myxedema coma can also be caused by a cerebrovascular accident, status epilepticus, or meningitis.1
Management Guidelines
Patients with myxedema coma require prompt admission to an intensive care unit (ICU) and aggressive hemodynamic support.1 Due to the mortality risks associated with delays in treatment, therapy should begin prior to laboratory confirmation.31 A three-tiered treatment plan is recommended: early initiation of thyroid replacement, correction of organ-specific dysfunction, and management of the inciting event (most commonly infection or hypothermia).47
Expert consensus is that early thyroid hormone replacement is vital for recovery in uncompensated myxedema coma, but a paucity of clinical trials and a lack of randomized controlled studies have yet to yield censuses on proper timing or dosing.4,40,41 Timely implementation of therapy should be balanced with close monitoring for the fatal arrhythmias and myocardial ischemia associated with increased oxygen demand from T3 and T4 replacement.1,42 Continuous hemodynamic monitoring is mandatory, as is early cessation of treatment at any signs of instability.1
Replacement agents include levothyroxine (LT4) or liothyronine (LT3).4 Parental administration of LT4 is preferred because unpredictable gastric absorption is common in myxedema coma.1 Compared to LT3, LT4 results in fewer cardiac complications; however, LT4 requires activation via peripheral 5′-deiodination, a process that can become depressed in severely decompensated patients.4 Additionally, LT4 is not well transported across the blood–brain barrier, resulting in slower resolution of neurologic symptoms.41 Therefore, in the case of a critically ill patient who may have depressed 5′-deiodination, LT3 is preferred for its immediate bioactivity, faster therapeutic effect, and blood–brain barrier penetration.1,40,41 Note, however, that LT3 increases risk of cardiac abnormalities, including ischemia and lethal arrhythmias, which are heightened in the setting of concomitant vasopressor therapy.42,48
Some authors have advocated for the use of both agents, combining the quick onset and increased bioavailability of LT3 with the relative cardiovascular stability associated with LT4.1,42 This combination permits lower doses than those would be used in solo therapy, often beginning with intravenous LT4 and LT3 and transitioning to oral LT4 for long-term therapy.4 Rates of cardiovascular complication are higher with parenteral delivery of either agent, so a prompt transition to oral dosing is advocated once the patient achieves clinical stability.41
Controversy over the optimal therapeutic strategy persists, especially regarding dosing.1 Regardless of therapy used, restoration of hemodynamic stability typically occurs within 24 hours and of thermoregulation within 2 to 3 days.50 Respiratory dysfunction and kidney injury may take weeks to months to fully resolve.4 A decline in TSH serves as a marker of clinical recovery and helps to guide further therapy.41
Supportive treatment of the patient with myxedema coma can include mechanical ventilation for correction of hypercarbia and support of diaphragmatic weakness; early broad-spectrum antibiotics; aggressive fluid resuscitation; and correction of associated electrolyte disorders (e.g., hyponatremia and hypoglycemia).48 Care must be taken with treatment of hypothermia, as rapid rewarming can cause peripheral vasodilation and worsening hypotension.42 Because thyroid hormone therapy results in increased cortisol clearance, all patients treated with LT3 and LT4 should be maintained on hydrocortisone until clinically stable.4 Moreover, the clinical features of myxedema coma and adrenal insufficiency may overlap; if an appropriate response to thyroid hormone therapy is not observed, the provider should assess for, and if present, treat, coexisting adrenal insufficiency.44,45 Hypotension typically resolves with initiation of thyroid hormone replacement; low-dose vasopressors may be added for additional support.44
Special Populations
A rare complication of Hashimoto thyroiditis is Hashimoto encephalopathy.51 This disease presents as subacute or acute encephalopathy with seizures, stroke-like episodes, myoclonus, and tremor similar to myxedema coma.52 Lab testing will reveal elevations in thyroid-specific antibodies, elevated cerebrospinal fluid protein without pleocytosis, and an abnormal electroencephalogram.53 The patient, however, is in a euthyroid state, and steroids are a first-line treatment.51
CONCLUSION
Thyroid storm and myxedema coma are disease processes representing the extremes of thyroid dysfunction.1 Nonspecific presentations and extremely high mortality rates make early recognition essential.4,7 With early clinical suspicion, prompt laboratory evaluation, and early administration of multifaceted therapies, the morbidity and mortality for both pathologic processes can be lessened substantially.50 Aggressive hemodynamic and respiratory support in the ED, coupled with referral to an intensive care setting, is imperative to assure successful treatment outcome.8,47
LITERATURE TABLE
RR, relative risk.
REFERENCES
1.Klubo-Gwiezdzinska J, Wartofsky L. Thyroid emergencies. Med Clin North Am. 2012;96:385–403.
2.Pimental L, Hansen KN. Thyroid disease in the emergency department: a clinical and laboratory review. J Emerg Med. 2005;28(2):201–209.
3.Franklyn JA, Boelaert K. Thyrotoxicosis. Lancet. 2012;379:1155–1166.
4.Wartofsky L. Myxedema coma. Endocrinol Metab Clin North Am. 2006;35:687–698.
5.Bahn RS, Burch HB, Cooper DS, et al. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21(6):593–649.
6.Hollowell JG, Staehling NW, Flanders WD, et al. Serum TSH, T4 and thyroid antibodies in the United States population (1988–1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab. 2002;87:489–499.
7.McKeown NJ, Tews MC, Gossain VV, et al. Hyperthyroidism. Emerg Med Clin North Am. 2005;23:669–685.
8.Migneco A, Ojetti V, Testa A, et al. Management of thyrotoxic crisis. Eur Rev Med Pharmacol Sci. 2005;9:69–74.
9.Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Thyroid storm. Endocrinol Metab Clin North Am. 1993;22(2):263–277.
10.Seigel SC, Hodak SP. Thyrotoxicosis. Med Clin North Am. 2012;96:175–201.
11.Cooper DS. Hyperthyroidism. Lancet. 2003;362:459–468.
12.Reid JR, Wheeler SF. Hyperthyroidism: diagnosis and treatment. Am Fam Physician. 2005;72(4):623–630.
13.Iglesias P, Devora O, Garcia-Arevalo J, et al. Severe hyperthyroidism: etiology, clinical features and treatment outcome. Clin Endocrinol (Oxf). 2010;72:551–557.
14.Veloski C, Brennan KJ. Critical care endocrinology. In: Criner GJ, ed. Critical Care Study Guide. 2nd ed. New York: Springer; 2010:638–661.
15.Nayak B, Burman K. Thyrotoxicosis and thyroid storm. Endocrinol Metab Clin North Am. 2006;35:663–686.
16.Akamizu T, Satoh T, Isozaki O, et al. Diagnostic criteria, clinical features, and incidence of thyroid storm based on nationwide surveys, Japan Thyroid Association, Thyroid. 2012;22(7):661.
17.Ross DS. Radioiodine therapy for hyperthyroidism. N Engl J Med. 2011;364:542–550.
18.Streetman DD, Khanderia U. Diagnosis and treatment of Graves' disease. Ann Pharmacother. 2003;37:1100–1109.
19.Homonick M, Gessl A, Ferlitsch A, et al. Altered platelet plug formation in hyperthyroidism and hypothyroidism. J Clin Endocrinol Metab. 2007;92:3006–3012.
20.Pothiwala P, Levine SN. Thyrotoxic periodic paralysis: a review. J Intensive Care Med. 2010;25(2):71–77.
21.Lin SH. Thyrotoxic periodic paralysis. Mayo Clin Proc. 2005;80(1)99–105.
22.McDermott MT. In the clinic: hyperthyroidism. Ann Intern Med. 2012;1:1–16.
23.Dabon-Almirante CL, Surks MI. Clinical and laboratory diagnosis of thyrotoxicosis. Endocrinol Metab Clin North Am. 1998;27:25–35.
24.Burch HB, Wartofsky L. Graves' ophthalmopathy: current concepts regarding pathogenesis and management. Endocr Rev. 1993;14:747–793.
25.Hadlow NC, Rothacker KM, Wardrop R, et al. The relationship between TSH and free T in a large population is complex and nonlinear and differs by age and sex. J Clin Endocrinol Metab. 2013;98(7):2936–2943.
26.Sarlis NJ, Gourgiotis L. Thyroid emergencies. Rev Endocr Metab Disord. 2003;4:129–136.
27.Pearce EN, Braverman LE. Hyperthyroidism: advantages and disadvantages of medical therapy. Surg Clin North Am. 2004;84:833–847.
28.Varon J, Acosta P. Handbook of Critical and Intensive Care Medicine. New York, NY: Springer; 2010.
29.Kusic Z, Becker DV, Saenger EL, et al. Comparison of technetium-99m and iodine 123 imaging of thyroid nodules: correlation with pathologic findings. J Nucl Med. 1990;31:393–399.
30.Raptis S, Fekete C, Sarkar S, et al. Cocaine- and amphetamine-regulated transcript co-contained in thyrotropin-releasing hormone (TRH) neurons of the hypothalamic paraventricular nucleus modulates TRH-induced prolactin secretion. Endocrinology. 2004;145:1695–1699.
31.Bondugulapati L, Adlan M, Premawardhana L. Review- thyroid emergencies. Sri Lanka J Crit Care. 2011;2(1):1–12.
32.Fumarola A, Di Fiore A, Dainelli G, et al. Medical treatment of hyperthyroidism: state of the art. Exp Clin Endocrinol Diabetes. 2010;118(10):678–684.
33.Reinwein D, Benker G, Lazarus JH, et al. A prospective randomized trial of antithyroid drug dose in Graves' disease therapy. European Multicenter Study Group on Antithyroid Drug Treatment. J Clin Endocrinol Metab 1993;76(6):1516–1521.
34.Nakamura H, Noh JY, Itoh K, et al. Comparison of methimazole and propylthiouracil in patients with hyperthyroidism caused by Graves' disease. J Clin Endocrinol Metab. 2007;92(6):2157–2162.
35.Walter MA, Briel M, Christ-Crain M, et al. Effects of antithyroid drugs on radioiodine treatment: systematic review and meta-analysis of randomised controlled trials. BMJ. 2007;334(7592):514.
36.Palacious A, Cohen MAA, Cobbs R. Apathetic hyperthyroidism in middle age. Int J Psychiatry. 1991;21(4):393–400.
37.Bogazzi F, Bartalena L, Martino E. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010;95(6):2529–2535.
38.Thomas Z, Bandali F, McCowen K, et al. Drug-induced endocrine disorders in the intensive care unit. Crit Care Med. 2010;38(6):S219–S230.
39.Vaidya B, Pearce SH. Management of hypothyroidism in adults. Br Med J. 2008;337:284–289.
40.Wall CR. Myxedema coma: diagnosis and treatment. Am Fam Physician. 2000;62(11):2485–2490.
41.Kwaku MP, Burman KD. Myxedema coma. J Intensive Care Med. 2007;22:224–231.
42.Fliers E, Wiersinga WM. Myxedema coma. Rev Endocr Metab Disord. 2003;4:137–141.
43.Dutta P, Bhansali A, Masoodi SR, et al. Predictors of outcome in myxedema coma: a study from a tertiary care center. Crit Care. 2008;12:R1.
44.Mathew V, Misgar RA, Ghosh S, et al. Myxedema coma: a new look into an old crisis. J Thyroid Res. 2011;2011:493462.
45.Beynon J, Akhtar S, Kearney T. Predictors of outcome in myxoedema coma. Crit Care. 2008;12:111.
46.Chu M, Seltzer TF. Myxedema coma induced by ingestion of raw bok choy. N Engl J Med. 2010;362(20):1945–1946.
47.Nicoloff JT, LoPresti JS. Myxedema coma. A form of decompensated hypothyroidism. Endocrinol Metab Clin North Am. 1993;22:279–290.
48.Bello F, Bakari AG. Hypothyroidism in adults: a review and recent advances in management. J Diabetes Endocrinol. 2012;3(5);57–69.
49.Gaitonde DY, Rowley KD, Sweeney LB. Hypothyroidism: an update. S Afr Fam Pract. 2012;54(5):384–390.
50.Goldberg PA, Inzucchi SE. Critical issues in endocrinology. Clini Chest Med. 2003;24:583–606.
51.Schiess N, Pardo CA. Hashimoto's encephalopathy. Ann N Y Acad Sci. 2008;1142:254–265.
52.Mocellin R, Walterfang M, Velakoulis D. Hashimoto's encephalopathy: epidemiology, pathogenesis and management. CNS Drugs. 2007;21(10):799–811.
53.Fatourechi V. Hashimoto's encephalopathy: myth or reality? An endocrinologist's perspective. Best Pract Res Clin Endocrinol Metab. 2005;19(1):53–66.