TABLE 47.1 Causes of Toxicologic Hyperthermia
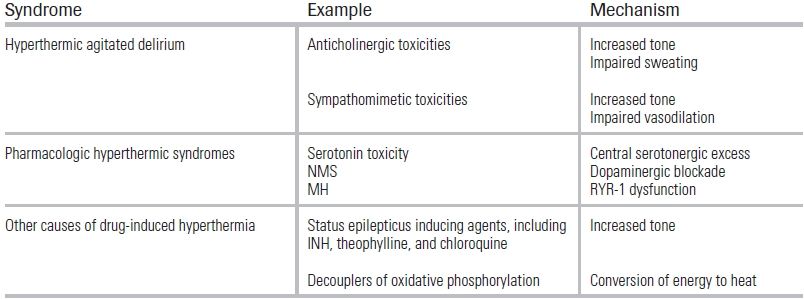
BACKGROUND
Elevations in body temperature may be caused by behavioral factors, exertion, infections, endocrinologic conditions, and environmental exposures; as well as by therapeutic and illicit drugs that disrupt the autonomic nervous system or impair the body's cooling capacity. Core body temperatures in excess of 106°F (41.1°C) precipitate life-threatening hyperthermia—termed heat stroke when the condition is accompanied by altered mental status.
Heat stroke is always a time-sensitive emergency that requires prompt diagnosis and treatment, as its mortality rate is directly related to delays in cooling.1 Patients subject to such delays are at risk for multisystem organ failure, often heralded by impaired liver synthetic function and disseminated intravascular coagulation (DIC). From 1999 to 2003, a total of 3,442 deaths from heat stroke were reported in the United States; while underlying illnesses contributed to the majority of these deaths, 4.2% were due to toxicologic causes.2 This chapter reviews some of the common toxicologic hyperthermic syndromes and their management (Table 47.1).
TABLE 47.1 Causes of Toxicologic Hyperthermia
HYPERTHERMIC AGITATED DELIRIUM
Patients with severe hyperthermia may present with agitated delirium—a difficult-to-manage form of heat stroke—that can impair the clinician's ability to obtain vital signs in a timely fashion. When presented with an agitated patient, especially during the summer months, the emergency physician should maintain a high degree of suspicion for a hyperthermic etiology.3
Agitated delirium has been used to describe patients with severe agitation who are unresponsive to verbal redirection, are combative, or have altered mental status. Cases are frequently associated with drug use; illicit sympathomimetic agents such as cocaine are common culprits, and patients classically present diaphoretic, tachycardic, hypertensive, and severely agitated (sympathomimetic toxidrome).4 The use of cocaine or other sympathomimetic agents causes vasoconstriction, which limits effective cooling and simultaneously increases motor tone, generating heat. Data suggest that mortality related to cocaine use increases when ambient temperatures are above 88°F (31.1°C).5 Centrally acting anticholinergic agents such as scopolamine can similarly impair cooling and may cause psychomotor agitation. These patients will present with altered mental status, tachycardia, dry skin, pupillary dilation, and urinary retention (anticholinergic toxidrome).6
The diagnosis of agitated delirium requires two factors: psychomotor agitation and a reduced ability to focus or shift attention.4 In addition to toxicologic etiologies, other causes of agitated delirium that should always be considered include infection, postictal states, and endocrinological emergencies. Laboratory assessments should include a basic metabolic panel and urinalysis; blood cultures if etiology of delirium is uncertain; and creatinine kinase to rule out the potential complication of rhabdomyolysis. In addition, liver function tests, a coagulation panel (PT/PTT/INR), and complete blood count should be used to screen for evidence of DIC due to hyperthermic liver injury as well as nonspecific tissue damage, which can lead to consumption of coagulation factors.7 Arterial/venous blood gas and serum lactate tests should be used to identify acidosis, which commonly accompanies heat stroke. If an intentional self-poisoning is suspected, serum salicylate and acetaminophen concentrations should be obtained.
Prompt sedation with benzodiazepines facilitates measurement of body temperature, hemodynamic stabilization, and the rapid cooling that is critical to patient survival. If intravenous access is unavailable, a rapidly sedating benzodiazepine such as midazolam may be administered intramuscularly (10 mg in a 70 kg adult). Lorazepam may also be administered intramuscularly, but its time to peak sedation is typically >15 minutes. In the hyperthermic patient with status epilepticus, however, lorazepam offers equivalent onset for seizure termination with a longer duration of action.
If intravenous access is available, then an adult patient may be administered diazepam in 10 mg aliquots intravenously every 5 minutes until adequate sedation is achieved, which allows for cooling and reduces psychomotor tone. Patients receiving repetitive doses of any intravenous or intramuscular benzodiazepine require close respiratory monitoring. Intramuscular ketamine has been reported for the patient presenting with an agitated delirium, but the data are limited.
Ideally, the hyperthermic patient is cooled using ice water immersion or cold, wet sheets with ice packed across the entire body, with continual fanning. Continuous core temperature monitoring using a rectal probe is preferable. Occasionally, interventions such as intubation and neuromuscular paralysis are required to reduce heat production. Patients can be removed from ice/ice water when the core temperature is 101.3°F (38.5°C) to avoid overshooting normothermia and provoking hypothermia.8 Cooling is ideally achieved within 15 minutes of presentation to reduce total hyperthermic time.
Invasive cooling methods—including ice water irrigation of the bladder and thoracic and peritoneal cavities—should be avoided, due to their inadequate rate of cooling in patients with hyperthermic emergencies. Similarly, cooling blankets, while low risk, are also inadequate and should also be avoided. Restoration of a patient's intravascular volume may be necessary, with serial evaluations for urine output, assessment of inferior vena cava (IVC) collapse, and lung examinations.9
Once cooled, patients should be admitted to an intensive care unit and have serial reassessment of basic laboratory tests. Renal dysfunction and coagulopathy are commonly seen within 24 hours of heat stroke onset and may worsen depending on the duration of hyperthermia; acute renal failure is seen in 30% to 50% of heat stroke patients.10 Liver function tests may also initially appear normal but worsen as organ dysfunction evolves.
TOXICOLOGIC HYPERTHERMIC SYNDROMES
Serotonin toxicity results from excessive stimulation of 5-HT1A and 5-HT2A receptors. It can develop in patients after a large overdose of a single serotonergic agent; in patients taking more than one serotonergic agent; or in individuals who initiate a new serotonergic agent without adequately timed discontinuation of another serotonergic agent11–15 (Table 47.2).
TABLE 47.2 Substances That Can Contribute to Serotonin Toxicity
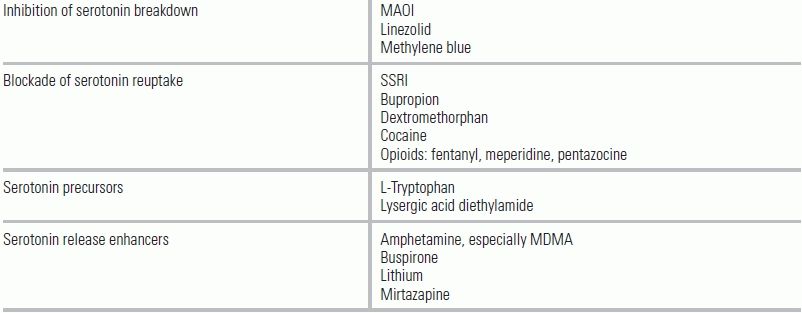
From Vasallo S, Delaney KA. Thermoregulatory principles. In: Nelson LS, Lewin NA, Howland MA, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill Medical Pub.; 2010:228–248.
Serotonin toxicity is often described as a clinical triad of altered mental status, autonomic instability, and muscular hyperactivity.16 Tremor, shivering, hyperreflexia, and clonus—prominent in the lower extremities and often described as the “dog shakes”—are classic manifestations of this syndrome. Clonus can also be seen in the ocular muscles as a “ping-pong gaze” in which the eyes make horizontal movements. In severe cases, muscle rigidity may lead to hyperthermia, which, when coupled with altered mental status, results in a toxicologic heat stroke.
Life-threatening serotonin toxicity is most often seen in the patient taking a monoamine oxidase inhibitor (MAOI) followed by the ingestion of or iatrogenic administration of a serotonergic drug.17–19 MAOIs inhibit the presynaptic intracellular breakdown of serotonin, enhancing the amount of serotonin released into the synapse. The subsequent administration of another serotonergic agent can cause excessive receptor stimulation. The onset of life-threatening serotonin syndrome is usually rapid, often within minutes to <2 hours of drug administration.
Serotonin toxicity is a diagnostic challenge; it lacks a specific biomarker, and patients may present within a spectrum of potential signs and symptoms. A milder serotonin toxicity without hyperthermia or autonomic instability may also be difficult to recognize.16 Such patients can exhibit hyperactivity of the extremities and delirium. Other minor manifestations of serotonin excess—such as diarrhea, hypertension, insomnia, or restlessness—may be present, but mistakenly be attributed to the patient's underlying psychiatric or medical condition.
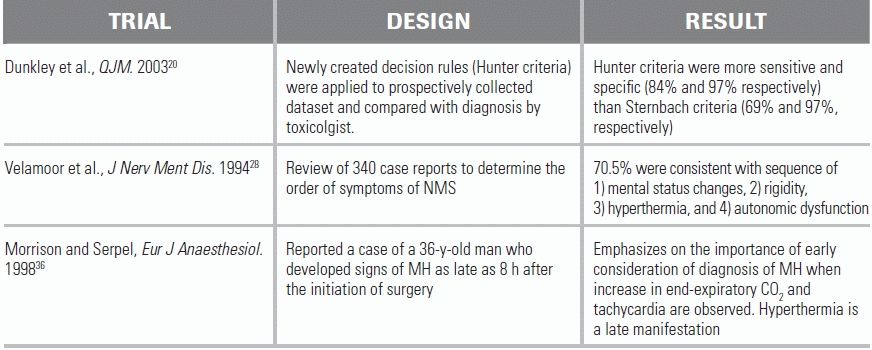
Unless the patient's presentation follows shortly after an interaction of medications known to precipitate the toxicity, the diagnosis is generally one of exclusion in the differential of hyperthermia. There is no universally accepted diagnostic test; however, the Hunter Serotonin Toxicity Criteria were demonstrated to have a sensitivity and specificity for detecting serotonin toxicity of 84% and 97% respectively. For the screen to be positive, the patient must have taken a serotonergic agent and have any of the five listed symptoms.20 (Table 47.3). In patients meeting these criteria, the emergency physician should suspect serotonin toxicity and evaluate for complications of hyperthermia, including rhabdomyolysis, renal failure, seizure, DIC, and abnormalities of liver function.8
TABLE 47.3 Hunter Serotonin Syndrome Criteria

When serotonin toxicity presents with severe hyperthermia, cooling should be started as soon as possible, and the offending serotonergic drug should be discontinued. Benzodiazepines will relieve most mild to moderate symptoms, including agitation and clonus; in patients with refractory muscle rigidity and hyperthermia, a neuromuscular blockade may be considered for muscle relaxation.1
Milder manifestations of serotonin toxicity are generally treated with supportive care and withdrawal or reduction of the responsible serotonergic agent. Evidence supports the use of cyproheptadine—an antihistamine with nonspecific antagonist effects at 5-HT1A and 5-HT2A receptors—for the targeted treatment of the serotonergic excess in patients with mild to moderate symptoms that are insufficiently controlled by sedation. The recommended initial dose is 12 mg followed by 2 mg every 2 hours with a maximum of 32 mg/day until symptoms resolve. A maintenance dose of 8 mg of cyproheptadine every 6 hours can be considered if mild symptoms persist. In case reports, patients responded to 4 mg of cyproheptadine within 2 hours with some requiring one repeat dose.21,22 There are, however, no definitive data regarding cyproheptadine's utility in severe cases due to the rarity of events and difficulty in randomization.
Neuroleptic malignant syndrome (NMS) is a rare but potentially fatal disorder caused by blockade of dopaminergic receptors in the striatum and hypothalamus—such as in patients taking therapeutic antipsychotics—or by withdrawal of therapeutic dopaminergic agents. Reduction of dopamine levels in the hypothalamus changes the core temperature set point, leading to hyperthermia, while blockade of striatal dopamine receptors contributes to muscle rigidity and tremor.23 Approximately 0.2% to 1.4 % of all patients receiving antipsychotics will develop NMS.24,25
Most cases of NMS occur in patients taking therapeutic antipsychotics. The condition is triggered by rapidly escalating dosage, use of high-potency agents such as haloperidol, parenteral administration, and use of depot preparations (intramuscular injections with slow release).23,24 Atypical antipsychotics may also cause NMS, but less commonly than typical antipsychotics.26 NMS risk is greatest during the first weeks to months of therapy but can occur anytime during use of neuroleptics. NMS may also be precipitated by cessation of dopamine agonists in patients being treated for Parkinson's, but this is less common.
The four main clinical findings of NMS are: changes in mental status (typically gradual-onset catatonia); increased muscle tone, described as “lead pipe rigidity” and “cogwheeling”; hyperthermia; and autonomic dysfunction presenting as tachycardia with alternating hypotension and hypertension. One study reviewing 340 patients with NMS showed 70.5% of patients developed symptoms in the following order: (1) mental status changes, (2) rigidity, (3) hyperthermia, and (4) autonomic dysfunction. In addition, 83.6% of individual patients demonstrated either altered mental status or rigidity before the onset of hyperthermia or autonomic instability.24,27,28 NMS is also frequently preceded by the onset of bradykinesis.
In vulnerable patients, NMS can be life threatening; clinicians should maintain a high index of suspicion for hyperthermia when presented with a catatonic, rigid patient, particularly one exposed to elevated ambient temperatures. Like serotonin toxicity, NMS has no diagnostic biomarker. Multiple diagnostic criteria, including the Levenson and Caroff criteria,27 have been proposed, but none is universally accepted. The most frequently referenced criteria are found in the Diagnostic and Statistical Manual of Mental Disorders, 4th ed., which requires the development of severe muscle rigidity and elevated temperature associated with the use of antipsychotic medication, as well as two or more of the following: diaphoresis, elevated blood pressure, tachycardia, incontinence, dysphagia, mutism, tremor, changes in the level of consciousness (ranging from confusion to coma), leukocytosis, and laboratory evidence of muscle injury (elevated creatinine kinase).29
Differentiating between NMS and serotonin syndrome can be difficult, since the two syndromes share many clinical features. The following clinical findings can help distinguish them (Table 47.4).
TABLE 47.4 Differentiation of Serotonin Toxicity from NMS
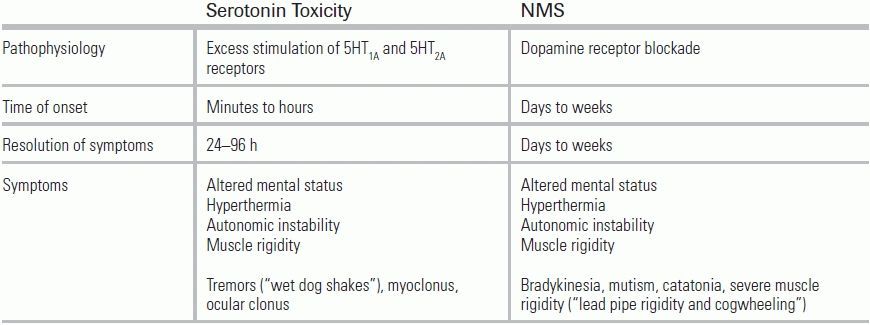
From Vasallo S, Delaney KA. Thermoregulatory principles. In: Nelson LS, Lewin NA, Howland MA, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill Medical Pub.; 2010:228–248.
Serotonin toxicity has a rapid onset, within minutes to hours; NMS, by comparison, evolves over days or weeks. Serotonin toxicity carries the additional features of tremors and myoclonus, while NMS is further characterized by bradykinesia, mutism, and gradual-onset catatonia. Finally, in most cases of serotonin syndrome, symptoms resolve within 24 to 72 hours after removal of the offending agent, while in NMS, symptoms may continue for weeks.
Patients with NMS who present with life-threatening hyperthermia—that is, heat stroke, or T > 41.1°C or 106°F—should be treated aggressively, as outlined in the management section of agitated delirium.8,30,31 If the syndrome was caused by discontinuation of a dopamine agonist, as with abrupt cessation of Parkinson's medications, the dopaminergic agent should be resumed as soon as possible.
Benzodiazepines are the first-line pharmacologic therapy used to provide sedation and muscle relaxation. Bromocriptine and, rarely, dantrolene may also be used. Bromocriptine is a central dopamine agonist that can be administered if supportive therapy with benzodiazepines and rapid cooling fail to improve the patient's symptoms. Bromocriptine, however, may lead to a worsening of a patient's underlying psychiatric illness due to dopamine agonism. This effect can be temporized by use of the same benzodiazepines used for muscle relaxation, until the NMS has resolved. Bromocriptine is only available in oral form, and the dose is 2.5 to 10 mg orally three to four times a day. The dosage can be increased with increments of 2.5 mg three times a day every 24 hours until a response is seen or up to a maximum 60 mg per day.23
Generally, supportive care, benzodiazepines, and bromocriptine are sufficient to manage muscle rigidity and autonomic dysfunction from NMS; however, patients with severe rigidity unresponsive to benzodiazepines or bromocriptine may require intubation followed by neuromuscular paralysis. Since the clearance of antipsychotics is slow, pharmacological dopaminergic therapies should be continued for at least 10 days after a return to baseline. Premature discontinuation of dopamine agonists may lead to recurrence of NMS. When NMS is related to depot neuroleptics, bromocriptine should be continued for 2 to 3 weeks.23 Full recovery may take weeks to return to baseline.
Some authors describe the use of dantrolene for treatment of NMS, although data supporting its use are limited, and it is rarely indicated. Dantrolene is more typically used to treat malignant hyperthermia (MH) by inhibiting calcium release from the sarcoplasmic reticulum (see more on “Malignant Hypothermia,” below).
Electroconvulsive therapy (ECT) has been suggested as treatment for NMS. ECT is thought to exert its therapeutic effect by enhancing central dopamine activity,32 and reports have shown that three to four sessions of ECT resolve symptoms of NMS with a mean time from initiation of treatment to resolution of symptoms of 6 days.33 However, data for ECT are limited, and patients with hyperthermia and organ dysfunction are not candidates for this therapy.
MH is a rare autosomal dominant disorder typically seen in patients who receive inhalational anesthetics or succinylcholine. The incidence of MH in patients exposed to general anesthesia has been reported to be anywhere from 1 in 5,000 to 1 in 62,000.34,35 In normal muscle, contraction occurs as action potentials open voltage-gated calcium channels, which in turn activate ryanodine receptors (RYR-1) that release calcium for myocyte contraction. The released calcium is recycled by sarcoplasmic Ca2+-ATPase. In MH, this cycling of calcium in the muscle cells becomes disordered.
MH is a form of drug-induced hypermetabolism of skeletal muscle. Patients who are predisposed to MH have mutations of the RYR-1. In susceptible persons, inhalational anesthetic agents and succinylcholine enhance the activity of the defective RYR-1, leading to accelerated release of Ca2+ inside the skeletal muscle. Intracellular ATP becomes depleted as the sarcoplasmic Ca2+-ATPase attempts to transport Ca2+ back into the sarcoplasmic reticulum of the muscle cells. The result is diffuse, sustained contraction of skeletal muscles, rigidity unresponsive to nondepolarizing neuromuscular blockade, and hyperthermia. This process also leads to anaerobic metabolism with resultant lactic acidosis and an increase in CO2 production.
Typically, MH symptoms manifest shortly after exposure to an inhalational anesthetic or succinylcholine; however, a delayed onset of up to 7 to 8 hours has been reported.36 Even among susceptible patients, MH does not always occur with anesthetic drug exposure, so a negative history cannot safely rule out the development of MH with subsequent exposures.37 The earliest signs of MH are tachycardia, an increase in end-tidal CO2 concentration, and generalized skeletal muscle rigidity and masseter spasm. Hyperthermia, which is the hallmark of MH, may not be seen until later in the process.
There are no readily available diagnostic tests for MH: diagnosis is suspected when administration of neuromuscular blockers fails to result in paralysis. The gold standard for diagnosing MH is an in vitro contracture test (IVCT). This tests the contracture of muscle fibers in the presence of halothane or caffeine and monitors for abnormal muscle contractility.35
The toxicologic differential diagnosis of MH is addressed in Table 47.1. The distinguishing features of MH are exposure to an implicated agent and failure to respond to neuromuscular blockade. Unlike in serotonin syndrome, rigidity in MH is not attended by muscle hyperactivity.
When hyperthermia does occur, temperatures will increase by 1°C to 2°C every 5 minutes, and that core temperatures should be continuously monitored.35 Similar to the treatment of hyperthermic agitated delirium and serotonin toxicity, it is crucial to stop administration of the offending agent. If hyperthermia is present or evolving, cooling should be initiated, as previously outlined.
Dantrolene can be lifesaving and is the definitive therapy for MH. Dantrolene blocks Ca2+ release from the skeletal muscle sarcoplasmic reticulum. After the introduction of dantrolene as a therapy, the mortality from MH decreased from 64% to <5%.38 The starting dose is 2.5 mg/kg IV bolus followed by 2 to 3 mg/kg IV every 15 minutes until resolution of symptoms or to a cumulative dose of 10 mg/kg. To prevent recrudescence, administration of 1 mg/kg IV every 4 to 6 hours for a minimum of 24 to 48 hours is recommended.35
Life-threatening hyperthermia may also be triggered when patients generate heat disproportionately to their cooling capacity. Drugs that cause status epilepticus, including isoniazid (INH) theophylline, and chloroquine, can cause excessive heat generation. Termination of convulsions, restoration of adequate ventilation, and rapid cooling are critical interventions. Most toxicologically caused seizures will not respond to traditional anticonvulsant agents such as phenytoin. Benzodiazepines or other sedative hypnotic agents are most efficacious. INH overdose with resultant status epilepticus may require pyridoxine therapy for adequate neurologic inhibition.
Agents that uncouple oxidative phosphorylation, such as salicylates or dinitrophenol (an illegal weight loss agent), may also cause life-threatening hyperthermia. Acidosis evolves as the potential energy unable to be transformed into ATP is dissipated as heat. In each of these cases, the primary intervention is rapid identification of hyperthermia, rapid cooling, and minimization of psychomotor agitation with benzodiazepines, supportive care, and, in extreme cases, intubation with a neuromuscular paralytic agent.
CONCLUSION
Hyperthermic agitated delirium, serotonin syndrome, NMS, and MH share many physical findings. Distinguishing between these syndromes can be difficult and depends on a clear understanding of the patient's pharmacologic history. Essential components of treatment include rapid cooling of life-threatening hyperthermia and supportive care with benzodiazepines. Secondary complications of toxicologic hyperthermic syndromes, which include rhabdomyolysis, acute kidney injury, hyperkalemia, liver injury, and DIC, should be identified and aggressively treated in an intensive care unit.
LITERATURE TABLE
1.Vasallo S, Delaney KA. Thermoregulatory principles. In: Nelson LS, Lewin NA, Howland MA, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill Medical Pub.; 2010:228–248.
2.Anonymous. Heat-related deaths—United States, 1999–2003. MMWR Morb Mort Wkly. 2006;55:796–798.
3.Marzuk PM, Tardiff K, Leon AC, et al. Ambient temperature and mortality from unintentional cocaine overdose. JAMA. 1998;279:1795–1800.
4.Vilke GM, DeBard ML, Chan TC, et al. Excited elirium Syndrome (ExDS): defining based on a review of the literature. J Emerg Med. 2012:897–905.
5.Marzuk PM, Tardiff K, Leon AC, et al. Ambient temperature and mortality from unintentional cocaine overdose. JAMA. 1998;279(22):1795–1800.
6.Anonymous. Scopolamine poisoning among heroin users—New York City, Newark, Philadelphia, and Baltimore, 1995 and 1996. MMWR Morb Mortal Wkly. 1996;45:457–460.
7.Khatim Y, Mustafa O, Omer M, et al. Blood coagulation and fibrinolysis in heat stroke. Br J Hematol. 1985;61:517–523.
8.Proulx CI, Ducharme MB, Kenny GP. Effect of water temperature on cooling efficiency during hyperthermia in humans. J Appl Physiol. 2003;94(4):1317–1323.
9.Seraj MA, Channa AB, al Harthi SS, et al. Are heat stroke patients fluid depleted? Importance of monitoring central venous pressure as a simple guideline for fluid therapy. Resuscitation. 1991;21:33–39.
10.Yeo TP. Heat stroke: a comprehensive review. AACN Clin Issues. 2004;15(2);280–293.
11.Ruiz F. Fluoxetine and the serotonin syndrome. Ann Emerg Med. 1994;24(5):983–985.
12.Safferman AZ, Masiar SJ. Central nervous system toxicity after abrupt monoamine oxidase inhibitor switch: a case report. Ann Pharmacother. 1992;26(3):337–338.
13.Daniels RJ. Serotonin syndrome due to venlafaxine overdose. J Accid Emerg Med. 1998;15(5):333–334.
14.Gill M, LoVecchio F, Selden B. Serotonin syndrome in a child after a single dose of fluvoxamine. Ann Emerg Med. 1999;33(4):457–459.
15.Paruchuri P, Godkar D, Anandacoomarswamy D, et al. Rare case of serotonin syndrome with therapeutic doses of paroxetine. Am J Ther. 2006;13(6):550–552.
16.Sampson E, Warner JP. Serotonin syndrome: potentially fatal but difficult to recognize. Br J Gen Pract. 1999;49(448):867–868.
17.Smilkstein MJ, Smolinske SC, Rumack BH. A case of MAO inhibitor/MDMA interaction: agony after ecstasy. J Toxicol Clin Toxicol. 1987;25:1–2.
18.Smith B, Prockop DJ. Central-nervous-system effects of ingestion of l-tryptophan by normal subjects. N Engl J Med. 1962;267:1338–1341.
19.Oates JA, Sjoerdsma A. Neurologic effects of tryptophan in patients receiving a monoamine oxidase inhibitor. Neurology. 1960;10:1076–1078.
20.Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635–642.
21.Graudins A, Stearman A, Chan B. Treatment of the serotonin syndrome with cyproheptadine. J Emerg Med. 1998;16(4):615–619.
22.Lappin RI, Auchincloss EL. Treatment of the serotonin syndrome with cyproheptadine. N Engl J Med. 1994;331(15):1021–1022.
23.Bhanushali MJ, Tuite PJ. The evaluation and management of patients with neuroleptic malignant syndrome. Neurol Clin. 2004;22(2):389–411.
24.Addonizio G, Susman VL, Roth SD. Neuroleptic malignant syndrome: review and analysis of 115 cases. Biol Psychiatry. 1987;22(8):1004–1020.
25.Delay J, Pichot P, Lemperiere T, et al. A non-phenothiazine and non-reserpine major neuroleptic, haloperidol, in the treatment of psychoses. Ann Med Psychol (Paris). 1960;118(1):145–152.
26.Kontaxakis VP, Havaki-Kontaxaki BJ, Christodoulou NG, et al. Olanzapine-associated neuroleptic malignant syndrome. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(5):897–902.
27.Caroff SN, Mann SC. Neuroleptic malignant syndrome and malignant hyperthermia. Med Clin North Am. 1993;21(4):477–478.
28.Velamoor VR, Norman RM, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis. 1994;182(3):168–173.
29.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC; 2000.
30.Armstrong LE, Crago AE, Adams R, et al. Whole-body cooling of hyperthermic runners: comparison of two field therapies. Am J Emerg Med. 1996;14(4):355–358.
31.Weiner JS, Khogali M. A physiological body-cooling unit for treatment of heat stroke. Lancet. 1980;1(8167):507–509.
32.Hermesh H, Aizenberg D, Weizman A. A successful electroconvulsive treatment of neuroleptic malignant syndrome. Acta Psychiatr Scand. 1987;75(3):237–239.
33.Nisijima K, Ishiguro T. Electroconvulsive therapy for the treatment of neuroleptic malignant syndrome with psychotic symptoms: a report of five cases. J ECT. 1999;15(2):158–163.
34.Ording H. Incidence of malignant hyperthermia in Denmark. Anesth Analg. 1985;64(7):700–704.
35.Rosenberg H, Davis M, James D, et al. Malignant hyperthermia. Orphanet J Rare Dis. 2007;2:21.
36.Morrison AG, Serpell MG. Malignant hyperthermia during prolonged surgery for tumour resection. Eur J Anaesthesiol. 1998;15(1):114–117.
37.Bendixen D, Skovgaard LT, Ording H, et al. Analysis of anaesthesia in patients suspected to be susceptible to malignant hyperthermia before diagnostic in vitro contracture test. Acta Anaesthesiol Scand. 1997;41(4):480–484.
38.Larach MG, Brandom BW, Allen GC, et al. Cardiac arrests and deaths associated with malignant hyperthermia in North America from 1987 to 2006. Anesthesiology. 2008;108(4):603–611.